
Autophosphorylation of the Bacterial Tyrosine-Kinase CpsD Connects Capsule Synthesis with the Cell Cycle in
Bacteria utilize a multi-protein membrane complex to synthesize and export the polysaccharide capsule that conceals and covers the cell. In bacterial pathogens, the capsule protects the cell form opsonophagocytosis and complement-mediated killing. The mechanisms allowing the bacterial cell to maintain this protective capsule during cell growth and division remain unknown. The capsule assembly machinery encompasses a particular type of tyrosine-kinases found only in bacteria, which are called BY-kinases. These kinases are involved in the regulation of several cellular functions including polysaccharide capsule production. Studying the role of BY-kinase represents thus an interesting approach to decipher the mechanisms of capsule synthesis and export. Here, we study the role of the BY-kinase CpsD in the human pathogen Streptococcus pneumoniae. We show that CpsD plays a dual function in the pneumococcus. Indeed, CpsD captures the capsule assembly machinery at the site of division, but we also show that CpsD coordinates capsule production with the cell cycle by interacting with the chromosome segregation system. These features provide a simple mechanism to cover the complete surface of the pneumococcal daughter cells. This finding further opens a new view of the function of BY-kinases in the bacterial cell notably in localizing protein complexes in subcellular regions over the cell cycle.
Published in the journal:
. PLoS Genet 11(9): e32767. doi:10.1371/journal.pgen.1005518
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005518
Summary
Bacteria utilize a multi-protein membrane complex to synthesize and export the polysaccharide capsule that conceals and covers the cell. In bacterial pathogens, the capsule protects the cell form opsonophagocytosis and complement-mediated killing. The mechanisms allowing the bacterial cell to maintain this protective capsule during cell growth and division remain unknown. The capsule assembly machinery encompasses a particular type of tyrosine-kinases found only in bacteria, which are called BY-kinases. These kinases are involved in the regulation of several cellular functions including polysaccharide capsule production. Studying the role of BY-kinase represents thus an interesting approach to decipher the mechanisms of capsule synthesis and export. Here, we study the role of the BY-kinase CpsD in the human pathogen Streptococcus pneumoniae. We show that CpsD plays a dual function in the pneumococcus. Indeed, CpsD captures the capsule assembly machinery at the site of division, but we also show that CpsD coordinates capsule production with the cell cycle by interacting with the chromosome segregation system. These features provide a simple mechanism to cover the complete surface of the pneumococcal daughter cells. This finding further opens a new view of the function of BY-kinases in the bacterial cell notably in localizing protein complexes in subcellular regions over the cell cycle.
Introduction
Streptococcus pneumoniae is a Gram-positive bacterium usually found as a commensal in healthy adults and children [1]. It does however have the potential to become pathogenic and is a frequent cause of community-acquired diseases. S. pneumoniae is associated with a variety of infections that can range in severity from otitis media to pneumonia or meningitis [2]. Despite the availability of antibiotics, pneumococcal infections still have high mortality rates and vaccine efficiency drops over time as new and infectious non-vaccine covered serotypes are emerging in clinical isolates [3]. Pneumococcal virulence is strictly dependent on the capsular polysaccharide (CPS) production: non-encapsulated mutants of clinical pneumococcal isolates are non-virulent [4]. The capsule plays a major role in both colonization and persistence of S. pneumoniae in the infected host due to its ability to form a shield that prevents antibodies and complement components from interacting with their receptors on the host phagocytic cells [5, 6].
In all serotypes, the cps operon includes serotype-specific genes, encoding enzymes required for the synthesis of specific sugar components, as well as conserved genes encoding proteins essential for capsular synthesis and export (Fig 1A) [7]. Export of the capsule across the plasma membrane occurs by a Wzy-dependent polymerization pathway, analogous to Group 1 CPS biosynthesis in Escherichia coli [8, 9] (Fig 1B). The 5’ region of the locus encodes the cpsA, cpsB, cpsC and cpsD genes, (also known as wzg, wzh, wzd and wze) (Fig 1A). CpsA was shown to interact with the pyrophosphoryl-lipid carrier of the polysaccharide precursor and is proposed to attach capsular polysaccharide to cell wall peptidoglycan [10]. cpsB, cpsC and cpsD constitute a phosphoregulatory system that controls the polysaccharide assembly machinery encompassing a glycosyl-transferase (CpsE), a flippase (CpsJ) and a polymerase (CpsH) (Fig 1B) [9]. CpsB is a metal-dependent phosphotyrosine-protein phosphatase of the PHP family [11] whereas CpsC and CpsD constitute a so-called BY-kinase, a particular type of tyrosine-autokinase, which shares no resemblance with eukaryotic tyrosine-kinase and is conserved among most bacterial phyla [12–14].

BY-kinases consist of two main structural domains: an N-terminal extracellular domain flanked by two transmembrane helices and a cytoplasmic C-terminal domain, harboring the kinase activity [15]. In Firmicutes, these domains are encoded by two successive genes, and are therefore present as separate polypeptide chains, one cytoplasmic and the other in the membrane (Fig 1C). The two polypeptides need to interact to form an active BY-kinase [16]. The crystal structure of the BY-kinase CapB from Staphylococcus aureus showed that the cytoplasmic C-terminal end of the transmembrane modulator CapA is required for the activation of the cytoplasmic kinase CapB [17]. More precisely, the C-terminal extremity of CapA forms a αA-ßA motif complementing the catalytic site of CapB and stabilizing the ATP molecule. The cytoplasmic domain of BY-kinases is able to autophosphorylate on several tyrosines forming a C-terminal tyrosine cluster motif (Fig 1C) [18, 19]. Although the detailed mechanisms by which BY-kinases promote CPS synthesis and export remain elusive, it has been proposed that cycling between phosphorylated and non-phosphorylated forms of the BY-kinase, regulated by the cognate phosphotyrosine-phosphatase, is required for proper synthesis and export of the polysaccharide polymer [20–23].
The single BY-kinase produced by most of the 93 S. pneumoniae serotypes [9] comprises the transmembrane modulator CpsC and the cytoplasmic kinase domain CpsD [12, 13]. Several studies reported that autophosphorylation of CpsD in the tyrosine cluster negatively regulates CPS production [18, 24]. Moreover, evidence was provided that CpsD tyrosine-kinase activity influences capsule production and modulates invasive pneumococcal disease [12, 25]. Interestingly, it was recently shown that CpsD and CpsC both localize at the division site in the serotype 14 strain ATCC6314 [13]. In addition, capsule is absent from the division site and detected only at the old cell halves in the absence of either CpsD or CpsC. Collectively, it is hypothesized that the CPS assembly machinery would adopt two distinct localizations: one around the cell and one at the division site captured by CpsC and CpsD for septal CPS production. In the latter, CpsC and CpsD would either act as activators of CPS export or function as the exporter [13].
Here, we have investigated the role of CpsD activation by CpsC as well as the impact of CpsD autophosphorylation on capsule production in the well-studied serotype 2 strain D39 [26]. We first show that the C-terminal cytoplasmic end of CpsC is required for CpsD autophosphorylation and localization at the division septum. Analysis of CPS production together with imaging of the polysaccharide polymerase CpsH localization demonstrates that CPS are exclusively produced at the division septum in WT cells and challenges the two-machine model for CPS assembly. Strikingly, we also observe that cells producing a non-phosphorylatable variant of CpsD display defective capsule production at the septum together with aberrant elongated shape with multiple non-constricted septa, nucleoid defects and reduced dynamics of the chromosome segregation protein ParB. In line with a role of CpsC and CpsD in controlling the pneumococcal cell cycle, it was shown that BY-kinases have homology to the large P-loop NTPase superfamily [27] that includes ParA-like proteins, which are involved in chromosome segregation by interacting with ParB [28]. Molecular modeling confirms that CpsD displays structural similarities with ParA. Interestingly, we found that CpsD interacts with ParB in vitro and that the stability of the CpsD/ParB complex is modulated by CpsD phosphorylation in vivo. These observations show that CPS production is tightly connected with the cell cycle and support a model wherein crosstalk between CpsD and ParB, modulated by CpsD autophosphorylation, signals the status of CPS production to the proteins in charge of chromosome segregation, thus ensuring coordination between encapsulation and cell division.
Results
The cytoplasmic C-terminal end of CpsC is required for CpsD phosphorylation and localization at the division site
Structural studies of BY-kinases have established that the C-terminal peptide of the transmembrane modulator specifically interacts and activates the cytoplasmic catalytic domain [17]. We first tested whether the C-terminal end of CpsC (CpsC-Cter) is required for CpsD autophosphorylation as it is the case for BY-kinases from other Firmicutes [16, 29, 30]. To do that, we first analyzed by yeast two-hybrid assays the ability of a derivative of CpsC lacking the C-terminal 30 amino acids (CpsC-ΔCter) to interact with CpsD. We observed that full-length CpsC interacts efficiently with CpsD while this interaction was abolished in the absence of CpsC-Cter (Fig 2A). As expected for a BY-kinase [17], CpsD interacted with itself. Next, we constructed a nonpolar markerless mutant strain expressing CpsC-ΔCter (cpsC-ΔCter strain) and analyzed CpsD autophosphorylation using anti-phosphotyrosine antibodies (Fig 2B). Non-polarity of the deletion of cpsC-Cter was confirmed by analyzing the expression of CpsD and CpsH fused to GFP (see below) in WT and cpsC-ΔCter strains (S1A and S2 Figs) and partial restoration of capsule production in the cpsC-ΔCter mutant carrying a copy of cpsC at the ectopic amiF/treR locus under the control of the maltose inducible promoter PM [31] (S1B and S1C Fig). No phosphorylation signal was detected for CpsD in the cpsC-ΔCter mutant (Fig 2B). As controls, CpsD was efficiently phosphorylated in the wild-type strain whereas no phosphorylation signal was detected in a mutant deficient for CpsD (ΔcpsD) or a strain producing CpsD mutated on the three tyrosines of its C-terminal tyrosine cluster motif. In addition, CpsD phosphorylation was partially restored in the cpsC-ΔCter strain complemented with PM-cpsC (Fig 2B). The partial restoration of CpsD phosphorylation and capsule synthesis observed for the cpsC-ΔCter PM-cpsC strain suggests that production of CpsC-ΔCter interferes with the ability of native CpsC to interact with CpsD and/or to function in the CPS assembly machinery. Next, we analyzed the effect of the CpsC C-terminal deletion (CpsC-ΔCter) on CpsD localization. For this purpose, we constructed a C-terminal monomeric GFP fusion to CpsD. The CpsD-GFP fusion is stable and functional since cells grew normally and displayed normal CPS production patterns (S3 and S4 Figs) [24]. As previously shown in a serotype 14 strain [13], CpsD-GFP also localized at midcell in serotype 2 D39 cells (Fig 2C). However, CpsD delocalized to the cytoplasm in cpsC-ΔCter cells whereas CpsC-ΔCter-GFP still localized at midcell (Fig 2C). Altogether, these observations show that the C-terminal end of CpsC is required to position CpsD at the division site and trigger its autophosphorylation.
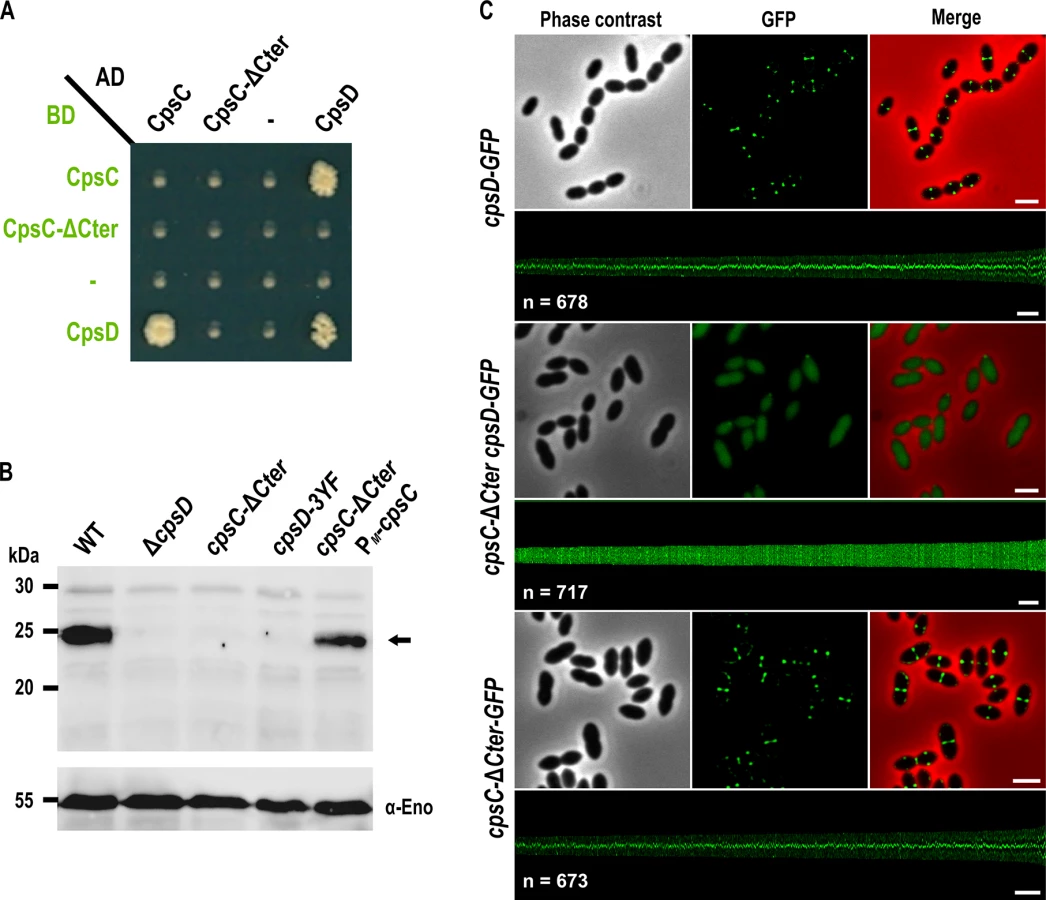
The cytoplasmic C-terminal end of CpsC is required for septal localization of the polysaccharide polymerase CpsH and CPS production
Our observations prompted us to analyze CPS synthesis in the cpsC-ΔCter mutant. For that, we used anti-serotype 2 capsule antibodies and localized CPS by immunofluorescence microscopy. Consistent with observations reported by Henriques and co-workers [13], CPS were not produced at the division site but only at the old cell halves in the ΔcpsD mutant while CPS were detected over the entire surface of wild-type cells (Fig 3A). We also observed that CPS were absent from the division septa in the cpsC-ΔCter mutant (Fig 3A). These data show that the absence of CpsC C-terminus, and consequently CpsD phosphorylation and localization at midcell, alters the localization and/or the activity of the capsule assembly machinery. To test these hypotheses, we quantified the CPS fluorescence signal in living cells and immunodetected the total fraction of CPS produced by our mutants using anti-type 2 capsule antibodies. We observed a striking CPS production and polymerization defect in both cpsC-ΔCter and ΔcpsD mutants compared to the WT strain (Fig 3B and 3C). This defect led to the accumulation of low molecular weight polysaccharides (Fig 3C). Next, we used the polymerase CpsH as a marker to localize the capsule assembly machinery. CpsH is a predicted membrane protein with both N - and C-terminal ends located outside the cell [9]. It was shown that superfolder GFP (sfGFP), in certain fusions, can fluoresce in the extracellular milieu [33]. We thus constructed a strain expressing CpsH fused to sfGFP at its C-terminus. WT cells producing CpsH-sfGFP as the only source of CpsH from their endogenous chromosomal locus grew and produced capsule similarly to WT cells attesting that the fusion CpsH-sfGFP is functional (S3 Fig). As shown in Fig 3D, we found that CpsH-sfGFP localized exclusively at the division septum in WT cells. Interestingly, CpsH partially delocalized in both cpsC-ΔCter and ΔcpsD mutants, and the signals were not exclusively present at the division septum (Fig 3D). CpsH-sfGFP was stable and produced at similar amount in WT and cpsC-ΔCter and ΔcpsD mutants (S2 Fig). These data suggest that the capsule assembly machinery localizes at the division site to assemble CPS. In addition, the localization and the activity of the capsule assembly machinery are both dependent on CpsD localization at the division site.

Non-phosphorylated CpsD hinders capsule production and cell division
To better understand the role of CpsD phosphorylation at the division site in CPS production, we constructed two mutant strains expressing either non-phosphorylated CpsD (cpsD-3YF) or its phosphomimetic form (cpsD-3YE). For that, each of the 3 tyrosines of the tyrosine cluster of CpsD was substituted either for phenylalanine or glutamic acid. Then, we analyzed these strains for capsule production and CpsH localization. As shown in Fig 3A and 3B, cpsD-3YE cells displayed CPS localization pattern and quantification indistinguishable from that of WT cells even if the capsular halo around WT cells suggested that CPS could be in less tighter association with the cells than in cpsD-3YE cells. CpsH-sfGFP was also found to localize properly at the septum in cpsD-3YE cells (Fig 3D). However, by measuring cell lengths, cpsD-3YE cells were significantly shorter (1.88 μm +/ - 0.33) than WT cells (2.02 μm +/ - 0.32, p<0.0001, Mann Whitney rank sum test) (Fig 4). Furthermore, cpsD-3YF cells displayed capsule mainly at the old cell poles as observed in ΔcpsD cells (Fig 3A). Strikingly, 26.6% of cpsD-3YF cells possessed an elongated cell shape (> 3 μm). This aberrant cell length of > 3 μm was observed in less than 1.4% of WT or cpsD-3YE cells (Fig 4). Furthermore, CpsH-sfGFP partially delocalized and formed several foci around the cell in the cpsD-3YF mutant although CpsH-sfGFP was stable and produced at similar amount in WT and cpsD-3YF and cpsD-3YE mutants (S2 Fig). In agreement with these observations, quantification of CPS fluorescence signal and western immunoblotting using anti-serotype 2 capsule antibodies showed that CPS production was hampered in the cpsD-3YF mutant compared to that of WT and cpsD-3YE cells (Fig 3B and 3C). However, CPS polymerization remained clearly more effective than in cpsC-ΔCter and ΔcpsD mutants suggesting that beyond CpsD phosphorylation, deletion of cpsD or cpsC-Cter further alters the global functioning of the CPS assembly machinery (Fig 3B and 3C). These data show that permanent dephosphorylation of CpsD is detrimental for localizing the capsule synthesis machinery at the division site and causes problems with cell division, thus suggesting a link between defective capsule synthesis and aberrant elongation of cells.

Septal localization of non-phosphorylated CpsD impairs cell division
We hypothesized that the absence of CpsD phosphorylation could be detrimental for its localization at midcell. To test whether the phosphorylation state of CpsD influences its localization, we constructed strains producing C-terminal GFP fusions to either CpsD-3YE (cpsD-3YE-GFP) or CpsD-3YF (cpsD-3YF-GFP). Both types of tyrosine substitutions did not affect the stability and the production of the CpsD-GFP fusion (S4 Fig). Fluorescence microscopy indicated that these mutations do not affect septal localization (Fig 5A). Therefore, we questioned why expression of cpsD-3YF induces cell elongation whereas cells expressing CpsC-ΔCter, in which CpsD autophosphorylation is prevented (Fig 2B), are not elongated (Figs 2B and 4). To reconcile this apparent contradiction, we constructed mutant strains expressing cpsC-ΔCter and cpsD-3YF fused or not to gfp (cpsC-ΔCter-cpsD-3YF-gfp and cpsC-ΔCter-cpsD-3YF). Consistent with our observations presented in Fig 2C, CpsD-3YF-GFP delocalized in the cytoplasm in the absence of the cytoplasmic C-terminal end of CpsC (Fig 5A). However, cpsC-ΔCter-cpsD-3YF cells did not display an aberrant elongated cell shape phenotype anymore indicating that the deletion of CpsC-Cter suppresses cell elongation of the cpsD-3YF mutant (Fig 4). Together, these data suggest that the presence of non-phosphorylated CpsD at the division site partially inhibits cell division, leading to an elongated cell shape phenotype.

cpsD-3YF cells display defective cell constriction and aberrant nucleoid morphology
To better analyze the ultrastructure of elongated cpsD-3YF cells, we examined these cells by transmission-electron microscopy (TEM). As controls, WT and cpsD-3YE cells were found to possess the characteristic ovoid cell shape (Figs 5B and S5). By contrast, examination of cpsD-3YF mutant confirmed the severe disturbed cell morphology observed by phase-contrast microscopy (Fig 5B). In addition, these images showed the presence of multiple septal initiations on each side of the long cell axis, indicative of impaired cell constriction (Fig 5B). In addition, and in agreement with CPS immunolabelling (Fig 3A), CPS were primarily detected at the pole in cpsD-3YF cells observed by electron microscopy (Fig 5B).
To further investigate the division defects of the cpsD-3YF mutant strain, we analyzed the localization of the major cell division protein FtsZ that forms contractile rings at mid-cell [34]. As expected, FtsZ-GFP [35] localized at mid-cell in WT cells as well as in cpsD-3YE cells (Fig 5C). In elongated cpsD-3YF cells, FtsZ-GFP fluorescence showed a ladder localization pattern indicating that it is properly recruited to each non-constricted septum (Fig 5C). This observation shows that the division defects of cpsD-3YF cells are not due to altered Z-ring assembly at the division site.
S. pneumoniae does not contain homologs of proteins involved in nucleoid occlusion, and it was shown that the cell division machinery assembles over the nucleoid and that septation occurs just after chromosomes splitting, indicating that DNA itself can act as a physical barrier for cell division [36]. In line with this idea, mutants affected in chromosome segregation are often elongated [36, 37]. To test whether the non-phosphorylated CpsD mutant induces chromosome segregation defects that could lead to the elongated cell phenotype, we analyzed nucleoid morphology using DAPI staining. As shown in Fig 6A, the nucleoid is properly condensed and segregated in WT and cpsD-3YE cells. However, DAPI staining revealed abnormal elongated nucleoids in cpsD-3YF cells (Fig 6A). To quantify these nucleoid defects, we used the histone-like HlpA-RFP fusion as a chromosome marker in live cells [36]. As expected, 93.7% of WT cells displayed normal, well-condensed nucleoid(s) (Fig 6B and 6C). The same observation (95.7% normal nucleoids) was made in the cpsD-3YE mutant confirming that the expression of CpsD-3YE as the sole source of CpsD does not impact the cell cycle. The situation differed in cpsD-3YF cells in which only 69.8% of cells displayed well condensed nucleoid(s). In addition, 3.2% of cpsD-3YF cells were either devoid of nucleoid or presented an uncondensed nucleoid (Fig 6B and 6C). These defects are significantly more abundant than in WT and cpsD-3YE cells in which they are observed in only 0.6% and 0.7% of cells, respectively. More importantly, we found that nucleoids are elongated and abnormally shaped in 27.0% of cpsD-3YF cells compared to 5.7% and 3.6% of WT and cpsD-3YE cells, respectively (Fig 6B and 6C). Altogether, these observations show that chromosome replication and/or segregation is impaired in the cpsD-3YF mutant, suggesting that expression of non-phosphorylated CpsD at the septum could interfere with these processes.

ParB mobility is modulated by CpsD phosphorylation
To test whether initiation of DNA-replication is perturbed when CpsD is not phosphorylated, we performed marker frequency analysis using quantitative real-time PCR [38]. Determining the origin-to-terminus ratios showed no significant difference between WT and cpsD-3YF cells (S6 Fig). Interestingly, the chimera CpsC/D composed by CpsD fused to the C-terminal end of CpsC exhibits 20% identity and 52.4% similarity to the Bacillus subtilis ParA protein Soj (Fig 7A) and molecular modeling using the calculated structure of the BY-kinase chimera CapA1/B2 from Staphylococcus aureus as template [17] suggests that the structure of the two proteins is similar (Fig 7B–7D). The accurate role of ParA in chromosome segregation remains unclear but it is proposed to interact with and assist the chromosome partitioning protein ParB during segregation of chromosomes to the daughter cells [28]. ParB specifically binds to centromere-like DNA sequences named parS that are located near the origin of replication [39, 40]. Therefore, to evaluate whether CpsD phosphorylation impact ParB dynamics in live cells, we constructed strains (WT, cpsD-3YE and cpsD-3YF) producing the functional fusion ParB-sfGFP [40] and we analyzed ParB dynamics by TIRF (Total Internal Reflection Fluorescence) microscopy, a technique in which only a thin section of the cell (approx. 100 nm) is excited, thus providing high axial resolution. ParB-sfGFP foci were categorized as stationary if they remained in TIRF focus during the 40 sec time-span of the experiment, while ParB-sfGFP foci appearing, disappearing or splitting were categorized as dynamic (Fig 8A and 8B). We observed that 66.9 +/ - 2.8% of ParB-sfGFP were stationary in WT cells (Fig 8C). Interestingly, this fraction increased up to 80.6 +/ - 2.4% in cpsD-3YF cells whereas it decreased down to 56.4 +/ - 2.4% in cpsD-3YE cells (Fig 8C). These experiments thus suggest that CpsD phosphorylation could play a role in chromosome segregation by modulating ParB mobility.


Interplay between CpsD and ParB
The latter observation, combined with the fact that CpsD is homologous to ParA-type proteins, prompted us to analyze the timing of CpsD and ParB localization in living cells. To do so, we constructed a double-labeled strain expressing both ParB-sfGFP and CpsD-RFP and performed time-lapse-microcopy (Fig 9A). As shown in Fig 9A and S1 Movie, and consistent with previous observations [40], ParB-sfGFP localizes as single foci at cell equators (future division site). In these predivisional cells, CpsD-RFP did not co-localize with ParB and was exclusively detected at the current division site (Fig 9A). As the cell cycle progresses, CpsD-RFP displayed a dual localization pattern; the fusion protein remained at the division site until cell constriction was completed, but also localized at the future-division site where it co-localizes for a short time with ParB (arrows in Fig 9A). Then, as the new cell cycle began, the oriC-localized ParB-sfGFP moved, due to chromosome segregation, toward the daughter cell equator while CpsD-RFP remained at the division site. This transient co-localization of CpsD with ParB suggests that the two proteins might interact physically. To investigate this, we applied the approach commonly used for the purification of BY-kinases from Firmicutes: we purified the active and fully autophosphorylated CpsC/D chimera in which the cytoplasmic C-terminal end of CpsC is fused to the N-terminal end of CpsD (Fig 1C and S7A and S7B Fig). As shown in S7A Fig, ParB and the non-phosphorylatable CpsC/D-YF chimera were also purified to homogeneity. Then, microscale thermophoresis was used to determine the binding affinity of the ParB-CpsC/D complex. While no binding of ParB could be detected with the BSA (Bovine Serum Albumin) control (S7C Fig), an affinity constant of 7 ± 0.8 μM (n = 5) was obtained for the ParB-CpsC/D complex (Fig 9B). A two-fold higher affinity constant was calculated for the ParB-CpsC/D-YF complex (14 ± 0.9 μM (n = 5) suggesting that CpsD autophosphorylation influenced the interaction between ParB and CpsC/D. Then, we investigated the interaction between ParB and CpsD in vivo. For that, we constructed strains producing ParB-sfGFP together with either CpsD, CpsD-3YE or CpsD-3YF fused to a 6his-tag. We checked that fusions were produced at similar levels (S7D Fig). After immunoprecipitation of ParB-sfGFP, the presence of CpsD variants in the immunoprecipitated fractions was probed by Western blot using anti-His antibodies (Fig 9C). Western blots were also performed using anti-GFP antibodies to confirm that similar amounts of ParB-sfGFP were loaded. We observed that all CpsD constructs co-immunoprecipitated with ParB-sfGFP. Yet, we observed that immunoprecipitation of CpsD-3YF by ParB-sfGFP was less efficient than that of CpsD or CpsD-3YE. Altogether, these observations indicate that CpsD interact with ParB and that this interaction is modulated by CpsD phosphorylation.

Discussion
Many reports have demonstrated that BY-kinases are key regulators of extracellular polysaccharides biosynthesis and export [22, 24, 41–44]. The current model proposes that they would function as co-polymerases assisting the polymerase of the capsule assembly machinery [8]. However, the detailed mechanism by which BY-kinases control this machinery remains unknown. It is proposed that BY-kinases could serve as a molecular scaffold for the other proteins of the machinery. Phosphorylation / dephosphorylation of BY-kinases would trigger a conformational switch affecting the functioning of the other protein components of the polysaccharide assembly machinery [17]. Alternatively, BY-kinases could form a channel across the cytoplasmic membrane, large and hydrophilic enough to allow the polysaccharide polymer to cross the membrane. Indeed, BY-kinases can form a ring-shaped octamer that upon autophosphorylation dissociates to monomers [21]. Recently, Henriques and co-workers have shown that the BY-kinase CpsD of S. pneumoniae ATCC6314 localizes at the bacterial division septum suggesting that BY-kinases might also function as spatial regulators of capsular polysaccharide biosynthesis [13]. They propose that in the pneumococcus, two types of CPS assembly machinery would allow to wrap the cell in CPS. The membrane machinery without CpsC and CpsD would allow production of CPS around the cell while the septal machinery, associated with CpsC and CpsD, would specifically produce CPS at the septum. Our finding that CpsH localizes exclusively at the septum in WT cells allows us to refine this model and strongly suggests that CPS are produced only at the division septum by a single machinery (Fig 3D). This observation is further supported by the septal localization of CpsJ (S8 Fig). The capsule detected at the old cell poles in the ΔcpsD mutant could thus reflect accumulation of basal amount of immature capsule produced by the defective and mislocalized CPS assembly machinery. To our knowledge, this represents the first study determining the cellular site of polysaccharide synthesis and export.
In this context, what is the contribution of CpsC and CpsD to CPS production at the division septum? Our data show that the C-terminal and cytoplasmic end of CpsC is required for the interaction between CpsC and CpsD and consequently, CpsD autophosphorylation and localization at the division septum (Fig 2). These data are in agreement with the existence of a conserved activation mechanism of BY-kinase autophosphorylation. We observe that both CpsD and CpsH delocalize in cells expressing CpsC devoid of its C-terminal cytoplasmic end (cpsC-ΔCter). Yet, CpsH also delocalizes in cells deficient for cpsD (Fig 3D). Collectively, these data are consistent with a sequential interaction model in which CpsC captures CpsD at the division septum allowing subsequent localization of CpsH at the septum. The absence of capsule at the division septum together with impaired polymerization of CPS in cpsC-ΔCter and ΔcpsD cells (Fig 3A–3C) support this model. One should, however, note that CPS are still detected at the old cell pole in cpsC-ΔCter and ΔcpsD cells (Fig 3A). This shows that the capsule assembly machinery is still able to export some polysaccharides at the surface of cells in these genetic backgrounds. This observation implies that some polysaccharide subunits produced in the cytoplasm are flipped across the membrane by the flippase CpsJ but not properly polymerized by CpsH. Supporting this, cells deficient for cpsJ do not produce CPS [45]. This indicates that CpsC and CpsD are unlikely to function themselves as the exporter as previously suggested by Henriques and co-workers and our previous work [13, 21]. More likely, CpsC together with CpsD capture the CPS assembly machinery at the division site and trigger CPS export and polymerization by the flippase CpsJ and the polymerase CpsH, respectively.
One may also wonder how CPS produced at the division site covers the whole cell. An interesting possibility lies in the mode of cell division and elongation of the pneumococcus. Contrary to rod-shaped bacteria, the pneumococcus does not perform lateral synthesis of peptidoglycan. Peptidoglycan is produced at mid-cell and serves both for cell elongation and division, resulting in its characteristic ovoid morphology [46]. More precisely, it was demonstrated that ongoing peptidoglycan synthesis pushes the previous synthesized peptidoglycan leading mechanically to cell elongation and then formation of the new cell pole [47]. Considering that CpsA localizes at midcell and likely ligates CPS to the cell wall [10, 48, 49], CPS concurrently produced with peptidoglycan at mid-cell could be shuttled by peptidoglycan as the cell elongates and constricts. Because CPS are required to avoid pneumococcus recognition by the host complement and immune systems, this would provide a very simple mechanism to conceal and cover the complete surface of the cell. To test this hypothesis, the generation of fluorescent CPS precursors might be useful, similar to the approaches used to image peptidoglycan synthesis [50].
An important feature of BY-kinases regulation of CPS synthesis and export is their phosphorylation on several tyrosine residues grouped in a C-terminal motif termed “tyrosine cluster” [19]. Here, we observe that the cpsD-3YF strain displays slightly reduced production of CPS (Fig 3C). This finding is consistent with previous observations made in E. coli and S. pneumoniae [20, 22, 23, 25]. In addition, we observe that the localization of CpsD remains unchanged whatever its phosphorylation state (Fig 5A). This suggests that CpsD autophosphorylation is not crucial for the activity of CPS assembly machinery per se. However, and strikingly, expression of CpsD-3YF hinders CPS production at mid-cell, alter CpsH septal localization and results in an elongated cell phenotype (Figs 3 and 4). Altogether, these data suggest that CpsD phosphorylation would coordinate CPS production with the pneumococcal cell cycle. In this context, the presence of permanently non-phosphorylated CpsD (CpsD-3YF) at the division site would interfere with cell constriction, leading to elongated cells. Our finding that the non-septal localization of CpsD-3YF (due to deletion of the CpsC C-terminus) (Fig 5A) suppresses the elongated phenotype of the cpsD-3YF mutant cells (Fig 4), together with the presence of non-constricted septa in cpsD-3YF cells (Fig 5B), support this hypothesis.
Our data also show that the cpsD-3YF elongated cells display nucleoid defects (Fig 6). Interestingly, BY-kinases have been grouped with ParA and MinD proteins in the same protein superfamily on the basis of sequence similarity-based clustering [27]. ParA and MinD are involved in chromosome segregation and positioning of the Z-ring at mid-cell, respectively [51]. The first structure of the BY-kinase CapB from S. aureus was solved by molecular replacement using the structure of MinD from Pyrococcus horikoshii, and the two structures are highly similar [17]. Interestingly, the pneumococcus lacks ParA and MinD proteins [51]. On this basis, it is tempting to speculate that CpsD could share some functional properties with either ParA or MinD. Positioning of the Z-ring at mid-cell has been recently elucidated in the pneumococcus and it relies on the protein MapZ [47, 52]. Together with our finding that Z-rings are recruited to each non-constricted septum of cpsD-3YF cells, it is unlikely that CpsD contributes to division site selection as is the case for MinD (Fig 5C). However, it would be interesting to see whether the CpsD-phosphorylation mutants (YE/YF), by affecting CPS production, also cause an imbalance in PG precursor levels and hence cell division defects. By contrast, the structural similarity between CpsD and ParA proteins (Fig 7) suggests that CpsD could behave as a ParA-like protein even if we did not detect any DNA-binding for CpsD (S9 Fig). The accurate role of ParA in chromosome segregation remains unclear but it is proposed to interact with and assist ParB during chromosome segregation [28]. Unencapsulated strains of pneumococcus do not possess CpsD, thus suggesting that CpsD is unlikely to represent an authentic ParA protein involved in chromosome segregation. However, Δcps unencapsulated cells are also significantly shorter (Fig 4), suggesting that the production of the large CPS structure may require additional checkpoints to ensure correct chromosome segregation and cell division. Several studies have reported that ParA-like proteins are actually involved in protein localization in connection with the cell cycle [53]. For instance, ParC and PpfA facilitate polar localization and segregation of chemotaxis proteins in Vibrio cholerae and Rhodobacter sphaeroides, respectively [54, 55]. Another example is the ParA-like protein FlhG that is required for the polar localization of flagellar assembly factors [56]. Considering the CPS and nucleoid defects of cpsD-3YF cells, together with the timing of localization of CpsD and ParB and the ability of CpsD to interact with ParB (Figs 6 and 9), it is tempting to speculate that a crosstalk between the ParA-like CpsD protein and ParB could coordinate septal CPS production with the cell cycle. Our finding that CpsD phosphorylation modulates the stability of the complex encompassing CpsD and ParB (Fig 9B and 9C) further suggests that CpsD phosphorylation constitutes a system for signaling the CPS synthesis status to chromosome segregation and ensuring that daughter cells are properly wrapped in CPS.
Collectively, these data fit into the model presented in Fig 10. ParB localizes first at the division site, rapidly followed by CpsD before chromosome segregation starts. When CpsD is not phosphorylated, reflecting that CpsD does not hydrolyze ATP and that the CPS assembly machinery is switched off, chromosome segregation is delayed due to reduced ParB mobility. By contrast, when the CPS assembly machinery is switched on, CpsD hydrolyzes ATP and autophosphorylates. Phosphorylation of CpsD then favors the mobility of ParB and chromosome segregation. CPS would thus cover the new cell halves as the cell elongates and constricts. Consistent with this model, cpsD-3YE cells are significantly shorter in length (Fig 4) reflecting that cell constriction likely occurs prematurely due to an enhanced crosstalk between CpsD and ParB.

Finally, our data question the raison d’être of such a phospho-regulatory process. While cpsD-3YE cells are slightly shorter in length, they divide properly and are covered by CPS (Fig 3A). An interesting and promising hypothesis concerns the life-style of the pneumococcus. CPS are required during infection for protection against the human immune system but they are also disadvantageous because of their inhibitory effects on adherence to the host cell [3, 6, 57, 58]. One can speculate that the presence of a regulatory mechanism based on CpsD phosphorylation coordinating CPS production with the cell cycle would allow modulating capsule production to satisfy optimal colonization and dissemination.
Materials and Methods
Strains and growth conditions
S. pneumoniae strains were cultivated at 37°C in Todd-Hewitt Yeast (THY) broth (Difco) or in C+Y medium [59]. S. pneumoniae mutants were constructed by transformation in D39 as previously described [60], using precompetent cells treated at 37°C with synthetic competence stimulating peptide 1 (CSP1) to induce competence. Transformants were selected on THY-agar or Columbia agar supplemented with either 3% (vol/vol) defibrinated horse blood or 2% defibrinated sheep blood and containing the appropriate antibiotic (streptomycin 200 μg.mL-1, kanamycin 250 μg.mL-1, spectinomycin 100 μg.mL-1, chloramphenicol 2 μg.mL-1). Strains complemented with cpsC or cpsD at the ectopic amiF/treR locus under the control of the maltose inducible promoter PM [31] were grown in C+Y medium containing 20% maltose. The E. coli XL1-Blue strain was used as a host for cloning. E. coli BL21 (DE3) strain was used as a host for overexpression. Luria-Bertani (LB) broth and agar supplemented with appropriate antibiotic (tetracycline 15 μg.mL-1, ampicillin 100 μg.mL-1) were used for routine growth at 37°C. Strains used in this study are listed in S1 Table.
Allelic replacement mutagenesis
To construct pneumococcus mutants (gene deletions, gfp/rfp/sfgfp fusions or site-directed mutagenesis), we used a two-step procedure based on a bicistronic kan-rpsL cassette called Janus [61], except for allelic replacement of parB by parB-sfGFP and hlpA by hlpA-RFP, where we used a one-step procedure with a spectinomycin or chloramphenicol resistance marker, respectively [36, 40]. Throughout this study, gene mutagenesis or fusion with fluorescent protein were constructed at each native chromosomal locus, expressed under the control of the native promoter and represented the only source of protein. Full description of primers used for the construction of strains is provided in S2 Table. The genes encoding monomeric sfGFP, monomeric GFP and RFP were from [33, 62, 63], respectively.
Construction of plasmids
DNA fragments coding for CpsC, CpsC-ΔCter, CpsD and ParB were obtained by PCR using chromosomal DNA from S. pneumoniae D39 as template. For the chimera CpsC/D, we used DNA from strain TIGR4. Oligonucleotides used are described in S2 Table. The chimera DNA fragment was constructed by fusion of fragments obtained using primer pairs I-III and IV-II. The obtained DNA fragment was cloned between the BamHI and HindIII cloning sites of the pQE30 plasmid (Qiagen). parB was cloned between the NdeI and PstI cloning sites of the pT7.7 plasmid [64]. To construct plasmids for yeast two-hybrid, the PCR DNA fragments were digested by EcoRI and PstI and ligated either into pGAD-C1 or pGBDU-C1 vectors [65]. The nucleotide sequences of all DNA fragments were checked to ensure error-free amplification. Plasmids and primers used in this study are listed in S1 and S2 Tables, respectively.
Protein purification
Recombinant plasmids overproducing the chimera CpsC/D and ParB were transformed into BL21 (DE3) E. coli strain. The transformants were grown at 37°C until the culture reached an OD600 = 0.5. Expression was induced by adding IPTG to a final concentration of 0.5 mM and incubation was continued for 3 h. After 3 h culture at 37°C, cells were harvested and resuspended in buffer A (Tris-HCl 50 mM, pH 7.5; NaCl 300 mM; DTT 1 mM; imidazole 10 mM; glycerol 10%) containing 10 mg.L-1 of lysozyme and 6 mg.L-1 of DNase I and RNase A and sonicated. After centrifugation at 15000 g for 30 min, the supernatant was applied to a Ni-NTA agarose column (Qiagen) and extensively washed with buffer A supplemented with 30 mM imidazole. Samples were eluted with buffer A supplemented with 300 mM imidazole. The fractions corresponding to the pure protein were pooled and dialyzed against the following buffer: HEPES 50 mM, pH 7.5; NaCl 100 mM; DTT 1 mM; MgCl2 1 mM; glycerol 10%. The protein concentrations were determined using a Coomassie Assay Protein Dosage Reagent (Uptima) and aliquots were stored at -80°C.
Co-immunoprecipitation
Cultures of S. pneumoniae cells were grown at 37°C in THY medium until OD550 = 0.4. Cells pellets were incubated at 4°C for 15 min in buffer B (Tris-HCl 50 mM, pH 8.0; NaCl 150 mM; MgCl2 5 mM) containing 0.1 mg.mL-1 of lysozyme, 800 units of mutanolysine and 0.2 mg.mL-1 of DNase I and RNase A and sonicated. After centrifugation, the supernatant was incubated with the GFP-Trap resin suspension (Chromotech). Resin was washed according to the manufacturer’s instructions. Protein-bounded GFP-Trap resins were eluted with Laemmli buffer at 95°C for 10 min and analyzed by SDS-PAGE followed by an immunoblot directed against 6-Histidines tag or GFP.
Yeast two-hybrid
The yeast two-hybrid phenotypic assays were performed as described previously [66]. Briefly, genes encoding for CpsD, CpsC and CpsC-ΔCter were fused to either the activating domain of Gal4 or the DNA-binding domain of Gal4. Resulting plasmids were inserted in yeast haploid cells and interactions were screened for ability to grow on the selective medium.
Immunoblot analysis
In vitro CpsC/D autophosphorylation and in vivo phosphorylated proteins in crude extract of S. pneumoniae were immunodetected using mouse anti-phosphotyrosine monoclonal antibody PY-20 (Sigma-Aldrich) at 1/2000. Detection of GFP fusions was performed using a rabbit anti-GFP polyclonal antibody (AMS Biotechnology) at 1/10000. Detection of Enolase was performed using a rabbit anti-enolase polyclonal antibody at 1/50000 [32]. Proteins fused to a 6-histidines tag were detected using a mouse anti-6His monoclonal antibody (Sigma-Aldrich) at 1/1500. Detection of capsular polysaccharides was performed using a rabbit anti-serotype 2 CPS polyclonal antibody (Statens serum Institute) at 1/2000. A goat anti-rabbit or anti-mouse secondary polyclonal antibody horseradish peroxidase (HRP) conjugated (Biorad) was used at 1/5000 to reveal immunoblots.
Preparation and analysis of CPS
CPS were prepared as previously described with minor modifications [67]. Briefly, S. pneumoniae cultures were grown until OD550 = 0.3 and cells were harvested by centrifugation at 14,000xg for 20 min at 4°C. Pellets were then washed once with PBS and resuspended in buffer A (Tris-HCl 50 mM, pH 7,4; sucrose 20%; MgSO4 50 mM) at 1/100 of the original culture volume. The solution was then added with 400 units of mutanolysin and 6 μg of DNase and RNase per milliliter of solution and incubated overnight at RT. After centrifugation at 16,000xg for 20 min at 4°C, pellets were resuspended in the same volume of buffer A. 10 μL of the mixture were then mixed with 5 μL of buffer B (Tris-HCl 50 mM, pH 8.0; EDTA 50 mM; Tween20 0.5%; Triton X100 0.5%) and 20 μg of proteinase K, incubated 30 min at 37°C and analyzed by immunoblotting as previously described [68].
Molecular modeling
Protein sequence alignments were obtained using ClustalW and ESPript3 [69, 70]. Predicted secondary structures of CpsC/D and the B. subtilis Soj were made using Jpred3 [71]. The three-dimensional model of the chimera CpsC/D, based on the structure of the CapA1/B2 protein (PDB code 3BFV) was built using I-Tasser [72]. The visualization of 3D-molecules was performed using PyMOL (Schrödinger).
Microscale thermophoretic analysis
Microscale thermophoresis was used to test the interaction of ParB with the chimeras CpsC/D and CpsC/D-YF [73]. BSA (Bovine Serum Albumin) was used as negative control. Binding experiments were carried out with a Monolith NT.115 Series instrument (Nano Temper Technologies GMBH). ParB was labeled with the red dye NT-647. Briefly, 4 μl of sample containing 100 nM of labeled ParB and increasing concentrations of CpsC/D (from 4 nM to 92 μM) or BSA (from 5 nM to 180 μM) were loaded on K003 Monolith NT.115 hydrophilic treated silicon capillaries and thermophoresis was measured for 30 s. Each measurement was made in triplicates. Experiments were carried out at 25°C in MST optimized buffer (50 mM Tris-HCl, 150 mM NaCl, 10 mM MgCl2, 0.05% Tween-20). Analysis was performed with the Monolith software. Affinity KD was quantified by analyzing the change in normalized fluorescence (Fnorm = fluorescence after thermophoresis/initial fluorescence) as a function of the concentration of the titrated CpsC/D protein. The fraction of ParB bound was plotted against the concentration of CpsC/D.
Microscopy techniques
Microscopy was performed on exponentially growing cells (OD550 = 0.3). For in vivo immunofluorescence microscopy, cells were mixed with an rabbit anti-serotype 2 CPS polyclonal antibody (Statens Serum Institute) at 1/1000, washed several times with THY at 37°C and then incubated with an anti-rabbit DyLight-549 (Jackson ImmunoResearch) at 1/2000. After a last wash with PBS, CPS were imaged. For DAPI, 10 μL of S. pneumoniae cell culture were mixed with 1 μL of DAPI at 2 mg.mL-1 (Molecular Probes) and incubated 5 min at room temperature. GFP, RFP and sfGFP fusions were visualized by fluorescence microscopy. Slides were visualized with a Zeiss AxioObserver Z1 microscope fitted with an Orca-R2 C10600 charge-coupled device (CCD) camera (Hamamatsu) with a 100× NA 1.46 objective. Images were collected with axiovision (Carl Zeiss), deconvolved with ImageJ (http://rsb.info.nih.gov/ij/) and analyzed with Coli-Inspector (detection was approved manually) running under the plugin ObjectJ (http://simon.bio.uva.nl/objectj/) to measure CPS fluorescence intensity or generate fluorescent intensity linescans sorted to cell length and MicrobeTracker suite [74] extended by custom MATLAB routines to generate cell length distribution histograms. Cell lengths were determined using MicrobeTracker. Images are representative of experiments made in triplicate.
Fluorescence time-lapse microscopy was performed on a DV Elite (Applied Precision) with a sCMOS camera using SSI Solid State Illumination (Applied Precision) through a 100× oil immersion objective (phase contrast), essentially as described before [36, 75]. During the experiment, cells were incubated on a slide of C+Y agarose and kept at 37°C in a temperature controlled chamber. Phase contrast and fluorescent (GFP and RFP) images were acquired every 5 min. Images were processed using softWoRx 5.5 (Applied Precision) and ImageJ.
For TEM, S. pneumoniae cells (wild type and mutants) were collected, centrifuged and pre-fixed 20 min on ice with the fixing mix (cacodylate 0.1 M, pH 7.4; glutaraldehyde 5%; lysine-HCl 0.075 M; ruthenium red 0.075%). After washing, cells were fixed overnight at 4°C with fixing mix without lysine-HCl. Cells were took onto 2% agarose and post-fixation with 1% osmium tetroxide in cacodylated buffer added to ruthenium red was carried out for 1 h at room temperature. These fixed cells were dehydrated using a graded series of ethanol and embedded in LR-White at 60°C for 48 h. Ultrathin sections (60 nm) were obtained using a Leica UC7 microtome and were counter-stained with uranyl acetate and lead citrate (Reichert Ultrostainer, Leica). Samples were examined with a Phillips CM120 transmission electron microscope equipped with a Gatan Orius SC200 CCD camera.
TIRF microscopy was performed as described before [36]. Cells were grown in C+Y until mid-exponential phase prior to examination on a DV Elite (Applied Precision) with a sCMOS camera using 50 mW laser illumination (488 nm and 561 nm) through a 100× oil 1.49 NA TIRF objective. Cells were imaged every 10 seconds for 40 seconds. The GFP foci in the cells were classified as stationary or dynamic (splitting, appearing or disappearing foci) using intensity line scans at different time points in Image J. Fluorescence foci were classified as “splitting” when one fluorescence peak splits in two peaks during the time span of the experiment and foci were classified as “disappearing” when the signal of a fluorescence peak was reduced >90% during the time span of the experiment.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Henriques-Normark B, Normark S. Commensal pathogens, with a focus on Streptococcus pneumoniae, and interactions with the human host. Exp Cell Res. 2010;316 : 1408–14. doi: 10.1016/j.yexcr.2010.03.003 20227406
2. van der Poll T, Opal SM. Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet. 2009;374 : 1543–56. doi: 10.1016/S0140-6736(09)61114-4 19880020
3. Kadioglu A, Weiser JN, Paton JC, Andrew PW. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol. 2008;6 : 288–301. doi: 10.1038/nrmicro1871 18340341
4. Magee AD, Yother J. Requirement for capsule in colonization by Streptococcus pneumoniae. Infect Immun. 2001;69 : 3755–61. 11349040
5. Winkelstein JA. The role of complement in the host's defense against Streptococcus pneumoniae. Rev Infect Dis. 1981;3 : 289–98. 7020046
6. Kjos M, Aprianto R, Fernandes VE, Andrew PW, van Strijp JA, Nijland R, et al. Bright Fluorescent Streptococcus pneumoniae for Live-Cell Imaging of Host-Pathogen Interactions. J Bacteriol. 2015;197 : 807–18. doi: 10.1128/JB.02221-14 25512311
7. Bentley SD, Aanensen DM, Mavroidi A, Saunders D, Rabbinowitsch E, Collins M, et al. Genetic analysis of the capsular biosynthetic locus from all 90 pneumococcal serotypes. PLoS genetics. 2006;2:e31. 16532061
8. Whitfield C. Biosynthesis and assembly of capsular polysaccharides in Escherichia coli. Annu Rev Biochem. 2006;75 : 39–68. 16756484
9. Yother J. Capsules of Streptococcus pneumoniae and other bacteria: paradigms for polysaccharide biosynthesis and regulation. Annu Rev Microbiol. 2011;65 : 563–81. doi: 10.1146/annurev.micro.62.081307.162944 21721938
10. Kawai Y, Marles-Wright J, Cleverley RM, Emmins R, Ishikawa S, Kuwano M, et al. A widespread family of bacterial cell wall assembly proteins. Embo J. 2011;30 : 4931–41. doi: 10.1038/emboj.2011.358 21964069
11. Morona JK, Morona R, Miller DC, Paton JC. Streptococcus pneumoniae capsule biosynthesis protein CpsB is a novel manganese-dependent phosphotyrosine-protein phosphatase. J Bacteriol. 2002;184 : 577–83. 11751838
12. Morona JK, Morona R, Paton JC. Attachment of capsular polysaccharide to the cell wall of Streptococcus pneumoniae type 2 is required for invasive disease. Proc Natl Acad Sci U S A. 2006;103 : 8505–10. 16707578
13. Henriques MX, Rodrigues T, Carido M, Ferreira L, Filipe SR. Synthesis of capsular polysaccharide at the division septum of Streptococcus pneumoniae is dependent on a bacterial tyrosine kinase. Mol Microbiol. 2011;82 : 515–34. doi: 10.1111/j.1365-2958.2011.07828.x 21929561
14. Grangeasse C, Nessler S, Mijakovic I. Bacterial tyrosine kinases: evolution, biological function and structural insights. Philos Trans R Soc Lond B Biol Sci. 2012;367 : 2640–55. doi: 10.1098/rstb.2011.0424 22889913
15. Grangeasse C, Cozzone AJ, Deutscher J, Mijakovic I. Tyrosine phosphorylation: an emerging regulatory device of bacterial physiology. Trends Biochem Sci. 2007;32 : 86–94. 17208443
16. Soulat D, Jault JM, Duclos B, Geourjon C, Cozzone AJ, Grangeasse C. Staphylococcus aureus operates protein-tyrosine phosphorylation through a specific mechanism. J Biol Chem. 2006;281 : 14048–56. 16565080
17. Olivares-Illana V, Meyer P, Bechet E, Gueguen-Chaignon V, Soulat D, Lazereg-Riquier S, et al. Structural basis for the regulation mechanism of the tyrosine kinase CapB from Staphylococcus aureus. PLoS Biol. 2008;6:e143. doi: 10.1371/journal.pbio.0060143 18547145
18. Morona JK, Morona R, Miller DC, Paton JC. Mutational Analysis of the Carboxy-Terminal (YGX)(4) Repeat Domain of CpsD, an Autophosphorylating Tyrosine Kinase Required for Capsule Biosynthesis in Streptococcus pneumoniae. J Bacteriol. 2003;185 : 3009–19. 12730159
19. Grangeasse C, Doublet P, Cozzone AJ. Tyrosine phosphorylation of protein kinase Wzc from Escherichia coli K12 occurs through a two-step process. J Biol Chem. 2002;277 : 7127–35. 11751920
20. Obadia B, Lacour S, Doublet P, Baubichon-Cortay H, Cozzone AJ, Grangeasse C. Influence of Tyrosine-Kinase Wzc Activity on Colanic Acid Production in Escherichia coli K12 Cells. J Mol Biol. 2007;367 : 42–53. 17254603
21. Bechet E, Gruszczyk J, Terreux R, Gueguen-Chaignon V, Vigouroux A, Obadia B, et al. Identification of structural and molecular determinants of the tyrosine-kinase Wzc and implications in capsular polysaccharide export. Mol Microbiol. 2010;77 : 1315–25. doi: 10.1111/j.1365-2958.2010.07291.x 20633230
22. Wugeditsch T, Paiment A, Hocking J, Drummelsmith J, Forrester C, Whitfield C. Phosphorylation of Wzc, a tyrosine autokinase, is essential for assembly of group 1 capsular polysaccharides in Escherichia coli. J Biol Chem. 2001;276 : 2361–71. 11053445
23. Paiment A, Hocking J, Whitfield C. Impact of phosphorylation of specific residues in the tyrosine autokinase, Wzc, on its activity in assembly of group 1 capsules in Escherichia coli. J Bacteriol. 2002;184 : 6437–47. 12426330
24. Morona JK, Paton JC, Miller DC, Morona R. Tyrosine phosphorylation of CpsD negatively regulates capsular polysaccharide biosynthesis in streptococcus pneumoniae. Mol Microbiol. 2000;35 : 1431–42. 10760144
25. Morona JK, Miller DC, Morona R, Paton JC. The effect that mutations in the conserved capsular polysaccharide biosynthesis genes cpsA, cpsB, and cpsD have on virulence of Streptococcus pneumoniae. J Infect Dis. 2004;189 : 1905–13. 15122528
26. Avery OT, Macleod CM, McCarty M. Studies on the Chemical Nature of the Substance Inducing Transformation of Pneumococcal Types: Induction of Transformation by a Desoxyribonucleic Acid Fraction Isolated from Pneumococcus Type Iii. J Exp Med. 1944;79 : 137–58. 19871359
27. Leipe DD, Wolf YI, Koonin EV, Aravind L. Classification and evolution of P-loop GTPases and related ATPases. J Mol Biol. 2002;317 : 41–72. 11916378
28. Gerdes K, Howard M, Szardenings F. Pushing and pulling in prokaryotic DNA segregation. Cell. 2010;141 : 927–42. doi: 10.1016/j.cell.2010.05.033 20550930
29. Mijakovic I, Poncet S, Boel G, Maze A, Gillet S, Jamet E, et al. Transmembrane modulator-dependent bacterial tyrosine kinase activates UDP-glucose dehydrogenases. Embo J. 2003;22 : 4709–18. 12970183
30. Toniolo C, Balducci E, Romano MR, Proietti D, Ferlenghi I, Grandi G, et al. Streptococcus agalactiae capsule polymer length and attachment is determined by the proteins CpsABCD. J Biol Chem. 2015.
31. Guiral S, Henard V, Laaberki MH, Granadel C, Prudhomme M, Martin B, et al. Construction and evaluation of a chromosomal expression platform (CEP) for ectopic, maltose-driven gene expression in Streptococcus pneumoniae. Microbiology. 2006;152 : 343–9. 16436422
32. Fleurie A, Cluzel C, Guiral S, Freton C, Galisson F, Zanella-Cleon I, et al. Mutational dissection of the S/T-kinase StkP reveals crucial roles in cell division of Streptococcus pneumoniae. Mol Microbiol. 2012;83 : 746–58. doi: 10.1111/j.1365-2958.2011.07962.x 22211696
33. Dinh T, Bernhardt TG. Using superfolder green fluorescent protein for periplasmic protein localization studies. J Bacteriol. 2011;193 : 4984–7. doi: 10.1128/JB.00315-11 21764912
34. Bi EF, Lutkenhaus J. FtsZ ring structure associated with division in Escherichia coli. Nature. 1991;354 : 161–4. 1944597
35. Fleurie A, Manuse S, Zhao C, Campo N, Cluzel C, Lavergne JP, et al. Interplay of the serine/threonine-kinase StkP and the paralogs DivIVA and GpsB in pneumococcal cell elongation and division. PLoS genetics. 2014;10:e1004275. doi: 10.1371/journal.pgen.1004275 24722178
36. Kjos M, Veening JW. Tracking of chromosome dynamics in live Streptococcus pneumoniae reveals that transcription promotes chromosome segregation. Mol Microbiol. 2014;91 : 1088–105. doi: 10.1111/mmi.12517 24417389
37. Land AD, Tsui HC, Kocaoglu O, Vella SA, Shaw SL, Keen SK, et al. Requirement of essential Pbp2x and GpsB for septal ring closure in Streptococcus pneumoniae D39. Mol Microbiol. 2013;90 : 939–55. doi: 10.1111/mmi.12408 24118410
38. Slager J, Kjos M, Attaiech L, Veening JW. Antibiotic-induced replication stress triggers bacterial competence by increasing gene dosage near the origin. Cell. 2014;157 : 395–406. doi: 10.1016/j.cell.2014.01.068 24725406
39. Livny J, Yamaichi Y, Waldor MK. Distribution of centromere-like parS sites in bacteria: insights from comparative genomics. J Bacteriol. 2007;189 : 8693–703. 17905987
40. Minnen A, Attaiech L, Thon M, Gruber S, Veening JW. SMC is recruited to oriC by ParB and promotes chromosome segregation in Streptococcus pneumoniae. Mol Microbiol. 2011;81 : 676–88. doi: 10.1111/j.1365-2958.2011.07722.x 21651626
41. Vincent C, Duclos B, Grangeasse C, Vaganay E, Riberty M, Cozzone AJ, et al. Relationship between exopolysaccharide production and protein-tyrosine phosphorylation in gram-negative bacteria. J Mol Biol. 2000;304 : 311–21. 11090276
42. Elsholz AK, Wacker SA, Losick R. Self-regulation of exopolysaccharide production in Bacillus subtilis by a tyrosine kinase. Genes Dev. 2014;28 : 1710–20. doi: 10.1101/gad.246397.114 25085422
43. Nakar D, Gutnick DL. Involvement of a protein tyrosine kinase in production of the polymeric bioemulsifier emulsan from the oil-degrading strain Acinetobacter lwoffii RAG-1. J Bacteriol. 2003;185 : 1001–9. 12533476
44. Niemeyer D, Becker A. The molecular weight distribution of succinoglycan produced by Sinorhizobium meliloti is influenced by specific tyrosine phosphorylation and ATPase activity of the cytoplasmic domain of the ExoP protein. J Bacteriol. 2001;183 : 5163–70. 11489870
45. Xayarath B, Yother J. Mutations blocking side chain assembly, polymerization, or transport of a Wzy-dependent Streptococcus pneumoniae capsule are lethal in the absence of suppressor mutations and can affect polymer transfer to the cell wall. J Bacteriol. 2007;189 : 3369–81. 17322316
46. Wheeler R, Mesnage S, Boneca IG, Hobbs JK, Foster SJ. Super-resolution microscopy reveals cell wall dynamics and peptidoglycan architecture in ovococcal bacteria. Mol Microbiol. 2011;82 : 1096–109. doi: 10.1111/j.1365-2958.2011.07871.x 22059678
47. Fleurie A, Lesterlin C, Manuse S, Zhao C, Cluzel C, Lavergne JP, et al. MapZ marks the division sites and positions FtsZ rings in Streptococcus pneumoniae. Nature. 2014;516 : 259–62. doi: 10.1038/nature13966 25470041
48. Morona R. Encapsulating bacteria. Structure. 2013;21 : 692–3. doi: 10.1016/j.str.2013.04.011 23664360
49. Eberhardt A, Hoyland CN, Vollmer D, Bisle S, Cleverley RM, Johnsborg O, et al. Attachment of capsular polysaccharide to the cell wall in Streptococcus pneumoniae. Microb Drug Resist. 2012;18 : 240–55. doi: 10.1089/mdr.2011.0232 22432711
50. Kuru E, Hughes HV, Brown PJ, Hall E, Tekkam S, Cava F, et al. In Situ probing of newly synthesized peptidoglycan in live bacteria with fluorescent D-amino acids. Angew Chem Int Ed Engl. 2012;51 : 12519–23. doi: 10.1002/anie.201206749 23055266
51. Pinho MG, Kjos M, Veening JW. How to get (a)round: mechanisms controlling growth and division of coccoid bacteria. Nat Rev Microbiol. 2013;11 : 601–14. doi: 10.1038/nrmicro3088 23949602
52. Holeckova N, Doubravova L, Massidda O, Molle V, Buriankova K, Benada O, et al. LocZ is a new cell division protein involved in proper septum placement in Streptococcus pneumoniae. mBio. 2015;6:e01700–14.
53. Lutkenhaus J. The ParA/MinD family puts things in their place. Trends Microbiol. 2012;20 : 411–8. doi: 10.1016/j.tim.2012.05.002 22672910
54. Ringgaard S, Schirner K, Davis BM, Waldor MK. A family of ParA-like ATPases promotes cell pole maturation by facilitating polar localization of chemotaxis proteins. Genes Dev. 2011;25 : 1544–55. doi: 10.1101/gad.2061811 21764856
55. Roberts MA, Wadhams GH, Hadfield KA, Tickner S, Armitage JP. ParA-like protein uses nonspecific chromosomal DNA binding to partition protein complexes. Proceedings of the National Academy of Sciences of the United States of America. 2012;109 : 6698–703. doi: 10.1073/pnas.1114000109 22496588
56. Correa NE, Peng F, Klose KE. Roles of the regulatory proteins FlhF and FlhG in the Vibrio cholerae flagellar transcription hierarchy. J Bacteriol. 2005;187 : 6324–32. 16159765
57. Weiser JN, Austrian R, Sreenivasan PK, Masure HR. Phase variation in pneumococcal opacity: relationship between colonial morphology and nasopharyngeal colonization. Infect Immun. 1994;62 : 2582–9. 8188381
58. Hyams C, Camberlein E, Cohen JM, Bax K, Brown JS. The Streptococcus pneumoniae capsule inhibits complement activity and neutrophil phagocytosis by multiple mechanisms. Infect Immun. 2010;78 : 704–15. doi: 10.1128/IAI.00881-09 19948837
59. Martin B, Garcia P, Castanie MP, Claverys JP. The recA gene of Streptococcus pneumoniae is part of a competence-induced operon and controls lysogenic induction. Mol Microbiol. 1995;15 : 367–79. 7538190
60. Martin B, Prudhomme M, Alloing G, Granadel C, Claverys JP. Cross-regulation of competence pheromone production and export in the early control of transformation in Streptococcus pneumoniae. Mol Microbiol. 2000;38 : 867–78. 11115120
61. Sung CK, Li H, Claverys JP, Morrison DA. An rpsL cassette, janus, for gene replacement through negative selection in Streptococcus pneumoniae. Appl Environ Microbiol. 2001;67 : 5190–6. 11679344
62. Martin B, Granadel C, Campo N, Henard V, Prudhomme M, Claverys JP. Expression and maintenance of ComD-ComE, the two-component signal-transduction system that controls competence of Streptococcus pneumoniae. Mol Microbiol. 2010;75 : 1513–28. doi: 10.1111/j.1365-2958.2010.07071.x 20180906
63. Beilharz K, Novakova L, Fadda D, Branny P, Massidda O, Veening JW. Control of cell division in Streptococcus pneumoniae by the conserved Ser/Thr protein kinase StkP. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E905–13. doi: 10.1073/pnas.1119172109 22431591
64. Cortay JC, Negre D, Scarabel M, Ramseier TM, Vartak NB, Reizer J, et al. In vitro asymmetric binding of the pleiotropic regulatory protein, FruR, to the ace operator controlling glyoxylate shunt enzyme synthesis. J Biol Chem. 1994;269 : 14885–91. 8195118
65. James P, Halladay J, Craig EA. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics. 1996;144 : 1425–36. 8978031
66. Noirot-Gros MF, Dervyn E, Wu LJ, Mervelet P, Errington J, Ehrlich SD, et al. An expanded view of bacterial DNA replication. Proceedings of the National Academy of Sciences of the United States of America. 2002;99 : 8342–7. 12060778
67. Yother J, White JM. Novel surface attachment mechanism of the Streptococcus pneumoniae protein PspA. J Bacteriol. 1994;176 : 2976–85. 7910604
68. Bender MH, Cartee RT, Yother J. Positive correlation between tyrosine phosphorylation of CpsD and capsular polysaccharide production in Streptococcus pneumoniae. J Bacteriol. 2003;185 : 6057–66. 14526017
69. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22 : 4673–80. 7984417
70. Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–4. doi: 10.1093/nar/gku316 24753421
71. Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36(Web Server issue):W197–201. doi: 10.1093/nar/gkn238 18463136
72. Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9 : 40. doi: 10.1186/1471-2105-9-40 18215316
73. Jerabek-Willemsen M, Wienken CJ, Braun D, Baaske P, Duhr S. Molecular interaction studies using microscale thermophoresis. Assay Drug Dev Technol. 2011;9 : 342–53. doi: 10.1089/adt.2011.0380 21812660
74. Sliusarenko O, Heinritz J, Emonet T, Jacobs-Wagner C. High-throughput, subpixel precision analysis of bacterial morphogenesis and intracellular spatio-temporal dynamics. Mol Microbiol. 2011;80 : 612–27. doi: 10.1111/j.1365-2958.2011.07579.x 21414037
75. de Jong IG, Beilharz K, Kuipers OP, Veening JW. Live Cell Imaging of Bacillus subtilis and Streptococcus pneumoniae using Automated Time-lapse Microscopy. J Vis Exp. 2011;(53).
76. Leonard TA, Butler PJ, Lowe J. Bacterial chromosome segregation: structure and DNA binding of the Soj dimer—a conserved biological switch. Embo J. 2005;24 : 270–82. 15635448
77. Hester CM, Lutkenhaus J. Soj (ParA) DNA binding is mediated by conserved arginines and is essential for plasmid segregation. Proc Natl Acad Sci U S A. 2007;104 : 20326–31. 18077387
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 9
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Arabidopsis AtPLC2 Is a Primary Phosphoinositide-Specific Phospholipase C in Phosphoinositide Metabolism and the Endoplasmic Reticulum Stress Response
- Bridges Meristem and Organ Primordia Boundaries through , , and during Flower Development in
- KLK5 Inactivation Reverses Cutaneous Hallmarks of Netherton Syndrome
- XBP1-Independent UPR Pathways Suppress C/EBP-β Mediated Chondrocyte Differentiation in ER-Stress Related Skeletal Disease
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy