
Critical Function of γH2A in S-Phase
ATM (ataxia telangiectasia mutated) and ATR (ATM and Rad3 related) are evolutionary conserved protein kinases that phosphorylate the carboxyl-tail of histone H2AX in chromatin flanking DNA lesions. Phosphorylated histone H2AX (aka γH2AX) tethers important DNA damage response (DDR) proteins to DNA double-strand breaks but its function during DNA replication is unclear. A novel genetic screen reveals that a partial defect in Replication Factor C (RFC) creates a critical requirement for γH2AX in fission yeast. These studies indicate that γH2AX stabilizes replication forks by recruiting Brc1 when RFC is unable to load the DNA clamp known as proliferating cell nuclear antigen (PCNA) onto duplex DNA. Surprisingly, this activity of γH2AX is more critical than ATM/ATR-mediated activation of the checkpoint kinase Chk1 and Chk2.
Published in the journal:
. PLoS Genet 11(9): e32767. doi:10.1371/journal.pgen.1005517
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005517
Summary
ATM (ataxia telangiectasia mutated) and ATR (ATM and Rad3 related) are evolutionary conserved protein kinases that phosphorylate the carboxyl-tail of histone H2AX in chromatin flanking DNA lesions. Phosphorylated histone H2AX (aka γH2AX) tethers important DNA damage response (DDR) proteins to DNA double-strand breaks but its function during DNA replication is unclear. A novel genetic screen reveals that a partial defect in Replication Factor C (RFC) creates a critical requirement for γH2AX in fission yeast. These studies indicate that γH2AX stabilizes replication forks by recruiting Brc1 when RFC is unable to load the DNA clamp known as proliferating cell nuclear antigen (PCNA) onto duplex DNA. Surprisingly, this activity of γH2AX is more critical than ATM/ATR-mediated activation of the checkpoint kinase Chk1 and Chk2.
Introduction
DNA lesions elicit highly orchestrated DNA damage responses (DDRs) controlled by the master checkpoint kinases ATM and ATR. These responses protect genome integrity and prevent diseases characterized by chromosome instability and cancer [1,2]. ATM and ATR have many substrates but none is more ubiquitous than the SQ motif at the carboxyl tail of histone H2AX or H2A [3]. Key DDR proteins such as mammalian MDC1 have C-terminal regions consisting of tandem BRCA1 C-terminus (BRCT) domains that form a highly sculpted binding pocket for the phosphorylated C-terminus of phospho-H2AX (γH2AX) [4]. These DDR proteins decorate large chromatin domains flanking DNA lesions. However, H2AX phospho-site mutations generally cause modest genotoxin sensitivity compared to eliminating γH2AX-binding proteins, suggesting that docking to γH2AX enhances but is not always essential for DDR protein functions [5–7]. Endogenous sources of DNA damage might create a more acute requirement for γH2AX to protect genome integrity.
Whilst γH2AX has been most intensively studied in the context of DNA double-strand breaks (DSBs) formed by exogenous clastogens, recent studies with fission yeast and budding yeast established that γH2AX (aka γH2A in yeast) increases every DNA synthesis (S)-phase [8,9]. Single-stranded DNA (ssDNA) at stalled or damaged replication forks appears to be the triggering DNA structure. Here, we investigate the function of γH2AX by using a genetic screen to identify DNA replication mutants whose viability critically depends on γH2A in Schizosaccharomyces pombe. These studies reveal that a defect in Replication Factor C (RFC), which loads the replicative DNA polymerase processivity factor known as proliferating cell nuclear antigen (PCNA) onto duplex DNA, creates an acute requirement for γH2A. Our studies track this requirement to Brc1, a γH2A-binding protein that functions in the replication stress response [10,11]. From our studies we propose that large-scale adornment of γH2A-marked chromatin with Brc1 prevents replication fork collapse when PCNA loading or DNA polymerase activity limit DNA synthesis.
Results
Mutation of Rfc3 creates a critical requirement for γH2A
We have constructed S. pombe “htaAQ” strains in which both histone H2A genes have been mutated to alter the C-terminal SQ phosphorylation site to AQ (hta1-S129A hta2-S128), thereby eliminating γH2A [7]. We sought to identify mutations having synthetic sick or lethal (SSL) genetic interactions with htaAQ. We used tetrad analysis to introduce htaAQ into strains having conditional mutations in genes that are essential for DNA replication. We initially chose mutations of genes encoding subunits of the pre-initiation complex (pre-IC; sld3-10 and cdc45-192), pre-replication complex (pre-RC; cdc18-K9), MCM replicative DNA helicase (mcm2-P1 and mcm6-568), Dpb11 replication and checkpoint scaffold protein (cut5-T401), replication factor C subunit 3 (rfc3-1), and an Schizosaccharomyces-specific gene whose product associates with Dna2 flap endonuclease/helicase that is required for Okazaki fragment processing (cdc24-M28). For all but one of these mutations the SSL interactions were undetectable or weak when tested in the absence of exogenous DNA damaging agents or replication inhibitors. The most obvious exception was rfc3-1 [12], which had a clear SSL interaction with htaAQ at the permissive temperature of 25°C (Fig 1A). γH2A is therefore critical when Rfc3 function is impaired.

The requirement for γH2A is specific for defects in RFC
Rfc3 is as an essential subunit of RFC, which is a heteropentameric AAA+ protein clamp loader for PCNA [13]. The ring-like PCNA homotrimer encircles DNA and slides spontaneously along the duplex as an essential subunit of the replisome [14]. RFC consists of the large subunit Rfc1 along with four smaller subunits: Rfc2, 3, 4 and 5. The smaller subunits are also present in alternative RFC-like complexes in which Rfc1 is replaced by Rad17, Ctf18 or Elg1 [15]. The Rad17-RFC complex has a well-characterized role in loading the Rad9-Hus1-Rad1 PCNA-like checkpoint clamp at DNA lesions and stalled replication forks, where it is essential for DNA damage and replication checkpoints enforced by Chk1 and Cds1/Chk2, respectively [16,17]. Ctf18 and Elg1 also play important but less well understood roles in maintaining genome integrity in response to replication-associated DNA damage [15,18].
As the rfc3-1 mutation potentially impairs the functions of the canonical and alternative RFCs, we tested whether htaAQ has genetic interactions with rad17∆, ctf18∆ or elg1∆. No obvious SSL interactions were detected (Fig 1B). To further test whether a defect in the canonical RFC creates a requirement for γH2A, we crossed htaAQ with the temperature sensitive rfc1-44 mutation [15]. We detected a SSL interaction at 25°C that was enhanced at 32°C (Fig 1C). From these data we conclude that γH2A is crucial when the canonical RFC is impaired but not when the alternative RFC complexes are each individually ablated.
Increased γH2A in rfc3-1 cells
Our data suggested that replication defects in rfc3-1 cells trigger a DNA damage response leading to formation of γH2A that is critical for maintaining viability. To test this idea we measured γH2A with anti-γH2A antisera [19] and found that it was increased in rfc3-1 cells (Fig 2), matching the levels seen in wild type cells treated with the topoisomerase I poison camptothecin (CPT) that collapses replication forks [20].

Brc1 binding to γH2A is crucial in rfc3-1 cells
Crb2, Brc1 and Mdb1 bind γH2A in fission yeast [7,10,21,22]. Crb2 and Brc1 are most critical for surviving genotoxins [11,23,24], therefore we investigated the requirements for Crb2 and Brc1 in rfc3-1 cells.
The tandem C-terminal BRCT domains of Crb2 that bind γH2A adjoin paired Tudor domains that bind dimethylated lysine-20 of histone H4 (H4-K20me2). Mutations that ablate these interactions are genetically epistatic and both interactions are required for large-scale localization of Crb2 at DSBs [25–27]. We found the elimination of the sole H4-K20 methyltransferase Set9 had no effect in rfc3-1 cells (Fig 3A). Similarly, we found that rfc3-1 cells were unaffected by the crb2-K619M mutation [26] that disrupts the γH2A-binding pocket (Fig 3B). As Crb2 retains partial function when γH2A and H4-K20me2 are simultaneously eliminated [26], we also tested the crb2∆ mutation and found that it only weakly impaired growth in rfc3-1 cells (Fig 3C). We conclude that Crb2 binding to γH2A and H4-K20me2 is not required in rfc3-1 cells, while complete loss of Crb2 has a minor effect.

We next examined Brc1 and found that brc1∆ rfc3-1 cells grew poorly compared to either single mutant (Fig 3D). We tested the brc1-T672A mutation that disrupts the γH2A binding pocket in Brc1 [10] and found a strong negative genetic interaction with rfc3-1 (Fig 3E). These results established the importance of Brc1 binding to γH2A in rfc3-1 cells.
Increased Brc1 foci in rfc3-1 cells
Our findings suggested that rfc3-1 cells experience replication difficulties that trigger formation of γH2A and recruitment of Brc1 that is critical for survival. To further test this model we monitored formation of green fluorescent protein (GFP)-Brc1 foci, which increases in response to replication stress [10]. As predicted we detected a significant increase in GFP-Brc1 foci in rfc3-1 cells incubated at 25°C (Fig 3F).
Hus1-independent activity of Rad3/ATR is crucial in rfc3-1 cells
Tel1/ATM and Rad3/ATR kinases create γH2A [7]. Eliminating Tel1 had no effect in rfc3-1 cells (Fig 3G), which is consistent with Tel1 acting specifically at DSBs and telomeres as opposed replication forks [28,29]. In contrast, we detected a strong requirement for Rad3 in rfc3-1 cells (Fig 3H), which supports evidence that Rad3 is critical for surviving replication stress [30].
Rad3 forms γH2A at stalled replication forks [8]. The dispensability of Rad17 in rfc3-1 cells suggested that Rad17-dependent loading of the Rad9-Hus1-Rad1 checkpoint clamp was not required for phosphorylation of H2A by Rad3 at stalled forks. This result was surprising because the Rad3 activator Cut5/Rad4 (TopBP1/Dpb11 ortholog) binds Rad9-Hus1-Rad1 [16,31]. We therefore investigated whether Rad9-Hus1-Rad1 regulates γH2A formation by Rad3 in S-phase. First, we used a synchronous culture to establish that γH2A in cycling cells occurs predominantly during S-phase (Fig 4A), confirming previous analyses performed by chromatin immunoprecipitation [8]. The large reduction of γH2A in untreated (-IR) rad3∆ cells confirmed that Rad3 is principally responsible for forming γH2A during S-phase (Fig 4B). In contrast, the basal level of γH2A was maintained in hus1∆ cells, showing that Rad3 activity towards histone H2A in S-phase does not require the checkpoint clamp (Fig 4B). Interestingly, eliminating Tel1 nearly abolished the IR-induced increase of γH2A in hus1∆ cells, indicating that Rad3 activity towards histone H2A does require Hus1 at DSBs.

We also examined the genetic requirements for γH2A formation in rfc3-1 cells grown at 25°C. In these assays the increase of γH2A in untreated rfc3-1 required Rad3 but not Hus1 (Fig 4C), which is consistent with Rad3 but not Rad17 being required in rfc3-1 cells (Figs 1B and 3H) Interestingly, IR induction of γH2A was largely abrogated in rfc3-1 tel1∆ cells, indicating that phosphorylation of histone H2A by Rad3 at DSBs is decreased by rfc3-1 at 25°C, presumably because of impaired loading of the Rad9-Hus1-Rad1 checkpoint clamp by Rad17-RFC. Indeed, Rad3-dependent phosphorylation of Chk1 was severely impaired in rfc3-1 cells irradiated at 25°C (Fig 4D), mirroring previous studies performed at 28°C [12].
To summarize, the crucial phosphorylation of histone H2A by Rad3 during S-phase in rfc3-1 cells does not require the Rad9-Hus1-Rad1 checkpoint clamp, which explains why neither Rad17 nor Rfc3 are required for Rad3 activity towards histone H2A in rfc3-1 cells.
Neither Cds1 nor Chk1 are required in rfc3-1 cells
Rad3 activates the checkpoint kinases Cds1/Chk2 and Chk1 by a mechanism that requires loading Rad9-Hus1-Rad1 checkpoint clamp onto DNA by Rad17-RFC [32]. Chk1 activation by Rad3 also requires Crb2. As Cds1 and Chk1 are amongst the most important and highly conserved Rad3 substrates it was surprising that neither Rad17 nor Crb2 are required in rfc3-1 cells. We confirmed that neither Cds1 nor Chk1 are required in rfc3-1 cells at 25°C (Fig 5A and 5B). The absence of a genetic interaction with cds1∆ is especially notable because Cds1 is crucial for survival of hydroxyurea (HU) treatment, which stalls replication forks by inhibiting ribonucleotide reductase. Indeed, our spot dilution assays showed that cds1∆ causes much greater HU sensitivity than htaAQ or brc1∆ (Fig 5A). These data establish that very different DNA damage responses are required for survival of RFC defects and dNTP starvation, with the former requiring γH2A and the latter Cds1/Chk2 activation.
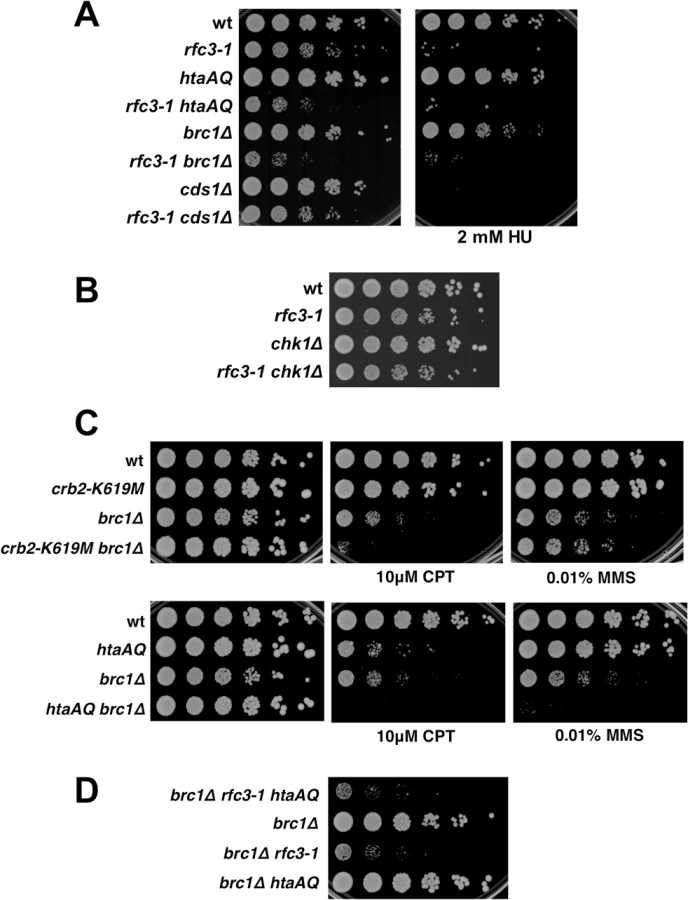
Brc1 does not have an important checkpoint dampening function
The Brc1 structural homolog Rtt107 in S. cerevisiae competes with the Crb2 homolog Rad9 for binding γH2A to prevent hyper-activation of the checkpoint kinase Rad53 [33,34]. An equivalent activity might explain why Brc1 binding to γH2A is critical in rfc3-1 cells. To test whether Brc1 has an important checkpoint dampening function we explored the effects of preventing Crb2 binding to γH2A in brc1∆ cells. We found that the crb2-K619M mutation, which prevents Crb2 binding to γH2A [26], did not suppress the CPT or methyl methanesulfonate (MMS) sensitivity of brc1∆ cells (Fig 5C). Indeed, crb2-K619M increased CPT sensitivity in the brc1∆ background. Similarly, the htaAQ genotype increased both CPT and MMS sensitivity in brc1∆ cells (Fig 5C). These data suggest that Brc1 is unlikely to have an important checkpoint dampening function in cells experiencing replication stress.
To investigate a potential anti-checkpoint activity of Brc1 in rfc3-1 cells we constructed a brc1∆ rfc3-1 htaAQ strain. If Brc1 binding to γH2A is needed to dampen Crb2-dependent checkpoint signaling we would expect htaAQ to suppress the SSL interactions between brc1∆ and rfc3-1. We observed no suppression; in fact, colony size appeared to be slightly smaller in brc1∆ rfc3-1 htaAQ cells compared to brc1∆ rfc3-1 (Fig 5D).
Taken together these data indicate that Brc1 does not have an important checkpoint dampening function that could explain why brc1∆ cells are sensitive to replication stress.
Homologous recombination repair of collapsed replication forks is essential in rfc3-1 cells
Our data suggested that rfc3-1 causes defects in DNA replication that may lead to the collapse of replication forks that are subsequently reestablished by homology directed repair (HDR) of the broken forks [35,36]. To investigate this possibility we first examined the Mre11-Rad50-Nbs1 (MRN) protein complex, which directly binds DSBs where it associates with Ctp1 to initiate 5’-to-3’ DNA end resection required for HDR [37,38]. Tetrad analysis revealed that rfc3-1 mre11∆ cells are inviable at 25°C (Fig 6A).

Whereas MRN is required for HDR of all DSBs, Mus81-Eme1 endonuclease is specifically required to resolve Holliday Junctions created during HDR of one-ended DSBs formed by replication fork breakage [35,39]. We found that Mus81 is essential in rfc3-1 cells germinated at 25°C, supporting the conclusion that the RFC defect in these cells leads to replication fork collapse (Fig 6B).
Brc1 binding to γH2A suppresses catastrophic formation of ssDNA
Replication fork collapse is typically associated with nuclear foci formed by Rad52 HDR protein [40]. As predicted by our results, we detected a large increase in Rad52-yellow fluorescent protein (YFP) foci in rfc3-1 cells grown at 25°C (Fig 7A). The rfc3-1 strain further differed in having a significant percentage of cells with an unusually large and bright Rad52 focus that is likely clusters of Rad52 foci. However, eliminating γH2A did not substantially alter the Rad52 foci pattern of rfc3-1 cells (Fig 7A).

We also monitored Ssb1 (aka Rad11), which is the largest subunit of Replication Protein A (RPA), the 3-subunit ssDNA-binding protein complex essential for DNA replication and most DNA repair mechanisms. RPA-green fluorescent protein (GFP) foci in rfc3-1 cells appeared similar to wild type, indicating that in this situation Rad52 foci are better indicator of replication fork collapse. However, there was a large increase of RPA foci in rfc3-1 htaAQ cells (Fig 7B). Moreover, ~15% of the rfc3-1 htaAQ cells contained a very bright focus or cluster of RPA foci, which was rarely observed in wild type, htaAQ or rfc3-1 cells. These results suggest Brc1 binding to γH2A suppresses catastrophic formation of ssDNA at replication forks in rfc3-1 cells.
γH2A is critical in a DNA polymerase epsilon mutant
RFC loads the PCNA clamp onto DNA, which facilitates the processivity of leading strand DNA replication through its interactions with DNA polymerase epsilon (Pol ε). We tested for genetic interactions between htaAQ and the cdc20-M10 temperature sensitive mutation of Pol ε [41]. At the intermediate permissive temperature of 33.5°C we detected an acute requirement for γH2A in cdc20-M10 cells (Fig 8A), mirroring the negative genetic interactions between htaAQ and rfc3-1 or rfc1-44 (Fig 1). These data indicate that a defect in tethering the leading strand DNA polymerase Pol ε at replication forks at least partially explains the requirement for γH2A in rfc3-1 cells. Other deficiencies in rfc3-1 cells, such as reduced tethering of additional DNA polymerases, might also be involved in creating the requirement for γH2A.

Discussion
Phosphorylation of histone H2AX/H2A by ATM and ATR orthologs has long been known as a ubiquitous response to DSBs and was more recently uncovered as a response to replication stress, yet its physiological significance has remained unclear. The ATM/ATR-regulated checkpoint effector kinases Cds1/Chk2 and Chk1 are generally more important in clastogen and genotoxin sensitivity assays, as is Rad17 that is required for Cds1/Chk2 and Chk1 activation. In the same assays the γH2A-binding proteins Crb2 and Brc1 appear to have more crucial functions than γH2A itself [10,26]. The key discovery to emerge from these studies is that γH2A, and specifically Brc1 binding to γH2A, is critical when RFC is defective. By contrast, neither Cds1 nor Chk1 are required in this situation. Similarly, ablating the Rad17-dependent Rad9-Hus1-Rad1 clamp loader causes acute genotoxin sensitivity but has little effect in rfc3-1 cells. This complete reversal of DDR mutant sensitivities when comparing rfc3-1 to exogenous DNA damaging agents and replication inhibitors is striking.
The picture that emerges from these studies is that most genotoxins fail to replicate the effects of impairing RFC, which presumably reduces PCNA loading and DNA polymerase tethering at the replication fork. This idea is supported by the requirement for γH2A when Pol ε is partially impaired, although the genetic interactions involving rfc3-1 and cdc20-M10 are not precisely identical [41]. If DNA polymerase activity is a rate-limiting step for DNA replication under some physiological conditions our studies suggest that Brc1 binding to γH2A is critical for protecting genome integrity in these situations. This model is consistent with increased Rad52 foci in htaAQ and brc1∆ cells [10,11].
Our findings call to mind a study in which reduced levels of DNA polymerase alpha were found to trigger chromosome translocations in budding yeast [42]. These translocations involved HDR events between long terminal repeats (LTRs) of Ty retrotransposons elements, which were proposed to be chromosome fragile sites. Another study found that LTRs are specifically enriched for γH2A during S-phase [9], as was also observed in fission yeast [8]. Our studies suggest that besides marking DSBs arising at chromosome fragile sites, γH2A also serves to stabilize replisomes at these sites and thereby prevent chromosome breakage, perhaps by binding the Brc1 structural homolog known as Rtt107 in budding yeast [43,44].
The unusual RPA foci in rfc3-1 htaAQ cells suggest to us either catastrophic DNA unwinding or massive resection of DSBs. Both events may occur but we favor the idea that DNA unwinding uncoupled from DNA synthesis most likely explains the critical requirement for γH2A in RFC and Pol ε defective cells (Fig 8B). We favor this model because elimination of Exo1 exonuclease, which is primarily responsible for long-range resection in fission yeast [37], does not suppress the poor growth of rfc3-1 htaAQ cells (Fig 8C). To the contrary, the exo1∆ mutation further impairs the growth of rfc3-1 htaAQ cells. This effect can be explained by a requirement for Exo1 in rfc3-1 cells regardless of whether these cells are able to form γH2A (Fig 8C). This genetic interaction might indicate that Exo1 contributes to HDR of broken replication forks in rfc3-1 cells, although we note that exo1∆ cells are largely insensitive to IR, CPT and MMS, all of which cause DNA damage that is repaired via HDR [45,46].
The massive accumulation of RPA foci in rfc3-1 htaAQ cells calls to mind a recent study with mammalian cells in which ATR was found to prevent global exhaustion of RPA to prevent replication catastrophe [47]. In this study ATR was proposed to prevent RPA exhaustion by restraining origin firing. Our studies suggest that stabilization of stalled replication forks may also play a role in this process, perhaps involving formation of γH2AX. The requirement for Mus81 in rfc3-1 cells suggests that Brc1 binding to γH2A does not completely prevent replication fork collapse. Brc1 binding to γH2A likely facilitates repair of broken replication forks, thereby compounding the requirement for γH2A in rfc3-1 cells. This proposal comports with the evidence that htaAQ mutants are sensitive to camptothecin [7,19]. We also note that post-translational modifications of PCNA promote post-replication repair (PRR) of DNA lesions [48,49]. Brc1 was proposed to function in conjunction with PRR proteins, including components of the HDR machinery, as well as with multiple structure-specific nucleases [11,50,51]. Thus a defect in PRR might also contribute to the requirement for γH2A in rfc3-1 cells, although it remains to be established whether Brc1 binding to γH2A promotes PRR.
If defective loading of PCNA leads to DNA polymerase uncoupling from MCM DNA helicase in rfc3-1 htaAQ cells it might be possible to suppress this defect by using a temperature sensitive mutation to partially impair MCM activity. We attempted this experiment with the mcm2-P1 (aka cdc19-P1) allele. Although we found that eliminating γH2A had no effect on the growth of mcm2-P1 cells at 25°C, combining htaAQ mcm2-P1 with rfc3-1 resulted in synthetic lethality. In an independent experiment we confirmed that mcm2-P1 rfc3-1 cells were inviable at 25°C (Fig 8D).
ATM and ATR use γH2AX to effect chromatin-specific responses to DNA damage. Multi-kilobase γH2AX domains have been detected in yeast and megabase domains in mammals [3]. Why are these responses so highly conserved? In fission yeast, coating chromatin with Crb2 likely serves to rapidly amplify and reliably maintain Chk1 activity during DSB repair [26]. These properties may be most critical when cells suffer a single DSB, which is the most common situation for endogenous sources of DSBs. The purpose of γH2A at stalled or damaged replication forks is probably quite different. From the insights provided by the current study we propose that mounting large-scale changes in chromatin by decorating it with Brc1 is well suited for coordinating the activities of the replicative DNA helicase with leading and lagging stand DNA polymerases (Fig 8B).
It remains unclear whether Brc1 activities are conserved with its structural homologs Rtt107 in budding yeast or PTIP in mammals [52,53]. Rtt107 binding to γH2A was most recently shown to be important for assembling Slx4 signaling protein complexes behind damaged replication forks [54]. However, this signaling activity does appear to be conserved in fission yeast, in which Slx4 function appears to solely involve forming an active structure-specific endonuclease with Slx1 [55,56]. Furthermore, the checkpoint dampening activity of Rtt107 [33,34] does not appear to be conserved in fission yeast or at least is not detectably important in our genetic assays (Fig 5). The function of PTIP, which shares the 6 BRCT domains arrangement with Brc1 and Rtt107, the C-terminal pair of which bind γH2AX [57], is also a matter of substantial interest. Functional relationships of Brc1 or Rtt107 to PTIP are currently unobvious. Recent studies suggest that PTIP functions with 53BP1 in inhibiting HDR [58–60]. Perhaps Brc1, Rtt107 and PTIP all modulate DNA end resection with varying effects, although we note the genetic interactions involving Exo1 do not support this idea for Brc1 (Fig 8C). Functional similarities may emerge with new functional insights into this class of genome protection proteins.
Materials and Methods
General methods
Standard genetic procedures and media for S. pombe were used as described [61]. Strains expressing GFP-Brc1 were constructed by inserting MluI digested pREP41-GFP-Brc1 [10] into the ars1 locus. For spot dilution assays log phase cultures were suspended at 0.4 OD600 and serially diluted five-fold onto YES (yeast extract, glucose and supplements) agar plates. Cell survival was determined after 5 days at 25°C, 3–4 days at 30°C and 2 days at temperatures over 30°C. Strains used in this study are listed in S1 Table.
Immunoblot analysis
The γH2A immunoblots were performed using acid protein extraction to obtain histone-enriched extracts [19] in Fig 2 or total cell extracts in Fig 4. Proteins were resolved by SDSPAGE on 4–20% tris-glycine gels (Life Technologies). Blocking and blotting were performed with Odyssey Blocking Buffer (Li-Cor) per manufacturer instructions and incubated with a rabbit polyclonal phospho-specific anti-γH2A antibody (courtesy of C. Redon). Total H2A was detected using polyclonal anti-H2A antibody 07–146 Millipore for Fig 2 or Active Motif 39235 for Fig 4. Blots were incubated with goat anti-rabbit antibody conjugated to an infrared dye (Li-Cor 827–11081) and scanned and quantified with Odyssey Infrared Imaging System (Li-Cor) with an intensity of 4.5, subtracting median (top/bottom) background.
Microscopy
Cells were photographed using a Nikon Eclipse E800 microscope equipped with a Photometrics Quantix CCD camera and IPlab Spectrum software. Rad52-YFP and Ssb1-GFP were expressed from endogenous loci. Ssb1 (aka Rad11) is the largest subunit of RPA. Rad52-YFP and Ssb1-GFP experiments used cells grown in YES at 25°C. GFP-Brc1 was expressed from the nmt1 promoter using EMM2 (Edinburgh Minimal Media) without thiamine. At least 300 nuclei were scored in three independent experiments. All microscopy was conducted with live (unfixed) cells.
Supporting Information
Zdroje
1. Cimprich KA, Cortez D (2008) ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 9 : 616–627. doi: 10.1038/nrm2450 18594563
2. Jackson SP, Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461 : 1071–1078. doi: 10.1038/nature08467 19847258
3. Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, et al. (2008) GammaH2AX and cancer. Nat Rev Cancer 8 : 957–967. doi: 10.1038/nrc2523 19005492
4. Stucki M, Jackson SP (2006) gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst) 5 : 534–543.
5. Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, et al. (2002) Genomic instability in mice lacking histone H2AX. Science 296 : 922–927. 11934988
6. Downs JA, Lowndes NF, Jackson SP (2000) A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 408 : 1001–1004. 11140636
7. Nakamura TM, Du LL, Redon C, Russell P (2004) Histone H2A phosphorylation controls Crb2 recruitment at DNA breaks, maintains checkpoint arrest, and influences DNA repair in fission yeast. Mol Cell Biol 24 : 6215–6230. 15226425
8. Rozenzhak S, Mejia-Ramirez E, Williams JS, Schaffer L, Hammond JA, et al. (2010) Rad3 decorates critical chromosomal domains with gammaH2A to protect genome integrity during S-Phase in fission yeast. PLoS Genet 6: e1001032. doi: 10.1371/journal.pgen.1001032 20661445
9. Szilard RK, Jacques PE, Laramee L, Cheng B, Galicia S, et al. (2010) Systematic identification of fragile sites via genome-wide location analysis of gamma-H2AX. Nat Struct Mol Biol 17 : 299–305. doi: 10.1038/nsmb.1754 20139982
10. Williams JS, Williams RS, Dovey CL, Guenther G, Tainer JA, et al. (2010) gammaH2A binds Brc1 to maintain genome integrity during S-phase. EMBO J 29 : 1136–1148. doi: 10.1038/emboj.2009.413 20094029
11. Bass KL, Murray JM, O'Connell MJ (2012) Brc1-dependent recovery from replication stress. J Cell Sci 125 : 2753–2764. doi: 10.1242/jcs.103119 22366461
12. Shimada M, Okuzaki D, Tanaka S, Tougan T, Tamai KK, et al. (1999) Replication factor C3 of Schizosaccharomyces pombe, a small subunit of replication factor C complex, plays a role in both replication and damage checkpoints. Mol Biol Cell 10 : 3991–4003. 10588638
13. Indiani C, O'Donnell M (2006) The replication clamp-loading machine at work in the three domains of life. Nature reviews Molecular cell biology 7 : 751–761. 16955075
14. Johnson A, O'Donnell M (2005) Cellular DNA replicases: components and dynamics at the replication fork. Annu Rev Biochem 74 : 283–315. 15952889
15. Kim J, Robertson K, Mylonas KJ, Gray FC, Charapitsa I, et al. (2005) Contrasting effects of Elg1-RFC and Ctf18-RFC inactivation in the absence of fully functional RFC in fission yeast. Nucleic Acids Res 33 : 4078–4089. 16040599
16. Navadgi-Patil VM, Burgers PM (2009) A tale of two tails: activation of DNA damage checkpoint kinase Mec1/ATR by the 9-1-1 clamp and by Dpb11/TopBP1. DNA Repair (Amst) 8 : 996–1003.
17. Griffiths DJ, Barbet NC, McCready S, Lehmann AR, Carr AM (1995) Fission yeast rad17: a homologue of budding yeast RAD24 that shares regions of sequence similarity with DNA polymerase accessory proteins. EMBO J 14 : 5812–5823. 8846774
18. Ansbach AB, Noguchi C, Klansek IW, Heidlebaugh M, Nakamura TM, et al. (2008) RFCCtf18 and the Swi1-Swi3 complex function in separate and redundant pathways required for the stabilization of replication forks to facilitate sister chromatid cohesion in Schizosaccharomyces pombe. Mol Biol Cell 19 : 595–607. 18045993
19. Redon C, Pilch DR, Rogakou EP, Orr AH, Lowndes NF, et al. (2003) Yeast histone 2A serine 129 is essential for the efficient repair of checkpoint-blind DNA damage. EMBO Rep 4 : 678–684. 12792653
20. Pommier Y (2006) Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer 6 : 789–802. 16990856
21. Wei Y, Wang HT, Zhai Y, Russell P, Du LL (2014) Mdb1, a fission yeast homolog of human MDC1, modulates DNA damage response and mitotic spindle function. PLoS One 9: e97028. doi: 10.1371/journal.pone.0097028 24806815
22. Kilkenny ML, Dore AS, Roe SM, Nestoras K, Ho JC, et al. (2008) Structural and functional analysis of the Crb2-BRCT2 domain reveals distinct roles in checkpoint signaling and DNA damage repair. Genes & development 22 : 2034–2047.
23. Saka Y, Esashi F, Matsusaka T, Mochida S, Yanagida M (1997) Damage and replication checkpoint control in fission yeast is ensured by interactions of Crb2, a protein with BRCT motif, with Cut5 and Chk1. Genes & development 11 : 3387–3400.
24. Willson J, Wilson S, Warr N, Watts FZ (1997) Isolation and characterization of the Schizosaccharomyces pombe rhp9 gene: a gene required for the DNA damage checkpoint but not the replication checkpoint. Nucleic Acids Res 25 : 2138–2146. 9153313
25. Sanders SL, Portoso M, Mata J, Bahler J, Allshire RC, et al. (2004) Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell 119 : 603–614. 15550243
26. Du LL, Nakamura TM, Russell P (2006) Histone modification-dependent and-independent pathways for recruitment of checkpoint protein Crb2 to double-strand breaks. Genes Dev 20 : 1583–1596. 16778077
27. Sofueva S, Du LL, Limbo O, Williams JS, Russell P (2010) BRCT domain interactions with phospho-histone H2A target Crb2 to chromatin at double-strand breaks and maintain the DNA damage checkpoint. Mol Cell Biol 30 : 4732–4743. doi: 10.1128/MCB.00413-10 20679485
28. Moser BA, Chang YT, Kosti J, Nakamura TM (2011) Tel1ATM and Rad3ATR kinases promote Ccq1-Est1 interaction to maintain telomeres in fission yeast. Nat Struct Mol Biol 18 : 1408–1413. doi: 10.1038/nsmb.2187 22101932
29. Limbo O, Porter-Goff ME, Rhind N, Russell P (2011) Mre11 nuclease activity and Ctp1 regulate Chk1 activation by Rad3ATR and Tel1ATM checkpoint kinases at double-strand breaks. Molecular and cellular biology 31 : 573–583. doi: 10.1128/MCB.00994-10 21098122
30. Enoch T, Carr AM, Nurse P (1992) Fission yeast genes involved in coupling mitosis to completion of DNA replication. Genes Dev 6 : 2035–2046. 1427071
31. Furuya K, Poitelea M, Guo L, Caspari T, Carr AM (2004) Chk1 activation requires Rad9 S/TQ-site phosphorylation to promote association with C-terminal BRCT domains of Rad4TOPBP1. Genes & development 18 : 1154–1164.
32. Rhind N, Russell P (2000) Chk1 and Cds1: linchpins of the DNA damage and replication checkpoint pathways. J Cell Sci 113 (Pt 22): 3889–3896. 11058076
33. Ohouo PY, Bastos de Oliveira FM, Liu Y, Ma CJ, Smolka MB (2013) DNA-repair scaffolds dampen checkpoint signalling by counteracting the adaptor Rad9. Nature 493 : 120–124. doi: 10.1038/nature11658 23160493
34. Cussiol JR, Jablonowski CM, Yimit A, Brown GW, Smolka MB (2015) Dampening DNA damage checkpoint signalling via coordinated BRCT domain interactions. EMBO J 34 : 1704–1717. doi: 10.15252/embj.201490834 25896509
35. Roseaulin L, Yamada Y, Tsutsui Y, Russell P, Iwasaki H, et al. (2008) Mus81 is essential for sister chromatid recombination at broken replication forks. EMBO J 27 : 1378–1387. doi: 10.1038/emboj.2008.65 18388861
36. McGlynn P, Lloyd RG (2002) Recombinational repair and restart of damaged replication forks. Nat Rev Mol Cell Biol 3 : 859–870. 12415303
37. Langerak P, Mejia-Ramirez E, Limbo O, Russell P (2011) Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genetics 7: e1002271. doi: 10.1371/journal.pgen.1002271 21931565
38. Williams RS, Dodson GE, Limbo O, Yamada Y, Williams JS, et al. (2009) Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell 139 : 87–99. doi: 10.1016/j.cell.2009.07.033 19804755
39. Boddy MN, Gaillard PH, McDonald WH, Shanahan P, Yates JR 3rd, et al. (2001) Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell 107 : 537–548. 11719193
40. Noguchi E, Noguchi C, Du LL, Russell P (2003) Swi1 prevents replication fork collapse and controls checkpoint kinase Cds1. Mol Cell Biol 23 : 7861–7874. 14560029
41. Yin L, Locovei AM, D'Urso G (2008) Activation of the DNA damage checkpoint in mutants defective in DNA replication initiation. Mol Biol Cell 19 : 4374–4382. doi: 10.1091/mbc.E08-01-0020 18667534
42. Lemoine FJ, Degtyareva NP, Lobachev K, Petes TD (2005) Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell 120 : 587–598. 15766523
43. Li X, Liu K, Li F, Wang J, Huang H, et al. (2012) Structure of C-terminal tandem BRCT repeats of Rtt107 protein reveals critical role in interaction with phosphorylated histone H2A during DNA damage repair. J Biol Chem 287 : 9137–9146. doi: 10.1074/jbc.M111.311860 22262834
44. Ohouo PY, Bastos de Oliveira FM, Almeida BS, Smolka MB (2010) DNA damage signaling recruits the Rtt107-Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol Cell 39 : 300–306. doi: 10.1016/j.molcel.2010.06.019 20670896
45. Tomita K, Matsuura A, Caspari T, Carr AM, Akamatsu Y, et al. (2003) Competition between the Rad50 complex and the Ku heterodimer reveals a role for Exo1 in processing double-strand breaks but not telomeres. Mol Cell Biol 23 : 5186–5197. 12861005
46. Limbo O, Chahwan C, Yamada Y, de Bruin RA, Wittenberg C, et al. (2007) Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol Cell 28 : 134–146. 17936710
47. Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, et al. (2013) ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155 : 1088–1103. doi: 10.1016/j.cell.2013.10.043 24267891
48. Lee KY, Myung K (2008) PCNA modifications for regulation of post-replication repair pathways. Mol Cells 26 : 5–11. 18525240
49. Ulrich HD (2009) Regulating post-translational modifications of the eukaryotic replication clamp PCNA. DNA Repair (Amst) 8 : 461–469.
50. Sheedy DM, Dimitrova D, Rankin JK, Bass KL, Lee KM, et al. (2005) Brc1-mediated DNA repair and damage tolerance. Genetics 171 : 457–468. 15972456
51. Lee KM, Nizza S, Hayes T, Bass KL, Irmisch A, et al. (2007) Brc1-mediated rescue of Smc5/6 deficiency: requirement for multiple nucleases and a novel Rad18 function. Genetics 175 : 1585–1595. 17277362
52. Rouse J (2004) Esc4p, a new target of Mec1p (ATR), promotes resumption of DNA synthesis after DNA damage. EMBO J 23 : 1188–1197. 14988729
53. Roberts TM, Kobor MS, Bastin-Shanower SA, Ii M, Horte SA, et al. (2006) Slx4 regulates DNA damage checkpoint-dependent phosphorylation of the BRCT domain protein Rtt107/Esc4. Mol Biol Cell 17 : 539–548. 16267268
54. Balint A, Kim T, Gallo D, Cussiol JR, Bastos de Oliveira FM, et al. (2015) Assembly of Slx4 signaling complexes behind DNA replication forks. EMBO J.
55. Coulon S, Gaillard PH, Chahwan C, McDonald WH, Yates JR 3rd, et al. (2004) Slx1-Slx4 are subunits of a structure-specific endonuclease that maintains ribosomal DNA in fission yeast. Mol Biol Cell 15 : 71–80. 14528010
56. Coulon S, Noguchi E, Noguchi C, Du LL, Nakamura TM, et al. (2006) Rad22Rad52-dependent repair of ribosomal DNA repeats cleaved by Slx1-Slx4 endonuclease. Mol Biol Cell 17 : 2081–2090. 16467377
57. Yan W, Shao Z, Li F, Niu L, Shi Y, et al. (2011) Structural basis of gammaH2AX recognition by human PTIP BRCT5-BRCT6 domains in the DNA damage response pathway. FEBS Lett 585 : 3874–3879. doi: 10.1016/j.febslet.2011.10.045 22064073
58. Wang J, Aroumougame A, Lobrich M, Li Y, Chen D, et al. (2014) PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev 28 : 2693–2698. doi: 10.1101/gad.252478.114 25512557
59. Callen E, Di Virgilio M, Kruhlak MJ, Nieto-Soler M, Wong N, et al. (2013) 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell 153 : 1266–1280. doi: 10.1016/j.cell.2013.05.023 23727112
60. Zimmermann M, de Lange T (2014) 53BP1: pro choice in DNA repair. Trends Cell Biol 24 : 108–117. doi: 10.1016/j.tcb.2013.09.003 24094932
61. Forsburg SL, Rhind N (2006) Basic methods for fission yeast. Yeast 23 : 173–183. 16498704
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 9
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Arabidopsis AtPLC2 Is a Primary Phosphoinositide-Specific Phospholipase C in Phosphoinositide Metabolism and the Endoplasmic Reticulum Stress Response
- Bridges Meristem and Organ Primordia Boundaries through , , and during Flower Development in
- KLK5 Inactivation Reverses Cutaneous Hallmarks of Netherton Syndrome
- XBP1-Independent UPR Pathways Suppress C/EBP-β Mediated Chondrocyte Differentiation in ER-Stress Related Skeletal Disease
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy