
The AMPK, Snf1, Negatively Regulates the Hog1 MAPK Pathway in ER Stress Response
All organisms are always exposed to several environmental stresses, including ultraviolet, heat, and chemical compounds. Therefore, every cell possesses defense mechanisms to maintain their survival under stressed conditions. Numerous studies have shown that a family of protein kinases plays a principal role in adaptive response to environmental stresses and perturbation of their regulation is implicated in a variety of human pathologies, such as cancer and neurodegenerative diseases. Elucidation of molecular mechanisms controlling their activities is still important not only for understanding how the organism acquires stress tolerance, but also for development of therapies for various diseases. In Saccharomyces cerevisiae, the Hog1 stress-responsive MAP kinase is activated by ER stress and coordinates a pleiotropic response to ER stress. However, the mechanisms for regulating Hog1 activity during ER stress response remain poorly understood. In this paper, we demonstrate that a Saccharomyces cerevisiae ortholog of mammalian AMP–activated protein kinase (AMPK), Snf1, negatively regulates Hog1 in ER stress response. ER stress induces expression of Ssk1, a specific activator of the Hog1 MAPK cascade. Snf1 lowers the expression level of Ssk1, thereby downregulating the signaling from upstream components to the Hog1 MAPK cascade. The activity of Snf1 is also enhanced by ER stress. Thus, our data suggest that Snf1 plays an important role in regulation of ER stress response signal mediated by Hog1.
Published in the journal:
. PLoS Genet 11(9): e32767. doi:10.1371/journal.pgen.1005491
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005491
Summary
All organisms are always exposed to several environmental stresses, including ultraviolet, heat, and chemical compounds. Therefore, every cell possesses defense mechanisms to maintain their survival under stressed conditions. Numerous studies have shown that a family of protein kinases plays a principal role in adaptive response to environmental stresses and perturbation of their regulation is implicated in a variety of human pathologies, such as cancer and neurodegenerative diseases. Elucidation of molecular mechanisms controlling their activities is still important not only for understanding how the organism acquires stress tolerance, but also for development of therapies for various diseases. In Saccharomyces cerevisiae, the Hog1 stress-responsive MAP kinase is activated by ER stress and coordinates a pleiotropic response to ER stress. However, the mechanisms for regulating Hog1 activity during ER stress response remain poorly understood. In this paper, we demonstrate that a Saccharomyces cerevisiae ortholog of mammalian AMP–activated protein kinase (AMPK), Snf1, negatively regulates Hog1 in ER stress response. ER stress induces expression of Ssk1, a specific activator of the Hog1 MAPK cascade. Snf1 lowers the expression level of Ssk1, thereby downregulating the signaling from upstream components to the Hog1 MAPK cascade. The activity of Snf1 is also enhanced by ER stress. Thus, our data suggest that Snf1 plays an important role in regulation of ER stress response signal mediated by Hog1.
Introduction
The endoplasmic reticulum (ER) is the cellular organelle responsible for the folding and modification of newly synthesized secretory or membrane proteins. Environmental or developmental changes which perturb ER homeostasis, or genetic alterations causing production of irreversibly misfolded proteins lead to an accumulation of unfolded and misfolded proteins within the ER. This condition, which is collectively termed ER stress, is toxic to cells and has been implicated in a variety of human pathologies, such as diabetes, cancer and neurodegeneration, including Alzheimer, Parkinson and Huntington disease [1, 2]. Therefore, when ER stress is sensed, cells actuate adaptive signaling pathways to alleviate ER stress [1, 3]. In the budding yeast Saccharomyces cerevisiae, the unfolded protein response (UPR) signaling pathway, composed of an ER transmembrane protein Ire1 and a transcriptional activator Hac1, plays a principal role in ER stress response [1, 3]. When activated by ER stress, Ire1 excises the translation-inhibitory intron from HAC1 mRNA, initiating splicing of HAC1 mRNA and consequent production of Hac1 protein. Hac1 induces expression of target genes, such as genes encoding chaperones and proteins functioning ER-associated degradation, thus increasing the protein folding capacity of the ER. Although the UPR is undoubtedly essential for yeast cells to alleviate ER stress, a previous genome-wide study [4] has predicted that not less than 100 genes are involved in response to ER stress. Therefore, it remains to be fully elucidated how ER stress response is precisely controlled.
AMPK is evolutionarily conserved in eukaryotic cells and a key sensor of cellular energy status [5–7]. In Saccharomyces cerevisiae, a catalytic subunit of AMPK is encoded by the SNF1 gene (S1 Fig). Similar to other members of the AMPK family, Snf1 forms a heterotrimeric complex with two regulatory subunits, the γ subunit Snf4 and one of the three alternative β subunits, Sip1, Sip2, or Gal83 [5]. The catalytic activity of Snf1 is regulated by phosphorylation at Thr-210 that is located in the activation loop of its kinase domain [8, 9]. Three upstream kinases, Sak1, Tos3, and Elm1, have been identified as kinases responsible for Snf1 activation [10–12]. Oppositely, Snf1 is inactivated by the Reg1-Glc7 protein phosphatase 1 complex; the catalytic subunit Glc7 is directed to Snf1 through the regulatory subunit Reg1 [13, 14]. Besides critical roles in adaptation to glucose deprivation and utilization of alternative carbon sources to glucose, the Snf1 complex is involved in the response to environmental stresses, such as heat and oxidative stresses [5, 15]. However, the role of Snf1 in ER stress response is as yet poorly understood.
The budding yeast Hog1, which is structurally highly similar to the mammalian p38 MAPK, was originally identified as a key protein kinase required for the adaptation of yeast cells to osmotic stress [16, 17]. In osmotic stress response, the Sln1-Ypd1-Ssk1 multistep phosphorelay system, which is homologous to bacterial two-component systems, regulates the Hog1 MAPK cascade (S2 Fig) [16, 17]. Under normal osmotic conditions, the membrane-associated histidine kinase Sln1 phosphorylates itself. The phosphate group is transferred to Ssk1 through the Ypd1 phosphotransmitter. Hyperosmotic stress inactivates Sln1, resulting in downregulation of the phosphorylation level of Ssk1. Dephosphorylated Ssk1 directly binds to and activates the Ssk2 and Ssk22 MAPKKKs, and consequently, leads to sequential activation of Pbs2 MAPKK and Hog1 MAPK. In addition to a pivotal role in osmotic stress response, Hog1 has been revealed as a regulator of a wide array of stress responses, including cold, heat and ER stresses [16–19]. In ER stress response, Hog1 is activated in an Ssk1-dependent manner [18]. However, the mechanisms that control Hog1 activity in ER stress response are still poorly understood.
In this study, we identified Snf1 as a negative regulator of Hog1 in ER stress response. Cells lacking Snf1 have elevated levels of active Hog1, whereas upregulation of Snf1 activity reduces Hog1 activation. ER stress induces expression of Ssk1, but this induction is counteracted by Snf1. These results indicate that Snf1 modulates Hog1 activation by controlling the expression level of its activator Ssk1. We also demonstrated that loss of Snf1 leads to upregulation of the UPR pathway, whereas the UPR activity is downregulated in Snf1-activated cells. Thus, Snf1 negatively regulates the Hog1 MAPK pathway and the UPR pathway in ER stress response.
Results
The Snf1 complex negatively regulates ER stress response
In order to test whether the Snf1 protein kinase regulates ER stress response, cells carrying snf1 deletion were plated on medium containing tunicamycin, a natural inhibitor of N-linked glycosylation that is widely employed as an inducer of ER stress. We unexpectedly found that compared to wild-type cells, the snf1 mutant was resistant to tunicamycin (Fig 1A). To confirm that snf1 mutation caused tunicamycin resistance, we transformed the snf1 mutant with the plasmid that expresses SNF1 and tested the transformants for growth on medium containing tunicamycin. Expression of SNF1 significantly rescued the tunicamycin-resistant phenotype associated with the snf1 mutation (Fig 1A and S3A Fig). To address the biological importance of Snf1 kinase activity, we generated a catalytically inactive form of Snf1 [Snf1(K84M)], in which Lys-84 in the ATP-binding motif was mutated to methionine [8]. When Snf1(K84M) was expressed in snf1 mutants, the tunicamycin resistance was not rescued (Fig 1A). To examine the effect of Snf1 hyperactivation on ER stress response, we generated Snf1(G53R), a catalytically active form in which Gly-53 in the kinase domain has been mutated to arginine [8]. Expression of Snf1(G53R) resulted in hypersensitivity to tunicamycin (Fig 1B). These results indicate that Snf1 negatively regulates the response to ER stress in a manner dependent on its kinase activity.

We next asked if the regulatory subunits of Snf1 are involved in ER stress response. We found that deletion of the SNF4 gene, which encodes the γ subunit of the Snf1 complex, caused increased resistance to tunicamycin (Fig 1C). Furthermore, cells harboring both snf1 and snf4 mutations displayed ER stress tolerance indistinguishable from that observed in the snf1 single mutants (Fig 1C), indicating that Snf1 and Snf4 act in the same pathway. We next examined whether the β subunits, Sip1, Sip2, and Gal83, regulate ER stress response. We found that the sip1 sip2 gal83 triple mutant was resistant to tunicamycin, although neither of their single mutants exhibited the obvious tunicamycin-resistant phenotype (Fig 1D). These results indicate that the Snf1 complex negatively regulates ER stress response.
Phosphorylation of Snf1 is required for its role in ER stress response
Snf1 is phosphorylated at Thr-210 and activated by exposure of cells to alkaline pH and oxidative stresses [15]. Therefore, we investigated whether Snf1 is activated by treatment with ER stress. Anti-phospho AMPK antibodies that recognize the phosphorylated, activated form of AMPK were used to monitor phosphorylation of Snf1 at Thr-210 in the budding yeast [12]. In wild-type cells, we could detect Snf1 phosphorylation under unstressed conditions (Fig 2A). Treatment of cells with tunicamycin stimulated Snf1 phosphorylation within 1.5–3 hr, and its phosphorylation was persisted for at least 7.5 hr (Fig 2A). Similar observation was seen when cells were exposed to dithiothreitol (DTT), which causes ER stress by blocking disulfide bond formation in the ER (Fig 2B). Thus, ER stress induces activation of Snf1 through phosphorylation at the Thr-210 residue. We next asked whether ER stress-induced Snf1 activation is mediated by the upstream kinases, Sak1, Tos3, and Elm1. We found that sak1 tos3 elm1 triple mutations completely abolished activation of Snf1 both in the presence or absence of ER stress (Fig 2C), although activated Snf1 levels were only slightly decreased in each single mutants (S4A and S4B Fig). This result indicates that ER stress induces Snf1 activation in a manner dependent on the three redundant kinases. Snf1 is inactivated through dephosphorylation mediated by the Reg1-Glc7 phosphatase complex. We next investigated the role of the Reg1-Glc7 complex in ER stress-induced Snf1 activation. Because the glc7 deletion strain is lethal [20, 21], we used reg1 deletion. Both in the presence or absence of tunicamycin treatment, phosphorylated Snf1 levels were clearly upregulated by reg1 deletion (Fig 2D), indicating that the Reg1-Glc7 protein phosphatase 1 acts to inactivate Snf1 in ER stress response.

We next asked whether phosphorylation of Snf1 at Thr-210 is important for its function in ER stress response. We first examined the ability of Snf1(T210A), which contains a mutation of Thr-210 to Ala, to complement ER stress resistance associated with snf1 deletion. The Snf1(T210A) mutant failed to complement the snf1 defect (Fig 2E). We investigated whether the ER stress response involves the three upstream kinases and the phosphatase complex. We found that the sak1 tos3 elm1 triple mutant cells, which were defective in Snf1 activation, were resistant to tunicamycin (Fig 2F). In contrast, reg1 mutant cells in which Snf1 activity is upregulated exhibited hypersensitivity to tunicamycin; however, the stress sensitivity of the reg1 mutant was completely suppressed by snf1 deletion (Fig 2G). Similar results were obtained when DTT was used as an ER stressor (Fig 2H). Taken together, these results demonstrate that Snf1 is activated by ER stress through phosphorylation at Thr-210 and then negatively regulates ER stress response.
Snf1 is involved in negative regulation of the UPR pathway
Next, we explored the mechanism underlying the effect of Snf1 on ER stress response. Previous analyses in Saccharomyces cerevisiae have revealed that the UPR, composed of Ire1 and Hac1, is at the center of ER stress response [1, 3]. Therefore, we investigated a potential role for Snf1 in regulating the UPR. Upon ER stress, activated Ire1 excises the translation-inhibitory intron from HAC1 mRNA, consequently producing Hac1 protein. Hac1 transcriptionally activates its target genes, including KAR2 and ERO1. We first examined the kinetics of HAC1 mRNA splicing (Fig 3A). In wild-type cells under unstressed conditions, the unspliced form of HAC1 mRNA (HAC1u) was robustly detected, but the spliced form (HAC1s) was rarely detectable. Treatment of cells with ER stress promoted splicing of HAC1 mRNA. The amount of HAC1s peaked 1.5 to 3 hr after DTT addition and gradually decreased thereafter. We next investigated the role of Snf1 in regulation of HAC1 mRNA splicing using the snf1 and reg1 mutant cells. We found that both in snf1 and reg1 mutant cells, HAC1 mRNA splicing was normally promoted in response to ER stress (Fig 3A). We found that downregulation of HAC1 mRNA splicing was unaffected by snf1 mutation. On the other hand, in the reg1 mutant cells, HAC1s was decreased rapidly within 3 hr of DTT addition. Therefore, we compared the protein level of Hac1 between wild-type and the reg1 mutant cells (Fig 3B). In wild-type cells, Hac1 protein was hardly detectable prior to ER stress treatment. Production of Hac1 was induced within 1.5 hr after exposure to DTT and subsequently downregulated. In the reg1 mutant cells, Hac1 production was induced at levels comparable to that of wild-type cells. However, the amount of Hac1 declined rapidly within 3 to 4.5 hr after DTT treatment. A rapid decrease in Hac1 protein was also seen when cells harboring reg1 mutation were exposed to tunicamycin (S5A Fig). Consistent with the protein level of Hac1, expression of the well-known Hac1 target genes, ERO1 and KAR2, was reduced by reg1 mutation (Fig 3E and S5B Fig). These UPR defects observed in the reg1 mutant could be significantly restored by snf1 mutation (Figs 3A, 3C and 3E and S5B). These results suggest that Snf1 participates in downregulation of the UPR pathway.

The observation that the UPR activity was downregulated by reg1 mutation prompted us to perform a detailed analysis of the UPR activity in the snf1 mutant cells. We found that the UPR activity under unstressed conditions was increased in the snf1 mutant cells. In the absence of ER stress, the level of HAC1s in cells harboring snf1 deletion was statistically higher than that in wild-type cells (Fig 3A). Consistent with this, the snf1 mutant cells expressed a small amount of Hac1 protein even prior to treatment with ER stress (Fig 3D). Furthermore, under unstressed conditions, both ERO1 and KAR2 mRNAs were statistically significantly increased in the snf1 mutant compared to wild-type cells (Fig 3E and S5B Fig). Taken together, these results indicate that Snf1 negatively regulates the UPR pathway.
Snf1 acts as a negative regulator of Hog1 in ER stress response
The snf1 mutation significantly enhanced resistance against ER stress, although UPR upregulation caused by snf1 mutation was detected only under unstressed conditions. Therefore, additional mechanisms may contribute to ER stress resistance caused by snf1 mutation. In the budding yeast, Hog1 MAPK is activated by ER stress through phosphorylation at critical threonine and tyrosine residues located in the activation loop [16, 17], and is in fact required for protecting cells against ER stress [18, 19]. Anti-phospho-p38 antibodies that recognize the phosphorylated form of mammalian p38 MAPK can be used to detect activated Hog1 in the budding yeast [22]. As observed previously [18], western blot analysis with anti-phospho-p38 antibody marginally detected the activated form of Hog1 in wild-type cells and its abundance was increased by treatment of cells with DTT (Fig 4A). To investigate the role of Snf1 in regulation of Hog1 activity, we monitored the activated form of Hog1 in the snf1 mutant following induction of ER stress. We found that activated Hog1 levels were increased in snf1 mutant cells both in the presence or absence of DTT treatment (Fig 4A). Similar results were obtained when cells were exposed to tunicamycin (S6A Fig). These results suggest that Snf1 has the inhibitory effect on Hog1 activation. The observation that Snf1 is activated by ER stress prompted us to test whether Snf1 acts to downregulate Hog1 activity during recovery from ER stress. We observed that Snf1 remained active even after removal of DTT from the medium (Fig 4C). In contrast, DTT removal allowed reduction of Hog1 activity (Fig 4B). However, Hog1 activation was prolonged in cells lacking Snf1 (Fig 4B). These results suggest that ER stress-activated Snf1 participates in the process that Hog1 activity returns to the basal level. Furthermore, it is suggested that additional mechanisms function in Hog1 inactivation during recovery from ER stress, since Hog1 dephosphorylation after DTT removal was delayed, but occurred in snf1 mutant cells. Next, we examined the effect of Snf1 hyperactivation on ER stress-induced Hog1 activation. Strikingly, activation of Hog1 in response to DTT was diminished by reg1 mutation which increases Snf1 activity (Fig 4D), and this defect could be entirely restored by snf1 mutation (Fig 4E). Similar results were obtained when cells were exposed to tunicamycin (Fig 4F and S6B Fig). Our finding that reg1 mutation interferes with Hog1 activation in response to ER stress strongly suggests that Snf1 acts as a negative regulator of Hog1 in ER stress response.

Previous report showed that ER stress-activated Hog1 accumulated in the nucleus [18]. To investigate the effect of Snf1 on nuclear accumulation of Hog1 in response to ER stress, we used the strain which expresses Hog1 carboxyl-terminally tagged with GFP (Fig 4G). As observed previously [18], Hog1 was uniformly distributed in the nucleus and cytosol under normal conditions and became enriched in the nucleus after ER stress treatment. Loss of Snf1 slightly but significantly increased nuclear localization of Hog1 even in the absence of ER stress. In contrast, nuclear accumulation of Hog1 in response to ER stress was obviously decreased in the reg1 mutant cells; however, this defect was clearly suppressed by snf1 deletion. These observations support a role of Snf1 in negative regulation of Hog1 in ER stress response.
It has been well-characterized that yeast cells activate Hog1 when exposed to hyperosmotic extracellular environments [16, 17]. We therefore examines whether Snf1 might be involved in the osmotic stress response mediated by Hog1. In wild-type cells, activated Hog1 is robustly detectable within 3 min of NaCl treatment and then rapidly decreases by 30 min (Fig 4H). Hog1 activation in response to hyperosmotic stress appeared to be enhanced and reduced by snf1 and reg1 mutations, respectively (Fig 4H). These alterations are probably attributed to a potential role of Snf1 in inhibiting Hog1 activation. Indeed, snf1 mutation elevated the basal activity of Hog1 (Fig 4A), and reg1 mutation partially suppressed the lethality of SLN1 - and YPD1-deleted cells in which Hog1 is constitutively activated (see below). However, we could not find that reg1 mutation resulted in hypersensitivity to osmotic stress (S6C Fig). Therefore, it remains obscure whether Snf1-mediated Hog1 regulation is physiologically important for osmotic stress response.
Relationship between the UPR, Hog1 and Snf1
We next examined whether enhanced ER stress resistance in the snf1 mutants is caused by Hog1 hyperactivation. We constructed the snf1 hog1 double mutants and test them for growth on medium containing tunicamycin (Fig 5A). The snf1 hog1 double mutant was sensitive to tunicamycin. However, we also found that ER stress sensitive phenotype of the hog1 mutant could be partially suppressed by snf1 mutation. As snf1 mutation leads to upregulation of the UPR, we compared the effect of snf1 deletion in cells having a wild-type, hac1, or hac1 hog1 background. The snf1 mutation modestly restored ER stress sensitivity caused by hac1 mutation (Fig 5B). In contrast, the snf1 hog1 hac1 triple mutants exhibited hypersensitivity to tunicamycin, similar to the hog1 hac1 double mutants (Fig 5C), indicating that Hog1 and UPR are key targets of Snf1 in ER stress response.

As shown above, activities of the UPR and Hog1 pathways are upregulated by snf1 deletion, but downregulated by reg1 mutation which leads to Snf1 hyperactivation. These observations raised the possibility that Snf1 continuously regulates the UPR and Hog1 pathways. If this is true, we can observe the diminished Hog1 activation in hac1 mutant cells or the reduced UPR activity in hog1 mutant cells. First, we measured Hog1 activity in cells lacking Hac1. However, we could not find that loss of Hac1 reduced Hog1 activation (Fig 5D). We next monitored HAC1 mRNA splicing in hog1 mutant cells. In hog1 mutant cells, HAC1 mRNA splicing was normally induced, but retained longer than wild-type cells (Fig 5E). This is consistent with a previous observation [18] and indicates that hog1 mutation does not reduce, but rather upregulates the UPR activity. Thus, the activities of the UPR and Hog1 pathways are independently regulated by Snf1.
Snf1 negatively regulates Ssk1 expression
We next investigated how Snf1 negatively regulates Hog1 in ER stress response. The dephosphorylation of MAPK by protein phosphatases is well-known as a common mechanism for the negative regulation of the signaling mediated by MAPK [23]. Hog1 is dephosphorylated and inactivated by Ptp2 tyrosine phosphatase [24, 25]. Previously, it has been shown that loss of Ptp2 results in enhanced resistance to ER stress in a HOG1-dependent manner [19]. Therefore, we examined the relationship between Snf1 and Ptp2. In the ptp2 mutant cells, basal activity of Hog1 was modestly increased and ER stress-induced Hog1 activation was significantly upregulated (S7A Fig). We found that Hog1 activation was enhanced in the ptp2 snf1 double mutants compared with the ptp2 mutant cells (S7A Fig), indicating that Snf1 negatively regulates Hog1 in ER stress response independently of Ptp2.
In ER stress response, signaling through the Hog1 pathway is controlled by the Sln1-Ypd1-Ssk1 phosphorelay system [18, 19]. Disruption of the SLN1 gene results in lethality due to constitutive activation of Hog1 and, indeed, mutations in any of the four downstream genes, SSK1, SSK2, PBS2, and HOG1, suppress the sln1 lethality by blocking activation of Hog1 [26, 27]. As shown above, Hog1 activity is considerably decreased in reg1 mutant cells in which Snf1 is hyperactivated. Therefore, we tested whether deletion of the REG1 gene suppresses the sln1 lethality. We found that reg1 mutation modestly suppressed the growth defect associated with sln1 deletion (Fig 6A). Similarly, the lethality caused by ypd1 deletion was partially suppressed by reg1 mutation (Fig 6B). However, loss of Snf1 interfered with the ability of reg1 mutation to suppress the ypd1 lethality (S8A and S8B Fig). These results suggest that Snf1 regulates the component functioning downstream of Ypd1 in the Hog1 pathway.
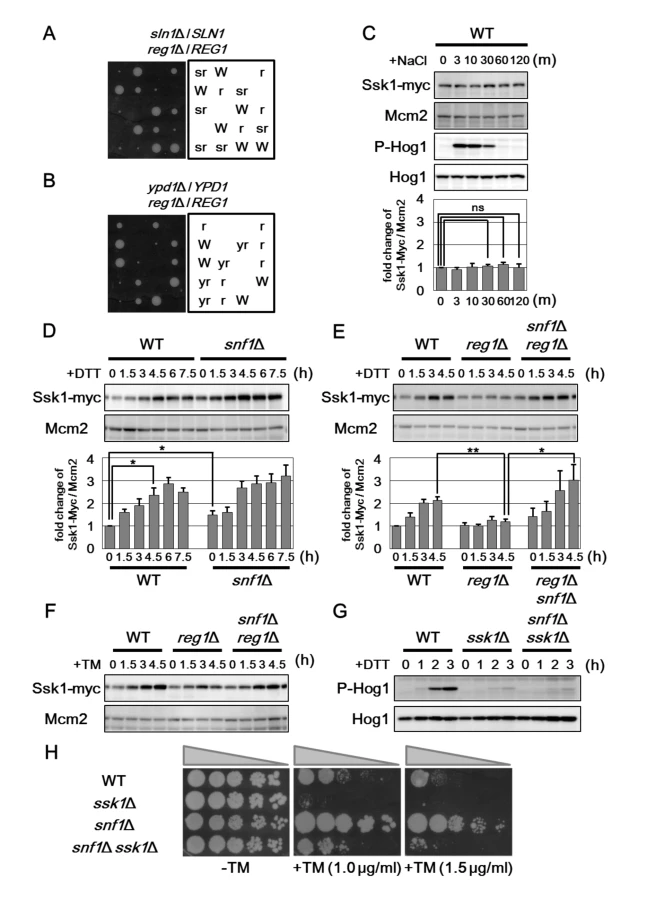
In order to identify the molecule that mediates the signaling from Snf1 to Hog1, we examined the expression levels of components that act in the Hog1 pathway. We generated yeast strains carrying the carboxyl-terminally Myc-tagged genes, including SSK1, SSK2, SSK22, and PBS2, and analyzed their expression levels (Figs 6C–6E and S8C–S8E). Among them, we found that Ssk1 expression is changed by treatment with ER stress and genetic modulation of Snf1 signaling. In wild-type cells, the protein abundance of Ssk1 is increased following exposure to DTT and tunicamycin, but not NaCl (Fig 6C–6F), suggesting that ER stress specifically affects Ssk1 expression. The snf1 mutation moderately increased Ssk1 expression (Fig 6D), suggesting that Ssk1 expression is negatively regulated by Snf1. Next, we examined the effect of Snf1 hyperactivation on the expression level of Ssk1. ER stress-mediated Ssk1 induction was effectively inhibited by reg1 mutation that leads to hyperactivation of Snf1 (Fig 6E). This defect could be significantly restored by snf1 mutation (Fig 6E). Similar results were obtained when cells were exposed to tunicamycin (Fig 6F). These results suggest that Ssk1 expression is negatively regulated by Snf1.
We next examined the functional importance of Ssk1 in Snf1-mediated regulation of Hog1 activity. As shown previously [18], activated Hog1 levels were significantly decreased in ssk1 mutant cells (Fig 6G). This defect could not be suppressed by snf1 mutation (Fig 6G), indicating that Ssk1 is important for Hog1 hyperactivation caused by snf1 mutation. We also asked whether Ssk1 is involved in enhanced ER stress resistance of the snf1 mutants. We found that ssk1 mutation rendered cells lacking Snf1 sensitive to tunicamycin (Fig 6H). We also observed that the ssk1 snf1 double mutant cells were more resistant to ER stress than the ssk1 single mutants. ER stress tolerance of the ssk1 snf1 double mutants is probably due to increased UPR activity caused by snf1 mutation. Taken together, these results indicate that Snf1 inhibits Hog1 activation in response to ER stress by negatively regulating the expression level of Ssk1.
Ssk1 expression is regulated by Snf1 at mRNA level
To explore the mechanism by which Snf1 regulates the expression level of Ssk1, we measured the amount of SSK1 mRNA by qRT-PCR (Fig 7A and S9A Fig). In wild-type cells, SSK1 mRNA is increased following exposure to ER stress. This induction seemed to be normal in snf1 mutant cells. On the other hand, reg1 deletion significantly inhibited the induction of SSK1 mRNA. This reg1 defect could be restored by snf1 mutation. These results indicate that Snf1 hyperactivation reduce the expression level of SSK1 mRNA.

Numerous studies have revealed that Snf1 regulates the gene expression at the transcriptional level through phosphorylation of transcription factors [5, 6]. This raised the possibility that Snf1 regulates SSK1 promoter activity. To test this possibility, we generated a PSSK1-GFP reporter, consisting of the 5' upstream region of the SSK1 gene to drive GFP expression (Fig 7B). Wild-type cells harboring the PSSK1-GFP reporter displayed GFP expression in the absence of ER stress (Fig 7C). GFP expression from the PSSK1-GFP reporter was increased following incubation with DTT (Fig 7C). On the other hand, we observed that DTT treatment had no obvious effect on expression of GFP derived from the PMCM2-GFP reporter, in which the 5' upstream region of the MCM2 gene is fused to GFP (Figs 7B and 7C and S9B). These results suggest that SSK1 promoter is activated by ER stress. Next, we tested whether PSSK1-GFP induction is regulated by the Snf1 pathway. In contrast to wild-type cells, PSSK1-GFP expression was barely induced by DTT in reg1 mutant cells (Fig 7D). This reg1 defect could be significantly restored by snf1 mutation (Fig 7D). These results strongly support the model in which Snf1 inhibits the activity of SSK1 promoter.
We next examined whether SSK1 induction in response to ER stress is important for resistance to ER stress. To test this, we generated two constructs, PSSK1-SSK1 and PMCM2-SSK1, which express SSK1 under the control of SSK1 and MCM2 promoters, respectively. Introduction of the PSSK1-SSK1 construct significantly rescued ER stress sensitive phenotype associated with ssk1 mutation (Fig 7E). On the other hand, when the PMCM2-SSK1 construct was introduced into ssk1 mutant cells, ER stress sensitivity was less effectively rescued (Fig 7E). This suggests that SSK1 induction via its promoter activation is important for protecting cells against ER stress.
We also attempted to compare the ability of the PSSK1-SSK1 and PMCM2-SSK1 constructs to rescue the osmotic stress sensitivity caused by ssk1 mutation. In osmotic stress response, the Hog1 pathway is activated by the membrane protein Sho1 in addition to Ssk1 [26]; hence, as shown in S9C Fig, the ssk1 sho1 double mutants was sensitive to osmotic stress, although neither of their single mutants exhibited the obvious sensitivity to osmotic stress. Therefore, we transformed the ssk1 sho1 double mutant cells with the PSSK1-SSK1 and PMCM2-SSK1 constructs and tested the transformants for growth under hyperosmotic conditions. We found that the PMCM2-SSK1 construct could rescue the osmotic stress sensitivity caused by ssk1 sho1 mutations to same extent as the PSSK1-SSK1 construct (Fig 7F). Thus, it is unlikely that the SSK1 promoter is involved in regulation of osmotic stress response. Taken together, the mechanisms underlying Hog1 activation mediated by Ssk1 are different between osmotic and ER stresses.
Discussion
Previous studies have revealed that the snf1 mutant cells exhibited hypersensitivity to a number of environmental stresses, including alkaline pH, heat shock, and genotoxic stress caused by hydroxyurea and methylmethane sulfonate [5]. Therefore, Snf1 was regarded as an essential regulator to confer resistance to environmental stresses. In this study, we unexpectedly found that the snf1 mutants were resistant to ER stress. This finding not only indicates that Snf1 negatively regulates ER stress response, but also reveal a novel inhibitory role for Snf1 in stress response.
The pleiotropic functions of Snf1 in ER stress response
Here, we revealed that Snf1 inhibits Hog1 activity by downregulation of the expression level of SSK1 mRNA encoding an upstream activator of the Hog1 MAPK cascade. It is well-known that the dephosphorylation of MAPK by protein phosphatases is crucial for the negative regulation of the signaling mediated by MAPK [23]. The protein phosphatases, such as Ptc1, Ptp2, and Ptp3, play an important role in Hog1 inactivation [16, 17, 24, 25]. Previous report showed that Ptp2 and Ptp3 play the main and minor role, respectively, in negative regulation of Hog1 during ER stress response [19]. Nevertheless, why is Snf1 needed to function in downregulation of Hog1 activity? It is possible that Snf1 coordinates ER stress response with other cellular responses, since Snf1 is activated in the response to various environmental stresses [5, 6, 15]. Indeed, it has been reported that ER stress sensitivity is enhanced under the extracellular environments in which Snf1 activity is known to be elevated [28]. Alternatively, Snf1 may function to inactivate Hog1 in the whole cell level. Previous study showed that upon exposure to ER stress, Hog1 not only translocates into the nucleus and regulates the gene expression, but also functions in activation of autophagy in the cytoplasm [18]. On the other hand, Ptp2 and Ptp3 phosphatases are localized in the nucleus and cytoplasm, respectively [29]. Therefore, it is anticipated that Hog1 activity and its related cellular responses are negatively regulated in a manner different from the nucleus and cytoplasm. In contrast, Snf1 interferes with the signal from Ssk1 to Hog1 MAPK cascade through negative regulation of Ssk1 expression. Therefore, Snf1 is expected to contribute to downregulation of Hog1 in the whole cell level. Our analyses showed that loss of Snf1 moderately increased Hog1 activity, while Snf1 hyperactivation caused by reg1 deletion effectively inhibited Hog1 activation. The existence of protein phosphatases for Hog1 may make apparently difficult to observe the effect of snf1 mutation on Hog1 activity. It is well-known that expression of protein phosphatases for MAPK is induced by environmental stresses [23]. In fact, we found that Ptp2 was induced in response to ER stress (S7C Fig). Consistent with this, Hog1 inactivation after removal of ER stress was modestly delayed, but occurred in snf1 mutant cells. Thus, it is likely that intricate signaling networks regulate Hog1 activity during ER stress response. Therefore, detailed analyses of the relationships between Hog1-mediated ER stress responses and the function of each negative regulator for Hog1 will be important for further understanding how Hog1 activity is controlled during ER stress response.
In this study, we observed that the snf1 mutant cells expressed Hac1 even in the absence of ER stress. On the other hand, the expression level of Hac1 in the presence of ER stress was rapidly decreased in Snf1-hyperactivated cells. These observations suggest that Snf1 acts as a negative regulator of the UPR pathway. Although the activation mechanisms of the UPR pathway has been well-studied, there are only a few reports about how the UPR is inactivated after ER stress. Previously, two groups demonstrated the importance of the phosphorylation state of Ire1 kinase domain in the attenuation of the UPR activity [30, 31]. However, their proposed models are significantly different from each other. Therefore, the mechanisms by which the UPR is finally attenuated have yet to be elucidated. In the course of preparation of this manuscript, Casamayor and colleagues also reported that Snf1 is involved in yeast ER stress response [28]. Consistent with our findings, they showed that reg1 mutation results in increased sensitivity to ER stress in a Snf1-dependent manner. Interestingly, they proposed the model in which Snf1 plays an inhibitory role in attenuation of the UPR by regulating the oligomerization of Ire1. In regard to the UPR activity in the reg1 mutant cells, their results are distinctly different from our observations: they showed that increased UPR activity after ER stress treatment was prolonged in the reg1 mutant cells; we found that in the reg1 mutant cells, the UPR activity declined rapidly during ER stress response. The reason for this discrepancy is not clear now. However, this phenotypic distinction may be attributed to the difference in genetic background: their strains were derivatives of BY4741 and DBY746; we used W303 derivatives. Indeed, we could observe that the snf1 mutant was resistant to tunicamycin, although they found no difference in ER stress sensitivity between wild-type and the snf1 mutant cells. Thus, further analyses should be needed to elucidate the molecular mechanism by which Snf1 regulates the UPR signaling pathway.
We found here that snf1 mutation increases the activities of the Hog1 and UPR pathways and leads to resistance to ER stress. Numerous studies have revealed that improper hyperactivation of stress-responsive signaling pathways is deleterious to cells and organisms [16, 23, 32]. In fact, constitutive activation of Hog1 under unstressed conditions causes cell lethality [26, 33]. We observed that cells deleted for both SNF1 and PTP2 genes showed a high basal activity of Hog1, but remains viable (S7B Fig). Thus, Hog1 activity in the ptp2 snf1 double mutant cells is not high enough to induce lethality. We found that compared with wild-type cells, the snf1 mutant cells possessed an increased Hog1 activity during ER stress response. However, it is noteworthy that a decline in the Hog1 activity occurred after removal of ER stress even in cells lacking Snf1. This indicates that the Hog1 activity in the snf1 mutant cells is upregulated, but still remains under the control of the regulatory mechanisms. Therefore, it is possible that Hog1 upregulation caused by snf1 mutation is preferable for yeast cells to survive in the presence of ER stress. Similar view may be applied to the UPR activity. Previous studies revealed that perturbation of the mechanism for properly attenuating Ire1 activity results in reduction of cell viability in the presence of ER stress [30, 31]. In the snf1 mutant cells, the basal activity of UPR is significantly higher than wild-type cells; however, attenuation of the UPR was nearly unaffected by snf1 mutation. Since the snf1 mutant cells possesses high, but adjustable, UPR activity, loss of Snf1 may be preferable for cells to survive under ER stress. Thus, Snf1 plays pleiotropic roles in negative regulation of ER stress response.
Snf1 negatively regulates Hog1 activation by downregulating Ssk1 expression
The signaling through the Hog1 pathway is controlled by the Sln1-Ypd1-Ssk1 phosphorelay system [16, 17]. In osmotic stress response, Sln1 inactivation is a key step of Hog1 activation. Under normal osmotic conditions, active Sln1 leads to Ssk1 phosphorylation. Hyperosmotic stress inactivates Sln1, causing an increase of the dephosphorylated form of Ssk1. This promotes the Ssk1-Ssk2/Ssk22 physical interaction and results in activation of the Hog1 MAPK cascade. Previous studies demonstrated that Ssk1 is implicated in regulation of Hog1 activity during ER stress response [18, 19]. Little is understood, however, about the mechanism by which ER stress activates Hog1. In this study, we demonstrated that the expression level of Ssk1 is increased during ER stress response, and that elevation of Ssk1 protein level is important for cells to survive under ER stress conditions. In contrast, Ssk1 expression remained unchanged upon osmotic stress. These findings suggest that the mechanisms for the regulation of Hog1 are different among these different types of stress. Based on our data, we propose the model in which Hog1 activation in response to ER stress involves upregulation of Ssk1 (Fig 8). In unstressed conditions, Ssk1 is phosphorylated and inactivated by the upstream phosphorelay system. In the presence of ER stress, increased expression of Ssk1 overwhelms the phosphorylation activity of upstream phosphorelay system, leading to accumulation of dephosphorylated Ssk1 and consequent activation of the Hog1 MAPK cascade.

It is well-known that many protein kinases of the MAPKKK family can be activated by binding of their activators, similar to the budding yeast Ssk2 and Ssk22 MAPKKKs [23]. In unstimulated cells, MAPKKK is kept catalytically inactive through an autoinhibitory interaction between the regulatory domain and the kinase domain. Upon stimulation, binding of an activator protein leads to the dissociation of the autoinhibitory domain from the kinase domain and a consequent activation of MAPKKK. Mammalian MTK1/MEKK4 is a stress responsive MAPKKK that is structurally highly similar to the yeast Ssk2/Ssk22 and locates upstream in the p38 pathway [34, 35]. Previous studies have identified the GADD45 family proteins (GADD45α, GADD45β and GADD45γ) as MTK1/MEKK4 activators [36]. GADD45 binds to the autoinhibitory domain of MTK1/MEKK4 and relieve autoinhibition. Furthermore, each GADD45 exhibits distinct tissue expression patterns and is induced by a certain subset of environmental stresses, showing functional distinction among the GADD45 isoforms in p38 activation [36]. Our data presented here suggest that ER stress activates the Hog1 MAPK cascade by induction of Ssk1, in a manner similar to activation of p38 MAPK cascade though stress-mediated induction of GADD45.
To date, there is little understanding of the mechanism that controls the expression level of Ssk1. In this study, we show that Snf1 acts as a negative regulator of SSK1 expression in ER stress response. We also demonstrate that the SSK1 promoter is important for Snf1 to negatively regulate the mRNA level of SSK1. Snf1 phosphorylates a large number of transcription factors, and influences the transcription of hundreds of genes, including those involved in the utilization of alternate carbon sources and the metabolism of amino acids [5, 6]. Therefore, Snf1 is the most likely to inhibit the promoter activity of the SSK1 gene through phosphorylation of the transcription factor. We also found that the expression level of Ssk1 protein was slightly different from that of SSK1 mRNA. Ssk1 protein is more abundant in the snf1 mutants than wild-type cells, although there was little difference in the expression level of SSK1 mRNA between wild-type and the snf1 mutant cells. This suggests that Snf1 inhibits Ssk1 expression at the translational or posttranslational levels. Intriguingly, it has been showed that the protein level of Ssk1 is negatively regulated by Ubc7 [37]. Ubc7 is an endoplasmic reticulum-associated ubiquitin-conjugating enzyme responsible for ER-associated degradation (ERAD). Snf1 may modulate Ssk1 degradation mediated by the ubiquitin-proteasome system involving Ubc7. Thus, identification of components downstream of Snf1, for example, which is responsible for induction of Ssk1 in response to ER stress, will provide valuable insights into the evolutionally conserved mechanism for regulation of the p38/Hog1 MAPK cascade.
Materials and Methods
Strains
Strains used in this study are listed in S1 Table. Yeast strains harboring the complete gene deletions and carboxyl-terminally Myc or GFP-tagged genes were generated by a PCR-based method as described previously [38]. All strains constructed by a PCR-based method were verified by PCR to confirm that replacement had occurred at the expected locus. Standard procedures were followed for yeast manipulations [39].
Plasmids
Plasmids used in this study are described in S2 Table. In-Fusion cloning kits (Takara) was used to construct plasmids. The PSSK1-GFP and PMCM2-GFP plasmids were constructed as follows. The DNA fragment encoding GFP followed by the ADH1 terminator (TADH1) was obtained by PCR using the pFA6a-GFP vector [38] as a template. The GFP-TADH1 DNA fragment was fused to 999-bp and 762-bp genomic fragments containing 5' upstream sequences of the SSK1 and MCM2 genes, yielding the PSSK1-GFP and PMCM2-GFP plasmids, respectively. Schemes detailing construction of plasmids and primer sequences are available on request.
Protein extraction, western blot analysis and antibodies
Protein extracts were prepared essentially as described previously [40]. Briefly, cells grown to exponential phase were incubated with YPD or SD medium containing 2 μg/ml tunicamycin, 4 mM DTT or 0.4 M sodium chloride, for the indicated times. Cells were transferred into test tubes, mixed 1 : 1 with boiled medium, submerged in the boiling water for 3 min, and harvested by centrifugation. Cells were then subjected to a mild alkali treatment-based protein extraction method [41]. Western blot analysis was performed using the immunoreaction enhancer solution Can Get Signal (Toyobo) according to the manufacturer's protocol. Anti-HA monoclonal antibody 16B12 (Covance), anti-Myc monoclonal antibody 9E10 (Santa Cruz), anti-GFP monoclonal antibody JL-8 (Clontech), anti-phospho-p38 MAPK monoclonal antibody 28B10 (Cell Signaling), anti-phospho-AMPKα monoclonal antibody 40H9 (Cell Signaling), anti-Hog1 polyclonal antibody y-215 (Santa Cruz), anti-Snf1 polyclonal antibody yk-16 (Santa Cruz), and anti-Mcm2 polyclonal antibody N-19 (Santa Cruz) were used. Detection was carried out by using a LAS-4000 (Fuji Film) with Immobilon Westren (Merck Millipore). Signal intensities were quantified by ImageQuant (GE Healthcare), and statistical analysis was performed with Excel (Microsoft).
RNA isolation and RT–PCR
Cells grown to exponential phase were incubated with YPD medium containing 2 μg/ml tunicamycin or 4 mM DTT, and harvested at the indicated times. Total RNA was then prepared using ISOGEN reagent (Nippon Gene) and the RNeasy Mini kit (Qiagen). First strands of cDNA were generated using the PrimeScript RT reagent Kit (Takara). The HAC1 cDNA was amplified from first strands of cDNA with Blend Taq (TOYOBO), and then analyzed by agarose gel electrophoresis. Detection, quantification, and statistical analysis was carried out by using a LAS-4000 (Fuji Film), ImageQuant (GE Healthcare), and Excel (Microsoft), respectively. The cDNAs of ERO1, KAR2, and SSK1, were quantitated by a quantitative real-time RT-PCR (qRT-PCR) method using a 7500 fast real-time RT-PCR system (Applied Biosystems) with SYBR Premix Ex Taq (Takara). A standard curve was generated from diluted RNA derived from wild-type cells, and levels of gene expression were normalized to ACT1 expression. HAC1 primers (CTGGCTGACCACGAAGACGC and TTGTCTTCATGAAGTGATGA) were used to monitor splicing of HAC1 mRNA. ERO1 primers (TAACAGCAAATCCGGAACG and ACCAAATTTGACCAGCTTGC), KAR2 primers (AGACTAAGCGCTGGCAAGCT and ACCACGAAAAGGGCGTACAG), SSK1 primers (AGCTGGAAGCAGGGAGAAAG and TGAGTGAGGGTTGGAAGGTG), and ACT1 primers (TGCCGAAAGAATGCAAAAGG and TCTGGAGGAGCAATGATCTTGA) were used to analyze the mRNA level of ERO1, KAR2, and SSK1.
Stress sensitivity
Assays for tunicamycin toxicity were carried out as follows. Cells were grown to exponential phase, and cultures were adjusted to an optical density of 0.5. Cell cultures were then serially diluted 5-fold, spotted onto normal plates or plates containing the indicated concentrations of tunicamycin, followed by incubation at 25°C for 3 days (for plates lacking or containing 0.1 μg/ml tunicamycin), 5 days (for plates containing 0.5 μg/ml tunicamycin) and 7 days (for plates containing above 1 μg/ml tunicamycin). Assays for DTT toxicity were carried out as follows. Cells were grown to exponential phase, and cultures adjusted to an optical density of 0.05 were incubated with YPD medium or YPD medium containing 4 mM DTT for 12 h at 25°C. The sensitivity to DTT was estimated by dividing absorbance units in the presence of DTT by absorbance units in the absence of DTT, and then the ratios of DTT sensitivities of the mutants/wild-type were calculated as the DTT sensitivity index.
Microscopy
To visualize GFP-tagged Hog1 in living cells, cells grown to exponential phase were incubated with SD medium containing 2 μg/ml tunicamycin or 4 mM DTT. Cells were then harvested at the indicated times, suspended in SD medium, and observed immediately using a Keyence BZ-X700 microscope (Keyence Corporation, Japan). Fluorescence intensities were quantified using Hybrid Cell Count BZ-H2C software (Keyence Corporation, Japan). To confirm nuclear localization of Hog1-GFP, cells were fixed for 10 min at 25°C by direct addition of 37% formaldehyde to a final concentration of 3.7%. Cells were then washed with PBS, stained with 4',6-diamidino-2-phenylindole (DAPI) and subjected to microscope observation. Images of Hog1-GFP in fixed cells were similar to those observed in living cells.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334 : 1081–1086. doi: 10.1126/science.1209038 22116877
2. Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006;13 : 385–392. 16397584
3. Mori K. Signalling pathways in the unfolded protein response: development from yeast to mammals. J Biochem. 2009;146 : 743–750. doi: 10.1093/jb/mvp166 19861400
4. Parsons AB, Brost RL, Ding H, Li Z, Zhang C, Sheikh B, et al. Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat Biotechnol. 2004;22 : 62–69. 14661025
5. Hedbacker K, Carlson M. SNF1/AMPK pathways in yeast. Front Biosci. 2008;13 : 2408–2420. 17981722
6. Broach JR. Nutritional control of growth and development in yeast. Genetics. 2012;192 : 73–105. doi: 10.1534/genetics.111.135731 22964838
7. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13 : 251–262. doi: 10.1038/nrm3311 22436748
8. Estruch F, Treitel MA, Yang X, Carlson M. N-terminal mutations modulate yeast SNF1 protein kinase function. Genetics. 1992;132 : 639–650. 1468623
9. McCartney RR, Schmidt MC. Regulation of Snf1 kinase. Activation requires phosphorylation of threonine 210 by an upstream kinase as well as a distinct step mediated by the Snf4 subunit. J Biol Chem. 2001;276 : 36460–36406. 11486005
10. Nath N, McCartney RR, Schmidt MC. Yeast Pak1 kinase associates with and activates Snf1. Mol Cell Biol. 2003;23 : 3909–3917. 12748292
11. Hong SP, Leiper FC, Woods A, Carling D, Carlson M. Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc Natl Acad Sci U S A. 2003;100 : 8839–8843. 12847291
12. Sutherland CM, Hawley SA, McCartney RR, Leech A, Stark MJ, Schmidt MC, et al. Elm1p is one of three upstream kinases for the Saccharomyces cerevisiae SNF1 complex. Curr Biol. 2003;13 : 1299–1305. 12906789
13. Tu J, Carlson M. REG1 binds to protein phosphatase type 1 and regulates glucose repression in Saccharomyces cerevisiae. EMBO J. 1995;14 : 5939–5946. 8846786
14. Sanz P, Alms GR, Haystead TA, Carlson M. Regulatory interactions between the Reg1-Glc7 protein phosphatase and the Snf1 protein kinase. Mol Cell Biol. 2000;20 : 1321–1328. 10648618
15. Hong SP, Carlson M. Regulation of Snf1 protein kinase in response to environmental stress. J Biol Chem. 2007;282 : 16838–16845. 17438333
16. Saito H, Posas F. Response to hyperosmotic stress. Genetics. 2012;192 : 289–318. doi: 10.1534/genetics.112.140863 23028184
17. Brewster JL, Gustin MC. Hog1 : 20 years of discovery and impact. Sci Signal. 2014;7: re7. doi: 10.1126/scisignal.2005458 25227612
18. Bicknell AA, Tourtellotte J, Niwa M. Late phase of the endoplasmic reticulum stress response pathway is regulated by Hog1 MAP kinase. J Biol Chem. 2010;285 : 17545–17555. doi: 10.1074/jbc.M109.084681 20382742
19. Torres-Quiroz F, García-Marqués S, Coria R, Randez-Gil F, Prieto JA. The activity of yeast Hog1 MAPK is required during endoplasmic reticulum stress induced by tunicamycin exposure. J Biol Chem. 2010;285 : 20088–20096. doi: 10.1074/jbc.M109.063578 20430884
20. Feng ZH, Wilson SE, Peng ZY, Schlender KK, Reimann EM, Trumbly RJ. The yeast GLC7 gene required for glycogen accumulation encodes a type 1 protein phosphatase. J Biol Chem. 1991;266 : 23796–23801. 1660885
21. Cannon JF, Pringle JR, Fiechter A, Khalil M. Characterization of glycogen-deficient glc mutants of Saccharomyces cerevisiae. Genetics. 1994;136 : 485–503. 8150278
22. Tatebayashi K, Takekawa M, Saito H. A docking site determining specificity of Pbs2 MAPKK for Ssk2/Ssk22 MAPKKKs in the yeast HOG pathway. EMBO J. 2003;22 : 3624–3634. 12853477
23. Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92 : 689–737. doi: 10.1152/physrev.00028.2011 22535895
24. Wurgler-Murphy SM, Maeda T, Witten EA, Saito H. Regulation of the Saccharomyces cerevisiae HOG1 mitogen-activated protein kinase by the PTP2 and PTP3 protein tyrosine phosphatases. Mol Cell Biol. 1997;17 : 1289–1297. 9032256
25. Jacoby T, Flanagan H, Faykin A, Seto AG, Mattison C, Ota I. Two protein-tyrosine phosphatases inactivate the osmotic stress response pathway in yeast by targeting the mitogen-activated protein kinase, Hog1. J Biol Chem. 1997;272 : 17749–17755. 9211927
26. Maeda T, Wurgler-Murphy SM, Saito H. A two-component system that regulates an osmosensing MAP kinase cascade in yeast. Nature. 1994;369 : 242–245. 8183345
27. Maeda T, Takekawa M, Saito H. Activation of yeast PBS2 MAPKK by MAPKKKs or by binding of an SH3-containing osmosensor. Science. 1995;269 : 554–558. 7624781
28. Ferrer-Dalmau J, Randez-Gil F, Marquina M, Prieto JA, Casamayor A. Protein kinase Snf1 is involved in the proper regulation of the unfolded protein response in Saccharomyces cerevisiae. Biochem J. 2015;468 : 33–47. doi: 10.1042/BJ20140734 25730376
29. Mattison CP, Spencer SS, Kresge KA, Lee J, Ota IM. Differential regulation of the cell wall integrity mitogen-activated protein kinase pathway in budding yeast by the protein tyrosine phosphatases Ptp2 and Ptp3. Mol Cell Biol. 1999;19 : 7651–760. 10523653
30. Chawla A, Chakrabarti S, Ghosh G, Niwa M. Attenuation of yeast UPR is essential for survival and is mediated by IRE1 kinase. J Cell Biol. 2011;193 : 41–50. doi: 10.1083/jcb.201008071 21444691
31. Rubio C, Pincus D, Korennykh A, Schuck S, El-Samad H, Walter P. Homeostatic adaptation to endoplasmic reticulum stress depends on Ire1 kinase activity. J Cell Biol. 2011;193 : 171–184. doi: 10.1083/jcb.201007077 21444684
32. Mizuno T, Hisamoto N, Terada T, Kondo T, Adachi M, Nishida E, et al. The Caenorhabditis elegans MAPK phosphatase VHP-1 mediates a novel JNK-like signaling pathway in stress response. EMBO J. 2004;23 : 2226–2234. 15116070
33. Maeda T, Tsai AY, Saito H. Mutations in a protein tyrosine phosphatase gene (PTP2) and a protein serine/threonine phosphatase gene (PTC1) cause a synthetic growth defect in Saccharomyces cerevisiae. Mol Cell Biol. 1993;13 : 5408–5417. 8395005
34. Gerwins P, Blank JL, Johnson GL. Cloning of a novel mitogen-activated protein kinase kinase kinase, MEKK4, that selectively regulates the c-Jun amino terminal kinase pathway. J Biol Chem. 1997;272 : 8288–8295. 9079650
35. Takekawa M, Posas F, Saito H. A human homolog of the yeast Ssk2/Ssk22 MAP kinase kinase kinases, MTK1, mediates stress-induced activation of the p38 and JNK pathways. EMBO J. 1997;16 : 4973–4982. 9305639
36. Takekawa M, Saito H. A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell. 1998;95 : 521–530. 9827804
37. Horie T, Tatebayashi K, Yamada R, Saito H. Phosphorylated Ssk1 prevents unphosphorylated Ssk1 from activating the Ssk2 mitogen-activated protein kinase kinase kinase in the yeast high-osmolarity glycerol osmoregulatory pathway. Mol Cell Biol. 2008;28 : 5172–5183. doi: 10.1128/MCB.00589-08 18573873
38. Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14 : 953–961. 9717241
39. Kaiser CA, Adams A, Gottschling DE. Methods in yeast genetics. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY; 1994.
40. Orlova M, Barrett L, Kuchin S. Detection of endogenous Snf1 and its activation state: application to Saccharomyces and Candida species. Yeast. 2008;25 : 745–754. doi: 10.1002/yea.1628 18949820
41. Kushnirov VV. Rapid and reliable protein extraction from yeast. Yeast. 2000;16 : 857–860. 10861908
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 9
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Arabidopsis AtPLC2 Is a Primary Phosphoinositide-Specific Phospholipase C in Phosphoinositide Metabolism and the Endoplasmic Reticulum Stress Response
- Bridges Meristem and Organ Primordia Boundaries through , , and during Flower Development in
- KLK5 Inactivation Reverses Cutaneous Hallmarks of Netherton Syndrome
- XBP1-Independent UPR Pathways Suppress C/EBP-β Mediated Chondrocyte Differentiation in ER-Stress Related Skeletal Disease
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy