
Synergistic and Dose-Controlled Regulation of Cellulase Gene Expression in
Cellulolytic fungi have evolved into sophisticated lignocellulolytic systems to adapt to their natural habitat. This trait is important for filamentous fungi, which are the main source of cellulases utilized to degrade lignocellulose to fermentable sugars. Penicillium oxalicum, which produces lignocellulolytic enzymes with more diverse components than Trichoderma reesei, has the capacity to secrete large amounts of cellulases. Meanwhile, cellulase expression is regulated by a complex network involved in many transcription factors in this organism. To better understand how cellulase genes are systematically regulated in P. oxalicum, we employed molecular genetics to uncover the cellulolytic transcription factors on a genome-wide scale. We discovered the synergistic and tunable regulation of cellulase expression by integrating cellulolytic regulators and their target genes, which refined our understanding of transcriptional-regulatory network as a “seesaw model” in which the coordinated regulation of cellulolytic genes is established by counteracting activators and repressors.
Published in the journal:
. PLoS Genet 11(9): e32767. doi:10.1371/journal.pgen.1005509
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005509
Summary
Cellulolytic fungi have evolved into sophisticated lignocellulolytic systems to adapt to their natural habitat. This trait is important for filamentous fungi, which are the main source of cellulases utilized to degrade lignocellulose to fermentable sugars. Penicillium oxalicum, which produces lignocellulolytic enzymes with more diverse components than Trichoderma reesei, has the capacity to secrete large amounts of cellulases. Meanwhile, cellulase expression is regulated by a complex network involved in many transcription factors in this organism. To better understand how cellulase genes are systematically regulated in P. oxalicum, we employed molecular genetics to uncover the cellulolytic transcription factors on a genome-wide scale. We discovered the synergistic and tunable regulation of cellulase expression by integrating cellulolytic regulators and their target genes, which refined our understanding of transcriptional-regulatory network as a “seesaw model” in which the coordinated regulation of cellulolytic genes is established by counteracting activators and repressors.
Introduction
Cellulolytic fungi have an inherent characteristic of cellulose deconstruction and can be used for bioconversion of insoluble plant cell wall polysaccharides into fermentable sugars [1–3]. The highly efficient production of their extracellular hydrolytic enzymes and other synergistic proteins [2,4], such as swollenin [5], plays a key role in reducing the cost of the biorefinery process [4]. However, incomplete knowledge of transcriptional regulatory networks for cellulolytic fungi has hampered the systematic improvement of cellulase production. These cellulolytic system genes are coordinately but differentially regulated in various cellulase producers [2,6]. Further characterization and manipulation of the cellulase regulatory network’s components will allow the rational engineering of cellulolytic fungi for improved enzyme production.
Transcriptional regulation of cellulolytic gene expression is central in controlling the carbohydrate hydrolysis process [6], and several positive or negative transcriptional factors of these degradative pathways were identified, such as the regulators encoded by the creA/cre1/cre-1 [7–9], xyr1/xlnr/xlr-1 [10–12], aceI [13], aceII [14], ace3 [15], clrB/clr-2/manR [16,17], and bglR [18] genes. The overexpression of these activators or deletion of some repressors is efficient in enhancing the cellulase and hemicellulase expressions [19,20]. However, the degree of cellulase induction differentially responds to these diverse regulator abundances. The transcription factor CreA, an ortholog of Migl from Saccharomyces cerevisiae [21], is a pivotal regulator mediating carbon catabolite repression (CCR) in filamentous fungi [7–9], and its deletion results in the obvious increase of cellulase expression and secretion. A transcriptional regulatory cascade that controls the xylanolytic genes between CreA and XlnR is also built in Aspergillus niger in response to preferred carbon sources [22]. In addition, the cre1 deletion mutant shows a conidiation formation defect [23]. Two novel zinc binuclear cluster transcription factors (CLR-1 and CLR-2) required for growth and enzymatic activity on cellulose were identified in Neurospora crassa [17]. The constitutive expression of clr-2 by the control of the promoter from ccg-1 is sufficient to drive cellulase gene expression when cultures are subjected to starvation [20]. In addition, the β-glucosidase regulator BglR and cellulase expression activator AceII were identified in Trichoderma reesei [18], but their orthologous encoding genes were absent in the Penicillium oxalicum genome [24]. Currently, the abilities to tune the expression abundance of just one transcription factor, as noted above, have profound effects on cellulase expression in these cellulolytic fungi [18–20,25]. However, whether such a simple mechanism could operate in the context of cellulolytic regulator combinations, including these characterized and novel transcription factors, remains unclear.
The P. oxalicum wild-type strain 114–2 was isolated from the soil in China more than 30 years ago [26]. A partially derepressed mutant JU-A10, which shows cellulolytic activity that is more than three times higher than that of its parent strain 114–2, was obtained after many rounds of mutagenesis and screening [26]. The mutant JU-A10 was further mutated to a cellulase hyper-producer JU-A10-T [26] and has been utilized in industrial processes for years. The clear genetic background provided by genome sequencing facilitated the rational improvement of these strains to enhance the expression of cellulolytic enzymes [24]. Currently, several structural genes associated with cellulase expression have been studied. The deletion of gene bgl2 (encoding the major intracellular β-glucosidase) [27] or PDE_01641 (the ortholog of N. crassa NCU05137) [28] results in the increase of cellulase production in P. oxalicum. In addition, three cellodextrin transporters (CdtC, CdtD, and CdtG) were identified, and their overexpression obviously increases the extracellular cellobiohydrolase activities [29]. However, evidence from diverse cellulolytic fungi showed that engineering cellulolytic transcription factors might have more efficacy in upregulating cellulase expression than merely manipulating the expression of structural genes for the major cellulases [1,19,20,30]. Subsequent studies to identify several specific regulators and their roles in regulating cellulase gene expression were conceptually appealing in cellulolytic fungi.
In this study, twenty transcription factors putatively involved in cellulase expression pathways were identified from a single-gene disruptant library. The single overexpression or deletion of these genes triggered cellulase expression to varying degrees, and synergistic and tunable cellulase expressions were observed in the combinations of the identified individual transcription factors. Furthermore, the responsiveness of the induction of cellulase expression by activator and relief from potential carbon catabolite repression to the internal signal cascades by the lack of the major intracellular β-glucosidase Bgl2 was also well-established. The suggested mechanisms of synergistic effects on cellulase expression might be general properties in cellulolytic fungi and broadly enable engineering strategies for the protein hyper-producers.
Results
Screening for novel transcription factors involved in P. oxalicum cellulose deconstruction
To decipher the transcriptional-regulatory network that governs cellulase expression in P. oxalicum, we first sought to identify the transcription factors (TF) that play roles in cellulolytic gene expression systematically. A total of 522 genes encoding sequence-specific regulators were predicted according to the protein sequence domain [24]. For the amplification of the flanking sequences of these TF-disrupting cassettes, primers were designed to meet the following criteria: GC-content 45%–60%, Tm: 50°C –60°C, and a length of 20 base pairs. The lengths of the 5’ and 3’ flanking regions ranged from 1.0 kb to 1.5 kb for each gene. The chimeric primers (S1 Table) for the amplification of upstream and downstream flanking fragments carried 25 bases of homologous sequence overlapping with the ends of ptra marker sequence [31]. A final fragment that contains target gene flanking sequences surrounding ptra was created by double-joint PCR [32] and transformed into the P. oxalicum Δpku70 mutant via protoplast transformation [33]. Pku70 and its homologs are involved in the non-homologous end joining (NHEJ) repair of double-strand breaks in diverse eukaryotes [32]. Considering the high homologous recombination frequency in the pku70 mutant (NHEJ-deficient background) [33], we selected three colonies per gene from these resulting transformants. The conidia from these primary transformants were purified by repeating the mono-spore isolation twice on the pyrithiamine resistance plates to obtain homokaryotic knockout mutants. The transcription factor gene replacement with ptra was verified by PCR-based screening. We found that the use of unpurified final amplicon of deletion cassettes resulted in almost 90% success in deleting the targeting genes in the Δpku70 mutant. Finally, a transcription factor mutant set, which bears a single deletion for 470 transcription factor genes in P. oxalicum, was successfully constructed.
The transcription factor deletion strains were screened and initially characterized for cellulose deconstruction on cellulose plates. According to the halos produced by the transformants on cellulose plates, 20 transcription factors that displayed putative roles in cellulase production were identified (Table 1). Twelve deletion strains exhibited increased cellulase activities and eight deletion strains exhibited decreased cellulase activities. These transcription factors represented negative and positive regulators of cellulose deconstruction, respectively. None of these transcription factors has been well characterized at the molecular level in P. oxalicum.

Among these transcription factor genes, PDE_05999, PDE_03168, PDE_07674 and PDE_03964 were previously known and encoded as ClrB, CreA, XlnR and AmyR regulators, respectively. The strongest effects on cellulose deconstruction observed in ΔclrB, ΔcreA, ΔxlnR and ΔamyR mutants indicated that these three genes encode the major regulators of some lignocellulolytic enzymes (Fig 1A). To clarify the mechanisms of lignocellulose deconstruction in P. oxalicum, we initially focused on the characterization of these central lignocellulolytic regulators ClrB, CreA, XlnR, and AmyR, and then identified their target genes involved in plant cell wall deconstruction. Up to date, mating assays in P. oxalicum have not been performed to remove the pku70 deletion through the crossing approach as T. reesei [34] and N. crassa [35] strains. We therefore constructed the corresponding mutants in the wild-type strain through the conventional transformation approach. Genomic DNA from putative transformants was analyzed by q-PCR (quantitative-PCR) and/or Southern blot (S1 Fig), and these transformants in which a single copy integration at the only a transcription factor gene locus were selected and further characterized.

Transcriptional regulation of cellulase genes by ClrB and the characterization of ClrB regulon
The function of clrB (PDE_05999) was identified independently in our lab. The regulator protein sequence has 39% (BioEdit, E Value = 1e-131) of identity to the homolog of N. crassa and 56% of identify (E Value = 0) to that of A. nidulans [17] (Table 1). The P. oxalicum clrB gene encodes a protein of 780 amino acid residues. Two introns of 76 and 64 nucleotides, which follow the characteristics of the clrB homolog in N. crassa, were identified [17]. The deduced transcription factor ClrB contains normal characteristics of Zn(II)2Cys6 binuclear cluster DNA binding motif near the N-terminus (residues 40–71) and the middle homology domain (residues 351–453) that is related to fungal specific transcription factors, including XlnR/XYR1 [10,11] and yeast regulatory protein GAL4 [36]. In this study, several putative cellulolytic transcription factors, such as PDE_03268, PDE_03964, and PDE_09881, also contain normal characteristics of these zinc binuclear cluster proteins (Table 1).
To investigate the influence of ClrB on cellulase expression, we constructed a ΔclrB strain from P. oxalicum wild-type strain 114–2 (CGMCC 5302). The ΔclrB strain displayed significantly reduced growth on cellulose plate, but identical phenotype on glucose, xylan, or potato dextrose agar (PDA) plates relative to wild-type strain (Fig 1A). The ΔclrB mutant exhibited dramatically reduced cellulase activities when compared with the wild-type strain (Fig 1B–1D), similar to the recent findings with clr-2/clrB in N. crassa/A. nidulans [17]. Northern blot analysis was used to study the cellulolytic gene cbh1 (PDE_07945), eg2 (PDE_09226), and xyn1 (PDE_08094) expressions in the wild-type and ΔclrB strains grown on cellulose. Fig 2A shows that the mRNA levels of cbh1 and eg2 in the ΔclrB mutant significantly decreased and could hardly be detected. As a result, a slight decrease in the xyn1 transcription level was observed in the ΔclrB strain compared with that in the wild-type control, while a low transient increase expression of xyn1 was observed at the fourth hour following the shift (Fig 2A). The introduction of a wild-type copy of clrB at the clrB locus (strain RclrB) completely restored the growth defects of the ΔclrB mutant in cellulose, as well as the cellulolytic enzyme activities of culture supernatants (Fig 1B–1D). These results demonstrated that ClrB might be in the central part of the transcriptional-regulatory network of cellulase expression by controlling the transcription of cellulolytic genes.

The P. oxalicum genome contains approximately 10,000 genes and is predicted to encode 18 cellulases, 51 hemicellulases, and other cellulolytic enzymes involved in plant biomass degradation [24]. Therefore, to build a comprehensive picture by which P. oxalicum responds to cellulose, we adopted RNA-Seq to measure genome-wide mRNA abundances in the P. oxalicum wild-type strain and ΔclrB mutant when exposed to Vogel’s minimal medium containing 2% cellulose for 4 hours. The three biological replicates of each strains showed a high Pearson correlation (S2 Fig). A total of 224 genes were differentially expressed between the ΔclrB and the wild-type strains on cellulose (S2 and S3 Tables). Of these genes, 103 genes showed lower transcription levels in the ΔclrB mutant than in the wild-type strain (S2 Table). These genes of decreased expression in clrB regulon were subjected to gene ontology enrichment analysis. Percentages of the genes distributed within each functional category are shown in Fig 3. Among these downregulated genes, 24 genes encoding transporters were enriched, including PDE_00607 (p = 7.99e-173), encoding cellodextrin transporter CdtC [29], and PDE_06576 (p = 2.96e-58), encoding putative maltose permease, which suggests that ClrB might also be involved in the cellodextrin and maltose metabolisms. In total, 32 genes encoding carbohydrate-active enzymes (CAZymes) were included, including 9 main cellulase genes and two of 11 β-glucosidases PDE_00579 (p = 1.98e-109) and PDE_04251 (p = 2.39e-42) (S2 Table), which demonstrates that the genes involved in plant cell wall deconstruction were significantly enriched. Only 6 of the 51 hemicellulase genes showed obvious reduction in transcription levels in the absence of ClrB, including PDE_02101 (p = 0.0033), PDE_06649 (p = 0.00046), PDE_01302 (p = 5.63e-12), PDE_09710 (p = 3.09e-09), PDE_05998 (p = 1.36e-37), and PDE_06023 (p = 1.87e-41) (S2 Table). These data demonstrated that ClrB might play a significant role in activating cellulase gene expression but has differential regulatory effects on cellulolytic and xylanolytic genes in the early inducing phase on cellulose (4 hours post-transfer).

In total, 121 genes showed higher transcription levels in the ΔclrB mutant than in the wild-type strain (S3 Table). Among these upregulated genes, only 7 genes encoding CAZy proteins, including two predicted hemicellulase genes PDE_07585 (p = 0.02) and PDE_08238 (p = 0.013), showed altered expressions. No cellulase, β-glucosidase, and xylanase genes were included. Genes in this set were enriched in oxidoreductase activity (p = 8.1e-6).
Overexpression of clrB increased cellulase expression
To assess whether the overexpression of clrB enhanced the cellulase expression, two clrB overexpression recombinants were constructed under the control of its native promoter (strain OEclrB) and the A. nidulans gpdA promoter (strain gpdA(p)::clrB) in P. oxalicum wild-type strain [37], respectively. Both the OEclrB and gpdA(p)::clrB strains showed varied halos on 1% cellulose plates (Fig 1A) and showed almost 2.5 - and 4.1-fold increases in filter paper enzyme activity (FPA), 2.5 - and 4.0-fold increases in cellobiohydrolase (pNPCase) activity, and 8.7 - and 16.5-fold increases in endoglucanase (CMCase) activity when grown on cellulose for 48 hours, respectively (Fig 1B–1E). Northern blot analyses also showed that the mRNA levels of cbh1 and eg2 in the gpdA(p)::clrB mutant were much higher than those in the wild-type strain on cellulose (Fig 2A).
To further test whether cellulase production was tightly responsive to clrB transcriptional abundance, we reconstructed the PDE_02864(p)::clrB expression cassette in which the clrB open reading frames and 3’ untranslated region were under the control of the novel promoter from the PDE_02864 encoding 40S ribosomal protein S8. The gpdA(p)::clrB-PDE_02864(p)::clrB strain was constructed and showed even higher cellulase expression than those in the single gpdA(p)::clrB and PDE_02864(p)::clrB mutants on cellulose (S3 Fig). These results demonstrated that dose effect of clrB transcriptional abundance is important for the high expression for cellulases, and tunable cellulase expression may be controlled by the ClrB concentration under cellulose conditions.
ClrB and XlnR additively regulated cellulolytic gene expression
In addition to ClrB, P. oxalicum XlnR is another cellulolytic activator in the Zn2Cys6 binuclear cluster motif superfamily that was identified previously along with the homologous N. crassa XLR-1 [12] and T. reesei XYR1 [10]. To demonstrate whether XlnR showed a differential role in cellulolytic gene expression regulation, the ΔxlnR and gpdA(p)::xlnR mutants with constitutive expression for xlnR under the control of the A. nidulans gpdA promoter in the wild-type strain were constructed. The ΔxlnR mutant showed a slightly decreased growth on both cellulose and xylan media, but not on glucose or PDA plates (Fig 1A). Northern blot results also showed that weak expressions for cbh1 and eg2 transcripts and invisible xyn1 were observed in the ΔxlnR strain compared with that in the wild-type strain in the cellulose-containing medium (Fig 2A). These data indicated that P. oxalicum XlnR is a general transcription factor that regulates cellulase and xylanase expressions but not like T. reesei XYR1, which is the essential regulator that governed both cellulolytic and xylanolytic gene expressions [10].
To further analyze whether synergistic or additive effects for these two major cellulolytic activators ClrB and XlnR existed, the clrB was also overexpressed in the gpdA(p)::xlnR mutant, and the gpdA(p)::clrB-gpdA(p)::xlnR mutant that contains simultaneously overexpressed ClrB and XlnR was obtained. The gpdA(p)::clrB-gpdA(p)::xlnR strain showed 1.3-, 1.7 - and 2.1-fold increased expressions in FPA, xylanase, and pNPCase activities compared with that in the gpdA(p)::clrB strain after shift to cellulose for 96 hours (Fig 4A–4C), but decreased production in the pNPGase activity (Fig 4D). Conversely, lack of both ClrB and XlnR (ΔclrB-ΔxlnR) led to a greater abrogation of cellulase and xylanase expression than each absence mutation under cellulose growth conditions (Figs 1B and 2A). These data revealed that ClrB and XlnR had additive effects on positively regulating the cellulase and hemicellulase gene expressions, and the high-abundance transcripts of ClrB and XlnR could facilitate the induction of cellulase expression under cellulose growth conditions.

CreA repressed the expression of cellulolytic genes and had a severe effect on morphology
PDE_03168 (CreA), as a homolog of the major carbon catabolite repressors in phylogenetically diverse fungi [7–9], played a negative role in the degradation of plant cell wall polymers. In this study, the ΔcreA mutant of P. oxalicum consumed cellulose faster than the wild-type strain (Fig 1A), similar to the findings in T. reesei cre1 [23] or N. crassa cre-1 deletion strains [38]. The ΔcreA strain grown on cellulose exhibited significantly increased cellulase activities compared with its parent strain, and showed almost 7.2-, 2.2-, 8.0-, and 4.4-fold increases in FPA, pNPCase, CMCase, and xylanase activities after shifting to cellulose for 96 hours, respectively (Fig 1B–1E). The ΔcreA mutant exhibited higher steady state amounts of cbh1 and eg2 mRNA than that in the wild-type strain by Northern blot analysis (Fig 2A) and q-PCR experiments (Fig 2B) under cellulose growth conditions. However, the gpdA(p)::creA mutant that contains an overexpression of creA showed significantly lower cellulase gene expression than that in the wild-type strain when grown on cellulose (Fig 2B), which indicates that cellulolytic gene expression under CCR mediated by the CreA in P. oxalicum was responsive to the creA transcript abundances.
Although the ΔcreA mutant produced higher cellulase expression than its parental strain P. oxalicum 114–2, we could not entirely exclude the possibilities that the increase of cellulolytic enzyme in ΔcreA mutant might be the partial cause of the enhancement of cellulolytic activators for ClrB and XlnR. To test these hypotheses, q-PCR was performed and the expression levels for clrB and xlnR in the ΔcreA mutant showed near to that in the wild-type strain on cellulose but obvious increase under glucose-repressing conditions (Fig 5A). These data indicated that CreA repressing the expression of clrB and xlnR was associated with the carbon source used in the medium, and CreA and ClrB, as well as CreA and XlnR might form a transcriptional cascade that regulates the cellulolytic gene expression in P. oxalicum.

Previously, we reported that a cellulase hyper-producer P. oxalicum mutant JU-A10, which bears a shift mutation at creA locus, has an extremely severe effect on morphology, including short and thicker hyphae [1]. The creA deletion in P. oxalicum wild-type strain 114–2 showed smaller colonies and reduced hyphae growth (Fig 1A). Under the microscope, the ΔcreA mutant on glucose plates exhibited considerably robust hyphae (Fig 5B). Thus, some hyphae morphology mutation in P. oxalicum mutant JU-A10 [1] might be specific results caused by the shift mutation at the creA location in the JU-A10 mutant by chemical mutagenesis. Similarly, the T. reesei Δcre1 mutant also displayed shorter and more robust hyphae than its parental strain [23]. We hypothesized that the morphology mutation caused by creA homologs may be general in filamentous fungi. However, whether hyphae morphology mutation was related to the increase in protein production and cellulase expression in these mutants containing deletion of creA needs to be further investigated in the future.
Simultaneous overexpression of ClrB and lack of CreA caused strong synergistic effects on cellulase expression
Cellulase genes were responsive to both major opposing regulators ClrB and CreA. First, whether the low cellulase expression exhibited by the ΔclrB mutant could be recovered when knocking out the CreA encoding gene remains unknown. To bypass this problem, we deleted the creA in the ΔclrB strain and obtained the ΔcreA-ΔclrB mutant. Northern blot, presented in Fig 2A, shows that ΔcreA-ΔclrB mutant exhibited a slightly higher amount of cbh1 mRNA than that in the ΔclrB strain. The ΔcreA-ΔclrB mutant also showed 3.1-, 0.5-, 3.1-, and 1.8-fold increases in FPA, pNPCase, CMCase, and xylanase activities relative to wild-type strain after shifting to cellulose for 96 hours, respectively (Fig 1B). In contrast to the ΔclrB mutant, the ΔcreA-ΔclrB strain produced visible halo in the cellulose medium plate when cultured for 9 days (S4 Fig). These data indicated that the lack of CreA partially rescued the cellulase expression defect in the ΔclrB mutant. Conversely, the gpdA(p)::creA-gpdA(p)::clrB strain that contains simultaneous overexpressions of CreA and ClrB also showed increases in the transcription expression levels for most of the cellulase genes compared with that in the gpdA(p)::creA mutant (Fig 2B).
The strong cellulase production observed in the ΔcreA and gpdA(p)::clrB mutants indicated that these two genes encoded the major transcription factors that oppositely regulate cellulolytic gene expression. Although, the triple-mutant RE-10 (Δbgl2-ΔcreA-gpdA(p)::clrB) was recently obtained [39], it is still not known why high cellulase expression in which occurs. Therefore, we deleted creA in the gpdA(p)::clrB strain to determine whether their functions in cellulase production were synergistic. To our knowledge, no reports have been described to perform this to determine their synergistic effects on cellulase induction expression in other cellulolytic fungi. Interestingly, the strain with the combination of creA deletion and overexpression of clrB displayed a much larger halo around its colony than that of the ΔcreA and gpdA(p)::clrB mutants on cellulose plate (Fig 1A). The differences in the FPA, pNPCase, CMCase, and xylanase activities were even more pronounced (11.6-, 11.6-, 58.6-, and 15.9-fold higher, respectively) between the ΔcreA-gpdA(p)::clrB and wild-type strains (Fig 1B). The ΔcreA-gpdA(p)::clrB mutant exhibited higher steady-state amounts of cbh1, eg2, and xyn1 mRNA than that of the ΔcreA and gpdA(p)::clrB strains under cellulose growth conditions by Northern blot analysis (Fig 2A). Q-PCR results also indicated that all three cellobiohydrolase and 10 endoglucanase genes showed strong synergistic increases in transcription levels with the exception of five endoglucanase genes for PDE_009267, PDE_00698, PDE_03711, PDE_09014, and PDE_06768 when grown on cellulose (Fig 2B). In addition, three of the 11 β-glucosidase genes (PDE_00579, PDE_04251, and PDE_04859) showed similar induction patterns as the above thirteen cellulase genes in the ΔcreA-gpdA(p)::clrB mutant on cellulose (S5 Fig). These data showed that the simultaneous overexpression of ClrB and lack of CreA have synergistic effects on cellulolytic and xylanolytic gene expressions.
Transcriptome analysis of the combinatorial control by ClrB and CreA
To build a comprehensive picture by which P. oxalicum responds to cellulose, the genome-wide mRNA abundance in P. oxalicum wild-type strain was first measured on Vogel’s medium with no carbon or 2% glucose for 4 hours as an alternative reference. A total of 581 genes were differentially expressed between Avicel and no carbon cultures at the fourth hour (|log2(fold change)| > 1 and probability ≥ 0.8), 155 and 117 of which showed greater and lower expression levels on cellulose than that under either glucose growth or no carbon conditions, respectively (S4 and S5 Tables). We refer to this gene set, including 272 genes, as a “cellulose regulon” for P. oxalicum. The cellulose regulon encompassed 10 of 18 predicted cellulase genes, 30 of 51 predicted hemicellulase genes, and 3 of 11 predicted β-glucosidase genes PDE_02736 (p = 1.65e-07), PDE_00579 (p = 9.24e-174), and PDE_04251 (p = 1.02e-80) (S4 and S5 Tables). To further elucidate the regulation mechanisms for the opposing regulators CreA and ClrB in the gene expression of P. oxalicum under cellulose growth conditions, three biological replicates of ΔcreA, ΔcreA-ΔclrB, and ΔcreA-gpdA(p)::clrB mutants were also subjected to transcriptional profiling at the same condition. The three biological replicates of each mutant showed a high Pearson correlation (S6 Fig) and demonstrated the reliability of RNA-Seq. The gene ontology enrichment analyses for these 117 decreased genes of cellulose regulon showed no statistically significant results with the threshold (FDR < 0.05).
The hierarchical clustering of expression patterns for the 155 increased genes of cellulose regulon in the wild-type strain and the ΔclrB, ΔcreA, ΔcreA-ΔclrB, and ΔcreA-gpdA(p)::clrB mutants displayed four classes of genes with similar expression patterns. A heat map that depicts the relative expression levels of selected genes from groups 1 to 4 is shown in Fig 6A.
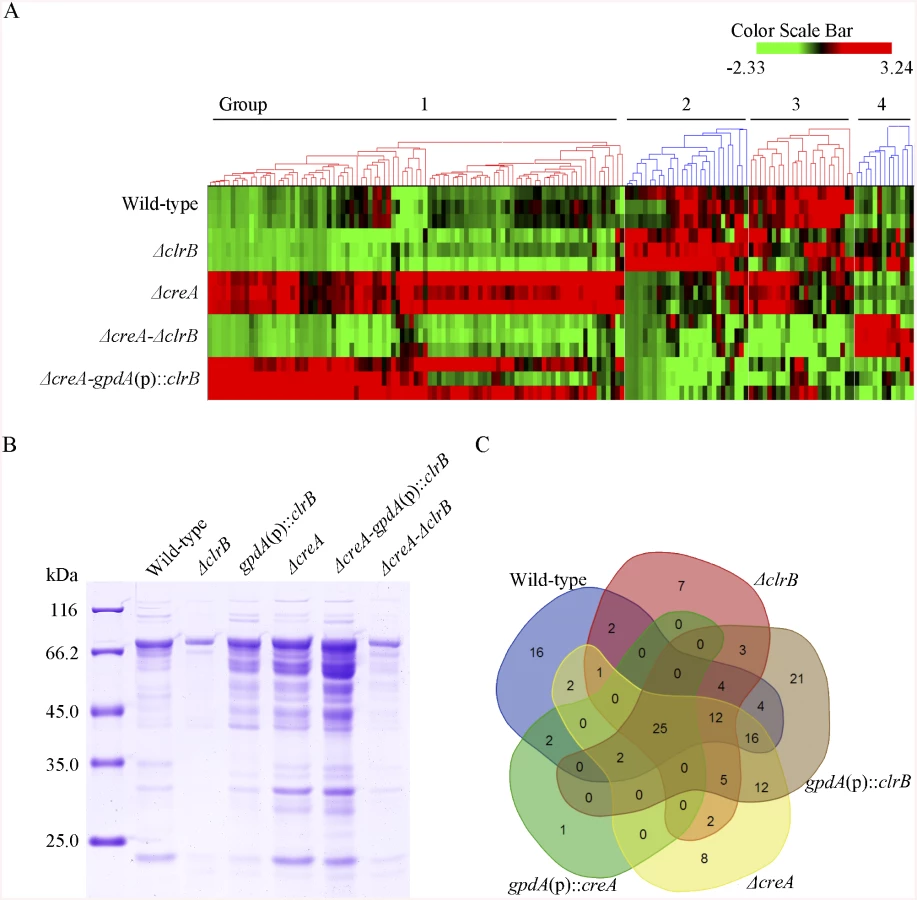
Group 1 consisted of 91 genes (S4 Table). Most of the genes in this set exhibited strong synergistic induction effects on transcriptional expression in the ΔcreA-gpdA(p)::clrB mutant (Fig 6A). In this cluster, we detected a significant enrichment of genes involved in hydrolase activities (p = 1.5e-30). This group included 9 of 18 cellulase genes (PDE_00507, PDE_09969, PDE_07929, PDE_07928, PDE_05633, PDE_09226, PDE_07124, PDE_07945, and PDE_01261), two β-glucosidase genes (PDE_00579 and PDE_04251), and three sugar transporter genes for cellodextrin transport-1 (PDE_00607), major facilitator superfamily (MFS) maltose transporter (PDE_06576) and monosaccharide transporter (PDE_04857), all of which showed significantly low levels in the ΔcreA-ΔclrB strain versus ΔcreA mutant, but similar transcription activities between the ΔcreA-ΔclrB and ΔclrB mutants, which indicates that the expression levels for these enriched cellulolytic genes were repressed by CreA but ClrB dependent under inducing conditions. Cellodextrin transport-2 genes (PDE_007257 and PDE_00753) were also CreA repressed and ClrB induced but not ClrB dependent. A total of 16 out of the 51 hemicellulase genes (PDE_06649, PDE_02101, PDE_00016, PDE_04478, PDE_01302, PDE_09278, PDE_06023, PDE_04182, PDE_08094, PDE_02514, PDE_00752, PDE_09710, PDE_06067, PDE_05998, PDE_02418, and PDE_07897) were enriched in this subset, and most of which were significantly repressed by CreA, but only four genes (PDE_05998, PDE_06023, PDE_01302, and PDE_09710) were ClrB induced. Noticeably, the expression levels for PDE_05998 (beta-mannosidase) and PDE_06023 (beta-1, 4-mannanase) were ClrB dependent. Interestingly, three genes involved in starch degradation, encoding starch binding domain-containing protein (PDE_01354), glucoamylase (PDE_05527), and α-amylase (PDE_01201) were also downregulated in the ΔclrB mutant and upregulated in the ΔcreA mutant, which suggests that the expression for these amylase genes were tightly co-regulated with the cellulase genes under cellulose growth conditions. A significant enrichment of genes (PDE_09832, PDE_04496, and PDE_09715) involved in the sporulation process (p = 8.2e-5) was also detected. In addition, 21 genes that encode hypothetical proteins were within this dataset (23%). The above data suggested that this cluster might represent the most central components associated with cellulose deconstruction, which indicates that this large synergistic activation by engineering CreA and ClrB might be specific to these cellulolytic target genes.
Group 2 consisted of 27 genes (S4 Table). The expression levels of this set were partially induced in the ΔclrB mutant and repressed in the ΔcreA mutant, but were significantly downregulated in the ΔcreA-gpdA(p)::clrB strain compared with that in the wild-type strain (Fig 6A). Although GO-term analyses revealed that no statistically significant results with the threshold (FDR < 0.05), genes that encode β-glucosidase (Bgl1, PDE_02736), endo-β-1, 4-glucanase (PDE_09267), and two β-xylosidases (PDE_07334 and PDE_08037) were included in this dataset.
Group 3 consisted of 23 genes (S4 Table), and the expression levels of which were partially repressed in the ΔcreA and ΔclrB mutants, and were cumulatively repressed in the ΔcreA-ΔclrB mutant, but were partially recovered in the ΔcreA-gpdA(p)::clrB mutant (Fig 6A). The GO-term analyses of the dataset of 23 genes showed a significant enrichment in the carbohydrate metabolic process (p = 3.6e-7). Five hemicellulase genes that encode PDE_06306, PDE_03572, PDE_07080, PDE_03573, and PDE_08036 and one α-glucosidase (PDE_00400) were also within this group.
Group 4 consisted of 13 genes (S4 Table). The group genes were slightly induced in the ΔclrB mutant and showed no induction in the ΔcreA mutant, but were significantly induced in the ΔcreA-ΔclrB mutant compared with that in the wild-type strain (Fig 6A). One gene that encodes β-xylosidase (PDE_00049) was within this dataset, and eight genes encoded hypothetical proteins.
Secretome analysis of the combinatorial control by ClrB and CreA on cellulose
Secreted proteins are expected to play a crucial role in cellulose degradation because of the nature of cellulolytic system for the deconstruction of plant cell walls. However, whether the cellulolytic protein concentration in secretome potentially correlates with their relative mRNA levels, and how this induction stimulus under cellulose conditions causes P. oxalicum to manipulate its secretome to facilitate cellulose degradation remain unclear. Thus, extensive secretome surveys using label-free LC-MS/MS were conducted to analyze the secretomes under cellulose conditions systematically. Herein, a supernatant from 4-day old wild-type culture grown on cellulose was digested with trypsin and analyzed by LC-MS-MS. For this purpose, 157 nonredundant proteins (P-value < 0.01) were identified based on a single or several peptide entries from the P. oxalicum protein database, the pI of which was concentrated in a pH range of 4–7, and 86 proteins were predicted to be secreted based on SignalP computational analysis (SignalP 4.1 Server, http://www.cbs.dtu.dk/services/SignalP/) (S6 Table). Proteins with predicted activities on carbohydrates in the P. oxalicum wild-type secretome dataset existed, including 10 of 18 predicted cellulases, one β-glucosidase Bgl1, and 10 of 51 predicted hemicellulases (S6 Table).
Subsequently, a question was posted with regard to the extent to which ClrB can be attributed to the regulation of extracellular protein abundances in the cellulolytic system. The total protein in the ΔclrB mutant culture supernatant was only 30% of that in the wild-type strain (Fig 6B). To characterize the secretome changes in response to the crucial regulator ClrB further, a total of 104 predicted secreted proteins in the gpdA(p)::clrB strain (S6 Table) and 61 predicted secreted proteins in the ΔclrB mutant (S6 Table) were identified after the shift to 2% cellulose for 4 days, respectively. These observations demonstrate that ClrB significantly increased the number of extracellular proteins on cellulose. The ΔclrB secretome dataset under cellulose growth condtions included only 5 of 18 predicted cellulases and 5 of 51 predicted hemicellulases in P. oxalicum genome. The gpdA(p)::clrB secretome dataset included 13 of 18 predicted cellulases and 10 of 51 predicted hemicellulases. A comparison between the secretomes of ΔclrB and those of gpdA(p)::clrB grown on cellulose showed that only 49 proteins overlapped (S6 Table), including 5 of 18 cellulases and 4 of 51 hemicellulases. These data indicate that ClrB enhanced various cellulolytic enzymes, including their secretion strength. More importantly, the observed changes of cellulolytic proteins in ΔclrB and gpdA(p)::clrB strains highly correlated with their corresponding mRNA abundances and broadly mirrored the ClrB-specific positive roles for the transcript expression of cellulolytic genes (S6 Table).
Concurrently, to evaluate the CreA-influenced extracellular proteins, we first used sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) to analyze the secretomes of ΔcreA and gpdA(p)::creA cultures under cellulose growth conditions (Fig 6B). The protein pattern of ΔcreA mutant showed more bands than that of gpdA(p)::creA mutant on SDS-PAGE (Fig 6B). The total protein concentration in ΔcreA mutant culture supernatant was 2.6-fold higher than that in the wild-type strain (Fig 6B). We further investigated the extracellular proteins influenced by CreA by adopting the label-free LC-MS/MS analysis to identify and quantify the proteins. A total of 85 and 30 predicted secretion proteins in ΔcreA and gpdA(p)::creA mutants were identified when grown on cellulose (S6 Table), respectively. The SDS-PAGE and LC-MS/MS analysis results for culture secretomes revealed that protein secretion, including protein abundance and distribution, was dramatically repressed by CreA under cellulose growth conditions.
The secretome of ΔcreA-gpdA(p)::clrB mutant was investigated under cellulose conditions to further identify the regulon with ClrB and CreA synergistic effects at the secretome levels. The total amount of the secreted protein in ΔcreA-gpdA(p)::clrB culture supernatant displayed a 7.5-fold increase compared with that in the wild-type strain (Fig 6B). Label-free LC-MS/MS analysis was used, and 97 predicted secretion proteins were identified in ΔcreA-gpdA(p)::clrB mutant (S6 Table). These proteins included 10 of 18 cellulases, and 15 of 51 hemicellulases. No β-glucosidase was detected in ΔcreA-gpdA(p)::clrB mutant.
To assess differences more accurately in protein distribution, the secretomes from ΔclrB, gpdA(p)::clrB, ΔcreA, gpdA(p)::creA, and ΔcreA-gpdA(p)::clrB mutants were combined to locate targets that were the basal components in P. oxalicum secretomes under cellulose growth conditions (Fig 6C and S6 Table). In these datasets, we identified that 25 proteins overlapped (S6 Table), including two cellobiohydrolases (i.e., PDE_07945 and PDE_07124), three endoglucanases (i.e., PDE_09969, PDE_07929, and PDE_09226), four hemicellulases (i.e., PDE_02101, PDE_06023, PDE_04182, and PDE_08094), two extracellular membrane proteins (i.e., PDE_08075 and PDE_02536) that contain common in fungal extracellular membranes domain, three amylases (i.e., PDE_01201 (alpha-amylase Amy13A), PDE_01354 (protein with starch binding domain), and PDE_09417 (glucoamylase GluA/Amy15A)), and five hypothetical proteins (i.e., PDE_03934, PDE_06089, PDE_07106, PDE_09289, and PDE_00667). All these proteins existed in the wild-type strain secretome under the same culture condition (S6 Table). After a 4-h shift from no carbon source, the transcription expression levels of 14 of these proteins increased in cellulose versus those in glucose (S6 Table).
Bgl2 expression level was ClrB-dependent and deletion of bgl2 facilitates the synergistic induction expression of cellulase genes in P. oxalicum
The above data imply that β-glucosidase gene bgl2 (PDE_00579) was the major object regulated by ClrB and CreA at the level of transcription (S5 Fig). However, lack of CreA in the ΔclrB mutant could not recapitulate bgl2 expression level (S5 Fig), suggesting that bgl2 expression level was strictly ClrB-dependent under induction conditions. Considering the upregulation of cellulolytic genes in Δbgl2 mutant [27], we speculated that the expression levels for cellulolytic genes were further enhanced via the overexpression of clrB or deletion of creA in a bgl2 deletion background. In support of this hypothesis, we first constructed Δbgl2-gpdA(p)::clrB and Δbgl2-ΔcreA mutants. The cellulase gene transcription levels and cellulase activities, which were greater than those in each single mutation strain on cellulose (Fig 7A and 7B, and S7A and S7B Fig), as well as the Δbgl2-ΔcreA strains exhibited even more cellulase productions than Δbgl2-gpdA(p)::clrB mutant on cellulose (Fig 7A and 7B, and S7A and S7B Fig). These results suggest that the decrease of intracellular β-glucosidase activity may facilitate the transcriptional induction of cellulolytic genes.

Cellulase gene expression depends on the presence of the inducers and on the positive regulation of activators in cellulolytic fungi [2]. Recent studies indicated that the constitutive expressions of N. crassa clr-2 [20] and T. reesei xyr1 [40] could recapitulate the response to cellulose when incubated without carbon. To assess whether the constitutive expression of clrB, deletions of bgl2 or creA, or combination of these genetic manipulations was sufficient for the induction of cellulase genes independent of inducers, the cellulase expression levels in Δbgl2, gpdA(p)::clrB, ΔcreA, Δbgl2-gpdA(p)::clrB, Δbgl2-ΔcreA, and Δbgl2-gpdA(p)::clrB-ΔcreA mutants were evaluated as opposed to the wild-type strain when cultures were shifted from a glucose medium to a carbon-free medium for 4 hours. The findings revealed that Δbgl2-gpdA(p)::clrB mutant exhibited even more transcriptional abundances on carbon-free medium than on cellulose (Fig 7C and S7C Fig), whereas the “starvation response” for cellulase expression also occurred in Δbgl2-ΔcreA and RE-10 mutants (Fig 7C and S7C Fig). However, such a response was significantly low compared with that in Δbgl2-gpdA(p)::clrB mutant (Fig 7C and S7C Fig). Consistent with these results, the pNPCase and CMCase activities were more than 10-fold higher in Δbgl2-gpdA(p)::clrB strain than those in each single mutant and wild-type strain under carbon-free conditions (S8A and S8B Fig). The creA, amyR and xlnR transcription abundances in Δbgl2-gpdA(p)::clrB, Δbgl2-ΔcreA, Δbgl2-gpdA(p)::clrB-ΔcreA and wild-type strains were assayed to test whether CreA, AmyR and XlnR mediated the synergistic induction in Δbgl2-gpdA(p)::clrB strain when subjected to starvation. The findings indicated that amyR had a 7.7-fold decrease, whereas creA had a 4.3-fold increase and clrB had a 17.5-fold increase in Δbgl2-gpdA(p)::clrB mutant versus that in the wild-type strain (Fig 7D). These data signify that AmyR may share a key role in the “starvation response” for cellulolytic genes and provide a novel insight into the cellulase gene regulatory mechanisms during energy abstinence.
Improvement of P. oxalicum cellulase production via reconstruction of expression regulation network (RERN)
Given the dose-controlled or additive regulation of cellulase genes by ClrB and XlnR presented in gpdA(p)::clrB-PDE_02864(p)::clrB, gpdA(p)::xlnR, and gpdA(p)::clrB-gpdA(p)::xlnR mutants (Fig 4A–4D and S3 Fig), and the synergistic transcriptional induction of cellulolytic genes in Bgl2-deficient background (Fig 7A and 7B, and S7A and S7B Fig), we assessed whether the dose effects of ClrB and XlnR transcriptional abundance were feasible in further enhancing the cellulase expression in triple-mutant RE-10 [39]. We examined this hypothesis by reconstructing two overexpression cassettes (i.e., PDE_02864(p)::clrB-sur and PDE_02864(p)::xlnR-sur), in which the sur cassette (conferring resistance to sulfonylurea) was used as a resistance marker. These overexpression cassettes for clrB and xlnR were separately transformed into RE-10 [39]. The quadruple mutants RE-27 (Δbgl2-ΔcreA-gpdA(p)::clrB-PDE_02864(p)::clrB) and RE-29 (Δbgl2-ΔcreA-gpdA(p)::clrB-PDE_02864(p)::xlnR) were obtained, and their cellulase expression abilities were separately evaluated on cellulose and wheat bran media. Although all these experiments were performed in flasks, both RE-27 and RE-29 mutants showed more cellulolytic and xylanolytic enzyme activities and secretion abilities than RE-10 (Fig 8A–8C and 8E, and S9A–S9F Fig). When grown on a medium with 2% of cellulose as a sole carbon source for 120 h, RE-27 mutant displayed 62.3%, 34.8%, 288.5% and 28.0% greater FPA, pNPCase activity, xylanase activity and total secreted protein level, but 26.3% lower pNPGase activity, respectively, than RE-10 (S9A–S9E Fig). Similarly, the RE-29 mutant showed 55.3%, 44.1%, 255.2% and 20.6% greater FPA, pNPCase activity, xylanase activity and total secreted protein level, but 39.8% lower pNPGase activity, respectively (S9A–S9E Fig). We also observed a significant decrease in amyR expression level for both RE-27 and RE-29 mutants as compared to the wild-type strain on cellulose by q-PCR (Fig 8F). The findings further signify that AmyR may share a key role in the regulatory network for cellulolytic genes. When grown on a wheat bran medium, RE-27 mutant exhibited FPA (8.85±0.66 U/mL), CMCase activity (31.25±0.77 U/mL), xylanase activity (1341.97±172.94 U/mL), amylase activity (125.07±1.32 U/mL) and total secreted protein concentration (16.40±1.08 g/L), and RE-29 mutant also displayed FPA activity (7.58±0.34 U/mL), CMCase activity (31.03±0.29 U/mL), xylanase activity (1285.93±11.12 U/mL), amylase activity (148.82±3.19 U/mL) and total secreted protein concentration (16.16±0.72 g/L), respectively (Fig 8A–8E). These data signify that the dose-controlled regulation mechanisms of the cellulolytic regulators are a promising strategy for cellulolytic fungi to develop enzyme hyper-producers via the RERN technology.

AmyR repressed cellulolytic gene expression and amyR expression was regulated by ClrB
In the above P. oxalicum cellulose regulon and basal secretome components, some enzymes involved in starch degradation were tightly associated with the cellulolytic protein expression on cellulose. The “starvation response” in Δbgl2-gpdA(p)::clrB mutant also dramatically decreased at the amyR expression level under carbon-free conditions. Therefore, P. oxalicum amyR (PDE_03964), an Aspergillus oryzae amyR homolog [41], was considered tightly associated with cellulolytic enzyme production. The strain with the deletion of amyR exhibited visible varying halos on cellulose and starch plates, as well as an identical phenotype on glucose relative to its parental strain (Fig 9A). As such, this strain demonstrated its differential roles in amylase and cellulase expressions. Moreover, this condition suggests that ΔamyR mutant has no defects in glucose uptake, sensing, or metabolism. The FPA in an amyR knockout mutant was about 1.6-fold higher than that in the wild-type strain of P. oxalicum (Fig 9B), while the amyR deletion reduced amylase under cellulose growth conditions (Fig 9C). ΔamyR mutant also displayed higher amounts of cbh1 and eg2 mRNA than that in the wild-type strain according to the results of northern blot (Fig 9D). Nonetheless, such mutant was deficient for transcribing the major glucoamylase gene gluA (PDE_09417) when grown on cellulose. This observation implies that AmyR was the main activator for amylase expression, and it repressed cellulase expression in response to the utilization of cellulose sources.
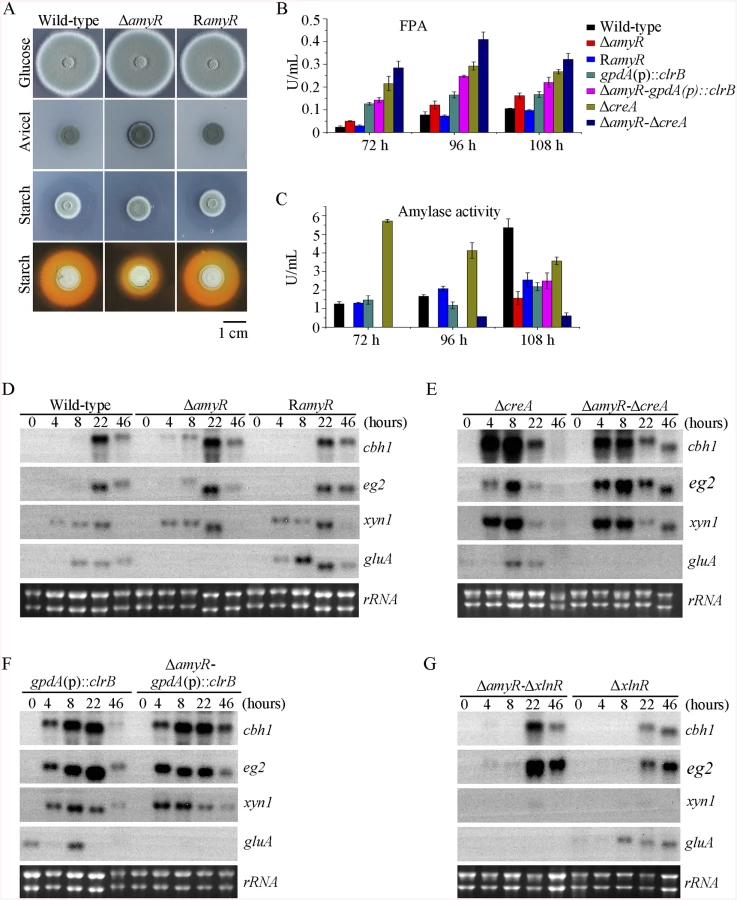
We constructed ΔamyR-gpdA(p)::clrB and ΔamyR-ΔcreA mutants to investigate whether AmyR plays a negative role in the synergistic/additive transcriptional activation of cellulolytic genes. As predicted, both ΔamyR-gpdA(p)::clrB and ΔamyR-ΔcreA mutants produced more cellulase activities than the strains that contain each individual mutation (Fig 9B) and showed higher transcription levels for cellulase genes under cellulose growth conditions (Fig 9E and 9F). The additive regulation for cellulase gene expression also existed in ΔamyR-ΔxlnR and ΔamyR-Δbgl2 mutants on cellulose (Fig 9G and S10A Fig). Therefore, the deletion of amyR in triple-mutant RE-10 (Δbgl2-ΔcreA-gpdA(p)::clrB) might further enhance cellulase expression under cellulose conditions. This premise also holds true in RE-27 and RE-29 mutants. Correspondingly, we constructed ΔamyR-Δbgl2-ΔcreA-gpdA(p)::clrB quadruple mutant (strain RE-30). However, the resulting strain RE-30 did not obtain greater cellulase expression than its parental triple-mutant RE-10 (S10B and S10C Fig), but AmyR still contributed to the functions of activating amylase expression in RE-10 on cellulose (S10D Fig). These results demonstrate that the deletions of amyR in RE-10 mutants were less effective for inducing cellulase expression than those in wild-type, gpdA(p)::clrB, ΔcreA, and Δbgl2 strains on cellulose.
The fact that RE-30 mutant produced cellulolytic enzymes near RE-10 implies that AmyR played a significantly different regulation activity for cellulase expression in RE-10 mutant than the wild-type strain under cellulose growth conditions. Considering the low expression of amyR in Δbgl2-gpdA(p)::clrB mutant under carbon-free conditions (Fig 7D), we hypothesized that the transcriptional abundance for amyR was also downregulated in RE-10 on cellulose. As predicted, we first observed a significant decrease in ΔcreA-gpdA(p)::clrB strain in RNA-seq data (RPKM: 124.5±5.8 in ΔcreA, 20.0±3.1 in ΔcreA-gpdA(p)::clrB for amyR versus 211.1±2.3 in the wild-type). The q-PCR experiments also revealed that the amyR expression was synergistically downregulated in the RE-10 mutant (Figs 7D and 10A). These data suggest that ClrB and CreA were supposed to participate in the control of the transcriptional response of amyR gene upon exposure to cellulose because the expression of amyR was decreased in gpdA(p)::clrB and ΔcreA mutants, and increased in ΔclrB and gpdA(p)::creA mutants (Fig 10A). The deletion of bgl2 also resulted in the decreased expression of amyR, which may also facilitate the decreased expression of amyR in ΔcreA-gpdA(p)::clrB (Fig 10A). In other words, no differential expression for cellulase expression between RE-30 and RE-10 mutants may be tightly related to the dramatic deregulation of amyR in RE-10 (Figs 7D and 10A). By contrast, the synergistic increase of cellulase induction in ΔcreA-gpdA(p)::clrB and RE-10 mutants may be partially a consequence of the decreased expression of amyR. The extent to which and how AmyR is involved in cellulase expression regulated by ClrB and CreA is still uncertain.

To gain insight into the molecular mechanism that underlies the AmyR-regulated cellulolytic gene expression on cellulose, we evaluated the global changes in ΔamyR mutants with three biological replicates by RNA-Seq. Consequently, 71% of the reads were mapped to the P. oxalicum 114–2 reference genome. The biological replicates of each ΔamyR mutant showed a high Pearson correlation (S6D Fig) and demonstrated the reliability of RNA-Seq. Given a |log2(fold change)|>1 and probability≥0.8 as the threshold, we determined that 131 genes (S7 Table) were upregulated and 579 genes (S8 Table) were downregulated in response to the deletion of amyR compared with the wild-type, respectively.
We then compared the RNA-Seq data for the 579 upregulated genes with that from the wild-type strain and ΔamyR, ΔclrB, ΔcreA, and ΔcreA-gpdA(p)::clrB mutants. The hierarchical clustering of these genes revealed nine groups of genes with similar expression patterns (S11 Fig). The expression levels for groups 1 and 2 increased in ΔcreA and ΔcreA-gpdA(p)::clrB mutants. Group 1 consisted of 195 genes. Within this subset, the proteins with dolichyl-diphosphooligosaccharide-protein glycotransferase activity (p = 3.0e-8) were enriched. Similarly, five genes in this group involved in starch degradation (i.e., PDE_04151, PDE_09417, PDE_05527, PDE_01201, and PDE_01354) were enriched. This case demonstrates that AmyR is the main activator for amylase gene expression. Group 2 composed of 122 genes. The GO-term analysis of these genes displayed a significant enrichment in the molecular function categories of the structural constituent of ribosome (p = 2.6e-101) and rRNA binding (p = 1.8e-5). These results signify that AmyR may play an important role in the translation process (p = 1.8e-38). Groups 3 to 6, 8, and 9 showed no statistically significant results with cutoff (FDR<0.05) via GO-term analyses. Group 7 contained 67 genes, in which 20 genes were enriched in the carboxylic acid metabolic process (p = 9.02e-12).
We also compared the RNA-Seq data for the 131 upregulated genes with that from the wild-type strain and ΔamyR, ΔclrB, ΔcreA, and ΔcreA-gpdA(p)::clrB mutants. The hierarchical clustering of these genes revealed four groups of genes with similar expression patterns (Fig 10B).
Group 1 included 84 genes (S7 Table), which were induced in ΔclrB mutant but repressed in ΔcreA mutant, particularly in ΔcreA-gpdA(p)::clrB mutant. The GO enrichment analysis revealed the induced expressions of the subsets of genes involved in the cellular amino acid metabolic (p = 5.4e-10) and organic acid biosynthetic processes (p = 1.6e-5). One gene cluster (from PDE_01212 to PDE_01220) encoding unclassified proteins was also involved in this dataset.
Group 2 consisted of 22 genes that were induced in ΔcreA mutant (S7 Table), especially in ΔcreA-gpdA(p)::clrB mutant. The GO function annotation of this subset genes revealed that the genes for hydrolase activity (p = 8.2e-12) constitute the largest group, including nine cellulase genes (i.e., PDE_07124, PDE_07945, PDE_05193, PDE_05633, PDE_09226, PDE_07929, PDE_00507, PDE_07928, and PDE_01261), five hemicellulase genes (i.e., PDE_06649, PDE_02101, PDE_09278, PDE_06023, and PDE_08094), β-glucosidase-encoding genes (i.e., PDE_00579 and bgl2), swollenin (i.e., PDE_02102), acetylesterase (i.e., PDE_05194), ABC multidrug transporter (i.e., PDE_07165), and formyltetrahydrofolate deformylase (i.e., PDE_07944). This group also contained cellodextrin transport-1-encoding genes (i.e., PDE_00607), a tetratricopeptide repeat protein (i.e., PDE_08095), and a hypothetical protein (i.e. PDE_06089).
Group 3 comprised 16 genes that were induced in ΔcreA mutant (S7 Table), particularly in ΔcreA-gpdA(p)::clrB and ΔamyR mutants. This gene set was categorized using GO terms. The results illustrated that the genes involved in the carbohydrate metabolic process (p = 1.06e-6) were enriched, including hemicellulases-encoding genes (i.e., PDE_01302, PDE_09710, and PDE_05998), endoglucanase (PDE_09969), β-glucosidase (PDE_04251), α-mannosyltransferase (PDE_09901), and α-xylosidase (PDE_06944). More importantly, a putative cellulose degradation regulator (PDE_05883) was observed within this set. The transcriptional expression for PDE_05883 was ClrB-dependent and repressed by CreA and AmyR. Moreover, this expression showed an additive increase in ΔcreA-gpdA(p)::clrB under cellulose conditions (RPKM: 6.9±0.8 in ΔclrB, 35.3±1.4 in ΔcreA, 3.4±1.1 in ΔcreA-ΔclrB, 62.9±9.6 in ΔcreA-gpdA(p)::clrB, and 51.4±3.8 in ΔamyR for PDE_05883 versus 16.1±0.6 in the wild-type strain). The variations in the expression levels of PDE_05883 in these mutants were further identified via q-PCR experiments (Fig 10C). PDE_05883 encodes a conserved fungal Zn2Cys6 binuclear cluster domain with a significant amino acid homology with N. crassa cellulase essential regulator CLR-2 (NCU08042) for cellulose degradation. PDE_05883 shares 37% identity with CLR-2 in N. crassa (BioEdit, Expect = 5e-096) and a 45% identity with P. oxalicum ClrB (BioEdit, Expect = 5e-0163). Therefore, the gene of PDE_05883 was named as clrB-2, and the corresponding protein was called ClrB-2. The P. oxalicum closest homolog ClrB (BioEdit, Expect = e-131) of N. crassa CLR-2 was identified in this study. The function of ClrB involved in cellulose degradation was also extensively characterized in the preceding discussion. Nonetheless, only limited information has been reported about the molecular mechanism of this ClrB-2. In sum, the above findings provide an additional objective assessment of the role of AmyR in regulating cellulase expression. Likewise, the preceding analyses suggest that the combinatorial cross-regulation of ClrB, CreA, AmyR, and PDE_05883 defining a regulatory network of cellulase expression must be further characterized.
Group 4 consisted of four genes that were repressed in ΔcreA mutant (S7 Table), but were significantly induced in ΔcreA-gpdA(p)::clrB and ΔamyR mutants. These genes involved β-1, 6-glucanase-encoding genes (PDE_02004), succinate semialdehyde dehydrogenase (PDE_05599), 5-nitroimidazole antibiotic resistance protein (PDE_08743), and 4-aminobutyrate aminotransferase (PDE_09301).
Direct interactions of the transcription factors ClrB, XlnR, CreA, and AmyR
The results of the transcriptome analyses, Northern blot, and q-PCR experiments specified above demonstrate that the core cellulolytic genes are tightly regulated by ClrB, CreA, AmyR, and XlnR transcription factors under inducing conditions. The fine-tuned regulation mechanisms allowed us to hypothesize that these central transcription factors may directly bind to the promoters of their core targets. This case was expected because the CreA and XlnR homologs in T. reesei [42] have been determined to be capable of binding to cellulase gene promoters, corresponding to cbh1 in P. oxalicum. To support this hypothesis, GST-tagged ClrB (GST-ClrB), CreA (GST-CreA), and XlnR (GST-XlnR) binding domains were separately expressed in Escherichia coli and were purified. The nucleotide sequences of the putative target gene corresponded to 2 kb cbh1 promoter fragment. The abilities of the recombinant proteins to bind to cbh1 were assessed via electrophoretic mobility shift assay (EMSA). When the concentration of GST-ClrB, GST-CreA, or GST-XlnR fusion proteins increased, slower migrating shifted bands were evident (Fig 11A). However, no shifted band could be observed only with a high-concentration GST (negative control). These findings signify that ClrB, CreA, and XlnR could directly bind to cbh1 promoter region, and the number of binding sites may be more than one.

An important question is whether the direct interactions associated with these regulators play important roles in regulating their target genes by assembling into active transcription complexes, in addition to their direct binding to DNA segments, as observed in T. reesei [42,43]. To investigate this possibility, the full-length open reading frames (ORFs) of the transcription factors ClrB, CreA, AmyR, and XlnR were PCR amplified using cDNA from P. oxalicum 114–2 as the templates. All amplicons were cloned into plasmid pGAD-T7 and separately obtained fusion proteins (i.e., AD-ClrB, AD-CreA, AD-XlnR, and AD-AmyR). Similarly, the full-length creA, amyR, and xlnR were cloned into the partner plasmid pGBK-T7 and resulted in BD-CreA, BD-AmyR, and BD-XlnR. Protein–protein interaction assay was performed. The results showed that the strains with interactions between ClrB and AmyR, XlnR and ClrB, XlnR and AmyR, and XlnR and CreA could grow on synthetic drop-out (SD) plates that lack Leu, Trp, and His (QDO, Clontech) (Fig 11B). Likewise, the results revealed that XlnR interacted directly with ClrB, CreA, and AmyR as well as with ClrB and AmyR in vitro.
Discussion
Several conserved and essential transcription factors for cellulase and hemicellulase genes have been recently described in cellulolytic fungi; yet, the regulatory patterns of these factors have not been thoroughly analyzed [10,15–17,23]. In this study, a single-gene disruptant mutant library of the transcription regulators in P. oxalicum was constructed, and 20 transcription factors that play a pivotal role in the activation or inhibition of cellulose deconstruction were identified. Of these screened transcription factors, ClrB, CreA, XlnR, and AmyR were selected for further analyses. The cellulase expression regulated by additive cellulolytic effectors was observed. The results correspondingly provided comprehensive information for deciphering and redesigning a cellulase expression regulation network for rational engineering of cellulase hyper-producers.
ClrB
In this study, a master transcription factor ClrB was exceptionally identified in P. oxalicum transcription factor mutant set screening. This key transcription factor positively regulated the cellulolytic gene expression, and its deletion strain exhibited dramatically reduced cellulase activities (Fig 1B). However, this factor was not strictly required for xylanase gene expression. The homology search of P. oxalicum ClrB within some fungal proteomes showed that the homologs of ClrB (i.e., Clr-2 in N.crassa, ClrB in A. nidulans, and ManR in A. oryzae) [16,17] were recently determined and were required to induce major cellulases, some major hemicellulases, and mannanolytic enzyme gene expression. The search of T. reesei protein databases via a Basic Local Alignment Search Tool using a ClrB/Clr-2 query revealed that a protein (Trire2 : 26163) with low sequence identity existed. However, the homology search of N.crassa Clr-2 within P. oxalicum proteome illustrated that the protein sequences for ClrB (BioEdit, Expect = 5e-0163) and PDE_05883 (BioEdit, Expect = 5e-096) have 45% and 37% identity with Clr-2 sequence [17], respectively. These phenomena for two Clr-2 homologs in P. oxalicum proteome were not observed in N. crassa and T. reesei proteomes. This case suggests that differential-inducing mechanisms for cellulase expression may exist among cellulolytic fungi.
XlnR
The homologs of regulator XlnR were the most conserved in cellulolytic fungi [10–12]. However, P. oxalicum XlnR does not have the same transcriptional inducing ability for cellulolytic and xylanolytic genes as in others [10–12]. Significant differences in these regulatory patterns for cellulase genes were observed in P. oxalicum ΔxlnR and T. reesei Δxyr1 mutants [10]. The lack of XlnR homolog in T. reesei eliminated cellulase expression, but not in P. oxalicum ΔxlnR mutant (Fig 2A and 2B). The deletion of P. oxalicum xlnR slightly reduced the transcript levels of some cellulases and abolished the major xylanase expression under induction conditions (Fig 2A), which were similar to that in N. crassa Δxlr-1 mutant [12]. These findings suggest that the transcriptional regulation of lignocellulose-degrading enzymes mediated by XlnR homologs may be highly conserved in various filamentous fungi, but may also have interesting differences.
CreA
CreA/CRE1/CRE-1 is a wide-domain master regulator of carbon metabolism identified in filamentous fungi [7–9]. This regulator allows an organism to utilize a preferred carbon source but hinders it from metabolizing complex carbon sources, including cellulose [7–9]. In this study, the function of CreA homologs in repressing cellulolytic and xylanolytic gene expressions was conserved among cellulolytic fungi (Fig 2A and 2B). CreA homologs generally play an important role among cellulolytic fungi by linking CCR to developmental programs, including the conidia formation and hyphal morphology in T. reesei [23] and P. oxalicum (Fig 5B). This study determined that some transcription factors (i.e., PDE_07199, PDE_04095, and PDE_08372) are involved in both developmental programs and cellulase induction expression (Table 1). These regulators are conserved in cellulolytic fungi (Table 1). Therefore, the possible existence of an intimate crosstalk among certain developmental processes, such as sporulation and cellulase production pathways, is mediated by some regulators in ascomycete fungi.
AmyR
By using RNA-seq data, we showed that expression of amyR was synergistically decreased in the ΔcreA-gpdA(p)::clrB mutant. The lack of AmyR significantly induced cellulase expression and decreased the expression for amylase genes involved in starch degradation (Fig 9A–9C). As such, AmyR may control the balance between starch and cellulose utilization by inducing and/or repressing cellulolytic and amylolytic gene expressions in P. oxalicum, respectively. The multiple-sequence alignment analysis showed that P. oxalicum AmyR shares a weak homology with N. crassa COL26 (NCU07788, 23% sequence identity, E value = 2e-038 by BioEdit) [44] and T. reesei BglR (Trire2 : 52368, 24% sequence identity, E value = 3e-031 by BioEdit) [18], but is highly homologous to T. reesei, a functionally uncharacterized Zn(II)2Cys6-type fungal-specific transcription factor (Trire2 : 55105, 38% sequence identity, E value = 9e-095 by BioEdit). Interestingly, the regulatory functions of AmyR gene were distinct from those of N. crassa COL26 [44] and T. reesei BglR [18]. During its initial response to cellulose, P. oxalicum ΔamyR mutant exhibited induction and did not decrease the cellulolytic gene expression as in N. crassa Δcol-26 mutant [44]. Moreover, N. crassa COL26 obviously repressed cre-1 transcription to promote the relief of CCR [44], but the creA transcript abundance only slightly increased in P. oxalicum ΔamyR mutant. N. crassa Δcol-26 mutant exhibited a severe growth defect on glucose, but not in P. oxalicum ΔamyR mutant. Although both T. reesei BglR [18] and P. oxalicum AmyR mutants displayed an elevated cellulase expression under inducing conditions, they demonstrated distinct regulatory trends to β-glucosidase expression. This difference might be related to the functional studies of BglR in T. reesei mutant PC-3-7 containing bgl2 mutation and other uncharacterized mutations [18]. Nevertheless, T. reesei 55105 (Trire2), which is much more homologous to P. oxalicum AmyR than T. reesei BglR (Trire2 : 52368), may be a candidate regulator involved in cellulase expression regulation.
Cascade regulation for cellulolytic genes
The cellulase expression in P. oxalicum is a highly coordinated process regulated by a suite of cellulolytic transcription factors (i.e., ClrB, CreA, XlnR, AmyR and ClrB-2) and other novel uncharacterized regulators. In this study, the cellulolytic regulators ClrB, XlnR, AmyR and ClrB-2 were significantly regulated at the transcriptional levels during their growth on glucose, but slightly at the early phase for under cellulose growth conditions (Figs 5A and 12A). The CreA tightly regulated the expression of clrB, xlnR, amyR and clrB-2 in response to environmental carbon. These data suggested that CreA might have a cascade regulation because it repressed the activator genes for AmyR, ClrB, ClrB-2 and XlnR as well as the structural genes whose expression was upregulated by ClrB, XlnR and ClrB-2 (Fig 12A). This “double-lock” regulation of cellulolytic genes mediated by regulator homologues in cellulolytic fungi might be general, which could facilitate the fast conversion of carbon metabolism from favored carbon sources to cellulose and hemicellulose utilization. This cascade regulation mechanism mediated by P. oxalicum CreA was similar to the pathway described Cre1-mediated double repression of xyr1 and xyn1 in H. jecorina [10]. Some similar situations with regard to the repression of the transcription of A. nidulans ethanol and xylan regulons have been previously reported [45,46]. The deletion of creA resulted in the slightly decreased expression of amyR (Fig 10A), whose absence further led to the upregulation of cellulase genes (Fig 12A). Similarly, the overexpression of ClrB led to a decreased expression of amyR, whose expression level was synergistically downregulated in RE-10 mutant, but increased in gpdA(p)::clrB-PDE_02864(p)::clrB mutant (Fig 10A). These data implied that regulatory function of ClrB and CreA on amyR expression may be important for cascade regulation for cellulolytic genes under cellulose growth conditions.

Another key finding of this study is that the transcriptional expression of ClrB-2, a novel regulator, is responsive to ClrB, XlnR, CreA and AmyR, which implied that ClrB-2 may mediate the cascade transcriptional regulation for cellulolytic genes by ClrB, XlnR, CreA and AmyR (Figs 5A, 10C and 12A). We did not systematically determine how cellulolytic genes could transform in clrB-2 mutant yet. Nonetheless, the variable expression levels on cellulose in these regulator mutants suggest that ClrB-2 is one of the most interesting target in P. oxalicum cellulolytic regulatory networks. Whether ClrB, CreA, XlnR, and AmyR converge to exert their partial regulatory function via ClrB-2 on cellulolytic gene expression must be urgently elucidated.
Synergistic and dose-controlled regulation by cellulolytic regulators
The synergistic and collective regulations of cellulase expression by cellulolytic regulators are still elusive and largely uncharacterized in cellulolytic fungi. Thus far, no research has systematically investigated whether or how the most central cellulolytic factors CreA and ClrB homologs perform the synergistic regulation of cellulase genes. In this study, ΔcreA-gpdA(p)::clrB strain yielded strong synergistic effects on cellulase expression (Figs 1A–1E, 2A and 2B). This observation indicates that the full induction of cellulase genes requires not only an exclusive inducer-induction and activation by positive regulators, but also the release of negative transcription factors. In accordance to these synergistic regulatory mechanisms in P. oxalicum, we constructed significantly higher cellulase hyper-producer RE-27 and RE-29 than the triple-mutant RE-10 (Fig 8A and 8B). We believe that the RERN technology presented here will be a valuable contribution in transforming some non-industrial model species (e.g., N. crassa and A. nidulans) into more industrially relevant species.
A very interesting finding in this study is that the cellulase expression increased evidently accompany with the increase of the copy number and the efficiency of its promoter of clrB or xlnR gene (Fig 8A and 8B, and S3A–S3D Fig), indicating that a tunable cellulase expression may be controlled by the activator concentration under cellulose conditions. These data signify that the cellulase expression is not only dependent on the presence of the activators ClrB and XlnR, but also severely dependent on their dose effects of ClrB and XlnR transcriptional abundances.
The other issue that must be explored is how cross-correlations occur between the cellulolytic regulators. To evaluate the role of CreA in CreA-mediated repression of cellulase and hemicellulase gene expressions, we developed three possible hypotheses. a) Putative CreA binding sites overlap with the putative ClrB or XlnR binding sites, and CreA could preferentially bind to the sites and/or block their binding to target promoters by competition. A recent study identified the presence of putative cis-regulatory elements recognized by both XYR1 and CRE1 and spaced in XYR1 - and CRE1-dependent cellulase gene promoters [42]. b) CreA could stably associate with ClrB or XlnR components to form a heterocomplex, thereby making ClrB and XlnR completely non-functional for the induction of cellulolytic and xylanolytic genes. In this study, CreA and XlnR were assumed to directly interact with each other according to the yeast two-hybrid assay (Fig 11B and 11C). c) The activity of transcription factor is influenced by intracellular protein post-translational modifications, such as phosphorylation. These modifications may influence the ability of the transcription factor to bind to its binding sites [47]. In sum, the findings presented above support our proposition that biological relevances may exist between CreA and ClrB as well as between CreA and XlnR, which tightly regulate the expression of cellulase and hemicellulase genes.
Cellulase regulatory network sensitive to inducers in the intracellular environments
Beta-glucosidases are the conserved components required in cellulose deconstruction, the number of which significantly varies among the genomes of cellulolytic fungi [24,48,49]. The lack of P. oxalicum intracellular Bgl2 contributes to the increase of cellulase expression [27], but not in T. reesei [50] and N. crassa [51]. These findings have raised the question as to how Bgl2 mediates the carbon metabolism involved in signal cascades in relation to the regulation of cellulase gene expression, which may reflect the general trend of cellobiose/cellodextrins-induced cellulase expression in cellulolytic fungi [27,50,51]. Some β-glucosidases in T. reesei [50] and N. crassa. [51] have been observed to play important roles in balancing cellobiose production and metabolism in intra - and extra-cellular environments. Given the general phenomenon that cellobiose induces cellulase expression in cellulolytic fungi [27,50,51], P. oxalicum Δbgl2-gpdA(p)::clrB mutant showed a strongly elevated cellulase expression (Fig 7A and 7B), which could be partially ascribed to the signal induction cascade mediated by cellobiose/cellodextrins from cellulose (Fig 12A). The robust induction of cellulase expression in Δbgl2-ΔcreA mutant was remarkably greater than that in each deletion mutant on cellulose (Fig 7A and 7B). This observation supports the premise that the CCR mediated by CreA increased when the major predicted intracellular β-glucosidase was absent under cellulose growth conditions. Although the lack of Cre-1 in N. crassa triple β-glucosidase mutant showed higher concentrations of secreted active cellulases than that in wild-type strain on cellobiose, it did not facilitate protein production and cellulase induction on cellulose [51]. In sum, ClrB positively regulated the transcriptional expression of bgl2 (S5 Fig), but its deletion conversely enhanced the signal cascade activation regulation by ClrB and/or the repression regulation of CCR mediated by CreA (Figs 7A, 7B and 12A). In addition, we also determined that the cellulase expression in Δbgl2-ΔamyR double mutant was further enhanced compared with each individual deletion strain (S10A Fig). These results revealed that the functional regulations for cellulase expression by these cellulolytic regulators may be sensitive to inducers in intracellular environments. This finding implies that the combination of intracellular cellodextrin induction and redesigned cellulolytic transcription factor regulation in P. oxalicum may be general for the full induction of cellulase expression on cellulose.
Inducer-free cellulase expression in cellulolytic fungi
Many recent studies have attempted to robustly produce cellulases without inducers, but the molecular mechanism of cellulase induction under non-inducing conditions has remained elusive in diverse cellulolytic species [20,40]. Recently published data also showed that the misexpression of N. crassa clr-2 through Pccg-1 [20] and Ptcu1-driven expression of xyr1 [40] was sufficient for inducer-free cellulase expression. However, the cellulolytic gene transcript between P. oxalicum gpdA(p)::clrB and wild-type strains had no obvious differences under non-inducing conditions. Such case was similar in the XlnR activator in the gpdA(p)::xlnR strain under CreA-mediated CCR, and cbh1 transcription was still remarkably in low-level expression in the ΔcreA mutant on a carbon-free medium, which indicates the full induction requirement of cellulase gene expression, as evident in T. reesei [25] or N. crassa [51]. However, the cellulolytic gene transcription levels were obviously upregulated in the ΔcreA-gpdA(p)::clrB mutant under repressing conditions. These findings implied that the lack of CreA contributed to the activating function for ClrB on cellulolytic genes even in glucose. Notably, the Δbgl2-gpdA(p)::clrB strain exhibited an induction transcription of core cellulase genes for several orders of magnitude increase than the wild-type strain that shifted to a carbon-free medium (Fig 7C and S7C Fig). No bulk of cellodextrins was apparently transported into the cell to induce cellulase gene expression when subjected to starvation, but the induction abilities of these core cellulase regulons in the Δbgl2-gpdA(p)::clrB mutant under non-inducing conditions were comparable to under cellulose conditions (Fig 7C and S7C Fig). The amyR expression level had a 7.7-fold decrease, clrB had a 17.5-fold increase, and clrB-2 had a 9.3-fold increase in the Δbgl2-gpdA(p)::clrB mutant versus wild-type strain under carbon-free conditions (Fig 7D), which implied that AmyR, ClrB, and ClrB-2 were possibly tightly involved in cellulase expression regulatory during energy abstinence, as well as that under cellulose growth conditions. These were novel findings that indicated the “starvation response” for cellulase genes in P. oxalicum and other diverse cellulolytic fungi.
Conclusion
Cellulase formation apparently occurred because of consistent respective regulators, including the characterized or novel transcription factors identified in P. oxalicum in this context. Cellulase synergistic and dose-controlled regulatory systems mediated by diverse cellulolytic effectors were observed in the system mutation of this study for P. oxalicum regulators. Using this model (Fig 12A), the accumulation of intracellular cellodextrins can trigger signaling cascades that include expression of cellulase genes repressed by CreA and AmyR and activated by ClrB and XlnR. However, our data also support that the transcriptional regulation for CreA, AmyR, ClrB and XlnR genes is a powerful part of the regulatory network of cellulase gene expression. In the early cellulolytic induction, ClrB functions to repress expression of amyR, whose expression level is activated by CreA and reduced in Bgl2-deficient background (Fig 12A). Moreover, transcriptional expression for ClrB, AmyR, XlnR and ClrB-2 genes is also significantly repressed by CCR mediated by CreA in the presence of glucose (Fig 12A). The data established ClrB as a focal point to regulate cellulase expression by integrating other regulators and the target genes of these regulators, which refined our understanding on transcriptional regulatory network as a “seesaw model” in which coordinated regulation of cellulolytic genes was established through activators and repressors counteraction (Fig 12B–12D). These observations also suggested the hypotheses that the rational design of cellulase or high-value protein super producers might be guided in the future for the combinatorial effects of diverse cellulolytic effectors.
Materials and Methods
Penicillium oxalicum strains, media, and growth conditions
All the strains used in this study are listed in the S9 Table and are grown on Vogel’s medium that contain 2% glucose (mass/volume percent), unless otherwise noted. The P. oxalicum wild-type strain 114–2 (CGMCC 5302) was used as parental strain throughout this study. Hygromycin B, pyrithiamine, phosphinothricin, and sulfonylurea were added to the media with final concentrations of 200, 300, 1.6, and 4 μg/mL used for transformant selection, respectively. Vogel’s 50x salts (1,000 mL) was used: 125 g Na3Citrate•2H2O, 250 g KH2PO4, 100 g NH4NO3, 10 g MgSO4•7H2O, 5 g CaCl2•2H2O, 0.25 mg biotin, and 5 ml trace element solution (5 g Citric acid•H2O, 5 g ZnSO4•7H2O, 1 g Fe(NH4)2(SO4)2•6H2O, 0.25 g CuSO4•5H2O, 0.05 g MnSO4•1H2O, 0.05 g H3BO3, and 0.05 g Na2MoO4•2H2O, which were dissolved in distilled water; the resulting total volume was 100 mL). The wheat bran medium (mass/volume percent) was composed of corn cob residue (2.0%), Avicel (0.6%), wheat bran (4.6%), soybean cake powder (1.0%), (NH4)2SO4 (0.2%), NaNO3 (0.28%), urea (0.1%), KH2PO4 (0.3%), and MgSO4 (0.05%). Vogel’s medium that contained 1 M sorbitol was used in all the transformation experiments. Vogel’s medium with 2% Avicel was used as a sole carbon source to induce cellulase expression for cellulase activity assays, q-PCR, Northern blot, or RNA-seq analyses. The fluid mediums of the P. oxalicum strains were all cultivated in Erlenmeyer flasks at 30°C in constant light and 200 rpm agitation rate. The cultivation was performed on plates by adding agar in the fluid medium as a solidifying agent at 30°C in constant light.
Protoplast preparation and transformation
P. oxalicum protoplast prepared according to modified methods, as described by Gruber et al. [52]. Seven to eight PDA plates were prepared, and 50 μL of fresh spore solution was streaked out on every cellophane-covered PDA plate. The plates were incubated at 30°C for 11–12 hours. Three milliliters of this protoplasting solution [0.075 g lysing enzymes up to 25 mL solution A (1.2 M sorbitol and 0.1 M KH2PO4, and pH = 5.6] were pipetted into a sterile petri dish, and one cellophane disc with freshly grown mycelium was added. Subsequently, 3 mL of protoplasting solution was readded. The petri dish was incubated at 30°C for 150 min. The final protoplast suspension was filtered into a sterile 50 mL centrifuge tube through a lens cleaning tissue in a glass funnel. The suspension was centrifuged for 10 min at 2000 rpm and 4°C in a swing-out rotor. The supernatant was cautiously decanted, and the pellet was resuspended in 10 mL of solution B (1 M sorbitol, 50 mM CaCl2, 10 mM TrisHCl, and pH = 7.5). The suspension was recentrifuged for 10 min at 2000 rpm and 4°C, and then the supernatant was cautiously decanted and the protoplasts were resuspended in 0.5 mL of solution B. The protoplasts were stored in ice. The following components: 200 μL protoplast suspension, 10 μL DNA fragment, and 50 μL of solution C (25% PEG 6000, 50 mM CaCl2, 10 mM TrisHCl, and pH = 7.5), were added into a 10 mL centrifuge tube and were mixed gently, and then the mixture was incubated on ice for 20 min. Two milliliters of solution C (room temperature) were added to the transformation mixture, which was mixed gently. The mixture was incubated for 5 min at room temperature, and then 4 mL of solution B was added and was mixed gently. All the transformation mixture was added to 30 mL Vogel’s medium (55°C) that contained hygromycin B, pyrithiamine, phosphinothricin, or sulfonylurea, and was then mixed shortly and was poured onto the bottom of the Vogel’s medium. Transformants would be visible for 3–4 days for hygromycin B, pyrithiamine, or phosphinothricin, and for 6–8 days for sulfonylurea at 30°C.
High throughput generation of 470 regulator deletion mutants
The P. oxalicum genome is available in DDBJ/EMBL/GenBank (under the accession number AGIH00000000) or at http://genome.jgi.doe.gov/Penox1/Penox1.home.html. Transcription factors were identified and annotated according to InterPro IDs in the Fungal Transcription Factor Database [53]. The transcription factor deletion strains, where each coding region of the transcription factor was substituted with a selective marker ptra gene [31], were constructed from the P. oxalicum strain Δpku70::hph [33] as follows. Deletion cassettes conferring resistance to pyrithiamine hydrobromide were constructed according to the double-joint PCR strategy [32]. Primer pairs for (x)-F1+(x)-ptraR and (x)-ptraF+(x)-R1 (S1 Table) were designed using the Primer 5 software, and the pairs were used to amplify the upstream and downstream fragments for 1000–1500 bp on either side of the encoding regions for each single target gene. The ptra selectable marker cassette was PCR-amplified from the pME2892 plasmid [54] with the PtraF1+PtraR1 (S1 Table) primer pair and was PCR product-purified. The upstream and downstream fragments contained 25 bp homology to the ptra cassette sequence. The primer pair (x)-F2+(x)-R2 (S1 Table) was used to produce final deletion cassettes through double-joint fusion PCR [the program used to fuse the three fragments: 94°C 2 min for 10 cycles (94°C for 30 s, 58°C for 10 min, and 72°C for 3 min), 72°C 5 min] [32]. PCR experiments were performed in final volumes of 50|μL that contained 1 unit of Trans HIFI DNA polymerase, 0.2|mM dNTP, and 0.4|μM of each primer. The program used to amplify the fused knockout cassettes was as follows: one cycle of 94°C (120 s), 30 cycles of 94°C (30 s), 58°C (30 s), and 72°C (1 min for every 1 kb of amplified product), followed by a final 10|min at 72°C. All the PCRs were performed in a Bio-Rad DNA Engine Peltier Thermal Cycler.
Each knockout cassette was independently transformed into P. oxalicum strain Δpku70::hph protoplasts [33]. The mature transformant conidia from the Vogel’s medium slant was streaked onto a Vogel’s medium plate that contained pyrithiamine hydrobromide. At least three Ptra-resistant colonies obtained from each assay were analyzed through diagnostic PCR to confirm the deletion using the primer pairs (x)-F1+ptraYZR or ptraYZF+(x)-R1 (S1 Table). Within the pair, the primer (x)-F1 or (x)-R1 (S1 Table) was located outside the transforming deletion fragment of the genome, and the primer ptraYZR or ptraYZF (S1 Table) was unique to the ptra sequence. These obtained strains constituted a stock of the P. oxalicum transcription factor deletion mutant.
Gene complementation
The complementation cassette that conferred resistance to hygromycin B was used to transform into the corresponding mutant to identify that the interesting phenotype(s) observed for the transcription factor gene deletion mutants was indeed caused by the deletion of a relevant gene. The upstream and downstream fragments contained 25 bp homology to the hph cassette sequence. clrB and amyR wild-type allele complementation cassettes were obtained by amplifying the upstream fragments (encompassing the 1.5 kb promoter, the open reading frame, and 0.5 kb 3’ untranslated region) using the primer pairs clrB-F1+clrBHPH-R and AmyR-F1+AmyRHPH-R (S1 Table), respectively. The 1.5 kb downstream flanking segments of the 3’ untranslated region from the P. oxalicum genome DNA were amplified through the primer pairs clrBHPH-F+clrB-R1 and AmyRHPH-F+AmyR-R1 (S1 Table). The 1.8|kb hph gene fragment was amplified from the pSilent-1 plasmid [55] with the primers Hphs-F and Hphs-R (S1 Table). These three PCR fragments were ligated through Double-joint PCR and were amplified through the nest primer pairs clrB-F2+clrB-R2 and AmyR-F2+AmyR-R2 (S1 Table). The resulting clrB and amyR complementation cassettes were transformed into ΔclrB::ptra and ΔamyR::ptra mutants, and the complementation strains RclrB and RamyR of these cassettes were obtained, respectively.
Construction of gpdA(p)::clrB, gpdA(p)::creA, gpdA(p)::creA-gpdA(p)::clrB, and gpdA(p)::xlnR mutants
The clrB promoter was replaced with the gpdA (glyceraldehyde-3-phosphate dehydrogenase) promoter from A. nidulans [37]. Moreover, 1,314 bp of the gpdA promoter was amplified from the plasmid pAN7-1 [56] using the primers PgpdA-F1 and PgpdA-R1 (S1 Table). Subsequently, 2,008 bp ptra selectable marker cassette was PCR-amplified with the primer pair PtraF1+PtraR1 (S1 Table) and was PCR product-purified. Furthermore, 3,148 bp of the clrB open reading frame and 3’ untranslated region were amplified with the primer pair clrB-Fa+clrB-Ra (S1 Table), and this fragment overlapped with the gpdA promoter and ptra fragment by 25|bp at the ends of this fragment. These 6375 bp PCR products were then ligated in the order of gpdA(p)::clrB-ptra by splicing through double-joint PCR with the nest primers PgpdA-F2 and PtraR1 (S1 Table).
A similar strategy was used to construct the creA overexpression cassette under the influence of the P. oxalicum gpdA promoter. Moreover, 1749 bp of the gpdA promoter was amplified from the P. oxalicum 114–2 genome DNA through the primers PGP-F1 and PGP-R (S1 Table). In addition, 1890 bp of the hph cassette was amplified from the plasmid pSilent-1 [55] with the primers Hphs-F and Hphs-R (S1 Table). Moreover, 1743 bp of the creA open reading frame and 3’ untranslated region was amplified using the primer pair GPcre-F+GPcre-R (S1 Table), and this fragment overlapped with the gpdA promoter and hph fragment by 25|bp at the ends of this fragment. These PCR products were then ligated in the order of gpdA(p)::creA-hph by splicing through double-joint PCR with the primers PGP-F2 and HPH-R1 (S1 Table). The resulting 5248 bp gpdA(p)::creA-hph overexpression cassette was transformed into P. oxalicum wild-type and gpdA(p)::clrB-ptra strain protoplasts, and the gpdA(p)::creA and gpdA(p)::creA-gpdA(p)::clrB mutants were obtained, respectively.
Similarly, the xlnR promoter was replaced with the gpdA promoter from A. nidulans. Moreover, 1314 bp of the gpdA promoter was amplified from the plasmid pAN7-1 [56] through the primer pair PgpdA-F1 and PgpdA-R1 (S1 Table). In addition, 1890 bp of the hph cassette was amplified from the plasmid pSilent-1 [55]. The xlnR open reading frames and 3’ untranslated region were amplified with the primer pair XlnR-Fa+XlnR-RH2 (S1 Table), and this fragment overlapped with the gpdA promoter and the hph fragment by 25|bp at the ends of this fragment. These PCR products were then ligated in the order of gpdA(p)-xlnR-hph by splicing through double-joint PCR with the primers PgpdA-F2 and Hphs-R1 (S1 Table). The resulting 6675 bp gpdA(p)::xlnR-hph overexpression cassette was transformed into gpdA(p)::clrB strain protoplasts.
The resulting gpdA(p)::clrB-ptra, gpdA(p)::creA-hph, and gpdA(p)::xlnR-hph overexpression cassettes were used to transform P. oxalicum wild-type strain protoplasts. The gpdA(p)::creA-ptra, gpdA(p)::clrB-ptra, and gpdA(p)::xlnR-hph overexpression mutants were selected on Vogel’s medium plate that contained hygromycin B or pyrithiamine hydrobromide.
Construction of the ΔcreA, ΔcreA-gpdA(p)::clrB, and ΔcreA-ΔclrB mutants
Double-joint PCR was performed to construct the creA knockout cassette, with the hph cassette flanked by 1.5 kb upstream (CreA-F1+Crehph-R) (S1 Table) and 1.5 kb downstream (Crehph-F+CreA-R1) (S1 Table) of the creA ORF. Moreover, 1.8 kb of the hph cassette was amplified from the plasmid pSilent-1 [55]. The ΔcreA::hph final deletion cassette fragment was obtained through the primer pair (CreA-F2+CreA-R2) (S1 Table) with the three fragments above used as a PCR template. The ΔcreA::hph cassettes were transformed into P. oxalicum wild-type, gpdA(p)::clrB-ptra, and ΔclrB strain protoplasts. The selection of the ΔcreA, ΔcreA-gpdA(p)::clrB, and ΔcreA-ΔclrB transformants was consistent with the previous study.
Construction of the ΔxlnR, ΔxlnR-ΔclrB, and gpdA(p)::clrB-gpdA(p)::xlnR mutants
The ΔxlnR cassettes conferred resistance to hygromycin B. The hph selectable marker cassette was PCR-amplified with the primers Hph-F1 and Hph-R1 (S1 Table) from the plasmid pAN7-1 [56]. The upstream and downstream flanking fragments were amplified with the primer pairs XlnR-F1+XyrHph-R and HphXyr-F+XlnR-R1 (S1 Table), which contained 25 bp homology to the hph cassette sequence. The primer pair XlnR-F2 and XlnR-R2 (S1 Table) was used to produce final deletion cassettes through a double-joint fusion PCR. The resulting ΔxlnR::hph knockout cassette was used to transform wild-type protoplasts, and the P. oxalicum ΔxlnR mutant was obtained. The ΔxlnR::hph knockout cassette was used to transform the ΔclrB protoplasts, and then the ΔxlnR-ΔclrB mutant was obtained. The above gpdA(p)::xlnR-hph overexpression cassette was used to transform the gpdA(p)::clrB-ptra strain protoplasts, and the gpdA(p)::clrB-gpdA(p)::xlnR mutant was constructed.
Construction of the Δbgl2-ΔcreA and Δbgl2-gpdA(p)::clrB mutants
The upstream and downstream fragments of bgl2 ORF were amplified with the primer pairs Bgl2-F1+Bgl2hph-R and Bgl2hph-F+Bgl2-R1 (S1 Table). Moreover, 1.8 kb of the hph cassette was amplified from the plasmid pSilent-1 [55]. The final Δbgl2::hph fragment was obtained through the primer pair Bgl2-F2+Bgl2-R2 (S1 Table), with the three fragments above used as a PCR template. The Δbgl2::hph cassette was used to transform the protoplasts of the ΔcreA and gpdA(p)::clrB mutants, and then the Δbgl2-ΔcreA and Δbgl2-gpdA(p)::clrB mutants were obtained, respectively.
Construction of PDE_02864(p)::clrB-hph and gpdA(p)::clrB-PDE_02864(p)::clrB mutants
PDE_02864(p)::clrB-hph overexpression cassettes that conferred resistance to hygromycin B was constructed. The hph selectable marker cassette was PCR-amplified with the primers Hphs-F and HPH-R1 from the plasmid pSilent-1 [55]. The PDE_02864 promoter, and clrB open reading frame and 3’ untranslated region were amplified with the primer pairs DF1+DClrB-R and ClrB-F+ClrBHPH-R (S1 Table) from the P. oxalicum genome DNA, respectively. The primer pair DF2+HPH-R1 (S1 Table) was used to produce the final PDE_02864(p)::clrB-hph overexpression cassette via double-joint fusion PCR. The resulting PDE_02864(p)::clrB-hph overexpression cassette was used to transform the wild-type and gpdA(p)::clrB strain protoplasts, and then the PDE_02864(p)::clrB-hph and gpdA(p)::clrB-PDE_02864(p)::clrB mutants were obtained, respectively.
Construction of the ΔamyR-gpdA(p)::clrB, ΔamyR-ΔcreA, ΔamyR-Δbgl2, ΔamyR-ΔxlnR mutants
The upstream and downstream flanking fragments of the amyR encoding region were amplified with the primer pairs PDE_03964-F1+amyRHph-R and amyRHph-F+PDE_03964-R1 (S1 Table), respectively. The final ΔamyR::hph fragment was obtained through the nest primer pair PDE_03964-F2+PDE_03964-R2 (S1 Table) with the three fragments (amyR flanking sequences and hph encoding cassette) used as templates via double-joint PCR. The ΔamyR::hph cassette was used to transform the gpdA(p)::clrB-ptra protoplasts, and the ΔamyR-gpdA(p)::clrB mutant was obtained. The ΔamyR::ptra (ΔPDE_03964::ptra, from TF knock-out cassette set) knockout cassette was used to transform the Δbgl2::hph, ΔcreA::hph, and ΔxlnR::hph mutants, and the ΔamyR-ΔcreA, ΔamyR-Δbgl2, ΔamyR-ΔxlnR mutants were obtained, respectively.
Construction of the RE-30, RE-27 and RE-29 mutants
First, the ΔamyR::sur knockout cassette, and PDE_02864(p)::clrB-sur and PDE_02864(p)::xlnR-sur overexpression cassettes that conferred resistance to sulfonylurea were constructed. The sur selectable marker cassette was PCR-amplified with the primers Sur-F1 and Sur-R1 (S1 Table) from the plasmid pCB1536 [57]. The upstream and downstream flanking fragments for the ΔamyR-sur knockout cassette were amplified with the primer pairs PDE_03964-F1+amyRsur-R and amyRsur-F+PDE_03964-R1 (S1 Table), which contained 25 bp homology to the sur cassette sequence, respectively. The primer pair PDE_03964-F2+PDE_03964-R2 (S1 Table) was used to produce the final deletion cassette ΔamyR-sur through a double-joint fusion PCR. The resulting ΔamyR-sur knockout cassette was used to transform the RE-10 (Δbgl2-ΔcreA-gpdA(p)::clrB) protoplasts, and the P. oxalicum and RE-30 (ΔamyR-Δbgl2-ΔcreA-gpdA(p)::clrB) mutants were obtained. The clrB promoter was replaced with the PDE_02864 (encoding 40S ribosomal protein S8) promoter from P. oxalicum. The PDE_02864 promoter sequence was amplified with the primer pair DF1+DP-R. In addition, the clrB and xlnR open reading frames, and the 3’ untranslated regions were amplified with the primer pairs DClrB-F+ClrBsur-R and DXlnR-F+XlnRSur-R, respectively. The primer pair DF2+Sur-R1 (S1 Table) was used to produce the final PDE_02864(p)::clrB-sur and PDE_02864(p)::xlnR-sur overexpression cassettes via double-joint fusion PCR. The resulting PDE_02864(p)::clrB-sur and PDE_02864(p)::xlnR-sur overexpression cassettes were used to transform the RE-10 protoplasts, and then the RE-27 (Δbgl2-ΔcreA-gpdA(p)::clrB-PDE_02864(p)::clrB) and RE-29 (Δbgl2-ΔcreA-gpdA(p)::clrB - PDE_02864(p)::xlnR) mutants were obtained.
Southern blot analysis
Fungal genomic DNA was isolated, as described previously [58]. The Primer 5 software was used to identify the appropriate restriction enzymes for the Southern blot analysis of the gene replacement mutants. The fragments used for the probes were amplified with the primers presented in the S1 Table. A DIG-High Prime labeling kit was used to label the knockout cassette flank fragment probes. The homokaryons and integration patterns of the transforming cassettes in the genome were confirmed through Southern blot analysis, as described in the manipulations.
Phenotypic observations
Colony morphology and conidiation were analyzed after inoculating the Vogel’s medium plates that contained 2% glucose, 2% xylan, 2% starch, or 1% cellulose as sole carbon source or potato dextrose agar (PDA) medium at 30°C for 5 days. The halo sizes that varied among the P. oxalicum strains were measured on cellulose or starch plates by adding Triton X-100 to a final concentration of 0.5%. Iodine solution (6 g of KI, 0.6 g of I2 in 100 mL of H2O) was used to indicate visualize starch-degrading colonies through the hydrolysis halo at room temperature for 10 min, which determined if the amylase expression was affected in the P. oxalicum strain. P. oxalicum hyphae and conidia were microscopically examined through lactophenol cotton blue staining (0.05 g cotton blue, 20 g phenol crystals, 40 mL glycerol, 20 mL lactic acid, and 20 mL distilled water).
Enzyme activity and protein concentration assays
Cellulase was produced in a 500 mL flask that contained 100 mL of fluid medium through a two-step cultivation procedure. Strains were first grown at 30°C in 100 mL of medium that contained 2 g of glucose as a carbon source and were then regulated at pH 5.5 and 200 rpm for 20 hours. The cultures were collected through vacuum drum filtration during this second step, and 0.5 g vegetative mycelia was added to 100 mL of Vogel’s medium that contained 2% cellulose as carbon source or wheat bran medium at an initial pH of 5.5 at 30°C and 200 rpm. Culture supernatants (crude enzyme) were diluted with sodium acetate buffer solution (SABF, 0.2 M, pH 4.8). Enzymatic hydrolyses of the polysaccharides were also performed in SABF (0.2 M, pH 4.8). The filter paper enzyme (FPA), endoglucanase (CMCase), xylanase, and amylase activities of the culture supernatants (diluted samples) were assayed using a DNS reagent (10 g 3, 5-dinitrosalicylic acid, 20 g sodium hydroxide, 200 g sodium potassium tartrate, 2.0 g redistilled phenol, and 0.50 g sodium sulfite anhydrous per 1000 mL DNS reagent) against Whatman No. 1 filter paper, carboxymethylcellulose sodium salt (CMC-Na), xylan (from beechwood), and soluble starch. CMC-Na, xylan, or starch was dissolved in SABF to a final concentration of 1% (mass/volume percent, m/v %), and then the mixture was left overnight and was shaken well before using. The following components were added in a 2.0 mL reaction mixture: 0.5 mL diluted culture supernatants and 1.5 mL CMC-Na, xylan, or starch solution for CMCase, xylanase, or amylase activity assays, respectively; and 2.0 mL diluted culture supernatants and 50 mg Whatman No. 1 filter paper for FPA assay into 25 mL colorimetric tube. The mixture was mixed gently and the reaction mixture was incubated for FPA measurement in a 50°C water bath for 1 hour, for CMCase and xylanase activity measurements at 50°C for 30 min, and for amylase activity measurement at 40°C for 10 min. Three milliliters of DNS reagent were then added to stop the reaction. A blank tube (with boiled crude enzyme) was used as control to correct any reducing sugar present in the crude enzyme samples. The tubes were placed in boiling water for 10 min, 20 mL distilled water was added, 200 μL of reaction mixture was pipetted, and the absorbance was determined at 540 nm. The cellobiohydrolase (pNPCase) and β-glucosidase (pNPGase) activities were measured by using 4-Nitrophenyl β-D-cellobioside (pNPC) and 4-Nitrophenyl β-D-glucopyranoside (pNPG) as substrates, respectively. The pNPC or pNPG was dissolved in SABF to a final concentration of 1 mg/mL. Moreover, 50 μL of pNPC solution (containing 1 mg/mL D-Glucono-δ-lactone) or 50 μL of pNPG solution and 100 μL of diluted culture supernatants were mixed, and then the mixtures were incubated in a 50°C water bath for 30 min. The reaction was stopped by adding 0.15 mL of sodium carbonate solution (10%, m/v), then 200 μL of these reaction mixtures was pipetted, and the absorbance was measured at 420 nm. One unit of enzyme activity was defined as the amount of enzyme required to release 1 μmol of glycoside bonds of the substrate per minute under defined assay conditions. Independent triplicate cultures were sampled and analyzed.
The total protein was determined using a Bradford assay kit according to the instructions of the manufacturer.
Total RNA extraction
Freshly harvested conidia of the wild-type strain or mutants were inoculated with 106 conidia/mL into 100 mL Vogel’s medium that contained 2% glucose, and then grown for 22 hours at 30°C. Mycelia were harvested via vacuum filtration, and then washed with Vogel’s medium without a carbon source, followed by 2 h growth in 100 mL Vogel’s medium without a carbon source. Subsequently, mycelia were harvested via vacuum filtration, and then transferred into Vogel’s medium that contained 2% cellulose for 4, 8, 22, and 46 hours, with 2% glucose for 4 hours, or into a medium without any carbon source for 4 hours. Mycelia were harvested via vacuum filtration, and then immediately finely ground under liquid nitrogen, and then 1 mL of TRIzol reagent was added per 50–100 mg powder. The total RNA was isolated according to the instructions of the manufacturer.
Northern blot analysis of cellulase gene expression
The total amount (2 μg) of mRNA loaded was normalized by using rRNA as a loading control. The probes used for the Northern blot analysis were the partial cDNAs of cbh1 (PDE_07945), eg2 (PDE_09226), and xyn1 (PDE_08094) that were cloned from the P. oxalicum genome DNA via PCR with primer pairs (S1 Table). The probes were labeled using a DIG Northern Starter kit, and Northern blot analysis was performed according to the instructions of the manufacturer.
Quantitative reverse transcription PCR (q-PCR)
Putative targets were validated through q-PCR. Each strain was cultured independently from the Northern blot and RNA-seq experiments. cDNA was synthesized from the total RNA by applying a reagent kit with a gDNA eraser according to the instructions of the manufacturer. The obtained cDNA was applied for quantitative reverse transcription-PCR experiments. All q-PCR amplification was performed in 20 μl total volume with 7.4 μl distilled water, 0.8 μl of each primer (10 mM), 10 μl SYBR Premix Ex TaqII, and 1 μl template cDNA through the following program [59]: 95°C for 2 min, 40 cycles at 95°C for 10 sec, and 30 s at 61°C. The fluorescence signal was measured at the end of each extension step at 80°C. A melting curve program with a temperature gradient of 0.1°C per second from 65°C to 95°C was performed. The corresponding primers are shown in the S1 Table. The quantity and copy number of each target gene were calculated using a standard curve. Six 10-fold serial dilutions of purified DNA template (0.5 kb–1.0 kb) were prepared for the target genes to determine the standard curve of each target gene. The correlation coefficient (R2) for each standard curve was verified to be 0.99 or greater. The transcriptional expression of transcription factor genes were measured by q-PCR, and their expression levels were normalized to wild-type. Gene expression levels for cellulase genes were measured by q-PCR using actin (PDE_01092) as a control and normalized to expression levels by actin values/10000. Three biological replicates were performed on the same 96-well plate by using cultures grown in parallel. Data processing and statistical analyses were performed using Microsoft Excel.
RNA sequencing and transcription expression analysis
A cDNA library prepared from mRNA was organized according to standard protocols. Quality control was implemented using the Real-Time PCR Systems. All the cDNA libraries were sequenced on the Illumina platform. Sequenced reads were mapped against predicted transcripts from the P. oxalicum 114–2 genome using the SOAP2 software for short oligonucleotide alignment (http://soap.genomics.org.cn/soapaligner.html) [60]. Transcript abundance (Reads Per Kb per Million reads, RPKM) [61] was estimated with RPKM = [# of mapped reads]/([length of transcript]/1000)/([total reads]/10^6). The differential gene expression was analyzed using DESeq software package [62] and NOIseq v2.10 (http://www.bioconductor.org/packages/release/bioc/html/NOISeq.html) with |log2(fold change)|>1 and probability≥0.8 as thresholds [63]. The biological replicates used for RNA-seq were highly reproducible. These datasets (RPKM) were subjected to hierarchical cluster analysis using the software HCE3.5 (http://www.cs.umd.edu/hcil/hce/) to determine the groups of genes with similar expression patterns for a different group of regulons. The Blast2go software v3.0 was used for the gene ontology analyses (https://www.blast2go.com/) [64]. Secreted proteins were predicted using SignalP 4.1 (http://www.cbs.dtu.dk/services/SignalP/). Secondary metabolism gene clusters were identified using annotated proteins in P. oxalicum [24]. Protein sequence alignments were performed among the P. oxalicum, T. reesei, and N. crassa proteomes using the BioEdit Sequence Alignment Editor software (http://www.mbio.ncsu.edu/BioEdit/bioedit.html).
Secretome analysis using label-free LC-MS/MS
Culture supernatants were collected after shifting to cellulose for 96 hours by filtrating using 0.22 μm PES membrane, and then the supernatants were desalted with 10 kDa molecular cut-off membrane, and were precipitated by acetone and trichloroacetic acid (20 : 1). The obtained protein powders were dissolved in denaturation buffer (0.5 M Tris-HCL, 2.75 mM EDTA, 6 M Guanadine-HCL), and were then reduced using 1 M DTT at 37°C for 1 hour. The following alkylation was performed using iodoacetamide for 2 hours away from light, and the samples were desalted and collected using a Microcon YM-10 Centrifugal Filter Unit. The obtained protein samples were digested thoroughly using trypsin for 12 hours, and these peptide mixtures were desalted with a ZipTip C18 column. These collected secretome samples were further separated on a C18-reversed phase column and then directly mounted on the electrospray ion source of a mass spectrometer. The peptides were subjected to nanoelectrospray ionization, followed by tandem mass spectrometry (MS/MS) in an LTQ Orbitrap Velos Pro coupled with high-performance liquid chromatography. Intact peptides were detected in the Orbitrap at 60000 resolution. Peptides were selected for MS/MS using a collision-induced dissociation operating mode with 35% normalized collision energy setting. Ion fragments were detected in the LTQ Orbitrap. A data-dependent procedure that alternated between one MS scan, followed by 10 MS/MS scans, was applied for the 10 most abundant precursor ions above the 5000 threshold ion count in the MS survey scan with the following Dynamic Exclusion settings: 2 repeat counts, 30 s repeat duration, and 120 s exclusion duration. An electrospray voltage of 2.2 kV was applied. For the MS scans, the m/z scan range was 350 Da to 1800 Da. Mass spectrometry data processing was performed using the Mass-Lynx software (version 4.1, Waters). LC-MS/MS analysis data were identified by searching the P. oxalicum protein database (http://genome.jgi.doe.gov/Penox1/Penox1.home.html).
Electrophoretic mobility shift assay
The DNA-binding domain of ClrB (1–163 amino acids) was PCR-amplified from the P. oxalicum 114–2 genome DNA using the primers ClrB-RTF and ClrB-RTR (S1 Table). The resulting amplicon was digested using EcoRI and BamHI, and then inserted into the expression vector pGEX-4T-1 at the GST downstream with corresponding restriction sites. The correct fusion plasmid was confirmed via nucleotide sequencing, and then transformed into E. coli BL21 (DE3). Parental and recombinant strains were cultured in a lysogeny broth medium and were induced by 0.05 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at 30°C and 150 rpm for 8 hours to induce the GST alone and the GST-ClrB binding-domain production. Protein purification was performed by using Glutathione Sepharose 4B beads after cell ultrasonic decomposition according to the product manual. The protein concentration was then measured using a Bradford Kit according to the instructions of the manufacturer. A 2 kb promoter of PDE_07945 was then amplified from the P. oxalicum 114–2 genome DNA using the primers PDE_07945-F and PDE_07945-R (S1 Table), and then purified using a gel extraction kit according to the instructions of the manufacturer. The DNA concentration was determined using a UV-Vis Spectrophotometer Q5000. GST alone or GST-ClrB binding-domain was mixed in the binding buffer (containing 100 mM Tris-HCl, 100 mM KCl, 10 mM EDTA, 2.5 mM DTT, and 20% Ficoll-400, supplementing 1 μg Poly(dI-dC) to avoid unspecific binding) with purified DNA (20 ng) at room temperature for 10 min for electrophoretic mobility shift assay. The protein-DNA mix was then separated via gel electrophoresis, stained by ethidium bromide, and then visualized. The protein and DNA complex were retardant relative to free DNA. Binding reaction was performed by gradient increasing the protein content to avoid artificial results. GST alone was used as negative control.
Yeast two-hybrid system (Y2H)
The Matchmaker GAL4 two-hybrid system 3 was used for yeast two-hybrid assays. Full-length ORFs of the transcription factors ClrB, CreA, AmyR, and XlnR were PCR-amplified using the P. oxalicum 114–2 cDNA as templates (with primers listed in S1 Table). All the amplicons were cloned into the plasmid pGAD-T7 with the corresponding restriction sites, leading to AD-ClrB, AD-CreA, AD-XlnR, and AD-AmyR. Similarly, the full-length creA, amyR, and xlnR were cloned into the partner plasmid pGBK-T7, which generated BD-CreA, BD-AmyR, and BD-XlnR, respectively. All the fused plasmids were confirmed via nucleotide sequencing. The plasmids pGAD-T7 or AD-X coupling with pGBK-T7 or BD-X in pair were co-transformed into S. cerevisiae AH109 and were cultivated on an SD medium without Trp and Leu for 3 days at 30°C. Protein–protein interaction assay was performed on SD plates without Leu, Trp, and His, and on YPD plates to avoid artificial results. pGBKT7-53/pGADT7-T and pGBKT7-Lam/pGADT7-T pairs were used as internal positive and negative controls, respectively. Moreover, AD-ClrB, AD-CreA, AD-xlnR, and AD-AmyR paired with an empty pGBK-T7 introduced into AH109 were used to eliminate false positive results.
The RNA-seq data have been deposited in NCBI's Gene Expression Omnibus with accession number GSE69298 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE69298).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Liu G, Zhang L, Qin Y, Zou G, Li Z, et al. (2013) Long-term strain improvements accumulate mutations in regulatory elements responsible for hyper-production of cellulolytic enzymes. Scientific reports 3 : 1569–1575. doi: 10.1038/srep01569 23535838
2. Glass NL, Schmoll M, Cate JH, Coradetti S (2013) Plant cell wall deconstruction by ascomycete fungi. Annu Rev Microbiol 67 : 477–498. doi: 10.1146/annurev-micro-092611-150044 23808333
3. Peterson R, Nevalainen H (2012) Trichoderma reesei RUT-C30—thirty years of strain improvement. Microbiology 158 : 58–68. doi: 10.1099/mic.0.054031-0 21998163
4. van den Brink J, de Vries RP (2011) Fungal enzyme sets for plant polysaccharide degradation. Appl Microbiol Biotechnol 91 : 1477–1492. doi: 10.1007/s00253-011-3473-2 21785931
5. Verbeke J, Coutinho P, Mathis H, Quenot A, Record E, et al. (2009) Transcriptional profiling of cellulase and expansin-related genes in a hypercellulolytic Trichoderma reesei. Biotechnol Lett 31 : 1399–1405. doi: 10.1007/s10529-009-0030-5 19479322
6. Amore A, Giacobbe S, Faraco V (2013) Regulation of cellulase and hemicellulase gene expression in fungi. Curr Genomics 14 : 230–249. doi: 10.2174/1389202911314040002 24294104
7. Ebbole DJ (1998) Carbon catabolite repression of gene expression and conidiation in Neurospora crassa. Fungal Genet Biol 25 : 15–21. 9806802
8. Ilmen M, Thrane C, Penttila M (1996) The glucose repressor gene cre1 of Trichoderma: isolation and expression of a full-length and a truncated mutant form. Mol Gen Genet 251 : 451–460. 8709949
9. Dowzer CE, Kelly JM (1991) Analysis of the creA gene, a regulator of carbon catabolite repression in Aspergillus nidulans. Mol Cell Biol 11 : 5701–5709. 1922072
10. Mach-Aigner AR, Pucher ME, Steiger MG, Bauer GE, Preis SJ, et al. (2008) Transcriptional regulation of xyr1, encoding the main regulator of the xylanolytic and cellulolytic enzyme system in Hypocrea jecorina. Appl Environ Microbiol 74 : 6554–6562. doi: 10.1128/AEM.01143-08 18791032
11. van Peij NN, Visser J, de Graaff LH (1998) Isolation and analysis of xlnR, encoding a transcriptional activator co-ordinating xylanolytic expression in Aspergillus niger. Mol Microbiol 27 : 131–142. 9466262
12. Sun J, Tian C, Diamond S, Glass NL (2012) Deciphering transcriptional regulatory mechanisms associated with hemicellulose degradation in Neurospora crassa. Eukaryot Cell 11 : 482–493. doi: 10.1128/EC.05327-11 22345350
13. Aro N, Ilmen M, Saloheimo A, Penttila M (2003) ACEI of Trichoderma reesei is a repressor of cellulase and xylanase expression. Appl Environ Microbiol 69 : 56–65. 12513977
14. Aro N, Saloheimo A, Ilmen M, Penttila M (2001) ACEII, a novel transcriptional activator involved in regulation of cellulase and xylanase genes of Trichoderma reesei. J Biol Chem 276 : 24309–24314. 11304525
15. Hakkinen M, Valkonen MJ, Westerholm-Parvinen A, Aro N, Arvas M, et al. (2014) Screening of candidate regulators for cellulase and hemicellulase production in Trichoderma reesei and identification of a factor essential for cellulase production. Biotechnol Biofuels 7 : 14. doi: 10.1186/1754-6834-7-14 24472375
16. Ogawa M, Kobayashi T, Koyama Y (2012) ManR, a novel Zn(II)2Cys6 transcriptional activator, controls the beta-mannan utilization system in Aspergillus oryzae. Fungal Genet Biol 49 : 987–995. doi: 10.1016/j.fgb.2012.09.006 23063954
17. Coradetti ST, Craig JP, Xiong Y, Shock T, Tian C, et al. (2012) Conserved and essential transcription factors for cellulase gene expression in ascomycete fungi. Proc Natl Acad Sci U S A doi: 10.1073/pnas.1200785109
18. Nitta M, Furukawa T, Shida Y, Mori K, Kuhara S, et al. (2012) A new Zn(II)(2)Cys(6)-type transcription factor BglR regulates beta-glucosidase expression in Trichoderma reesei. Fungal Genet Biol 49 : 388–397. doi: 10.1016/j.fgb.2012.02.009 22425594
19. Wang S, Liu G, Wang J, Yu J, Huang B, et al. (2013) Enhancing cellulase production in Trichoderma reesei RUT C30 through combined manipulation of activating and repressing genes. J Ind Microbiol Biotechnol 40 : 633–641. doi: 10.1007/s10295-013-1253-y 23467998
20. Coradetti ST, Xiong Y, Louise Glass N (2013) Analysis of a conserved cellulase transcriptional regulator reveals inducer-independent production of cellulolytic enzymes in Neurospora crassa. MicrobiologyOpen doi: 10.1002/mbo3.94
21. Nehlin JO, Ronne H (1990) Yeast MIG1 repressor is related to the mammalian early growth response and Wilms' tumour finger proteins. EMBO J 9 : 2891–2898. 2167835
22. de Vries RP, Visser J, de Graaff LH (1999) CreA modulates the XlnR-induced expression on xylose of Aspergillus niger genes involved in xylan degradation. Res Microbiol 150 : 281–285. 10376490
23. Kubicek CP, Portnoy T, Margeot A, Linke R, Atanasova L, et al. (2011) The CRE1 carbon catabolite repressor of the fungus Trichoderma reesei: a master regulator of carbon assimilation. BMC Genomics 12
24. Liu G, Zhang L, Wei X, Zou G, Qin Y, et al. (2013) Genomic and secretomic analyses reveal unique features of the lignocellulolytic enzyme system of Penicillium decumbens. PLoS One 8: e55185. doi: 10.1371/journal.pone.0055185 23383313
25. Kubicek CP, Mikus M, Schuster A, Schmoll M, Seiboth B (2009) Metabolic engineering strategies for the improvement of cellulase production by Hypocrea jecorina. Biotechnol Biofuels 2 : 19. doi: 10.1186/1754-6834-2-19 19723296
26. Y Qu G L Y Q Z L (2013) Improving lignocellulolytic enzyme production with Penicillium: from strain screening to systems biology. Adv Biochem Eng Biot 4 : 12.
27. Chen M, Qin Y, Cao Q, Liu G, Li J, et al. (2013) Promotion of extracellular lignocellulolytic enzymes production by restraining the intracellular beta-glucosidase in Penicillium decumbens. Bioresour Technol 137 : 33–40. doi: 10.1016/j.biortech.2013.03.099 23584406
28. Zhang Y, Zhang J, Xiao P, Wang T, Qu Y (2012) Improved cellulase production via disruption of PDE01641 in cellulolytic fungus Penicillium decumbens. Bioresour Technol 123 : 733–737. doi: 10.1016/j.biortech.2012.07.101 22981621
29. Li J, Liu G, Chen M, Li Z, Qin Y, et al. (2013) Cellodextrin transporters play important roles in cellulase induction in the cellulolytic fungus Penicillium oxalicum. Appl Microbiol Biotechnol doi: 10.1007/s00253-013-5301-3
30. Herpoel-Gimbert I, Margeot A, Dolla A, Jan G, Molle D, et al. (2008) Comparative secretome analyses of two Trichoderma reesei RUT-C30 and CL847 hypersecretory strains. Biotechnol Biofuels 1 : 18. doi: 10.1186/1754-6834-1-18 19105830
31. Kubodera T, Yamashita N, Nishimura A (2000) Pyrithiamine resistance gene (ptrA) of Aspergillus oryzae: Cloning, characterization and application as a dominant selectable marker for transformation. Biosci Biotech Bioch 64 : 1416–1421.
32. Yu JH, Hamari Z, Han KH, Seo JA, Reyes-Dominguez Y, et al. (2004) Double-joint PCR: a PCR-based molecular tool for gene manipulations in filamentous fungi. Fungal Genet Biol 41 : 973–981. 15465386
33. Li ZH, Du CM, Zhong YH, Wang TH (2010) Development of a highly efficient gene targeting system allowing rapid genetic manipulations in Penicillium decumbens. Appl Microbiol Biotechnol 87 : 1065–1076. doi: 10.1007/s00253-010-2566-7 20393703
34. Druzhinina IS, Seidl-Seiboth V, Herrera-Estrella A, Horwitz BA, Kenerley CM, et al. (2011) Trichoderma: the genomics of opportunistic success. Nat Rev Microbiol 9 : 749–759. doi: 10.1038/nrmicro2637 21921934
35. Colot HV, Park G, Turner GE, Ringelberg C, Crew CM, et al. (2006) A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc Natl Acad Sci U S A 103 : 10352–10357. 16801547
36. Johnston SA, Zavortink MJ, Debouck C, Hopper JE (1986) Functional domains of the yeast regulatory protein GAL4. Proc Natl Acad Sci U S A 83 : 6553–6557. 2944111
37. Punt PJ, Dingemanse MA, Jacobs-Meijsing BJ, Pouwels PH, van den Hondel CA (1988) Isolation and characterization of the glyceraldehyde-3-phosphate dehydrogenase gene of Aspergillus nidulans. Gene 69 : 49–57. 3066699
38. Sun J, Glass NL (2011) Identification of the CRE-1 Cellulolytic Regulon in Neurospora crassa. PLoS One 6: e25654. doi: 10.1371/journal.pone.0025654 21980519
39. Yao G, Li Z, Gao L, Wu R, Kan Q, et al. (2015) Redesigning the regulatory pathway to enhance cellulase production in Penicillium oxalicum. Biotechnol Biofuels 8 : 71. doi: 10.1186/s13068-015-0253-8 25949521
40. Lv X, Zheng F, Li C, Zhang W, Chen G, et al. (2015) Characterization of a copper responsive promoter and its mediated overexpression of the xylanase regulator 1 results in an induction-independent production of cellulases in Trichoderma reesei. Biotechnol Biofuels 8 : 67. doi: 10.1186/s13068-015-0249-4 25926888
41. Watanabe J, Tanaka H, Mogi Y, Yamazaki T, Suzuki K, et al. (2011) Loss of Aspergillus oryzae amyR function indirectly affects hemicellulolytic and cellulolytic enzyme production. J Biosci Bioeng 111 : 408–413. doi: 10.1016/j.jbiosc.2010.12.006 21193346
42. Silva-Rocha R, Castro LD, Antonieto AC, Guazzaroni ME, Persinoti GF, et al. (2014) Deciphering the Cis-Regulatory Elements for XYR1 and CRE1 Regulators in Trichoderma reesei. PLoS One 9: e99366. doi: 10.1371/journal.pone.0099366 24941042
43. Ling M, Qin Y, Li N, Liang Z (2009) Binding of two transcriptional factors, Xyr1 and ACEI, in the promoter region of cellulase cbh1 gene. Biotechnol Lett 31 : 227–231. doi: 10.1007/s10529-008-9857-4 18854952
44. Xiong Y, Sun J, Glass NL (2014) VIB1, a Link between Glucose Signaling and Carbon Catabolite Repression, Is Essential for Plant Cell Wall Degradation by Neurospora crassa. PLoS Genet 10: e1004500. doi: 10.1371/journal.pgen.1004500 25144221
45. Tamayo EN, Villanueva A, Hasper AA, de Graaff LH, Ramon D, et al. (2008) CreA mediates repression of the regulatory gene xlnR which controls the production of xylanolytic enzymes in Aspergillus nidulans. Fungal Genet Biol 45 : 984–993. doi: 10.1016/j.fgb.2008.03.002 18420433
46. Kulmburg P, Mathieu M, Dowzer C, Kelly J, Felenbok B (1993) Specific binding sites in the alcR and alcA promoters of the ethanol regulon for the CREA repressor mediating carbon catabolite repression in Aspergillus nidulans. Mol Microbiol 7 : 847–857. 8483416
47. Cziferszky A, Mach RL, Kubicek CP (2002) Phosphorylation positively regulates DNA binding of the carbon catabolite repressor Cre1 of Hypocrea jecorina (Trichoderma reesei). J Biol Chem 277 : 14688–14694. 11850429
48. Martinez D, Berka RM, Henrissat B, Saloheimo M, Arvas M, et al. (2008) Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat Biotechnol 26 : 553–560. doi: 10.1038/nbt1403 18454138
49. Tian C, Beeson WT, Iavarone AT, Sun J, Marletta MA, et al. (2009) Systems analysis of plant cell wall degradation by the model filamentous fungus Neurospora crassa. Proc Natl Acad Sci U S A 106 : 22157–22162. doi: 10.1073/pnas.0906810106 20018766
50. Zhou Q, Xu J, Kou Y, Lv X, Zhang X, et al. (2012) Differential involvement of beta-glucosidases from Hypocrea jecorina in rapid induction of cellulase genes by cellulose and cellobiose. Eukaryot Cell doi: 10.1128/EC.00170-12
51. Znameroski EA, Coradetti ST, Roche CM, Tsai JC, Iavarone AT, et al. (2012) Induction of lignocellulose-degrading enzymes in Neurospora crassa by cellodextrins. Proc Natl Acad Sci U S A 109 : 6012–6017. doi: 10.1073/pnas.1118440109 22474347
52. Gruber F, Visser J, Kubicek CP, de Graaff LH (1990) The development of a heterologous transformation system for the cellulolytic fungus Trichoderma reesei based on a pyrG-negative mutant strain. Curr Genet 18 : 71–76. 2245476
53. Park J, Park J, Jang S, Kim S, Kong S, et al. (2008) FTFD: an informatics pipeline supporting phylogenomic analysis of fungal transcription factors. Bioinformatics 24 : 1024–1025. doi: 10.1093/bioinformatics/btn058 18304934
54. Krappmann S, Bayram O, Braus GH (2005) Deletion and allelic exchange of the Aspergillus fumigatus veA locus via a novel recyclable marker module. Eukaryot Cell 4 : 1298–1307. 16002655
55. Nakayashiki H, Hanada S, Quoc NB, Kadotani N, Tosa Y, et al. (2005) RNA silencing as a tool for exploring gene function in ascomycete fungi. Fungal Genetics and Biology 42 : 275–283. 15749047
56. Punt PJ, Dingemanse MA, Kuyvenhoven A, Soede RD, Pouwels PH, et al. (1990) Functional elements in the promoter region of the Aspergillus nidulans gpdA gene encoding glyceraldehyde-3-phosphate dehydrogenase. Gene 93 : 101–109. 2121607
57. Sweigard J., Chumley F., Carroll A., Farrall L., V B. (1997) A series of vectors for fungal transformation. Fungal Genet Newsl 44 : 52–53.
58. Penttila M, Nevalainen H, Ratto M, Salminen E, Knowles J (1987) A versatile transformation system for the cellulolytic filamentous fungus Trichoderma reesei. Gene 61 : 155–164. 3127274
59. Wei X, Zheng K, Chen M, Liu G, Li J, et al. (2011) Transcription analysis of lignocellulolytic enzymes of Penicillium decumbens 114–2 and its catabolite-repression-resistant mutant. C R Biol 334 : 806–811. doi: 10.1016/j.crvi.2011.06.002 22078737
60. Li R, Yu C, Li Y, Lam TW, Yiu SM, et al. (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25 : 1966–1967. doi: 10.1093/bioinformatics/btp336 19497933
61. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5 : 621–628. doi: 10.1038/nmeth.1226 18516045
62. Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106. doi: 10.1186/gb-2010-11-10-r106 20979621
63. Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Conesa A (2011) Differential expression in RNA-seq: a matter of depth. Genome Res 21 : 2213–2223. doi: 10.1101/gr.124321.111 21903743
64. Conesa A, Gotz S (2008) Blast2GO: A comprehensive suite for functional analysis in plant genomics. International journal of plant genomics 2008 : 619832. doi: 10.1155/2008/619832 18483572
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 9
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Arabidopsis AtPLC2 Is a Primary Phosphoinositide-Specific Phospholipase C in Phosphoinositide Metabolism and the Endoplasmic Reticulum Stress Response
- Bridges Meristem and Organ Primordia Boundaries through , , and during Flower Development in
- KLK5 Inactivation Reverses Cutaneous Hallmarks of Netherton Syndrome
- XBP1-Independent UPR Pathways Suppress C/EBP-β Mediated Chondrocyte Differentiation in ER-Stress Related Skeletal Disease
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy