
Bioequivalence between innovator and generic tacrolimus in liver and kidney transplant recipients: A randomized, crossover clinical trial
In a randomized 3-treatment crossover clinical trial, Rita Alloway of the University of Cincinnati and colleagues establish bioequivalence between 3 versions of the drug tracolimus (a brand name and 2 disparate generics) in kidney and liver transplant recipients.
Published in the journal:
. PLoS Med 14(11): e32767. doi:10.1371/journal.pmed.1002428
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pmed.1002428
Summary
In a randomized 3-treatment crossover clinical trial, Rita Alloway of the University of Cincinnati and colleagues establish bioequivalence between 3 versions of the drug tracolimus (a brand name and 2 disparate generics) in kidney and liver transplant recipients.
Introduction
Most individuals receiving a solid organ transplant require lifelong immunosuppression. Switching to generic immunosuppressants may lead to significant savings and improved adherence [1,2], which is essential for long-term graft survival [3]. The current US Food and Drug Administration (FDA) generic drug approval process has performed well [4]. However, concerns persist regarding whether 2-way crossover studies in healthy individuals using conventional average bioequivalence (ABE) acceptance criteria of a 90% confidence interval contained within the confidence limits of 80.00% to 125.00% are a valid approach for generic immunosuppressant approval for use after transplantation [5,6]. This debate started when cyclosporine generics were developed over 15 years ago [7,8] and was reinvigorated when tacrolimus generics were approved. Consensus documents developed by professional societies from the US, Europe, and Canada [9–12] have cautioned against generic immunosuppressant use, citing (1) the lack of data in transplant recipients, especially “high risk” transplant recipients; (2) the need to implement stricter bioequivalence standards, as tacrolimus is a narrow therapeutic index (NTI) drug for which small changes in dose or exposure can result in therapeutic failure or toxicity; and (3) the lack of bioequivalence data between generics. Molnar et al. published a systematic review and meta-analysis to compare the clinical efficacy and bioequivalence of generic immunosuppressive drugs in individuals with a transplant and concluded that high-quality data were lacking. The authors went further to state that given the serious consequences of rejection and allograft failure, well-designed studies on the bioequivalence and safety of generic immunosuppression in individuals with a transplant are needed [13].
Differing worldwide bioequivalence regulatory standards for NTI drugs make it difficult to interpret bioequivalence study results [14–16]. For NTI drugs, the European Medicines Agency (EMA) requires a narrower 90.00%–111.11% acceptance criterion for the area under the curve (AUC, a measure of actual body exposure to a drug) but uses the usual 80.00%–125.00% acceptance criterion for the concentration maximum (Cmax) for NTI drugs [14]. Health Canada has adopted standards similar to those of EMA, with an AUC acceptance criterion of 90.00%–112.00% [15]. The FDA has classified tacrolimus as an NTI drug and recommended the scaled average bioequivalence (SCABE) approach to determine bioequivalence [16,17]. With this SCABE approach, both generic and innovator products are given twice with fully replicating measurements in each individual. An innovator pharmaceutical product is the one that was first authorized for marketing on the basis of quality, safety and efficacy. This allows for determination of within-subject variability, which is then used for scaling the bioequivalence acceptance limits based on the reference product for all products tested. This approach creates more stringent bioequivalence criteria: (1) the ABE limits for both AUC and Cmax are narrowed based on the within-subject variability of the reference product and are never wider than 80.00%–125.00%, and (2) the within-subject variabilities of all products are compared to each other.
Tacrolimus has a complex pharmacokinetic profile, as it is metabolized mainly by hepatic and intestinal cytochrome (CYP) P4503A enzymes and over 90% is eliminated as metabolites. CYP3A5 expressers (CYP3A5 *1/*1 and CYP3A5 *1/*3) are considered patients who “poorly absorb” and may exhibit higher within-subject variability of tacrolimus pharmacokinetics than nonexpressers (CYP3A5 *3/*3). These genetic differences have been associated with poorer outcomes. Tacrolimus is also a substrate of the drug efflux protein p-glycoprotein, ABCB1, thus impacting tacrolimus exposure [17–24]. Because of these complex metabolic and transport processes, stringent ABE testing is used to ensure product excipients do not impact these processes.
Given the aforementioned public concerns [9–12], we hypothesized that 2 generic tacrolimus products currently on the US market meet both FDA ABE and SCABE limits in individuals with a kidney or liver transplant when compared to innovator tacrolimus and when compared to each other in a high-quality study. All products met these bioequivalence criteria. In addition, we applied EMA NTI ABE criteria, and all products met the criteria except for one that narrowly fell above the AUC limit.
Methods
Study conduct and oversight
The study design was developed in collaboration with the American Society of Transplantation (Mount Laurel, New Jersey, US), the American Society of Transplant Surgeons (Arlington, Virginia, US), and the FDA. Individuals were recruited from the University of Cincinnati Medical Center and The Christ Hospital in Cincinnati. This trial adhered to the Declaration of Helsinki and was approved by local institutions’ review boards (2012–4891) and the FDA Research Involving Human Subjects Committee (13-018D). All individuals provided written informed consent. The study was monitored locally and by the FDA and registered on clinicaltrials.gov (NCT-01889758). Methodologies for tacrolimus quantification in whole blood (Tables A–F in S1 Appendix and Fig A in S1 Appendix) and genetic polymorphism testing (Table G in S1 Appendix) are described. The study protocol and statistical analysis plan are also included in the supporting information as S1 Text and S2 Text. Changes from the prespecified analysis plan included the analysis of the minimum concentration (Cmin) in lieu of C0 and C12 tacrolimus concentrations as appropriate based upon guidance documents, and dose normalization was not performed because each individual received the same dose in all treatment periods. No interim analyses were conducted prior to these data analyses.
Test product selection
At study initiation, 5 FDA-approved generic tacrolimus products were available in addition to the innovator product, Prograf (Astellas, Northbrook, Illinois) [25]. Abbreviated New Drug Applications (ANDAs) are submitted to the FDA for all generic products and represent the only data readily available for all products. ANDA data include, but are not limited to, pharmacokinetic data in healthy volunteers that demonstrate bioequivalence between the innovator and generic products by evaluating the pharmacokinetic parameters of AUC and Cmax. Product composition, manufacturing, and pharmacokinetic data for all approved generics were reviewed to identify the 2 most disparate generics. ANDA pharmacokinetic data from each product are provided in Table H-I in S1 Appendix. Additional manufacturing comparisons are summarized in Table J in S1 Appendix [26–31]. One tacrolimus product (Panacea; Baddi, India) was FDA-approved but not commercially available, and it was therefore excluded. Pharmacokinetic parameters of AUC and Cmax were examined for the greatest difference between the generic and the innovator product as being the most disparate and named Generic Hi and Generic Lo. Sandoz tacrolimus (Sandoz, Princeton, New Jersey, US) was identified as Generic Hi based upon higher point estimates and higher upper 90% confidence interval compared to innovator. Dr. Reddy tacrolimus (Dr. Reddy, Bachupally, India) was identified as Generic Lo based upon lower point estimates and lowest lower 90% confidence interval compared to innovator. Single tacrolimus 1 mg capsule lots (the most frequently prescribed dosage strength) of Innovator (Prograf), Generic Hi (Sandoz, Princeton, New Jersey, US), and Generic Lo (Dr. Reddy, Bachupally, India) were purchased from a pharmacy wholesaler and controlled by the University of Cincinnati Medical Center Investigational Drug Services. The University of Iowa Pharmaceuticals (Iowa City, Iowa, US), iC42 Clinical Research and Development (Aurora, Colorado, US), and the FDA independently performed dissolution, purity, and content uniformity testing according to applicable US Pharmacopeia Convention guidelines [29]. Similar results were obtained by both groups. The FDA results are reported in Tables K–O in S1 Appendix.
Study population, randomization and blinding
Individuals with a kidney or liver transplant included in the present study were at least 18 years old, with stable organ function and no evidence of rejection. Said individuals were at steady state and on stable doses of immunosuppressants including tacrolimus with no expected changes to their immunosuppressive drug regimens to eliminate confounders that occur early post-transplant or during times of rejection. Other eligibility criteria are listed in Section F of S1 Appendix. Study participants were stratified by organ type and randomized to 1 of 3 treatment sequences, each including 2 periods of Innovator (Prograf), Generic Hi (Sandoz), and Generic Lo (Dr. Reddy) (Fig 1). The replicate dosing design of administering each product twice allowed for analysis of within-subject variability by product. An independent statistician generated a randomization list using SAS (version 9.03, SAS Institute, Cary, North Carolina, US) and provided it to the investigational drug pharmacist. Eligible transplant recipients were recruited from 2 clinical sites, but all screening visits occurred at the University of Cincinnati Medical Center. The investigational drug pharmacist consecutively assigned individuals to a treatment sequence as received, independent of site. All parties were blinded to the randomization sequence allocation until after the pharmacokinetics (PK) analysis was completed. (Additional blinding information is located in Section G of S1 Appendix.)

Treatment protocol
Eligible transplant recipients were screened via telephone. Potential study participants completed a baseline visit, including written informed consent and laboratory, physical, and genetic polymorphism testing (including CYP3A5*3, CYP3A4*1B, CYP3A4*22, POR*28, and 3 ABCB1 SNPs) [18–24]. The study assessment schedule is shown in Table P in S1 Appendix. Study participants were seen 2 weeks later for baseline laboratory examination and randomization and were provided with study drug. Medications were dispensed with a Medication Event Monitoring System (MEMS, AARDEX, Palo Alto, California) bottle cap for electronic monitoring of study medication access. Pill counts were recorded at each pharmacokinetic visit. Individuals were excluded from the analysis if they were nonadherent within 48 hours of PK assessment (Section I in S1 Appendix).
Tacrolimus doses remained constant during the entire study period. After a 7-day treatment period, individuals underwent a 12-hour tacrolimus pharmacokinetic assessment with dosing and sampling times strictly controlled and monitored. The 7-day treatment period was adequate to reach steady state based upon observed half-life in individuals receiving a kidney or liver transplant. Fifteen tacrolimus samples were collected at C0 (before the morning dose) and at 20, 40, 60, 80, 100, 120, 140, and 160 minutes and 3, 4, 5, 6, 8, and 12 hours following dosing. Six 12-hour pharmacokinetic assessments were completed after 7 days of administration of each tacrolimus product. All samples were analyzed for tacrolimus and metabolites using a validated high-performance liquid chromatography-tandem mass spectrometry (LC-MS/MS) assay (Sections A and B in S1 Appendix). All individuals fasted until a standardized meal was allowed after the 4-hour and 12-hour sample collection. At each visit, study participants were assessed for safety lab tests, adverse events, medication adherence, and medication regimen changes (Table P in S1 Appendix).
Statistical, pharmacokinetic, and bioequivalence analysis
Data were stored electronically using a REDCap database [32], including all laboratory, patient diary, and bioanalytical data. Data monitoring and analysis plans defined a priori were executed. After data were monitored and all queries resolved, the database was locked. Only individuals completing all 6 pharmacokinetic study periods were analyzed. Actual sample collection times were used for analysis. For each type of organ transplant, a sample size of 24 individuals was required to achieve 90% statistical power for concluding bioequivalence in crossover trials at an alpha of 0.05 using standard bioequivalence limits of 80%–125% and assuming a true difference of 0 [16,33,34].
The primary outcome was to determine whether Innovator (Prograf), Generic Hi (Sandoz), and Generic Lo (Dr. Reddy) tacrolimus products were bioequivalent with each other by comparing their AUC and Cmax using conventional ABE limits (the 90% CIs of the ratio of geometric means of the 2 products for Cmax and AUC were within the range of 80%–125%) [34] and reference SCABE limits [16]. The observed Cmin represented the minimum concentration and was analyzed in lieu of C0 or C12. Observed Tmax represented the actual time at which the maximum concentration was measured. Each patient served as his or her own control; therefore, dose normalization was not required. The investigators and the FDA independently analyzed the primary end-point data using WinNonlin software (version 6.4. Phoenix, Certara, Princeton, New Jersey, US) and SAS (version 9.3, SAS Institute, Cary, North Carolina), respectively. The investigators’ analysis results are reported.
Secondary outcomes included ABE and SCABE assessment in prespecified subgroups and assessments for safety and efficacy. Subgroups in which there were at least 6 recipients and the statistical model converged are reported. The prespecified subgroups included recipient sex; age; African-American race; diabetes; CYP3A4/5, POR 28, and ABCB1 polymorphisms; and donor CYP3A5, as these subgroups are reported to strongly influence tacrolimus concentrations [18]. The study was not powered to identify differences by subgroup. In addition, pharmacokinetic parameters of the primary tacrolimus metabolite, 13-O-desmethyl tacrolimus, were compared.
Safety was assessed at baseline and weekly throughout the study by complete metabolic panels and complete blood cell count with differential. Baseline and weekly assessments included markers of transplant function in individuals with a kidney transplant (creatinine) or liver transplant (aspartate aminotransferase [AST], alanine aminotransferase [ALT], and alkaline phosphatase). To compare kidney and liver function tests among products, a mixed effects model was run with a term designating the product received in each period and a random subject term and using the Tukey’s honest significant difference (HSD) test for multiple comparisons, where needed [35]. The study was not powered to assess differences in transplant organ function. Total daily tacrolimus dose data were summarized using means and standard deviation, and between-group differences were analyzed using the t test. Reports of adverse events were collected at each visit and coded utilizing the Common Terminology Criteria for Adverse Events (CTCAE v4.0) [36]. The Exact McNemar test for paired categorical data with adjustments for multiple comparisons was used to compare adverse event rates among the products [37]. No statistically significant differences among any pairs of products were found for any adverse event code or for adverse events overall.
ABE was assessed within each transplant organ group (i.e., kidney and liver) by using a mixed effects analysis of variance model for a 6-period crossover design with the loge-transformed pharmacokinetic parameter estimates (AUC, Cmax, and Cmin) as the response variable. Fixed effect terms in the model included sequence, period, and treatment. Random effects included subject nested within sequence. The error variance structure accounted for the repeated measures of treatments within each subject. Two-sided 90% confidence intervals using the differences in least square means and the appropriate error terms from the model were calculated for each pairwise assessment of bioequivalence. The estimates and end points of the confidence intervals were back-transformed to obtain the ratios of the parameters being assessed for bioequivalence and the corresponding 90% confidence interval for the ratios. If the entire confidence interval was contained within the range of 80% to 125%, then ABE was established.
To assess SCABE, the estimate of within-subject variability for each treatment was obtained by using a mixed effects model within each organ type and treatment group. Fixed effect terms included sequence, replication (i.e., first or second), and sequence-by-replication interaction. Random effect terms include subject nested within sequence. This model provided estimates of the within-subject variability for each treatment, and these were then used to adjust the bioequivalence end points to obtain the SCABE limits and calculate the criterion bound in accordance with FDA guidance for NTI drugs [16]. SCABE was concluded if each of the following criteria were met: (1) the 2-sided 90% confidence interval calculated for the ABE assessment must fall entirely within the SCABE limits, (2) the criterion bound must be less than 0, and (3) the upper 90% confidence limit for the ratio of the within-subject variabilities for the 2 treatments being assessed must be less than 2.5 [16].
Results
Study individuals
From December 2013 through October 2014, 42 individuals with a kidney transplant and 40 individuals with a liver transplant were consented and followed as per the study protocol. Seventy-one individuals were analyzable (kidney, n = 35; liver, n = 36). The most frequent causes of noneligibility during screening were (1) a greater than 3-hour drive from the study center, (2) receiving the 0.5-mg tacrolimus dosage form, (3) renal function < 35 ml/min, (4) not receiving tacrolimus, (5) a history of multiorgan transplant (i.e., kidney and pancreas or liver and kidney), (6) documented nonadherence, or (7) a history of cancer. Moreover, individuals with a liver transplant and active hepatitis C were not eligible. A complete list of inclusion and exclusion criteria is in Section F in S1 Appendix. Consolidated Standards Of Reporting Trials (CONSORT) flow diagrams (Fig 2) and checklist (S1 CONSORT Checklist) are provided.

The demographic and baseline characteristics reported to impact the tacrolimus pharmacokinetics of the analyzed individuals are summarized in Table 1 and were similar to intent-to-treat individuals (Table Q in S2 Appendix). Most individuals received tacrolimus, mycophenolate, and corticosteroid-free immunosuppression. Immunosuppressive regimens, including tacrolimus doses, remained constant throughout the 6-week study. Individuals with a kidney or liver transplant received a median mg/day (IQR) tacrolimus dose of 5.0 (4.0–8.0) or 4.0 (3.0–6.0), respectively. During the study, no patient initiated, discontinued, or changed doses of known CYP3A inhibitors or inducers that could impact pharmacokinetic observations.

Adherence monitoring
Adherence was evaluated using the MEMS system to insure the quality of the pharmacokinetic evaluations. Adherence was defined as the degree to which the number of medication doses taken each day matched the number of prescribed doses. Over 6 weeks, MEMS-based adherence was 99.75% (range: 97.67%–100%). Three individuals were excluded from the analysis due to nonadherence (kidney, n = 1; liver, n = 2) (Fig 2). MEMS-based adherence rates are reported in Tables R and S in S2 Appendix.
ABE, SCABE, and within-subject variability comparison
Tacrolimus 12-hour concentrations following chronic dosing are presented for individuals with a kidney (Fig 3A) or liver transplant (Fig 3B). Two tacrolimus concentration-time curves for each product are depicted, representing the first and second exposures to the product. Values for the mean and the standard deviation (SD) for each pharmacokinetic time point are reported in Table 2. There were no statistically significant differences observed by product between time points as assessed by Kruskal-Wallis [38].


Pharmacokinetic parameters for individuals with a kidney or liver transplant by product are summarized in Table 3. Point estimates of the geometric means with the resulting 2-sided 95% confidence intervals are presented for AUC, Cmax, Cmin, and Tmax by product. Product selection was based upon pharmacokinetic studies in healthy individuals as previously described. The Generic Hi (Sandoz) observed exposure was numerically higher than that of Innovator in both individuals with a kidney transplant and those with a liver transplant. In this study, the point estimate for Generic Lo exposure was numerically higher than that of both Innovator and Generic Hi in both individuals with a kidney transplant and those with a liver transplant. However, this can be expected to be caused by random variability since these products were shown to be bioequivalent. (Table 3)

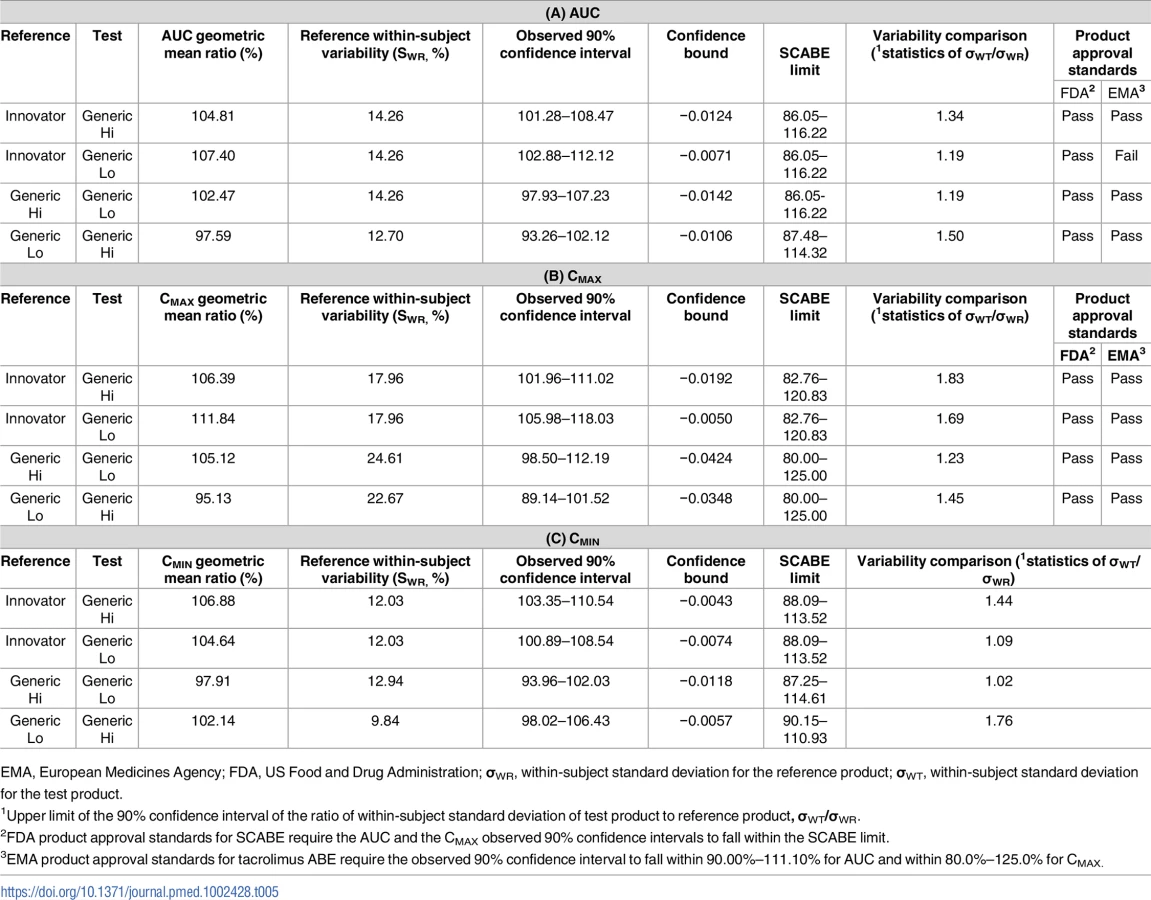
The primary end point of bioequivalence of these pharmacokinetic parameters was tested using SCABE on log-transformed data. When comparing Innovator with Generic Hi and Generic Lo, and Generic Hi with Generic Lo, AUC, Cmax, and Cmin fell within conventional ABE limits of 80%–125%, as well as within the tighter SCABE acceptance limits. Comparing interindividual variability of systemic tacrolimus exposure (AUC), within-subject variability ranged from 12.11% to 15.81%. Similarly, Cmax within-subject variability ranged from 17.96% to 24.72% for all products. Per the FDA guidance for SCABE testing, all products exhibited similar pharmacokinetic variability since the upper limit of the 90% confidence interval for the ratio of within-subject variability was equal to or less than 2.5 (Tables 4 and 5). SCABE criteria were met for all product comparisons for AUC and Cmax in both individuals with a kidney and those with a liver transplant. In reference to the EMA bioequivalence acceptance range for AUC of 90.00%–111.11%, these limits were met, with the only exception being in the case of Innovator versus Generic Lo. Here the upper limits of the 90% -confidence intervals—111.30% (kidney) and 112.12% (liver)—were slightly above the upper EMA AUC acceptance criterion [14], whereas only Innovator versus Generic Lo in liver transplant recipients was also above the Health Canada AUC bioequivalence acceptance limits for critical dose drugs of 90%–112%[15].

Individual pharmacokinetic tacrolimus time-concentration curves for each pharmacokinetic period by product are presented (Figs D and E in S2 Appendix) [39]. Upon visual inspection of individual pharmacokinetic curves, differences can be observed upon comparison between products and between replicate administration of the same product.
Subgroup analysis
The study was not powered to show a difference by any subgroup analyzed. A majority of individuals with a kidney transplant were genotyped as nonexpressers with CYP3A5 *3/*3 (n = 23), and 12 were expressers carrying *1/*3 (n = 10), and *1/*1(n = 2) variants (Figs F and G in S2 Appendix). Most individuals with a liver transplant were genotyped as nonexpressers (n = 30); however, 6 expressed the *1/*3 variant (Figs H and I in S2 Appendix). Donor samples were available for 17 individuals with a kidney transplant and for 24 with a liver transplant. Most kidney donors were genotyped as nonexpressers (n = 12); however, 5 were expressers with *1/*3 (n = 4) and *1/*1 (n = 1) variants (Figs J and K in S2 Appendix). Most liver individual donors were genotyped as CYP3A5 nonexpressers (n = 14); however, 10 were expressers with *1/*3 (n = 5) and *1/*1 (n = 5) variants (Figs L and M in S2 Appendix).
Individuals with a kidney transplant expressing CYP3A5 required significantly higher tacrolimus doses to achieve target tacrolimus trough blood levels (8.17 ± 2.5 versus 4.26 ± 1.9 mg/day [p = 0.0002]). In individuals with a liver transplant, recipient CYP3A5 genotype had no effect on the tacrolimus doses required to achieve target trough blood levels (4.67 ± 2.2 versus 4.5 ± 1.2 mg/day [p = 0.80]). When assessing the donor variant status, donor CYP3A5 expression had no impact on tacrolimus dose requirements for either individuals with a kidney transplant (6.25 ± 2.1 versus 5.40 ± 4.2 mg/day [p = 0.69]) or those with a liver transplant (4.71 ± 1.9 versus 5.70 ± 2.4 mg/day [p = 0.29]). No differences in within-subject variability were observed by CYP3A5 genotype in individuals with a kidney or liver transplant. No association between AUC and ABCB1 genotype as well as no impact on dosing requirements for individuals with a kidney (ANOVA p = 0.08) or liver transplant (ANOVA p = 0.35) was found (Figs N and O in S2 Appendix for AUCs).
ABE and SCABE limits for AUC were calculated for subgroups to assess for consistency of results (Figs PA–PC in S2 Appendix [kidney] and Figs QA–QC in S2 Appendix [liver]). In general, all FDA ABE limits were met, with most also meeting the stricter SCABE limits; however, most exceeded the EMA upper limit of 111.11%. For subgroups not meeting the ABE or SCABE criteria, the number of observations was 10 or fewer, resulting in low statistical power to conclude ABE or SCABE, except for POR*28. ABE was concluded for both POR*28 carriers and noncarriers for individuals with a kidney or liver transplant. For POR*28 carriers, SCABE was demonstrated except in the case of Generic Lo versus Innovator in individuals with a kidney transplant (n = 17) and Generic Hi versus Generic Lo in individuals with a liver transplant (n = 17). For POR*28 noncarriers, SCABE was demonstrated except for Generic Hi versus Innovator in individuals with a kidney transplant (n = 18) and individuals with a liver transplant (n = 19) and for Generic Lo versus Innovator in individuals with a liver transplant (n = 19). ABE criteria were met for most subgroups, except for individuals with a kidney transplant and a CYP3A4*1B genotype and individuals with a liver transplant and a CYP3A5 *1/*1 or *1/*3 genotype (Figs PA-C in S2 Appendix [kidney], Figs QA–C in S2 Appendix [liver]).
Tacrolimus metabolite exposure
The blood concentrations of the major tacrolimus metabolite, 13-O-desmethyl tacrolimus, were also found to meet FDA ABE and SCABE AUC bioequivalence acceptance criteria in individuals with a kidney or liver transplant, but they failed EMA AUC acceptance limits (Tables T–U in S2 Appendix).
Safety
One serious adverse event of pyelonephritis was reported in an individual with a kidney transplant, resulting in hospitalization prior to study drug administration. This event resolved with treatment, and the individual was withdrawn. Fifty-two percent and 65% of all individuals with a kidney or liver transplant, respectively, reported an adverse event. The adverse events are sorted by formulation and by individuals with a kidney or liver transplant and CTCAE disorder classification. The Exact McNemar test for paired categorical data with adjustments for multiple comparisons was used to compare adverse event rates among the products. No statistically significant differences among any pairs of products were found for any adverse event code or for adverse events overall (Table V in S2 Appendix).
No acute rejections occurred during the study period of 6 weeks. Baseline and weekly assessments included markers of transplant function in individuals with a kidney (creatinine) or liver (AST, ALT, and alkaline phosphatase) transplant. To compare kidney and liver function tests among products, a mixed effects model was run with a term designating the product received in each period and a random subject term and using the Tukey HSD test for multiple comparisons, where needed. No statistically significant differences were found among products (Fig R–W in S2 Appendix).
Discussion
Public concerns remain regarding generic tacrolimus use in individuals with a kidney or liver transplant despite the significant market penetration of generic tacrolimus in the US. Historically, concerns were generated by a lack of definitive clinical evidence with properly controlled trials in target populations [13]. Limitations of previous studies [40–45] include retrospective evaluations, case reports, poor study design (underpowered or without appropriate controls), analysis of trough concentrations only, lack of analysis of confounders such as comedications and comorbidities, incorrect pharmacokinetic analysis, and use of nonspecific immunoassays in which metabolites may interfere with tacrolimus concentration measurements, thus leading to considerable bias and limited conclusions [13].
The present randomized, prospective, 3-treatment, 6-period, crossover, replicate-dosing study in stable individuals with a kidney or liver transplant systematically addresses the aforementioned public concerns regarding generic tacrolimus. The replicate dosing study design allowed the analysis of tacrolimus products using the tighter SCABE standards required by the FDA for NTI drugs. Clinically, the present study represents a scenario in which an individual with a kidney or liver transplant is randomly switched between 3 tacrolimus products every week for 6 weeks. The pharmacokinetic parameters (AUC and Cmax) demonstrated bioequivalence by SCABE criteria, implying similar tacrolimus exposure is achieved when individuals with a kidney or liver transplant are switched between these tacrolimus products. Although not required for bioequivalence testing, Cmin also met the SCABE criteria.
These results support a previous prospective, multicenter, open-label, randomized, 2-period, crossover, pharmacokinetic study comparing twice-daily generic tacrolimus (Sandoz) versus reference tacrolimus (Prograf) in stable kidney transplant recipients [30]. In 68 kidney transplant recipients, there were no significant differences in AUC C0, Cmax, or Tmax between the generic and reference products, resulting in ratios of the geometric mean and 90% CI for AUC and Cmax that were reported as 102% (97%–108%) and 109% (101%–118%), respectively [46]. Post hoc analysis revealed the products also met SCABE acceptance criteria [47]. In contrast to the present study, the latter did not include comparisons to other generic tacrolimus products or the comparison of 2 generics, and liver transplant recipients and relevant genetic polymorphisms were not analyzed.
Pharmacogenomic profiling of individuals was performed, specifically, genotyping of CYP3A5 polymorphisms to identify the “poor absorber” [20,23]. The genetic polymorphisms most important for tacrolimus pharmacokinetics were assessed and, in general, had no effect on bioequivalence. The only exceptions were that bioequivalence of tacrolimus AUC was not found for individuals with a kidney transplant expressing CYP3A4*1B and individuals with a liver transplant expressing CYP3A5, but this analysis was underpowered for said subgroups. Moreover, an influence of POR*28 polymorphism on bioequivalence using SCABE metrics could not be excluded, although ABE criteria were met. Dosing differences by genotype were similar across products [48]. The major tacrolimus metabolite concentrations were bioequivalent for AUC. In contrast to tacrolimus, 13-O-desmethy tacrolimus is not directly administered but formed from tacrolimus, mostly by intestinal and liver cytochrome P4503A enzymes. Thus, its formation is greatly influenced by the aforementioned polymorphisms and its pharmacokinetics more variable than that of tacrolimus.
In this study, the 2 tacrolimus generic products met US FDA SCABE criteria when compared to the innovator product and with each other in individuals after a kidney or liver transplant. However, when applying the more rigid EMA criteria, the Generic Lo product failed AUC testing when compared to Innovator in individuals receiving a kidney or liver transplant. The EMA requires a narrow 90% confidence interval contained within the confidence limits of 90.00%–111.11% acceptance criterion for AUC, but not for Cmax, for which the usual 80.00%–125.00% acceptance limit applies [14]. As shown in Table 4, in general these acceptance criteria were met, with the only exception being that of Innovator versus Generic Lo, for which the upper limits of the 90% confidence intervals—111.30% (kidney) and 112.11% (liver)—were slightly above the upper EMA acceptance criterion for AUC. The conflicting approval guidelines lead to different interpretations of the bioequivalence data of the same study. In this context, it should be considered that the EMA and Health Canada bioequivalence limits for NTI drugs were set with single-dose healthy volunteer studies in mind, a population that is inherently less variable than individuals receiving a transplant after multiple doses [4]. While the FDA SCABE limits adjust based on the pharmacokinetic variability of the innovator in the study population, the EMA and Health Canada bioequivalence limits for NTI drugs are fixed, which explains the different conclusions when FDA, EMA, or Health Canada limits are employed. This study highlights the need for global harmonization of bioequivalence approval standards of NTI drugs to prevent different interpretations of bioequivalence study results.
Individual pharmacokinetic time concentration curves for all products are provided in Section C in S2 Appendix. These data visually depict the intraindividual variability that can be observed within the same product despite administering the same lot at the same dose in a controlled setting. Such observed intraindividual variability led the FDA to require repeat crossover study designs for NTI drugs, which requires that each individual receives each of the tested products twice to assess and to compare intraindividual variability between tacrolimus innovator and generic products. Upon visual inspection of individual pharmacokinetic curves, differences can be observed between products and between replicate administration of the same product.
Safety was similar across products over the observation period of 6 weeks. Although we did not evaluate the long-term impact of generic tacrolimus on acute rejection and graft survival, this study evaluates pharmacokinetic parameters as a surrogate for safety exposure.
Several design elements strengthen the findings, such as validated, specific, and sensitive high-performance liquid chromatography (HPLC)-tandem mass spectrometry analysis of tacrolimus and metabolites, quality control of the tacrolimus test batches, independent parallel analysis by the study team and the FDA, genotypic analysis of individuals donating or receiving a kidney or liver transplant, and close adherence monitoring using a combination of diaries, MEMS caps, and pill counts. Finally, this study was adequately powered to assess bioequivalence in both kidney and liver transplant recipients. However, our study also had certain limitations. A differential carryover or sequence effect cannot be fully excluded, even though we did not detect any statistically significant sequence effects in statistical modeling. When considering the half-life of immediate-release tacrolimus and the length of the treatment period, this effect is unlikely to occur. The pharmacokinetic profiling occurred in strictly controlled conditions with recipients who were highly adherent, which did not allow the evaluation about the impact of nonadherence or the impact of feeding on possible pharmacokinetic differences between products. The study design also did not allow for assessment of the potential impact of differing appearances of the 3 tacrolimus products on adherence. The safety data should be interpreted cautiously in the light of the small number of participants and short observation periods. Lastly, only the 1 mg tacrolimus dosage strength was utilized, limiting generalizability to the 0.5 mg and 5 mg dosage strengths; however, the 1 mg capsule is the most common clinically utilized dosage strength.
The present study was specifically designed to address lingering concerns in the transplant community [8–11]. While typical single-dose healthy volunteer bioequivalence studies mainly assess prescribability, our study in steady-state, stable individuals with a kidney or liver transplant mainly assessed the more important switchability between innovator and generics and between generics [5,6]. The present study suggests that tacrolimus and the tested generic products in healthy volunteers were also bioequivalent in individuals with a kidney or liver transplant. Moreover, the generics were bioequivalent to each other. Even the tighter FDA SCABE criteria were met, and there was no difference between the different tacrolimus products in terms of within-subject variability.
Supporting Information
Zdroje
1. Ensor CR, Trofe-Clark J, Gabardi S, McDevitt-Potter LM, Shullo MA. Generic maintenance immunosuppression in solid organ transplant recipients. Pharmacotherapy. 2011;31(11):1111–29. Epub 2011/10/27. doi: 10.1592/phco.31.11.1111 22026398.
2. O’Neil J. How increased competition from generic drugs has affected prices and returns in the pharmaceutical industry. Congressional Budget Office 1988 [cited 2012 May 29]. Available from: http://www.fda.gov/ohrms/DOCKETS/dailys/04/June04/061404/03p-0029-bkg0001-Ref-15-vol3.pdf
3. Sellares J, de Freitas DG, Mengel M, Reeve J, Einecke G, Sis B, et al. Understanding the causes of kidney transplant failure: the dominant role of antibody-mediated rejection and nonadherence. Am J Transplant. 2012;12(2):388–99. Epub 2011/11/16. doi: 10.1111/j.1600-6143.2011.03840.x 22081892.
4. Davit BM, Nwakama PE, Buehler GJ, Conner DP, Haidar SH, Patel DT, et al. Comparing generic and innovator drugs: a review of 12 years of bioequivalence data from the United States Food and Drug Administration. Ann Pharmacother. 2009;43(10):1583–97. Epub 2009/09/25. doi: 10.1345/aph.1M141 19776300.
5. Christians U, Klawitter J, Clavijo CF. Bioequivalence testing of immunosuppressants: concepts and misconceptions. Kidney Int Suppl. 2010;(115):S1–7. Epub 2010/02/13. doi: 10.1038/ki.2009.504 20150904.
6. Hauck WW, Anderson S. Measuring switchability and prescribability: when is average bioequivalence sufficient? J Pharmacokinet Biopharm. 1994;22(6):551–64. Epub 1994/12/01. 7473081.
7. Pollard S, Nashan B, Johnston A, Hoyer P, Belitsky P, Keown P, et al. A pharmacokinetic and clinical review of the potential clinical impact of using different formulations of cyclosporin A. Berlin, Germany, November 19, 2001. Clin Ther. 2003;25(6):1654–69. Epub 2003/07/16. 12860490.
8. Alloway RR, Isaacs R, Lake K, Hoyer P, First R, Helderman H, et al. Report of the American Society of Transplantation conference on immunosuppressive drugs and the use of generic immunosuppressants. Am J Transplant. 2003;3(10):1211–5. Epub 2003/09/27. 14510694.
9. Harrison JJ, Schiff JR, Coursol CJ, Daley CJ, Dipchand AI, Heywood NM, et al. Generic immunosuppression in solid organ transplantation: a Canadian perspective. Transplantation. 2012;93(7):657–65. Epub 2012/01/24. doi: 10.1097/TP.0b013e3182445e9d 22267158.
10. van Gelder T. European Society for Organ Transplantation Advisory Committee recommendations on generic substitution of immunosuppressive drugs. Transpl Int. 2011;24(12):1135–41. Epub 2011/10/29. 10.1111/j.1432-2277.2011.01378.x. 22032583. doi: 10.1111/j.1432-2277.2011.01378.x 22032583
11. Uber PA, Ross HJ, Zuckermann AO, Sweet SC, Corris PA, McNeil K, et al. Generic drug immunosuppression in thoracic transplantation: an ISHLT educational advisory. J Heart Lung Transplant. 2009;28(7):655–60. Epub 2009/06/30. 10.1016/j.healun.2009.05.001. 19560691. doi: 10.1016/j.healun.2009.05.001 19560691
12. Klintmalm GB. Immunosuppression, generic drugs and the FDA. Am J Transplant. 2011;11(9):1765–6. Epub 2011/07/29. doi: 10.1111/j.1600-6143.2011.03616.x 21794082.
13. Molnar AO, Fergusson D, Tsampalieros AK, Bennett A, Fergusson N, Ramsay T, et al. Generic immunosuppression in solid organ transplantation: systematic review and meta-analysis. BMJ. 2015;350:h3163. Epub 2015/06/24. doi: 10.1136/bmj.h3163 26101226; PubMed Central PMCID: PMCPMC4476317.
14. European Medicines Agency. European Medicines Agency. Guideline on the Investigation of Bioequivalence London, UK2010 [cited 2016 October 26]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf
15. Health Canada. Guidance Document—Comparative Bioavailability Standards: Formulations Used for Systemic Effects Ottawa, Ontario: Health Canada; 2012 [cited 2016 October 26]. Available from: http://www.hc-sc.gc.ca/dhp-mps/alt_formats/pdf/prodpharma/applic-demande/guide-ld/bio/gd_standards_ld_normes-eng.pdf
16. Food and Drug Administration. Draft Guidance on Tacrolimus 2014 [cited 2015 December 2]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM406344.pdf
17. Yu LX, Jiang W, Zhang X, Lionberger R, Makhlouf F, Schuirmann DJ, et al. Novel bioequivalence approach for narrow therapeutic index drugs. Clin Pharmacol Ther. 2015;97(3):286–91. Epub 2015/02/12. doi: 10.1002/cpt.28 25669762.
18. Birdwell KA, Decker B, Barbarino JM, Peterson JF, Stein CM, Sadee W, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines for CYP3A5 Genotype and Tacrolimus Dosing. Clin Pharmacol Ther. 2015;98(1):19–24. Epub 2015/03/25. doi: 10.1002/cpt.113 25801146; PubMed Central PMCID: PMCPMC4481158.
19. Picard N, Bergan S, Marquet P, van Gelder T, Wallemacq P, Hesselink DA, et al. Pharmacogenetic Biomarkers Predictive of the Pharmacokinetics and Pharmacodynamics of Immunosuppressive Drugs. Ther Drug Monit. 2016;38 Suppl 1:S57–69. Epub 2015/10/16. doi: 10.1097/ftd.0000000000000255 26469711.
20. Kuypers DR, de Jonge H, Naesens M, Lerut E, Verbeke K, Vanrenterghem Y. CYP3A5 and CYP3A4 but not MDR1 single-nucleotide polymorphisms determine long-term tacrolimus disposition and drug-related nephrotoxicity in renal recipients. Clin Pharmacol Ther. 2007;82(6):711–25. Epub 2007/05/15. doi: 10.1038/sj.clpt.6100216 17495880.
21. Pallet N, Jannot AS, El Bahri M, Etienne I, Buchler M, de Ligny BH, et al. Kidney transplant recipients carrying the CYP3A4*22 allelic variant have reduced tacrolimus clearance and often reach supratherapeutic tacrolimus concentrations. Am J Transplant. 2015;15(3):800–5. Epub 2015/01/16. doi: 10.1111/ajt.13059 25588704.
22. Elens L, Hesselink DA, Bouamar R, Budde K, de Fijter JW, De Meyer M, et al. Impact of POR*28 on the pharmacokinetics of tacrolimus and cyclosporine A in renal transplant patients. Ther Drug Monit. 2014;36(1):71–9. Epub 2013/09/26. doi: 10.1097/FTD.0b013e31829da6dd 24061445.
23. Stefanovic NZ, Cvetkovic TP, Jevtovic-Stoimenov TM, Ignjatovic AM, Paunovic GJ, Velickovic RM. Investigation of CYP 3A5 and ABCB1 gene polymorphisms in the long-term following renal transplantation: Effects on tacrolimus exposure and kidney function. Exp Ther Med. 2015;10(3):1149–56. Epub 2015/12/02. doi: 10.3892/etm.2015.2598 26622455; PubMed Central PMCID: PMCPMC4533232.
24. Christians U, Jacobsen W, Benet LZ, Lampen A. Mechanisms of clinically relevant drug interactions associated with tacrolimus. Clin Pharmacokinet. 2002;41(11):813–51. Epub 2002/08/23. doi: 10.2165/00003088-200241110-00003 12190331.
25. Food and Drug Administration. Orange Book. Approved Drug Products with Therapeutic Equivalence Evaluations 2015 [cited 2015 December 1]. Available from: http://www.accessdata.fda.gov/scripts/cder/ob/docs/queryai.cfm
26. Sandoz Tacrolimus Abbreviated New Drug Application Number A065461 [cited 2015 December 1]. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=065461
27. Dr Reddy Tacrolimus Abbreviated New Drug Application Number A090509 [cited 2015 December 1]. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=090509
28. Mylan Tacrolimus Abbreviated New Drug Application Number A090596 [cited 2015 December 1]. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=090596
29. Accord Tacrolimus Abbreviated New Drug Application Number A091195 [cited 2015 December 1]. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=091195
30. Panacea Tacrolimus Abbreviated New Drug Application Number A09082 [cited 2015 December 1]. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=090802
31. Convention United SP. Tacrolimus Revision Bulletin, April 1, 2013 Rockville, MD2013 [cited 2015 December 1]. Available from: http://www.usp.org/sites/default/files/usp_pdf/EN/USPNF/revisions/tacrolimus_capsulesm.pdf
32. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)—a metadata-driven methodology and workflow process for providing translational research informatics support. Journal of biomedical informatics. 2009;42(2):377–81. Epub 2008/10/22. doi: 10.1016/j.jbi.2008.08.010 18929686; PubMed Central PMCID: PMCPMC2700030.
33. Diletti E, Hauschke D, Steinijans VW. Sample size determination for bioequivalence assessment by means of confidence intervals. Int J Clin Pharmacol Ther Toxicol. 1992;30 Suppl 1:S51–8. Epub 1992/01/01. 1601532.
34. Administration FAD. Food And Drug Administration. Guidance for Industry—Statistical Approaches to Establishing Bioequivalence Rockville, MD2001 [cited 2012 June 7]. Available from: https://www.fda.gov/downloads/drugs/guidances/ucm070244.pdf
35. Montgomery DC. Design and Analysis of Experiments. 1st ed. Wiley. 2013.
36. National Cancer Instiute. Common Toxicty Criteria for Adverse Events, v 4.0 2010 [cited 2015 December 1]. Available from: http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcae_4_with_lay_terms.pdf
37. Agresti A. Categorical Data Analysis (PDF). Hooken, New Jersey: John Wiley & Sons, Inc. 2002. ISBN 0-471-36093-7
38. Daniel WW. Kruskal–Wallis one-way analysis of variance by ranks. Applied Nonparametric Statistics. 2nd ed. Boston: PWS-Kent. pp. 226–234. 1990. ISBN 0-534-91976-6
39. Sauter R, Steinijans VW, Diletti E, Bohm A, Schulz HU. Presentation of results from bioequivalence studies. Int J Clin Pharmacol Ther Toxicol. 1992;30(7):233–56. Epub 1992/07/01. 1506127.
40. Duong SQ, Lal AK, Joshi R, Feingold B, Venkataramanan R. Transition from brand to generic tacrolimus is associated with a decrease in trough blood concentration in pediatric heart transplant recipients. Pediatr Transplant. 2015;19(8):911–7. Epub 2015/10/27. doi: 10.1111/petr.12608 26497983.
41. Robertsen I, Asberg A, Ingero AO, Vethe NT, Bremer S, Bergan S, et al. Use of generic tacrolimus in elderly renal transplant recipients: precaution is needed. Transplantation. 2015;99(3):528–32. Epub 2014/08/26. doi: 10.1097/TP.0000000000000384 25148382.
42. Min SI, Ha J, Kim YS, Ahn SH, Park T, Park DD, et al. Therapeutic equivalence and pharmacokinetics of generic tacrolimus formulation in de novo kidney transplant patients. Nephrol Dial Transplant. 2013;28(12):3110–9. Epub 2013/10/03. doi: 10.1093/ndt/gft300 24084327.
43. Spence MM, Nguyen LM, Hui RL, Chan J. Evaluation of clinical and safety outcomes associated with conversion from brand-name to generic tacrolimus in transplant recipients enrolled in an integrated health care system. Pharmacotherapy. 2012;32(11):981–7. Epub 2012/10/18. doi: 10.1002/phar.1130 23074134.
44. Momper JD, Ridenour TA, Schonder KS, Shapiro R, Humar A, Venkataramanan R. The impact of conversion from prograf to generic tacrolimus in liver and kidney transplant recipients with stable graft function. Am J Transplant. 2011;11(9):1861–7. Epub 2011/07/01. doi: 10.1111/j.1600-6143.2011.03615.x 21714845.
45. McDevitt-Potter LM, Sadaka B, Tichy EM, Rogers CC, Gabardi S. A multicenter experience with generic tacrolimus conversion. Transplantation. 2011;92(6):653–7. Epub 2011/07/27. doi: 10.1097/TP.0b013e31822a79ad 21788920.
46. Alloway RR, Sadaka B, Trofe-Clark J, Wiland A, Bloom RD. A randomized pharmacokinetic study of generic tacrolimus versus reference tacrolimus in kidney transplant recipients. Am J Transplant. 2012;12(10):2825–31. Epub 2012/07/05. doi: 10.1111/j.1600-6143.2012.04174.x 22759200; PubMed Central PMCID: PMCPMC3472020.
47. Bloom RD, Trofe-Clark J, Wiland A, Alloway RR. A randomized, crossover pharmacokinetic study comparing generic tacrolimus vs. the reference formulation in subpopulations of kidney transplant patients. Clin Transplant. 2013;27(6):E685–93. Epub 2013/10/15. doi: 10.1111/ctr.12256 24118450.
48. Macphee IA, Fredericks S, Tai T, Syrris P, Carter ND, Johnston A, et al. Tacrolimus pharmacogenetics: polymorphisms associated with expression of cytochrome p4503A5 and P-glycoprotein correlate with dose requirement. Transplantation. 2002;74(11):1486–9. Epub 2002/12/20. doi: 10.1097/01.TP.0000045761.71385.9F 12490779.
Štítky
Interní lékařstvíČlánek vyšel v časopise
PLOS Medicine
2017 Číslo 11
- Ivabradin zlepšuje kvalitu života starších pacientů se srdečním selháním
- Fixní kombinace kandesartan/amlodipin v terapii arteriální hypertenze
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
- Rána vizitkou (nejen) chirurga
- Patogeneze vzniku keloidní jizvy
Nejčtenější v tomto čísle
- Postmenopausal hormone therapy and risk of stroke: A pooled analysis of data from population-based cohort studies
- Bioequivalence between innovator and generic tacrolimus in liver and kidney transplant recipients: A randomized, crossover clinical trial
- HIV pre-exposure prophylaxis and early antiretroviral treatment among female sex workers in South Africa: Results from a prospective observational demonstration project
- Bioequivalence of twice-daily oral tacrolimus in transplant recipients: More evidence for consensus?
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy