
Suppressor of Cytokine Signaling 4 (SOCS4) Protects against Severe Cytokine Storm and Enhances Viral Clearance during Influenza Infection
The suppressor of cytokine signaling proteins are key regulators of immunity. As yet there is no described biological role for SOCS4, despite its broad expression in cells of the immune system. Given the important role of other SOCS proteins in controlling the immune response, we have generated SOCS4-mutant mice and used a mouse influenza infection model to investigate the biological function of SOCS4. We demonstrate that mice lacking SOCS4 rapidly succumb to infection with a pathogenic H1N1 influenza virus and are hypersusceptible to infection with the less virulent H3N2 strain. This is the first demonstration of a functional phenotype in SOCS4-deficient mice. Our study reveals that in SOCS4-deficient animals, there is a dysregulated pro-inflammatory cytokine and chemokine production in the lungs and delayed viral clearance. This is associated with impaired trafficking of virus-specific CD8 T cells to the site of infection and linked to defects in T cell receptor activation. These results demonstrate that SOCS4 is a critical regulator of anti-viral immunity. Understanding the regulation of the inflammatory response to influenza is particularly relevant given the current climate concerning pandemic influenza outbreaks.
Published in the journal:
. PLoS Pathog 10(5): e32767. doi:10.1371/journal.ppat.1004134
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004134
Summary
The suppressor of cytokine signaling proteins are key regulators of immunity. As yet there is no described biological role for SOCS4, despite its broad expression in cells of the immune system. Given the important role of other SOCS proteins in controlling the immune response, we have generated SOCS4-mutant mice and used a mouse influenza infection model to investigate the biological function of SOCS4. We demonstrate that mice lacking SOCS4 rapidly succumb to infection with a pathogenic H1N1 influenza virus and are hypersusceptible to infection with the less virulent H3N2 strain. This is the first demonstration of a functional phenotype in SOCS4-deficient mice. Our study reveals that in SOCS4-deficient animals, there is a dysregulated pro-inflammatory cytokine and chemokine production in the lungs and delayed viral clearance. This is associated with impaired trafficking of virus-specific CD8 T cells to the site of infection and linked to defects in T cell receptor activation. These results demonstrate that SOCS4 is a critical regulator of anti-viral immunity. Understanding the regulation of the inflammatory response to influenza is particularly relevant given the current climate concerning pandemic influenza outbreaks.
Introduction
Influenza is a highly infectious, acute respiratory disease that causes profound morbidity and mortality. Annual seasonal influenza epidemics result in ∼500,000 deaths worldwide and substantial losses to global economies [1]. The development of a “cytokine storm” coupled with damage to pulmonary epithelium has been consistently observed in severe cases of influenza infection in humans. The mechanisms underlying this pathology, and an understanding of why some individuals respond excessively to virus, to an extent that results in hospitalisation or death, remains relatively unexplored.
The initial innate immune response to viral infection is characterized by an influx of neutrophils, monocytes and macrophages into the lung parenchyma and alveolar spaces, with the elevated levels of inflammatory cytokines/chemokines correlating strongly with pathogenesis and viral load [2]. However, exaggerated cytokine and chemokine responses have been observed in the lungs of critically ill patients in the absence of high viral load [3], suggesting that inflammation-driven pathology can occur independently of viral load.
The adaptive response subsequently results in the generation of strain-specific B cells and cross-strain protective CD4 and CD8 T cells. Influenza-specific CD8 T cells are largely responsible for host immunity to primary influenza infection and operate to promote the efficient elimination of virus, and host recovery, via the production of pro-inflammatory cytokines and direct killing of virus-infected cells [4]. Acquisition of these effector functions occurs in the draining lymph nodes where upon T cell receptor (TCR) recognition of the influenza-specific peptide:MHC complex, CD8 T cells become activated and then migrate to the infected lungs.
Cytokine binding to the cognate receptor complexes triggers an intracellular signaling cascade, most often coupled to the JAK-STAT pathway, which orchestrates an intricate series of transcriptional changes leading to the appropriate cellular response. The suppressors of cytokine signaling (SOCS) proteins are key negative regulators of the JAK/STAT pathway and are thus involved in the fine-tuning of the cytokine networks responsible for an adequate and efficient innate and adaptive immune response [5]. The family is composed of eight members, SOCS1 to 7 and cytokine-inducible Src-homology 2 protein (CIS) [6]. All proteins share a central SH2 domain and carboxyl-terminal SOCS box, but differ in their amino termini. SOCS4 to 7 are particularly distinguished by a long N-terminal region, which bears little homology to other SOCS proteins [7]. The SOCS box interacts with elongins B and C, and together with Rbx2 and Cullin-5 forms an E3 ubiquitin ligase [8]. The SOCS proteins therefore act as adaptors to target substrates bound to their SH2 or N-terminal regions for ubiquitination and proteasomal degradation [9]. In addition, SOCS1 and 3 can bind directly to JAK via their kinase inhibitory region (KIR) and SH2 domains, inhibiting JAK phosphorylation of substrates and downstream signaling [10], [11]. CIS and SOCS2 are thought to bind to phosphotyrosine residues within the receptor cytoplasmic domains to block recruitment of other signalling intermediates [12], [13], [14].
The generation of knockout mice has proven a powerful tool in defining the physiological role of the SOCS proteins. SOCS1 for instance, was revealed as a critical regulator of IFNγ signaling and γc-cytokine-dependent T cell homeostasis [15], SOCS2 as a regulator of growth hormone signaling [16] and conditional deletion of the Socs3 gene has identified a role for SOCS3 in regulating IL-6 and G-CSF signaling [17], [18]. Although a wealth of information is available on the role of CIS and SOCS1-3, there is much less data regarding the targets and pathways regulated by the remaining family members, including SOCS4.
In vitro studies have suggested that SOCS4 is involved in regulating epidermal growth factor (EGF) signaling [19], and indeed the SOCS4-SH2 domain binds with high affinity to a phosphopeptide corresponding to an EGF receptor (EGFR) autophosphorylation site (Tyr1092; 0.5 µM) [20]. This later study provides some information as to the binding preferences of the SOCS4-SH2 domain [20], however the relevance of the interaction with the EGFR remains to be elucidated. Other studies have suggested that SOCS4 may be regulated by parasitic infection [21], is linked to better outcomes in cancer patients [22], or may regulate pre-granulosa cells during folliculogenesis [23]. Thus far, none of these studies present a compelling case for a physiological role for SOCS4.
In the current study, we have generated the first loss of function allele of murine Socs4, a point mutation identified in a library of ENU-mutagenised mice [24]. At the protein level, this mutation is predicted to cause the substitution of a stop codon for arginine 108, resulting in deletion of the remaining 90 residues of the N-terminal domain, as well as the entire SH2 domain and SOCS box. Given that SOCS proteins play a critical role in regulating immune responses, and expression of SOCS4 in lymphocytes, we investigated the role of SOCS4 in a defined viral infection model in which T cells regulate pathogen clearance. We found that Socs4R108X/R108X mice were highly susceptible to infection with influenza A, showing increased morbidity and a delay in viral clearance comparable to that observed for mice lacking CD8 T cells [25] or IL-18 [26]. The increased lethality appeared to result from an elevation in key pro-inflammatory cytokines and chemokines, such as IL-6, IFNγ and MCP-1; whilst in the latter phase of the infection, Socs4R108X/R108X mice displayed impaired trafficking of virus-specific CD8 T cells to the lungs. The defect in trafficking appeared to be qualitatively linked to the activation status of these cytotoxic T lymphocytes and reveals a novel role for SOCS4 as a positive regulator of TCR signaling.
Results
SOCS4 protects mice against virulent influenza infection
Socs4R108X/R108X mice were viable, fertile and showed no overt phenotype under steady-state conditions, including normal thymocyte development and composition of peripheral immune cells (Fig. S1). To investigate the role of SOCS4 in the response to viral challenge, homozygous Socs4R108X/R108X mice and age-matched littermates or Balb/c controls were inoculated intranasally (i.n.) with 20 pfu of the virulent H1N1 influenza strain A/Puerto Rico/8/34 (PR8) and monitored for weight loss. In accordance with ethical guidelines, mice were considered moribund upon losing greater than 20% of their initial body weight, and removed from the study. At this relatively low challenge dose, Socs4R108X/R108X mice exhibited significantly enhanced disease progression and mortality compared to wild-type controls (Fig. 1A and B) and this correlated with an increased viral load in the lungs (Fig. 1C).

To dissect the defective response in more detail, Socs4R108X/R108X mice and wild-type controls were subsequently infected i.n. with 103 pfu of the less virulent H3N2 A/Hong Kong x31 (X31) influenza virus. Similarly to infection with PR8, Socs4R108X/R108X mice showed enhanced susceptibility to X31 infection with significantly greater weight loss (P = 0.005; Fig. 1D) and higher lung viral loads (Fig. 1E) when compared to controls. Initially (day 1 and 2 post-infection) there were no differences in viral titers between the groups, indicating that viral uptake and replication are not affected by SOCS4 deficiency. However, Socs4R108X/R108X mice displayed higher viral titers (0.5 to 1.8 log higher) on day 3 and day 5 post-infection and this difference was greatly exacerbated by day 6 (2 log difference), when the majority of control mice had cleared the infection. Virus was undetectable in the lungs of wild-type controls on day 7, however some of the Socs4R108X/R108X mice still retained low viral loads (Fig. 1E). The augmented susceptibility to influenza infection was independent of genetic background, with similar results observed in congenic Socs4R108X/R108X mice on the C3H/He and C57BL/6 backgrounds (data not shown).
To investigate whether the increased susceptibility to influenza derives from a defect in the hematopoietic compartment, we generated chimeric mice by transplanting bone marrow cells from Thy1.2 Socs4R108X/R108X or Thy1.2 wild-type mice into lethally irradiated Thy1.1 wild-type recipients. The chimeric mice were subsequently infected i.n. with 103 pfu of X31 virus and disease progression monitored for 6 days. Although not significantly different, weight loss and particularly viral load were both increased in Socs4R108X/R108X mice compared to controls (Fig. 1F and G), indicating that a defect in the hematopoietic compartment most likely contributes to these aspects of the phenotype.
Morbidity in Socs4R108X/R108X mice correlates with elevated proinflammatory cytokines
The poorer outcomes associated with virulent influenza strains are thought to be due to excessive production of proinflammatory cytokines and chemokines. This dysregulated immune response and the resulting lung inflammation and damage is one mechanism by which pandemic infections cause high mortality [27], [28]. Since Socs4R108X/R108X mice showed greater susceptibility to infection, we compared cytokine and chemokine profiles in lung homogenates from Socs4R108X/R108X and wild-type mice infected with X31 virus. In general, we did not observe significant differences in cytokine and chemokine levels in the latter phase of the infection (days 5–7; data not shown). However, at day 3 post-infection, the Socs4R108X/R108X mice showed significantly higher levels of cytokines and chemokines, such as IL-1β, IL-4, IL-5, IL-6, IL-12p40, IL-13, IFN-γ, and KC (CXCL1), MIP-2 (CXCL2) and MCP-1 (CCL2), respectively (Fig. 2A). There were elevated but not significantly different, levels of TGF-β and IL-10, and no difference was detected in levels of several other cytokines and chemokines (data not shown). The elevated levels of pro-inflammatory cytokines, in particular IL-6, IFNγ and IL-1β, are likely to account for the increased morbidity observed in the Socs4 mutant mice. No difference was observed between Socs4R108X/R108X and control mice in cytokine and chemokine production in either spleen or lungs on d3 following systemic administration of polyinosinic-polycytidylic acid (poly I:C), a non-dynamic, virus-like stimulus (Fig. S2). This indicates that the increased cytokine/chemokine production (Fig. 2A) is a specific response to infection with influenza virus, and may reflect the modestly increased viral titres on d3 (Fig. 1E). It further suggests that that the Toll-like receptor (TLR)3 pathway is not perturbed and therefore not directly regulated by SOCS4. This is consistent with our data suggesting a hematopoietic, rather than an innate epithelial defect.
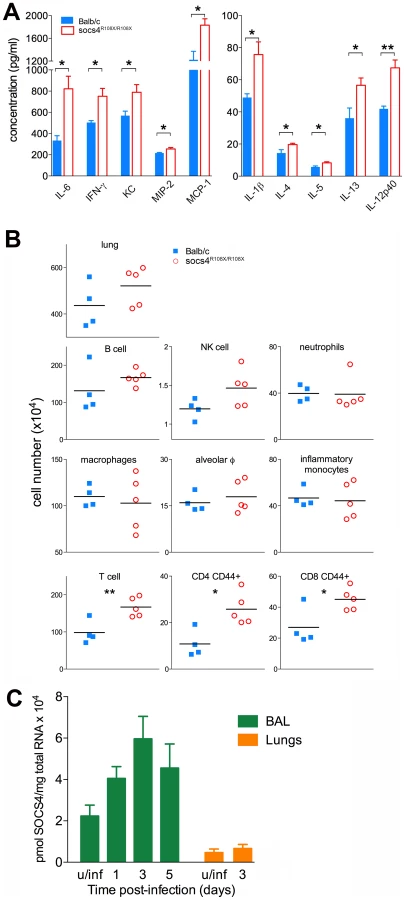
In order to determine whether the elevated cytokine levels in the lungs were caused by increased production per infiltrating cell or increased numbers of a particular subset of cells, we profiled infected lungs on day 3 post-infection. While total cell numbers in Socs4R108X/R108X-infected lungs were slightly elevated compared to wild-type controls, this difference did not reach statistical significance (Fig. 2B, top panel). Similarly, we observed slightly elevated levels of B cells and NK cells in Socs4R108X/R108X-infected lungs and no difference in neutrophil or macrophage numbers, including alveolar macrophages (CD11c+ Ly6Cint F4/80+) and inflammatory monocytes (CD11c- Ly6Glow Ly6C+ F4/80+) (Fig. 2B, middle panels). Surprisingly, modest but significantly higher levels of total T cells (TCRβ+) including higher levels of CD4 (CD44+) and CD8 (CD44+) T cells were detected in the lungs on day 3 post-infection (Fig. 2B, bottom panels). No difference was observed on day 2 post-infection, whereas on day 6 the cellularity of lungs of wild-type controls was higher than in SOCS4 mutant mice (Fig. S3). We also confirmed Socs4 expression in the infiltrating immune cells recovered by bronchioalveolar lavage (BAL). Socs4 mRNA expression increased over time, peaking at day 3 post-infection (due either to the changing cellular composition or to up-regulation within the infiltrating cells). In contrast, Socs4 mRNA was ∼15-fold lower in equivalent amounts of total lung RNA (Fig. 2C).
Together, these data suggest that activated T cells (not necessarily virus-specific, but CD44hi) accumulate in the lungs during the initial phase of infection in response to the elevated cytokine and chemokine levels. This may be a response to elevated chemoattractant levels or may potentially result from cytokine-driven proliferation (a bystander reaction rather than antigen-driven expansion). In summary, there is an increased net production of pro-inflammatory cytokines and chemokines, and while the initiating defect remains unclear, SOCS4 appears to have a classical role as a negative regulator of cytokine production and/or response in the innate immune reaction to influenza infection.
Socs4R108X/R108X mice display altered tissue distribution of influenza-specific CD8 T cells
Following intranasal inoculation with virus, CD8 T cells are primed and activated in the mediastinal lymph node (MLN) [29] and the anti-viral competence of these cells then depends on their ability to migrate to the site of infection [30]. We therefore used MHC class I tetramer staining to track and quantify influenza-specific (KdNP147) CD8 T cells in MLN, BAL and spleen, at days 5, 6 and 7 post-infection. Overall, the expansion of KdNP147-CD8 T cells did not differ between Socs4R108X/R108X mice and wild-type controls (Fig. 3A). However, at days 5 and 6 post-infection Socs4R108X/R108X mice had significantly less virus-specific cells at the site of infection (BAL) when compared to controls and instead, these cells appeared to localise to the spleen. No difference was observed on day 7 post-infection (Fig. 3B), at which time-point, viral titers in the majority of Socs4R108X/R108X mice were below detection levels (Fig. 1E). No differences were observed in MLN during the course of experiment (data not shown).
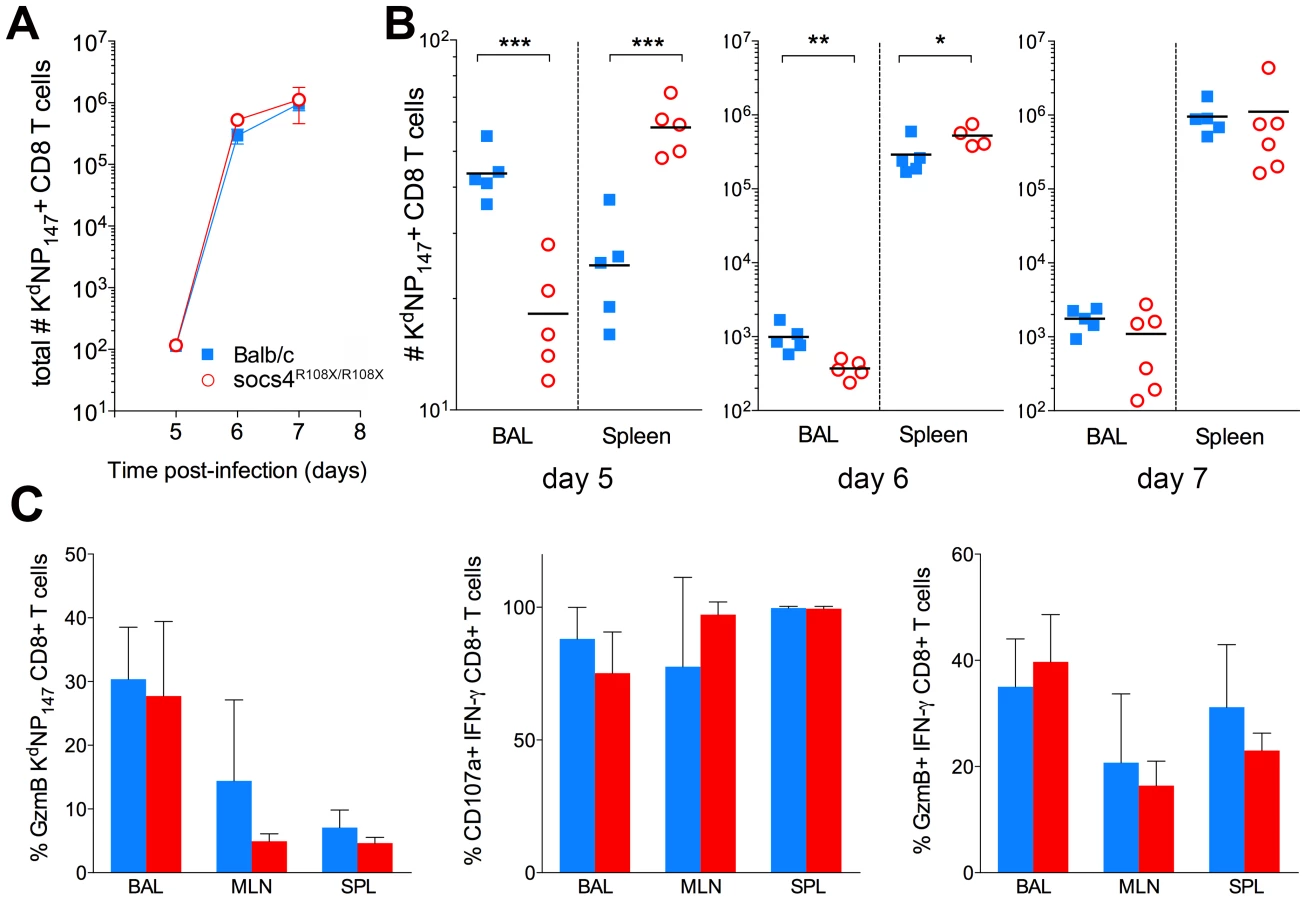
Although, virus-specific cells did not traffic efficiently to the site of infection, the clearance of virus on day 7 post-infection indicates that the loss of SOCS4 does not affect the ability of CD8 T cells to kill the virus. To confirm this, CTLs were analysed for markers of cytotoxic degranulation and function. Mice were primed with X31 and cells were harvested from lungs, draining lymph nodes and spleen (day 10 post-infection). KdNP147-CD8 T cells were analysed for granzyme B (GzmB) production as well as IFN-γ, TNF and IL-2. Alternatively, cells were stimulated ex vivo with KdNP147 peptide and stained with antibodies against lysosome-associated membrane protein 1 (LAMP-1, CD107a). No differences in GzmB production, cytokine production or CD107a mobilisation were observed between Socs4R108X/R108X mice and wild-type controls (Fig. 3C; data not shown).
Socs4 mutant T cells exhibit defects in activation
The defect in trafficking of KdNP147-CD8 cells to the site of infection could result from reduced activation of the T cells following MHC-antigen presentation or alternatively, from altered homing signals. To investigate this further, we examined a panel of T cell activation markers (CD62L, CD69, CD44, CD25) and homing receptors (CCR5, CXCR3, CCR7) at days 5, 6 and 7 post-infection. The majority of markers examined did not differ between wild-type and Socs4R108X/R108X CD8 T cells (data not shown). However, at day 5 post-infection Socs4R108X/R108X CD8 cells in the draining lymph nodes (MLN) showed comparatively higher levels of CD62L expression (Fig. 4), reflecting reduced activation. By day 6 there were reduced numbers of CD62L positive Socs4R108X/R108X CD8 cells in the lymph nodes (MLN), with no differences in the percentage of CD62Lhi versus CD62Llo expressing cells (Fig. 4B and C), and no differences observed at day 7 post-infection (Fig. 4B and C).
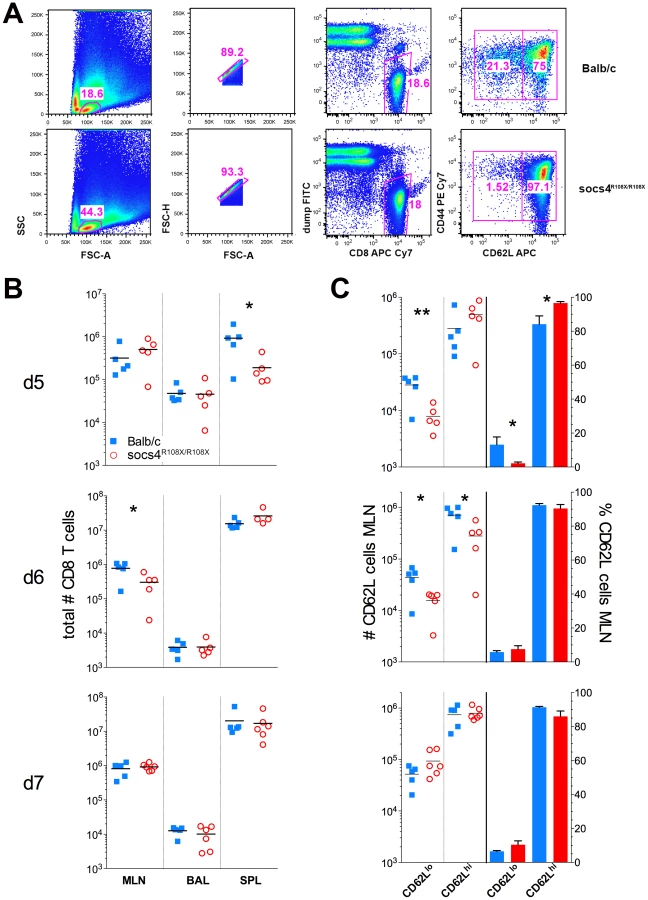
We also reconstituted Socs4R108X/R108X mice with wild-type Thy1.1 donor bone marrow. When analysed 8 weeks post-reconstitution for expression of congenic markers, we observed a much lower degree of chimerism (50–84% in CD4 and 43–78% in CD8 compartments) (Fig. S4A), compared to reconstitution of wild-type mice with Socs4R108X/R108X or wild-type donor cells. This result suggests that SOCS4 may regulate stromal signals, and although unlikely to contribute to the defective anti-viral response described here, warrants further independent investigation. Nevertheless, this competitive reconstitution enabled us to investigate the contribution of both donor and recipient cells to anti-influenza responses. Consistent with the level of chimerism, a higher proportion of wild-type donor cells were detected in the spleens and MLNs of all Socs4R108X/R108X recipient mice at day 6 post-infection. In contrast, the ratios of donor and recipient CD4 and CD8 T cells in the lungs (BAL) were approximately equal (Fig. S4B). Interestingly, the Socs4R108X/R108X Thy1.2 T cells in the lungs appeared to be significantly less activated than wild-type Thy1.1 cells, as evidenced by lower CD69 levels (Fig. S4C), whilst all T cells showed similar levels of CD44 on their surface (Fig. S4D).
Together, these data point to influenza-specific T cells receiving inadequate and/or incorrect signals in the lymph nodes, which in the Socs4R108X/R108X mice, results in their inability to migrate to the site of infection. In turn, this results in a reduced ability to clear the virus. To investigate this further, CD4 and CD8 T cells were isolated from Socs4R108X/R108X and wild-type mice, and signaling responses analysed following CD3 engagement.
SOCS4 regulates signaling downstream of TCR stimulation
We initially examined SOCS4 expression in purified splenic CD4 and CD8 cells following activation with anti-CD3 antibodies. SOCS4 expression was compatible with a role in TCR signaling, with Socs4 mRNA induced within 24 h of TCR stimulation with anti-CD3 antibodies, and peaking at 72 h. In addition, the magnitude of Socs4 expression was 4-fold higher in CD8 versus CD4 cells (Fig. 5A). To investigate T cell responses in the absence of functional SOCS4, purified CD8 T cells from Socs4R108X/R108X or wild-type mice were stimulated with anti-CD3 antibodies and analysed by flow cytometry for the expression of the T cell activation markers CD69, CD44, CD25 and CD62L. In comparison to wild-type cells, surface expression of CD62L remained high, indicating that the Socs4 mutant cells responded poorly to TCR activation (Fig. 5B). CD69, CD44 and CD25 expression levels were comparable in wild-type and Socs4R108X/R108X cells (data not shown). To further investigate the consequences of reduced TCR responses, cells were labelled with Cell Trace Violet (CTV) dye, and proliferation measured in response to stimulation with anti-CD3 antibodies. As shown in Fig. 5C and D, anti-CD3-induced proliferation was impaired in Socs4R108X/R108X CD8, but not in CD4, T cells. The defective proliferation seemed to result from a decrease in the number of cells that were mobilizing per division (Fig.5C, left panel) and resulted in a corresponding decrease in the total number of cells by day 4 of culture (Fig. 5D, middle panel). These differences resulted from a proliferative defect as opposed to differences in the rate of cell death, since the percentage of propidium iodide positive cells was equivalent for both Socs4R108X/R108X and wild-type cells (data not shown).
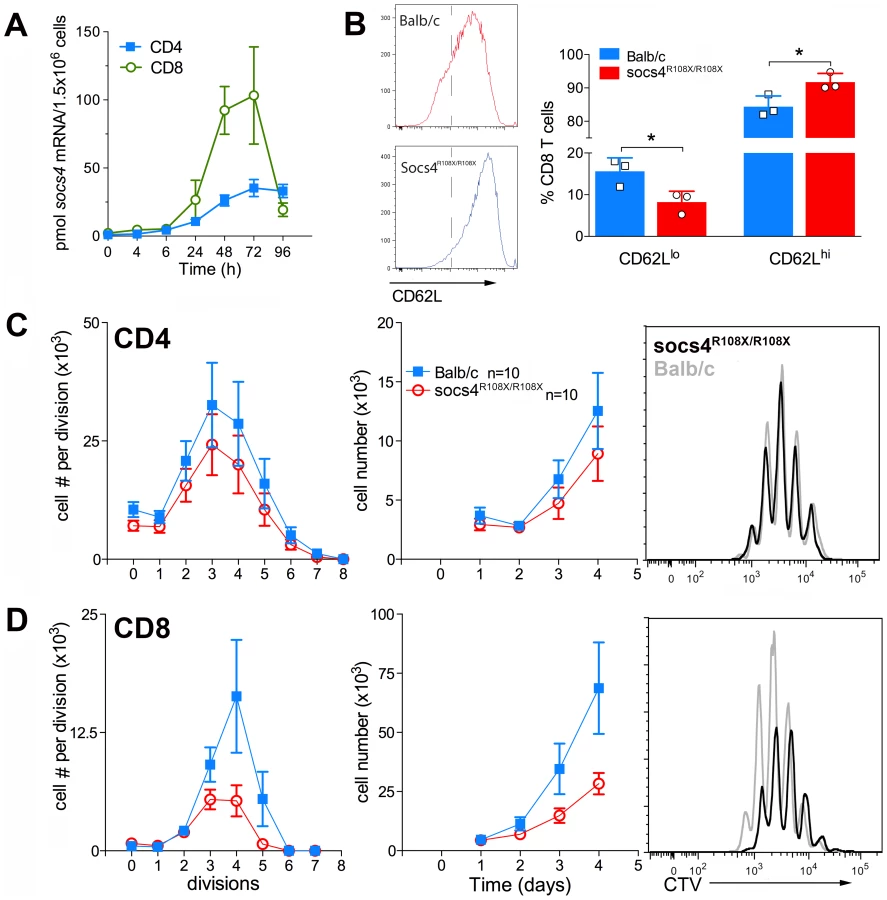
These results indicate a defect that is intrinsic to the Socs4R108X/R108X T cells and further suggest that signaling through the TCR may be qualitatively different, thus accounting for the reduced activation observed in the lymph nodes during infection and in vitro following TCR engagement.
Discussion
Loss of functional SOCS4 protein led to a dramatic phenotype following influenza A infection. Socs4R108X/R108X mice were highly susceptible to primary infection with the virulent PR8 H1N1 strain exhibiting increased mortality associated with weight loss and delayed viral clearance. Similar results were observed following infection with the less virulent X31 H3N2 strain, however the anorexia was somewhat ameliorated, enabling us to dissect the underlying pathology in greater detail. The transient weight loss induced by influenza infection is known to reflect viral pathogenicity and as in this study, correlates with increased cytokine/chemokine levels in the lungs [2]. This is the first description of SOCS4-deficient mice and suggests that SOCS4 will play an important role in immune regulation during infection. Deletion of the C-terminal 328 amino acids removes the main functional domains, the SH2 domain and SOCS box, and the conserved SOCS4/5 N-terminal motif [7] is no longer intact. There is no described biological function for the remaining 108 amino acids of SOCS4. The long N-terminal regions of SOCS proteins are predicted to be largely disordered [7], and although such regions within a full-length molecule can play a role in multi-protein complex formation, it is unlikely that a short, disordered fragment will be functional. We have however, been able to express the truncated 108-residue fragment in 293T cells (data not shown). It is possible that, if expressed in vivo, the 108-residue fragment could retain binding to its endogenous target and compete off other signaling intermediates, acting as a dominant negative. It might be speculated that residues 1–108 are involved in binding to a receptor subunit in a similar fashion to SOCS5 binding to the IL-4Rα [31]but the identity of such a receptor complex is currently unknown. Regardless, given that expression of this putative fragment is under the control of the SOCS4 promoter, the observed phenotype indicates that normal SOCS4 function has been disrupted at the endogenous level and reflects a biological role for SOCS4 in regulating anti-viral immunity to influenza A.
So far very little is known regarding the role of different SOCS proteins in influenza infection, outside of the ability of SOCS1 and SOCS3 to regulate specific cytokine receptor complexes. SOCS1 and SOCS3 have been implicated as negative regulators of innate immune responses (type I interferons) via a RIG-I dependent pathway [32], [33], [34]. SOCS3, but not SOCS1, has also been shown to inhibit type I interferon signaling via an NF-κB-dependent pathway [35]. SOCS1 and SOCS3 expression has been associated with symptomatic influenza infection, whereas SOCS2 and SOCS5 have been linked to asymptomatic disease [36].
The pathogenicity of virulent influenza infections is not well understood, but it is accepted that pathogenic strains (such as PR8) cause dysregulation of innate immunity resulting in aberrant cytokine and chemokine production (also known as a cytokine storm), which results in the lung tissue damage. Following infection with a viral strain of relatively moderate virulence, the absence of a functional SOCS4 protein led to dysregulated cytokine production akin to a cytokine storm. This uncontrolled inflammatory response is most likely responsible for the excessive weight loss in Socs4R108X/R108X mice, but it is unclear whether this increased cytokine production also contributes to the delayed viral clearance. Given that the delay in clearance is most pronounced during the adaptive phase of the immune response we postulate that the two phenomena (weight loss and delayed viral clearance) are independent of each other and result from multiple defects caused by the absence of a functional SOCS4.
Airway epithelial cells are the primary targets for influenza infection and are capable of producing cytokines as early as 3–6 hours post-infection [37]. Similarly, a variety of immunomodulators are produced by infected monocytes and macrophages [38]. IL-6 and IL-1β were among the proinflammatory cytokines elevated in the lungs of Socs4R108X/R108X mice (Fig. 2A). IL-6 is known to promote pulmonary inflammation and is a potential biomarker for patients at risk following infection with H1N1 [39]. In addition to its anti-viral activity [40], IL-1 has also been shown to contribute to acute inflammatory lung pathology following influenza infection [41]. The effects of IL-1 and IL-6 are synergistic [42] with both hyper-induced in H5N1 infected human macrophages leading to acute respiratory distress [43]. Although IL-6 expression is dysregulated in Socs4R108X/R108X mice, it does not follow that SOCS4 regulates IL-6 directly. We have tested this theory (data not shown) and are confident that SOCS4 does not directly inhibit gp130 signaling. The elevated levels of MCP-1 and IL-12p40 are also of interest. IL-12p40 is produced by epithelial cells [44] and forms a homodimer (IL-12p80) with both monomeric and dimeric forms implicated in instigating the inflammatory response in lungs [45]. High MCP-1 levels have been associated with both the profound inflammatory responses observed in Ifitm3-/- mice infected with influenza [46], and with the increased severity of disease observed in humans with a variant Ifitm3 gene [47].
Surprisingly, we did not detect major differences in the inflammatory cell infiltrates in the lungs of Socs4R108X/R108X and control mice. Numbers of neutrophils, NK cells, macrophages and inflammatory monocytes were comparable between the two groups. We do not understand the mechanism by which the lack of functional SOCS4 protein results in enhanced cytokine/chemokine production, but given the negligible expression of Socs4 in lung tissue and the relatively equal number of infiltrating cells, we propose that multiple immune cell types are producing greater amounts of cytokine per cell. This is also consistent with retention of the weight loss phenotype in wild-type mice reconstituted with Socs4R108X/R108X bone marrow.
However, our analysis of infected lungs did reveal significantly elevated numbers of T cells on day 3 post-infection, with both CD4 and CD8 showing an activated CD44hi phenotype. This increased number of T cells in the lungs of Socs4R108X/R108X mice is suggestive of proliferation due to bystander activation driven by cytokines rather than antigen-driven expansion of specific T cells or stimulation of cell division by cross-reactive antigens [48], [49], [50]. Interestingly, IL-6 appears to be one of the major factors driving the spontaneous proliferation of naïve T cells (both CD8 and CD4) [51], [52], [53]. Geginat et al. [52] further demonstrated that IL-4 in combination with IL-6 (and other cytokines such as IL-10, IL-12 and TNF) selectively induced the proliferation of naïve CD4 T cells. In our Socs4R108X/R108X mice we observed significantly increased levels of IL-4 and IL-6 as well as elevated levels of IL-10, which combined with other cytokines in a complex lung milieu might lead to the activation and proliferation of T cells already present in the lungs [54].
In addition to the early excessive inflammation, we observed a clear difference in the distribution of virus-specific CD8 T cells in Socs4R108X/R108X mice, with tetramer positive cells appearing to accumulate in the spleen of Socs4R108X/R108X mice instead of migrating to the site of infection, and this discrepancy was particularly apparent by day 5 post-infection (Fig. 3B). Activation of CD8 T cells occurs in response to TCR engagement by antigen in the draining lymph nodes. CD62L expression on naïve T cells is downregulated, with concomitant upregulation of the surface markers CD44, CD25 and CD69 [55]. The most highly activated T cells are then thought to traffic to the lungs, whereas less activated T cells migrate to the spleen and peripheral LNs [29]. Thus, cells migrating to the spleen display a CD69 low/negative phenotype, whilst CD8 T cells in the lungs show a CD69 high phenotype [29]. Consistent with this concept, CD62L down-regulation was less pronounced on Socs4R108X/R108X CD8 cells in the draining lymph nodes at day 5, suggesting that despite the high levels of virus, these cells were not being activated to the same extent as wild-type cells. On day 6 post-infection, Socs4R108X/R108X (Thy1.2) CD8 T cells migrating to the lungs had significantly lower levels of CD69 expression than wild-type (Thy1.1) CD8 cells. Although both subsets appear to be recruited to the lungs in similar ratios, their activation status differed significantly in an identical environment. Given that this result is in the context of a chimeric bone marrow reconstitution where up to 60% of the hematopoietic cells were wild-type, it strongly indicates an intrinsic CD8 T cell defect. CD69 expression declines in the absence of antigenic stimulation [56], however, again the high viral load in the lungs indicates that adequate levels of antigen were available for presentation in the lymph nodes, so that even in the presence of antigenic stimulation Socs4R108X/R108X T cells showed a much weaker activation compared to the wild-type cells.
We conclude that this defect in activation and trafficking of Socs4R108X/R108X CD8 cells to the lungs is likely to be linked to the reduced activation of the virus-specific cells, which in turn suggests impaired TCR activation and signaling. The significantly lower numbers of influenza-specific effector cells available in the Socs4R108X/R108X lungs no doubt result in the much higher viral loads observed (Fig. 1).
A defect in TCR signaling is supported by our in vitro data showing differences in the activation marker CD62L and the reduced capacity of Socs4R108X/R108X CD8 T cells to proliferate in response to anti-CD3-activation. Similar proliferative deficiencies have been observed previously in various settings where TCR signaling was affected [57], [58]. The in vitro proliferative defect (Fig. 5C) is in contrast to the normal expansion of CD8 T cells in response to influenza A infection, as indicated by total numbers of virus-specific CD8 T cells (Fig. 3A). However, such a discrepancy might be due to differences in CD3 versus peptide/MHC stimulation and/or several factors present in vivo that are not accounted for in the in vitro setting, including cytokine milieu or co-receptor stimulation [57]. In fact, in vitro co-stimulation alone can overcome a proliferative defect in cells stimulated only with anti-CD3 antibodies [59]. Therefore, in vivo viral challenge might be sufficient to induce a normal level of T cell expansion and overcome a proliferative impairment detected in vitro. CD62L is an L-selectin that plays a major role in directing lymphocytes to the site of infection and inflammation. Downregulation of CD62L reflects the activation status of T cells and is linked to gene transcription, mRNA stability and shedding from the surface due to increased activity of membrane proteases[60]. We suggest that loss of SOCS4 may affect TCR signalling resulting in maintained CD62L expression. We have also identified differences in CD69 expression between Socs4R108X/R108X and control CD8 T cells supporting this conclusion and indicating that other activation markers can also be affected, depending on the experimental context. We currently postulate that SOCS4 regulates TCR signalling rather than CD62L shedding, although at this stage we cannot rule out the involvement of SOCS4 in ubiquitylation of membrane proteases involved in CD62L shedding.
Our data describe for the first time a phenotype for Socs4R108X/R108X mice in the context of a viral challenge. While SOCS4 plays a negative regulatory role in inflammatory responses to influenza, it appears to be a positive regulator of TCR signaling. The former is consistent with the traditional role of SOCS proteins and a plausible mechanism of action is through inhibition of the JAK/STAT pathways, although the exact target/s are currently unknown. The positive involvement in TCR signaling is a novel function for a SOCS protein.
Materials and Methods
Ethics statement
All mice were bred at the Walter and Eliza Hall Institute's animal facility. Animal experiments followed the NHMRC Code of Practice for the Care and Use of Animals for Scientific Purposes guidelines and were approved by the Walter and Eliza Hall Institute's Animal Ethics Committee (AEC 2008.032 and 2011.031).
Animals
SOCS4 mutant mice (Socs4R108X/R108X) were generated through ENU mutagenesis by Ingenium Pharmaceuticals AG (Martinsried, Germany) on the C3HeB/FeJ background. The Socs4R108X mutation was subsequently backcrossed onto both the Balb/c and C57BL/6 backgrounds for 10 generations.
Virus infection
Mice were lightly anaesthetized by inhalation of methoxyflurane, and infected intranasally (i.n.) with 20 plaque forming units (pfu) of PR8 (A/Puerto Rico/8/1934 (H1N1)) or 103-104 pfu of X31 (A/X31(H3N2)) influenza virus in 30 µL PBS. Virus stocks were grown in the allantoic cavity of 10 day old embryonated chicken eggs, from which the viral titre was determined by plaque assay on monolayers of Madin derby canine kidney (MDCK) cells. The weight of mice was monitored daily from day 3 post-infection. Mice were euthanised at various time points following infection and tissues collected for analysis.
Determination of viral titres
Lungs taken from mice after primary viral infection were homogenised and the virus-containing supernatant above the cell debris was harvested and stored at −80°C. Titres of infectious virus in the lung supernatants were determined by plaque assay on monolayers of MDCK cells.
Tissue sampling and cell preparation
Spleen, mediastinal lymph node (MLN), and bronchoalveolar lavage (BAL) samples were recovered from mice at different stages during the acute phase of the primary infection. BAL samples were incubated on plastic petri-dishes for 1 h at 37°C to remove macrophages for tetramer experiments or used for phenotypic staining without adherent cell depletion. BAL fluid was collected and stored at −80°C for cytokine analysis. The spleens and MLN were disrupted, processed to single-cell suspensions and enriched for CD8 T cells by negative depletion using goat anti-mouse IgG and IgM antibodies to non-CD8 cell lineages (Jackson ImmunoResearch Labs, West Grove, PA, USA). Lungs collected from infected mice were minced, incubated in 2 mg/mL collagenase for 30 min at 37°C, and processed to a single-cell suspension.
Tetramer and phenotypic staining
Cells from BAL, MLN and spleen were stained with a tetramer conjugated to Strepavidin-PE (Molecular Probes, Eugene, OR, USA) at an optimal staining dilution (1∶100 KdNP147) for 1 h at room temperature (RT). All batches of tetramer used in this study were titrated and the optimal dilution was based on both the percentage of epitope-specific CD8 T cells and the mean fluorescence intensity (MFI) of tetramer staining. Cells were washed twice in FACS buffer, and stained with antibodies to CD4, B220 and F4/80 (all FITC conjugated, dump gate), CD8-APC Cy7, CD3ε-PerCP Cy5.5, CD62L-APC, CD44-PE Cy7 (BD Biosciences or BioLegend) for 30 min on ice, washed twice and analysed by flow cytometry on the FACS Canto (BD Biosciences) and analysed by FlowJo software (Tree Star). For phenotypic staining, different combinations of antibodies were used as indicated in the text.
Granzyme B and CD107a staining
Cells stained for tetramer and CD8α were fixed and permeabilized using a BD Cytofix/Cytoperm kit (BD Pharmingen), then stained for intracellular GzmB using anti-human GzmB-APC (clone GB12; Caltag Laboratories) as previously described [61]. Acquisition of cell surface CD107a was used to measure activation-induced degranulation by antigen-specific CD8+ T cells [62]. Briefly, cells were incubated for 1 h at 37°C with 1 µM NP147–155 peptide, 10 U/ml hrIL-2 and FITC-conjugated anti-CD107a (clone 1D4B; BD Pharmingen). Monensin (1 µl/ml; BD Pharmingen) was then added and cells were incubated for a further 4 h. Cells were stained for surface CD8α expression, then fixed and permeabilized before intracellular staining with anti-IFN-γ FITC (clone XMG1.2; BioLegend) and anti-GzmB APC. Negative controls incubated in the absence of peptide were used to control for spontaneous production of cytokine or expression of CD107a.
Bone marrow chimeras
To establish chimeras, recipient mice (Balb/c Thy1.1) were irradiated with two doses of 550R 3 h apart. At 3–5 h after the final irradiation, recipients were reconstituted with 3×106 T cell-depleted bone marrow (BM) cells isolated from femurs and tibias of donor mice (Thy1.2 Socs4R108X/R108X or Balb/c). Briefly, bone marrow was flushed from the femurs of 5–7 week old mice, washed once and incubated in complement-fixing antibodies anti-CD4 (RL172), anti-CD8 (3.186) and anti-Thy-1 (J1j) for 30 min on ice. After washing the cells once in HEPES Earle's medium containing 2.5% fetal calf serum (FCS) (HEM2.5), antibody-binding cells were incubated in rabbit complement at 37°C for 20 min. The mice were allowed to reconstitute for at least 8 weeks prior to use, blood samples were collected (by retro-orbital bleeding), and reconstitution of T cell compartment assessed by FACS analysis.
Cytokine analyses
Cytokine levels were assessed by capture ELISA or BioPlex Pro Assay (BioRad, CA, USA). Lung homogenates were stored at −80°C prior to analysis. BioPlex detection was performed according to the manufacturer's instructions. For ELISAs, U-bottom 96-well plates (Costar, NY, USA) were coated overnight at 4°C with capture antibodies against IL-4, IL-6, IL-10, IFN-γ, GM-CSF, KC, MIP-1α or MIP-2 (BD Pharmingen, CA or RD Systems, MN, USA) and were then incubated with serial dilutions of samples for 2 h at RT, washed and incubated with the appropriate biotinylated detection antibody for 2 h at RT, followed by incubation with streptavidin-horseradish peroxidase. Plates were developed with 3,3′,5,5′-tetramethylbenzidine (TMB) in 0.1 M sodium acetate pH 6, and colour development terminated by the addition of 2N H2SO4. Absorbance values were read at 450 nm. TGF-β ELISA was performed according to the manufacturer's instructions (RD Systems, MN, USA).
In vitro T cell proliferation assay
Single-cell suspensions were generated from spleens, and CD8 or CD4 T cells purified by negative selection using magnetic beads (Dynabeads, Invitrogen or BioMag, Qiagen). Enrichment of cells was verified by flow cytometry. Purified T cells were labelled with Cell Trace Violet (CTV) (Molecular Probes, OR, USA) according to the manufacturer's instructions. In vitro stimulation assays were performed by plating cells at 104 cells per well in RPMI 1640 medium containing 10% (v/v) heat-inactivated FCS (Sigma-Aldrich, MO, USA), 5×10−5 M 2-ME (Sigma-Aldrich), 100 µg/mL streptomycin and 100 U/mL penicillin (Invitrogen Life Technologies) into anti-CD3-coated (10 µg/mL, clone KT3-1-1) 96-well plates. Recombinant mouse IL-2 (20 ng/mL) was also added to wells. Cells were harvested at different timepoints, propidium iodide (2 µg/mL) and 5000 of Sphero Nile Red Fluorescent Particles (BD Biosciences) added per well and cell division analysed by flow cytometry. Each sample was analysed in duplicate.
Real-time quantitative PCR (Q-PCR)
Q-PCR analysis of Socs4 mRNA expression was performed essentially as described [63].
Statistical analyses
Statistical analyses were performed using the unpaired t-test provided within GraphPad Prism 5 software and Compare Groups of Growth Curves software package available on the Walter and Eliza Hall Institute Bioinformatics Division's website http://bioinf.wehi.edu.au/.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Newall AT, Scuttham PA, Hodgkinson B (2007) Economic Report into the cost of influenza to the Australian Health System. http://www.influenzaspecialistgrouporgau/images/stories/docs/isg_cost_influenza_report_30_2007pdf.
2. La GrutaNL, KedzierskaK, StambasJ, DohertyPC (2007) A question of self-preservation: immunopathology in influenza virus infection. Immunol Cell Biol 85 : 85–92.
3. ArankalleVA, LoleKS, AryaRP, TripathyAS, RamdasiAY, et al. (2010) Role of host immune response and viral load in the differential outcome of pandemic H1N1 (2009) influenza virus infection in Indian patients. PLoS One 5: e13099.
4. ThomasP, KeatingR, Hulse-PostD, DohertyP (2006) Cell-mediated protection in influenza infection. Emerg Infect Dis 12 : 48–54.
5. AlexanderWS (2002) Suppressors of cytokine signalling (SOCS) in the immune system. Nat Rev Immunol 2 : 410–416.
6. HiltonDJ, RichardsonRT, AlexanderWS, VineyEM, WillsonTA, et al. (1998) Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc Natl Acad Sci U S A 95 : 114–119.
7. FengZP, ChandrashekaranIR, LowA, SpeedTP, NicholsonSE, et al. (2012) The N-terminal domains of SOCS proteins: a conserved region in the disordered N-termini of SOCS4 and 5. Proteins 80 : 946–957.
8. ZhangJG, FarleyA, NicholsonSE, WillsonTA, ZugaroLM, et al. (1999) The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc Natl Acad Sci U S A 96 : 2071–2076.
9. LinossiEM, NicholsonSE (2012) The SOCS box-adapting proteins for ubiquitination and proteasomal degradation. IUBMB Life 64 : 316–323.
10. KershawNJ, MurphyJM, LiauNP, VargheseLN, LaktyushinA, et al. (2013) SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat Struct Mol Biol 20 : 469–476.
11. BabonJJ, KershawNJ, MurphyJM, VargheseLN, LaktyushinA, et al. (2012) Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity 36 : 239–250.
12. GreenhalghCJ, Rico-BautistaE, LorentzonM, ThausAL, MorganPO, et al. (2005) SOCS2 negatively regulates growth hormone action in vitro and in vivo. J Clin Invest 115 : 397–406.
13. EndoT, SasakiA, MinoguchiM, JooA, YoshimuraA (2003) CIS1 interacts with the Y532 of the prolactin receptor and suppresses prolactin-dependent STAT5 activation. J Biochem 133 : 109–113.
14. LavensD, MontoyeT, PiessevauxJ, ZabeauL, VandekerckhoveJ, et al. (2006) A complex interaction pattern of CIS and SOCS2 with the leptin receptor. J Cell Sci 119 : 2214–2224.
15. AlexanderWS, StarrR, FennerJE, ScottCL, HandmanE, et al. (1999) SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell 98 : 597–608.
16. MetcalfD, GreenhalghCJ, VineyE, WillsonTA, StarrR, et al. (2000) Gigantism in mice lacking suppressor of cytokine signalling-2. Nature 405 : 1069–1073.
17. CrokerBA, MetcalfD, RobbL, WeiW, MifsudS, et al. (2004) SOCS3 is a critical physiological negative regulator of G-CSF signaling and emergency granulopoiesis. Immunity 20 : 153–165.
18. CrokerBA, KrebsDL, ZhangJG, WormaldS, WillsonTA, et al. (2003) SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol 4 : 540–545.
19. KarioE, MarmorMD, AdamskyK, CitriA, AmitI, et al. (2005) Suppressors of cytokine signaling 4 and 5 regulate epidermal growth factor receptor signaling. J Biol Chem 280 : 7038–7048.
20. BullockAN, RodriguezMC, DebreczeniJE, SongyangZ, KnappS (2007) Structure of the SOCS4-ElonginB/C complex reveals a distinct SOCS box interface and the molecular basis for SOCS-dependent EGFR degradation. Structure 15 : 1493–1504.
21. HuG, ZhouR, LiuJ, GongAY, ChenXM (2010) MicroRNA-98 and let-7 regulate expression of suppressor of cytokine signaling 4 in biliary epithelial cells in response to Cryptosporidium parvum infection. J Infect Dis 202 : 125–135.
22. SasiW, JiangWG, SharmaA, MokbelK (2010) Higher expression levels of SOCS 1,3,4,7 are associated with earlier tumour stage and better clinical outcome in human breast cancer. BMC Cancer 10 : 178.
23. SutherlandJM, KeightleyRA, NixonB, RomanSD, RobkerRL, et al. (2012) Suppressor of cytokine signaling 4 (SOCS4): moderator of ovarian primordial follicle activation. J Cell Physiol 227 : 1188–1198.
24. AugustinM, SedlmeierR, PetersT, HuffstadtU, KochmannE, et al. (2005) Efficient and fast targeted production of murine models based on ENU mutagenesis. Mamm Genome 16 : 405–413.
25. BenderBS, CroghanT, ZhangL, SmallPAJr (1992) Transgenic mice lacking class I major histocompatibility complex-restricted T cells have delayed viral clearance and increased mortality after influenza virus challenge. J Exp Med 175 : 1143–1145.
26. DentonAE, DohertyPC, TurnerSJ, La GrutaNL (2007) IL-18, but not IL-12, is required for optimal cytokine production by influenza virus-specific CD8+ T cells. Eur J Immunol 37 : 368–375.
27. ImaiY, KubaK, NeelyGG, Yaghubian-MalhamiR, PerkmannT, et al. (2008) Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133 : 235–249.
28. PeirisJS, HuiKP, YenHL (2010) Host response to influenza virus: protection versus immunopathology. Curr Opin Immunol 22 : 475–481.
29. LawrenceCW, BracialeTJ (2004) Activation, differentiation, and migration of naive virus-specific CD8+ T cells during pulmonary influenza virus infection. J Immunol 173 : 1209–1218.
30. LawrenceCW, ReamRM, BracialeTJ (2005) Frequency, specificity, and sites of expansion of CD8+ T cells during primary pulmonary influenza virus infection. J Immunol 174 : 5332–5340.
31. SekiY, HayashiK, MatsumotoA, SekiN, TsukadaJ, et al. (2002) Expression of the suppressor of cytokine signaling-5 (SOCS5) negatively regulates IL-4-dependent STAT6 activation and Th2 differentiation. Proc Natl Acad Sci U S A 99 : 13003–13008.
32. PothlichetJ, ChignardM, Si-TaharM (2008) Cutting edge: innate immune response triggered by influenza A virus is negatively regulated by SOCS1 and SOCS3 through a RIG-I/IFNAR1-dependent pathway. J Immunol 180 : 2034–2038.
33. WeiH, WangS, ChenQ, ChenY, ChiX, et al. (2014) Suppression of Interferon Lambda Signaling by SOCS-1 Results in Their Excessive Production during Influenza Virus Infection. PLoS Pathog 10: e1003845.
34. Ramirez-MartinezG, Cruz-LagunasA, Jimenez-AlvarezL, EspinosaE, Ortiz-QuinteroB, et al. (2013) Seasonal and pandemic influenza H1N1 viruses induce differential expression of SOCS-1 and RIG-I genes and cytokine/chemokine production in macrophages. Cytokine 62 : 151–159.
35. PauliEK, SchmolkeM, WolffT, ViemannD, RothJ, et al. (2008) Influenza A virus inhibits type I IFN signaling via NF-kappaB-dependent induction of SOCS-3 expression. PLoS Pathog 4: e1000196.
36. HuangY, ZaasAK, RaoA, DobigeonN, WoolfPJ, et al. (2011) Temporal dynamics of host molecular responses differentiate symptomatic and asymptomatic influenza A infection. PLoS Genet 7: e1002234.
37. ChanMC, CheungCY, ChuiWH, TsaoSW, NichollsJM, et al. (2005) Proinflammatory cytokine responses induced by influenza A (H5N1) viruses in primary human alveolar and bronchial epithelial cells. Respir Res 6 : 135.
38. JulkunenI, SarenevaT, PirhonenJ, RonniT, MelenK, et al. (2001) Molecular pathogenesis of influenza A virus infection and virus-induced regulation of cytokine gene expression. Cytokine Growth Factor Rev 12 : 171–180.
39. PaquetteSG, BannerD, ZhaoZ, FangY, HuangSS, et al. (2012) Interleukin-6 is a potential biomarker for severe pandemic H1N1 influenza A infection. PLoS One 7: e38214.
40. JanewayCJr, MedzhitovR (2000) Viral interference with IL-1 and toll signaling. Proc Natl Acad Sci U S A 97 : 10682–10683.
41. SchmitzN, KurrerM, BachmannMF, KopfM (2005) Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J Virol 79 : 6441–6448.
42. LeJM, FredricksonG, ReisLF, DiamantsteinT, HiranoT, et al. (1988) Interleukin 2-dependent and interleukin 2-independent pathways of regulation of thymocyte function by interleukin 6. Proc Natl Acad Sci U S A 85 : 8643–8647.
43. CheungCY, PoonLL, LauAS, LukW, LauYL, et al. (2002) Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet 360 : 1831–1837.
44. MikolsCL, YanL, NorrisJY, RussellTD, KhalifahAP, et al. (2006) IL-12 p80 is an innate epithelial cell effector that mediates chronic allograft dysfunction. Am J Respir Crit Care Med 174 : 461–470.
45. CooperAM, KhaderSA (2007) IL-12p40: an inherently agonistic cytokine. Trends Immunol 28 : 33–38.
46. EverittAR, ClareS, PertelT, JohnSP, WashRS, et al. (2012) IFITM3 restricts the morbidity and mortality associated with influenza. Nature 484 : 519–523.
47. ZhangYH, ZhaoY, LiN, PengYC, GiannoulatouE, et al. (2013) Interferon-induced transmembrane protein-3 genetic variant rs12252-C is associated with severe influenza in Chinese individuals. Nat Commun 4 : 1418.
48. Murali-KrishnaK, AltmanJD, SureshM, SourdiveDJ, ZajacAJ, et al. (1998) Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity 8 : 177–187.
49. ToughDF, BorrowP, SprentJ (1996) Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science 272 : 1947–1950.
50. Di GenovaG, SavelyevaN, SuchackiA, ThirdboroughSM, StevensonFK (2010) Bystander stimulation of activated CD4+ T cells of unrelated specificity following a booster vaccination with tetanus toxoid. Eur J Immunol 40 : 976–985.
51. TajimaM, WakitaD, NoguchiD, ChamotoK, YueZ, et al. (2008) IL-6-dependent spontaneous proliferation is required for the induction of colitogenic IL-17-producing CD8+ T cells. J Exp Med 205 : 1019–1027.
52. GeginatJ, SallustoF, LanzavecchiaA (2001) Cytokine-driven proliferation and differentiation of human naive, central memory, and effector memory CD4(+) T cells. J Exp Med 194 : 1711–1719.
53. UnutmazD, PileriP, AbrignaniS (1994) Antigen-independent activation of naive and memory resting T cells by a cytokine combination. J Exp Med 180 : 1159–1164.
54. CoseS, BrammerC, KhannaKM, MasopustD, LefrancoisL (2006) Evidence that a significant number of naive T cells enter non-lymphoid organs as part of a normal migratory pathway. Eur J Immunol 36 : 1423–1433.
55. MarzioR, MauelJ, Betz-CorradinS (1999) CD69 and regulation of the immune function. Immunopharmacol Immunotoxicol 21 : 565–582.
56. TestiR, D'AmbrosioD, De MariaR, SantoniA (1994) The CD69 receptor: a multipurpose cell-surface trigger for hematopoietic cells. Immunol Today 15 : 479–483.
57. Smith-GarvinJE, BurnsJC, GohilM, ZouT, KimJS, et al. (2010) T-cell receptor signals direct the composition and function of the memory CD8+ T-cell pool. Blood 116 : 5548–5559.
58. HanJ, ShuiJW, ZhangX, ZhengB, HanS, et al. (2005) HIP-55 is important for T-cell proliferation, cytokine production, and immune responses. Mol Cell Biol 25 : 6869–6878.
59. D'SouzaWN, ChangCF, FischerAM, LiM, HedrickSM (2008) The Erk2 MAPK regulates CD8 T cell proliferation and survival. J Immunol 181 : 7617–7629.
60. RainerTH (2002) L-selectin in health and disease. Resuscitation 52 : 127–141.
61. JenkinsMR, KedzierskaK, DohertyPC, TurnerSJ (2007) Heterogeneity of effector phenotype for acute phase and memory influenza A virus-specific CTL. J Immunol 179 : 64–70.
62. BettsMR, BrenchleyJM, PriceDA, De RosaSC, DouekDC, et al. (2003) Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods 281 : 65–78.
63. LeeC, KolesnikTB, CaminschiI, ChakravortyA, CarterW, et al. (2009) Suppressor of cytokine signalling 1 (SOCS1) is a physiological regulator of the asthma response. Clin Exp Allergy 39 : 897–907.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 5
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Venus Kinase Receptors Control Reproduction in the Platyhelminth Parasite
- Dual-Site Phosphorylation of the Control of Virulence Regulator Impacts Group A Streptococcal Global Gene Expression and Pathogenesis
- Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Ion Channel Activity Promotes Virus Fitness and Pathogenesis
- High-Efficiency Targeted Editing of Large Viral Genomes by RNA-Guided Nucleases
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy