
The Exosome Component Rrp6 Is Required for RNA Polymerase II Termination at Specific Targets of the Nrd1-Nab3 Pathway
RNAPII is responsible for transcription of protein-coding genes and short, regulatory RNAs. In Saccharomyces cerevisiae, termination of RNAPII-transcribed RNAs ≤1000 bases requires the NNS complex (comprised of Nrd1, Nab3, and Sen1), processing by the exosome, and the nuclear specific catalytic subunit, Rrp6. It has been shown that Rrp6 interacts directly with Nrd1, but whether or not Rrp6 is required for NNS-dependent termination is unclear. Loss of Rrp6 function may result in extension (or inhibition of termination) of NNS-dependent transcripts, or Rrp6 may only function after the fact to carry out RNA 3’ end processing. Here, we performed in-depth differential expression analyses and compare RNA-sequencing data of transcript length and abundance in cells lacking RRP6 to ChIP-exo analysis of RNAPII localization. We find many transcripts that were defined as unterminated upon loss of Nrd1 activity are of similar length in rrp6Δ, and expression levels of downstream genes are significantly decreased. This suggests a similar transcription interference mechanism occurs in cells lacking either Nrd1 or Rrp6. Indeed we find increased RNAPII located downstream of its termination site at many know Nrd1-regulated transcripts. Overall, our findings clearly demonstrate that Rrp6 activity is required for efficient NNS termination in vivo.
Published in the journal:
. PLoS Genet 11(2): e32767. doi:10.1371/journal.pgen.1004999
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004999
Summary
RNAPII is responsible for transcription of protein-coding genes and short, regulatory RNAs. In Saccharomyces cerevisiae, termination of RNAPII-transcribed RNAs ≤1000 bases requires the NNS complex (comprised of Nrd1, Nab3, and Sen1), processing by the exosome, and the nuclear specific catalytic subunit, Rrp6. It has been shown that Rrp6 interacts directly with Nrd1, but whether or not Rrp6 is required for NNS-dependent termination is unclear. Loss of Rrp6 function may result in extension (or inhibition of termination) of NNS-dependent transcripts, or Rrp6 may only function after the fact to carry out RNA 3’ end processing. Here, we performed in-depth differential expression analyses and compare RNA-sequencing data of transcript length and abundance in cells lacking RRP6 to ChIP-exo analysis of RNAPII localization. We find many transcripts that were defined as unterminated upon loss of Nrd1 activity are of similar length in rrp6Δ, and expression levels of downstream genes are significantly decreased. This suggests a similar transcription interference mechanism occurs in cells lacking either Nrd1 or Rrp6. Indeed we find increased RNAPII located downstream of its termination site at many know Nrd1-regulated transcripts. Overall, our findings clearly demonstrate that Rrp6 activity is required for efficient NNS termination in vivo.
Introduction
The exosome is a 3’-5’ exonuclease complex involved in termination of short RNAs, RNA quality control surveillance, and RNA degradation in both the cytoplasm and the nucleus. The core RNA exosome is a multi-subunit complex that has been proposed to channel the substrate to the catalytic subunit Dis3 (also known as Rrp44), similar in structure to the proteasome [1,2,3,4]. Dis3 has both an endonucleolytic PIN domain and 3’-5’ exonuclease activity and is essential for viability of the yeast Saccharomyces cerevisiae [5,6]. In the nucleus, the exosome aquires a second exonuclease subunit, Rrp6. Although Rrp6 is not essential for viability, it has been shown to be required for proper 3’ end trimming of primary small nucleolar/nuclear RNAs (snRNAs) [7,8], degradation of short-lived, non-coding cryptic unstable transcripts (CUTs) [9,10,11,12,13] and improperly terminated RNAs [7,14], as well as regulation of polyA tail length through Nab2 and the non-canonical polyA polymerases Trf4 and Trf5 [15,16]. Recent studies using high-resolution tiling arrays found that Dis3 and Rrp6 have both shared and distinct roles in the degradation of various RNAs [17,18]. For instance, Rrp6 is largely responsible for snRNA processing whereas both Rrp6 and Dis3 seem to be responsible for the degradation of unspliced pre-mRNAs [17,18]. Rrp6 has also been implicated in the termination and 3’ end processing of a variety of noncoding RNAs, most of which are not terminated by traditional polyA-dependent termination mechanisms [12,19].
RNA polymerase II (RNAPII) transcription termination is coupled to 3’ end processing and carried out by two pathways [20]. The recruitment of factors involved in these pathways is orchestrated by post-translational modifications along the repetitive C-terminal domain (CTD) of Rpb1, the largest subunit of RNAPII (reviewed in [21]). Generally, long (>1000 bp) polyadenylated mRNAs are terminated by the Cleavage and Polyadenylation Factor (CPF) and Cleavage Factor (CF) complexes in a polyadenylation dependent mechanism [22,23,24,25,26]. The second pathway predominates on short (<1000 bp) RNAPII transcripts including snRNAs, CUTs, and stable unannotated transcripts (SUTs) that are terminated by an Nrd1, Nab3, and Sen1 (NNS) dependent mechanism [20,27,28,29,30]. Termination by NNS is coupled to RNA 3’ end processing and/or degradation by the Rrp6-containing nuclear exosome [8,31,32]. In the current model of NNS termination, Nrd1 can be recruited to the RNAPII CTD by its CTD interacting domain (CID) [8,33,34,35,36,37]. Nrd1 and Nab3 also contain RNA binding domains that facilitate their recruitment to specific sequences in the nascent RNA [38,39]. The helicase Sen1 participates through unwinding RNA:DNA hybrids formed by the nascent RNA and template strand promoting RNAPII termination in yeast and mammals [40,41,42,43]. RNAPII transcribes beyond the 3’ end of the functional RNA into a “termination zone” before it is terminated by Sen1 [40]. The RNA is then polyadenylated by the TRAMP complex, and the nuclear exosome trims the ends of stable transcripts (such as snRNAs and SUTs) or completely degrades the transcript in the case of CUTs [19,20,29,32,40,44,45,46,47].
Here, we focus on the role of Rrp6 in termination and processing of RNAPII-transcribed noncoding RNAs (ncRNAs) and specific mRNAs that are likely regulated through the NNS pathway. Recent work suggests that Nrd1-dependent termination of noncoding RNAs is necessary for transcriptome surveillance by regulating promoter directionality and suppressing antisense transcription [48]. Nrd1 interacts with the exosome and the Nrd1 CTD-interacting domain (CID) has been shown to be important for coupling of NNS-termination with RNA processing [7,8]. The interaction between Nrd1 and Rrp6 is direct and independent of the Nrd1 CID [49]. It has previously been shown that nuclear depletion of Nrd1 causes defective termination of many NNS pathway targets resulting in the identification of 1,526 extended ncRNAs referred to as NUTs (Nrd1 unterminated transcripts) [48]. The generation of NUTs upon Nrd1 nuclear depletion caused transcriptome-wide changes in gene expression predominately through transcription interference caused by defective termination. However, it was reported that deletion of RRP6 does not change ncRNA transcription termination and disruption of Nrd1 and Rrp6 activities did not cause differential expression of the same genes, suggesting that Rrp6 is not required for NNS-dependent termination [48]. Interestingly, deletion of RRP6 has recently been shown to both stabilize and promote elongation of CUT281. CUT281 is an antisense long noncoding RNA that regulates the expression of PHO84 through antisense transcription past the PHO84 promoter [50]. The catalytic subunits of the exosome (Rrp6 and Dis3) have also been implicated in termination at snR4 and snR34 [17]. These data suggest that Rrp6 can regulate both degradation and the efficiency of NNS-dependent termination of ncRNA transcripts such as CUTs that are implicated in mRNA regulation through transcription interference. Together, these studies suggest that Rrp6 may be required for NNS termination at some specific targets while dispensable at others. Our primary objective was to determine if Rrp6 is required for NNS termination through analysis of global changes in the transcriptome via high throughput RNA-sequencing (RNA-Seq) and analysis of global RNAPII occupancy changes using high-resolution chromatin IP followed by exonuclease treatment (ChIP-exo).
In this study, we show that deletion of RRP6 disrupts 3’ end processing and/or termination at multiple NNS-dependent transcripts resulting in extended 3’ RNA ends. Many of the rrp6Δ RNA extensions cause transcription interference from extended transcription, and closely match the transcripts observed following nuclear depletion of Nrd1 further supporting that they are NNS-dependent transcripts. We find altered transcript lengths all classes of NNS-terminated transcripts including snRNAs, CUTs, SUTs, and a subset of mRNAs resulting in transcriptome-wide changes in gene expression. These data suggest that Rrp6 acts with the NNS complex to promote transcription termination in addition to coupled 3’ end RNA processing (for mRNAs, snRNAs, and select SUTs) and/or degradation (for CUTs).
Results
Genome-wide analysis of RRP6 deletion strains by RNA-Seq
To identify classes of transcripts affected by the loss of Rrp6, we utilized previously published annotations for yeast transcripts and performed differential expression analysis using normalized sequencing reads through EdgeR (S1 Table). To accurately represent the entire transcribed region for mRNAs including the coding region, we employed annotations that include both the 5’ and 3’ untranslated region (UTR) for the majority of the yeast transcriptome [51]. In summary, the annotations used for our study include 5792 open reading frames (ORF-Ts), 658 CUTs, 648 SUTs, 1215 NUTs, 844 Rrp6-regulated antisense transcripts, 80 snRNAs, and 78 snRNA extended transcripts (ETs, manually annotated in this study). In rrp6Δ cells, 136 up-regulated and 734 down-regulated open reading frame transcripts were identified (ORF-Ts, fold change cut-off = +/ - 1.5, p-value < 0.05, FDR ≤ 0.1, Fig. 1A-B, S1 Table). We also identified 622 up-regulated CUTs out of 733, in agreement with previous reports that CUT expression and stability is increased in the absence of Rrp6 ([9,11,13], Fig. 1A, S1 Table). Importantly, using deep sequencing technology many CUTs were detected that have not previously been detected in WT cells using microarrays (S1 Table) [9,10,11,13]. Analysis of the NUTs revealed that the majority of that class of transcripts, 887/1215, were also up-regulated in rrp6Δ (S1 Table, S1A Fig.). Additionally, our analysis found that 223/648 SUTs were significantly up-regulated in rrp6Δ uncovering the specific SUT transcripts that are likely terminated by the NNS pathway (S1 Table). In rrp6Δ cells, a total of 54 sn/snoRNAs showed significant transcript extension (S1 Table, annotated as ETs—extended transcripts).

Considering that Rrp6 is an exonuclease, it is expected that the majority of RNA expression changes in rrp6Δ cells would be due to accumulation. However, we were surprised to see 734 mRNAs significantly decreased ≤ 1.5-fold in rrp6Δ (Fig. 1B, ORFs in yellow, S1 Table). To better understand the classes of mRNAs down-regulated in rrp6Δ, we performed GO-term enrichment analysis to determine if any cellular pathways showed significant enrichment within the set of decreased mRNAs. Surprisingly, the most enriched GOterm was GO:0005830 for the cytosolic ribosome with a p-value of 6.43E-82 (S2 Table). In total, nine of the 30 statistically significant enriched GO-terms related to ribosomal protein coding genes with p-values less than or equal to 7.85E-19 (S2 Table). Differential expression analysis determined that transcript levels of 109 out of 137 ribosomal protein coding genes were decreased more than 1.5-fold (Fig. 1C, S3 Table). Interestingly, only 3 ribosomal protein-coding genes had more than 1.5-fold increase in expression (Fig. 1C). It is also interesting to note that the average transcript length from the down-regulated ribosomal protein-coding mRNAs is 916 nucleotides putting many of the transcripts within the approximate length limits for the NNS pathway.
Pearson correlation coefficients were calculated comparing the average log2 fold-change values obtained in our RNA-Seq dataset (n = 4) and in a recent tiling array study by Castelnuovo et al 2014 [52,53]. The correlation coefficient when comparing these two datasets is 0.751 (Fig. 2A), a value indicating that there is a strong positive correlation between the two datasets similar to the extent of correlation previously found when comparing RNA-Seq to the tiling array platform [54]. Interestingly, the highly abundant sn/snoRNA transcripts had poor correlation across the two platforms, represented as red dots in Fig. 2A (values also given in S4 Table). It has previously been shown that RNA-Seq has a capacity for much greater dynamic range than microarrays, which can be saturated by very abundant transcripts and may not be able to quantitatively detect transcripts expressed at low levels [56]. For this reason, we propose that the differences seen between our dataset and previously published tiling array data are due to the increased dynamic range obtained with deep transcriptome coverage obtained by our RNA-Seq experiments. Considering that sn/snoRNAs are highly abundant and that they are one of the primary cellular targets of Rrp6 activity [14–16], the ability to accurately measure the abundance of snRNAs in this instance is a distinct advantage of using RNA-Seq. To compare the detection and quantification of extended 3’ ends of a selection of snRNAs in WT versus rrp6Δ cells, area-under-the-curve values were calculated for snR33 and snR37 using our RNA-Seq data compared to data from previously published tiling array dataset (Fig. 2B-D, [52]). To estimate the degree of extended transcript accumulation in rrp6Δ, the ET area was normalized against the area under the curve for the entire transcript resulting in a percent of extended transcript value (Fig. 1B). For snR37 and snR33, which have extended 3’ ends, there are significantly more extended reads in the RRP6 mutant than in wild-type in both the sequencing and array data sets (Fig. 2). This agrees with the known role of Rrp6 to trim 3’ ends of snRNAs after termination. However, the relative ratios of these extended transcripts to the sense transcript appear to be much higher in the tiling array data than in the sequencing data in RNA isolated from RRP6 deletion strains. For instance, approximately 11% of snR37 transcripts appeared extended in the tiling arrays dataset as opposed to 0.3% when analyzed by RNA-Seq (Fig. 2B). RNA sequencing read count values in our study span 5 orders of magnitude while the tiling array data only covers 3–4 orders of magnitude, likely causing a loss of accurate detection of highly abundant fully processed sn/snoRNAs (compare Fig. 2C and Fig. 2D). The highly abundant processed snR37 or snR33 peaks are clearly distinguished from extended products using RNA-Seq (Fig. 2). Our data suggests that the relative effects of the loss of RRP6 on the steady state levels of these extended transcripts is much less dramatic than previously described and that accurate quantitation of steady state levels of the processed snRNA upon loss of RRP6 was not previously obtained due to limited dynamic range.
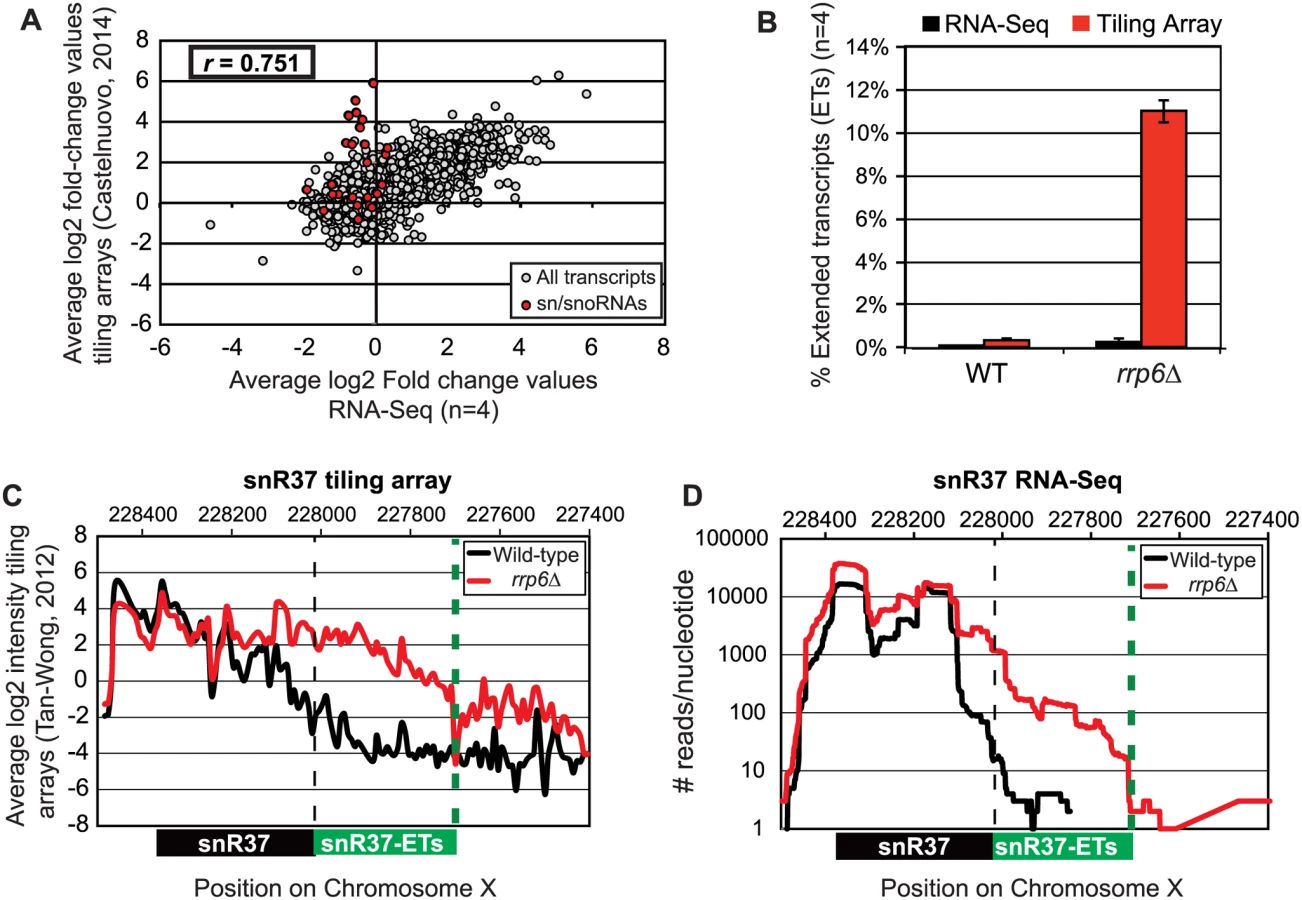
Analysis of snRNA termination in RRP6 deletion mutants
Loss of RRP6 results in improper 3’ end processing of multiple sn/snoRNAs. Such 3’ end processing defects could cause instability of snRNAs that leads to changes in their overall expression levels in cells [57,58]. Based on our differential expression analysis, 24 of the 78 sn/snoRNAs show decreased expression of at least 1.5-fold (log2 ≥ -0.6) in rrp6Δ versus wild-type cells with false discovery rates of ≤ 0.1 (S2 Table). To better understand the role of Rrp6 in global snRNA 3’ end processing, the length and intensity of snRNA extended transcripts was analyzed in rrp6Δ cells through manual annotation of the extended transcripts (annotated as “ETs”) observed in our RNA-Seq dataset (S1 Table). The length of snRNA transcript extension was compared to the annotated length of Nrd1-unterminated transcripts (NUTs) and cryptic unstable transcripts (CUTs) [11,48]. Additionally, we performed ChIP-exo according to established protocols to generate high-resolution maps of RNAPII localization throughout the yeast genomes in wild-type and RRP6 deletion cells with Rpb3-FLAG strains made for this study (S1B Fig.) [55]. It was recently reported that NUT annotations were significantly longer than CUT annotations, suggesting that Rrp6 is required for 3’ end processing of NNS terminated transcripts but is not directly involved in NNS termination [48]. Comparison of our datasets from RNA sequencing and ChIP-exo to NUT annotations as well as direct comparison of specific transcripts in rrp6Δ cells to NNS pathway mutants via northern blot analysis allowed us to discover a requirement for Rrp6 in Nrd1-dependent RNAPII termination.
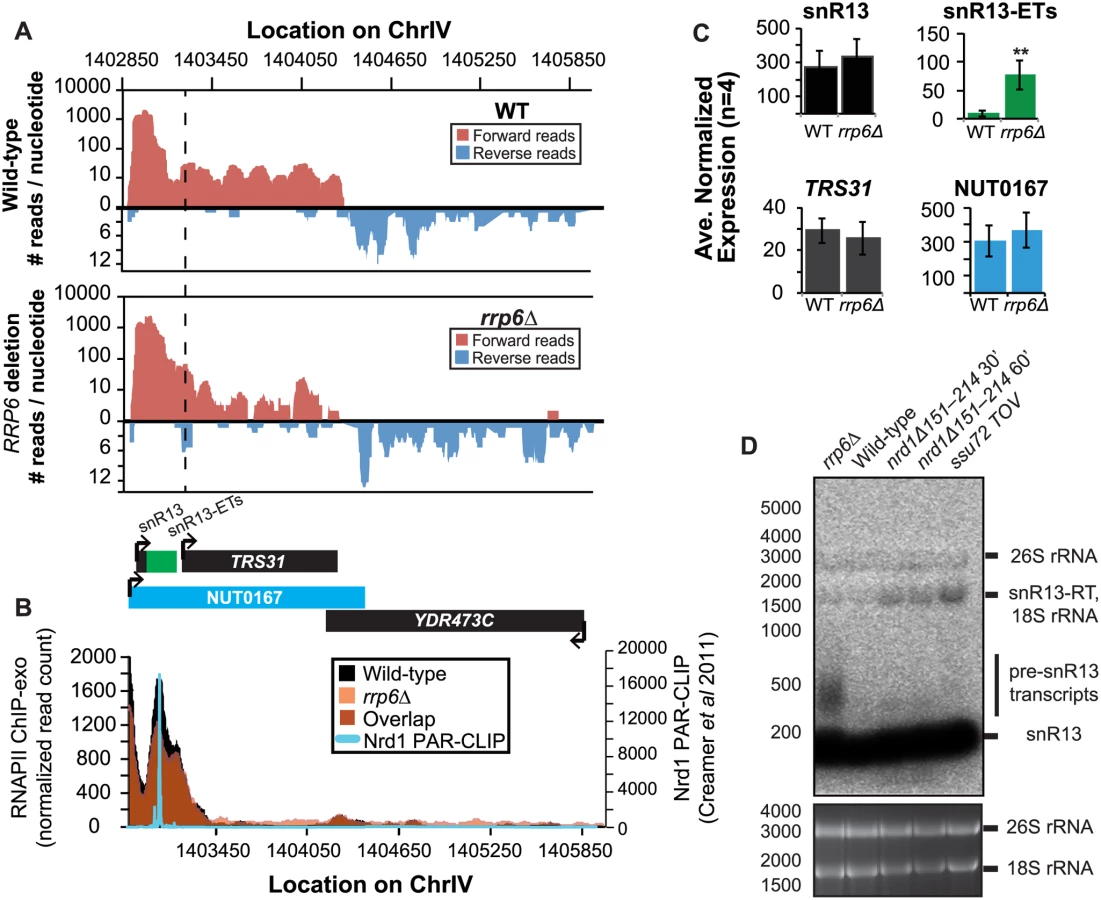
It has previously been reported that some snRNAs are significantly longer in Nrd1-depleted cells than in rrp6Δ, and our dataset confirms these findings. Previously published data by Kim et al. has shown a large extension for snR13 in SEN1 and NRD1 mutant strains, with the snR13 transcript extended across the coding region of TRS31 [20]. This extension at snR13 has also been reported in SSU72 mutant strains [52,56,57]. NUT0167 was annotated as an Nrd1-dependent unterminated transcript at snR13 following Nrd1 nuclear depletion. Kim et al. identified both normal length and an extended snR13 transcript in rrp6∆ cells, which corresponds to the pre-snRNA transcript that is not correctly processed in the absence of Rrp6 by northern blotting. For direct comparison of our dataset to previous work, we compared our RNA-Seq and ChIP-exo results to northern blot analysis using a strand specific probe against the processed version of snR13 (Fig. 3). The extended transcript detected in Nrd1 nuclear depletion experiments was annotated as NUT0167 and is 1378 nucleotides longer than the pre-snR13 transcript observed in our rrp6∆ RNA-Seq samples (Fig. 3A). In agreement, we observed a transcript approximately 1500 nucleotides long by northern blotting in the NRD1-temperature sensitive (ts) mutant (nrd1Δ151–214 that lacks the Nab3 interaction domain [34,58,59]) following 30 or 60 minute heat shock. Long extended transcripts were also observed in ssu72 TOV, a “terminator override” mutant previously shown to be deficient in Nrd1-dependent termination (kindly provided from the Reines lab) [60]. However, no such read-through transcript is observed in the rrp6∆ cells by RNA-Seq or by northern blotting, only the unprocessed pre-snR13 (Fig. 3A, D). Supporting the RNA transcript data, we see no shift in RNAPII localization in this region as detected by Rpb3-FLAG ChIP-exo sequencing (Fig. 3B). Previous studies to identify Nrd1 RNA binding sites using PAR-CLIP sequencing revealed a strong Nrd1 signal just downstream of the annotation for the mature snR13 transcript supporting the role of the NNS pathway in termination in this region ([39], Fig. 3B). No significant changes were observed in the expression levels of snR13 or TRS31, which also suggests that snR13 is correctly terminated in rrp6∆ (Fig. 2C). Taken together, these data clearly show that Rrp6 is not required for termination through the NNS pathway at snR13 in agreement with previous findings.
However, in contrast to previous conclusions that Rrp6 is dispensable for snRNA termination, our data indicates that a subset of snRNA transcripts require Rrp6 for proper RNAPII termination in vivo [20,48]. As shown in Fig. 4A, the apparent length of snR3 in rrp6Δ cells as revealed by RNA-Seq is 1396 nucleotides whereas the annotation for NUT0426 is 3363 nucleotides long (Fig. 4A, S1 Table). However, the Rrp6-dependent snR3 extension is 246 nucleotides longer than the three tandem CUTs (CUT221, 222, and 223) previously annotated from tiling array data [52]. All three CUTs were significantly up-regulated in rrp6Δ as expected (Fig. 4B and S2 Fig.). Interestingly, a comparison of the 4tU-Seq data used to annotated the NUTs with our RNA-Seq data clearly shows a difference in the length of the Nrd1-unterminated transcript NUT0426 resulting from snR3 read-through than that observed upon deletion of RRP6 (S2 Fig.). However, it is interesting to note that the overall read count for the snR3 read-through product in the 4tU-Seq dataset decreases dramatically just 3’ to the read-through observed in rrp6Δ, suggesting that the majority of transcripts are terminated at the location indicated in the rrp6∆ RNA-Seq (Fig. 4, S2 Fig.). ChIP-exo analysis of RNAPII localization in this region revealed an increase in RNAPII density just downstream of snR3 in the rrp6Δ cells when compared to WT (Fig. 4B, arrow 1). This increase in downstream RNAPII corresponds to the increased level of RNA detected downstream of snR3 in Fig. 4A past the pre-snRNA transcript and previously mapped Nrd1 and Nab3 binding sites located ~400bp from the start of the snR3 transcript [39,47]. Although the RNAPII density decreases just before the 3’ end of the snR3-ETs annotation, it remains slightly elevated in rrp6Δ compared to wild-type throughout the NUT0426 annotation region, indicated by arrow #2 (Fig. 4B). Differential expression analysis also revealed highly significant down-regulation of the snR3 convergent, “tail-to-tail,” gene YJR129C likely as a result of extended snR3 transcripts in rrp6Δ cells (Fig. 4C). These data suggest that extension of snR3 causes transcription interference at YJR129C as was also reported in Nrd1 nuclear depletion experiments [46]. To definitively determine the length of the snR3 transcripts and compare rrp6∆ cells directly to an NRD1 mutant, we also performed northern blot analysis with a probe that recognizes the short, processed snR3. A transcript that is approximately 4000 nucleotides long was detected in rrp6Δ, nrd1-ts mutants, and the ssu72 TOV (Fig. 4D). This transcript is approximately the length expected for NUT0426. We also detected two shorter transcripts approximately 500 and 1000 nucleotides long in the nrd1-ts and rrp6Δ cells. Together, these data clearly show that Rrp6 is required for proper RNAPII termination at snR3.

RRP6 deletion results in extension of snRNA transcripts leading to down-regulation of neighboring genes
Several genes downstream of snRNAs transcribed from the opposite DNA strand were found to be significantly down-regulated when transcript extension was high in rrp6Δ cells. We focused on snRNAs with “tail-to-tail” or convergent genes because the strand specific sequencing data allows us to distinguish between reads mapped to the two different transcripts. As mentioned in the example of snR3, the expression of neighboring downstream gene YJR129C is down almost three-fold (Fig. 4C). The detectable snR3 transcript maps beyond the transcription start site (TSS) for YJR129C suggesting that transcription interference is the likely mode of YJR129C repression as diagrammed in S3A Fig. Additional examples of snRNA termination defects causing transcription interference at downstream genes are shown in Fig. 5 and S3 Fig. As shown in S3 Fig., snR11 has a downstream gene transcribed on the opposite strand, CMC4 (S3C Fig.). snR11 has a long region of RNA-seq reads in rrp6Δ cells that extends beyond the CMC4 TSS, and CMC4 expression is decreased by more than half (S3C Fig. and S1 Table). However, northern blot analysis with a single stranded oligonucleotide probe detecting the short, processed snR11 does not show read-through transcripts >1000 bp in any of the samples, including the mutants known to have defective Nrd1 termination (S3B Fig.). Since there is no detectable read-through snR11 transcript in the nrd1-ts mutant, the transcript encoded in the region annotated “snR11-ETs” and “NUT0607” is likely initiated at start site downstream of the snR11 sequence recognized by our probe. The lack of highly extended transcripts, readily distinguishable by northern blot, shows the limitation of using short sequencing reads for transcript annotation (S3 Fig.). Interestingly, Rpb3-FLAG ChIP-exo data shows that the peak of polymerase localization at snR11 is extended farther 3’ in rrp6Δ cells than in wild type cells indicated by arrow #1, and this extension does indeed overlap the TSS of CMC4 (S3D Fig.). Polymerase occupancy quickly decreases just beyond the CMC4 TSS but remains higher than in wild type cells. The increase in polymerase localization in rrp6Δ downstream of the native termination site for snR11 suggests that loss of rrp6Δ decreases the efficiency of snR11 termination leading to a small degree of read-through that is sufficient to cause transcription interference at CMC4.

A substantial read-through transcript (1442 nucleotides) was detected at snR71, a C/D box small nucleolar RNA gene, in rrp6Δ cells that extends well beyond the TSS of downstream gene LIN1, which is significantly down-regulated (Fig. 5A, C) [61,62]. There is a 2281 nucleotide long NUT annotated at this locus (NUT0349) that overlaps with a second downstream gene, REC104, which is on the same strand as snR71 (Fig. 5A). With strand specific sequencing, it cannot be absolutely determined if reads mapped to the REC104 locus are from REC104 transcripts, from a much longer snR71 read-through transcript, or from a combination of both. To answer this question and compare the effects of RRP6 deletion to mutants with defective Nrd1-termination directly, we performed northern blot analysis with a short probe specific to the processed snR71 transcript (Fig. 5D). We see a striking band at approximately 1700 nucleotides in rrp6Δ, nrd1-ts, and Ssu72 TOV cells. This suggests that a percentage of RNAPIIs transcribing snR71 in all of these mutant strands can escape the Nrd1-termination pathway and are hence terminated much farther downstream (Fig. 5). This is supported by the Rpb3-FLAG ChIP-exo data that shows an increase in polymerase localization downstream of snR71 in rrp6Δ cells compared to wild type cells along the full length of the NUT0349 annotation (Fig. 5B). Interestingly, the expression level of the annotated mature snR71 region is also significantly decreased (Fig. 5C), suggesting there may be some instability or other defects caused by improper 3’ processing and/or termination caused by loss of Rrp6.
Overall these data show that multiple transcripts downstream of snRNAs have significant changes in expression following deletion of RRP6. ChIP-exo analysis of RNAPII (Rpb3-FLAG) clearly show that RNAPII terminates downstream of its normal stopping point in WT cells at specific snRNAs leading to mislocalization and causing transcription interference at downstream genes (Figs. 4 5, and S3 Fig.). In this model, ineffective termination of the snRNAs results in transcription interference at the downstream gene as previously described following inactivation of Nrd1 (S3A Fig. [46]). These findings, taken together with the similar lengths of Rrp6-dependent snRNA transcripts and Nrd1-dependent transcript annotations (NUTs), support the hypothesis that Rrp6 serves as important regulator of NNS-dependent termination at specific sn/snoRNAs.
RRP6 is required for proper RNAPII termination of NNS-dependent regulatory non-coding RNAs
In addition to extension of snRNA transcripts, changes in expression and/or apparent length of other previously described noncoding RNAs, such as CUTs, SUTs, and NUTs, were also detected with corresponding down-regulation of neighboring genes in cells lacking Rrp6 suggesting transcription interference. A number of these transcripts have been well-characterized as extended following depletion or genetic inactivation of NNS components but not the exonuclease RRP6. An early and well-characterized example of a gene that is regulated by Nrd1-dependent termination is the NRD1 gene itself [30,63,64]. There are a cluster of multiple Nrd1 and Nab3 binding sites in the 5’ UTR of the Nrd1 mRNA leading to early termination of the transcript and autoregulation of Nrd1 expression levels through a mechanism that also requires Sen1 [19,38,39,64]. In expression analysis of the RNA-seq data, we see nearly a 2-fold increase in the expression of the NRD1 transcript in rrp6Δ, as well as an increase in CUT320, a noncoding transcript near the NRD1 promoter on the opposite strand (Fig. 6A). For comparison, Nab3 mRNA levels were not significantly changed by RRP6 deletion (S1 Table). Since Rrp6 is responsible for the degradation of the short NRD1 transcript, this increase in expression could be attributed to stabilization of terminated transcript. Alternatively, increased expression of NRD1 in rrp6Δ cells could suggest that the early termination sites are not being utilized by the Nrd1 pathway as often in the rrp6Δ cells leading to transcription of the full-length mRNA. Analysis of RNAPII occupancy at the NRD1 gene in the ChIP-exo dataset can distinguish these two possibilities (Fig. 6B). In wild type cells, there are several peaks of RNAPII at the 5’ end of the NRD1 gene that quickly decreases to low levels along the NRD1 coding region in agreement with previous findings in an Nrd1 mutant [64]. In fact, comparison of the ChIP-exo data with Nrd1 RNA binding sites mapped by PAR-CLIP reveal that the majority of RNAPII terminates just downstream of the final Nrd1 binding site in WT cells (Fig. 6B). In rrp6Δ cells, the intense peaks of RNAPII localization at the 5’UTR are shifted 3’ and RNAPII localization is higher along the entire length of the gene, including a higher peak at the poly-A dependent termination site of the full-length transcript (Fig. 6B). This supports the hypothesis that the NNS-termination pathway is not terminating the short 5’UTR transcript efficiently in rrp6Δ cells and that RNAPII continues down the length of the gene producing the full transcript and increasing overall NRD1 expression levels (Fig. 6). The requirement for multiple Nrd1-Nab3 RNA binding sites at NRD1 when compared to RNAPII ChIP-exo data suggest that NNS-dependent termination requires multiple Nrd1 and/or Nab3 binding sites to effectively terminate RNAPII as has been previously proposed (Fig. 6B) [27,39]. The 3’ shift in RNAPII localization observed at NRD1 supports this hypothesis.

Changes in RNAPII occupancy downstream of known NNS-dependent early termination (also known as attenuated) targets was also observed at HRP1 and SRG1-SER3 (S4 Fig. and S5 Fig.). The RNAPII occupancy at HRP1 shows similar changes as seen at NRD1 with the majority of RNAPII terminating early in WT cells while showing a 3’ shift (arrow #1) and persistence through the coding region (arrow #2) in rrp6Δ (S4A Fig.). RNA-Seq analysis also revealed an increase in downstream HRP1 RNA levels relative to WT (S4B Fig.). Another well-characterized example of upstream noncoding RNA regulation occurs at the SRG1-SER3 region. SRG1 (SER3 regulatory gene 1) is a known non-coding RNA whose transcription down-regulates expression of SER3 [65]. SRG1 RNA is bound by both Nrd1 and Nab3 just prior to the SER3 transcribed region, which could then terminate SRG1 transcription to prevent interference with SER3 [31,47]. SRG1 can also be terminated through a polyA-dependent pathway at a site downstream of the NNS termination site(s) [31]. ChIP-exo revealed that RNAPII occupancy also shifts 3’ in rrp6Δ cells with increased RNAPII occupancy in the SER3 coding region (S5A Fig.). An increase in specific SRG1 transcripts including a SRG1:SER3 chimeric transcript has previously been observed in Nrd1-depletion rrp6Δ cells [31]. Our data suggests that defective termination of SRG1 can occur in rrp6Δ cells even in the absence of Nrd1 disruption (S5 Fig.). These data clearly show that Rrp6 is required for NNS-dependent termination of regulatory non-coding RNAs that participate in gene expression attenuation.
There are multiple published examples of transcription interference by CUTs in which an antisense CUT regulates the expression of the sense gene in cis through transcript extension across the sense gene promoter [50,66,67,68,69]. These lncRNA-type transcripts have also been shown to be required for efficient gene activation [70]. Expression of the FMP40 transcript has been previously shown by several groups to be regulated by an Nrd1-terminated antisense transcript initiating at the 3’ end of FMP40 [28,39,71]. The FMP40 antisense transcript (also known as CUT882 and YPL222C-A) is readily detectable in our RNA-sequencing data from WT cells and it is significantly upregulated in rrp6Δ cells as has been previously reported (Fig. 7A and C) [10,11,72,73]. Mapped sequencing reads suggest that the antisense transcript was significantly longer in rrp6Δ than in wild type (specifically in the region of CUT882), supporting the hypothesis that Nrd1-termination is not as efficient in rrp6Δ mutants. To determine the length of the antisense transcript in rrp6Δ and compare to an nrd1-ts mutant, we performed northern blot analysis with strand specific oligonucleotide probes to FMP40 and its antisense transcript YPL222C-A (Fig. 7D). By northern blot, YPL222C-A transcripts are detected as smears extending into the region of the CUT882 annotation suggesting that these transcripts terminate at multiple 3’ end locations as has previously been shown for other specific CUTs including NEL025c [9]. The YPL222C-A antisense transcripts were not detected in WT cells by northern blot even after an extended exposure time (Fig. 7D). In the nrd1-ts mutant, there is an accumulation of a strong band ~4500 nucleotides long in the YPL222C-A blot, and the FMP40 transcript is undetectable as a consequence of transcription interference. There is a band of the same size in the rrp6Δ, although it is not as abundant as observed in the Nrd1 mutant. There are also shorter transcripts present in YPL222C-A blots in the nrd1-ts mutant that are also present in the rrp6Δ cells (Fig. 7D). RNAPII localization in the FMP40 region was determined by ChIP-exo and clearly shows that the majority of polymerase was localized at the 5’ end of the antisense YPL222C-A transcript, even in wild-type cells (Fig. 7B). The highest peaks of RNAPII localization at YPL222C-A are of similar intensity in WT and rrp6Δ cells, but polymerase spreads 3’ in rrp6Δ and continues to be higher through the CUT882 annotation and past the FMP40 promoter. These data indicate that the antisense transcript is terminated less efficiently in rrp6Δ cells leading to increased RNAPII occupancy downstream of the normal YPL222C-A/CUT882 termination site. The RNAPII occupancy data also suggest that CUT882 is an extended transcript of YPL222C-A that occurs as a consequence of inefficient RNAPII termination in rrp6Δ (Fig. 7B, red arrows). In nrd1Δ151–214 temperature sensitive mutants, YPL222C-A termination rarely occurs resulting in a 4500-nucleotide antisense transcript that is also seen at low levels in rrp6Δ (Fig. 7D).

Discussion
Through comparison of RNA-Seq and RNAPII ChIP-exo datasets, these data clearly show that NNS-terminator read-through occurs at a significant number of NNS-target genes in the absence of the 3–5’ exonuclease Rrp6. These findings support the hypothesis that the NNS pathway requires Rrp6 function for both 3’ end processing and NNS-dependent termination of specific transcripts. Overall, these data show a striking similarity between the annotations for Nrd1 unterminated transcripts (NUTs) at many locations and the transcripts observed following deletion of the nuclear specific 3’-5’ exonuclease subunit Rrp6 genome-wide. The tight coupling of the NNS pathway with exosome function has been previously characterized; however our findings are the first to demonstrate on a large scale that Rrp6 function is required for termination through the NNS pathway at specific transcripts. We have also shown that deletion of RRP6 causes changes in RNAPII localization that are indicative of termination defects. The comparison of changes in RNA transcript signals to actual changes in RNAPII occupancy allowed us to distinguish between effects of RNAPII termination events versus exonuclease-dependent RNA processing, which was of particular importance for this study. Although it has been proposed that Nrd1-unterminated transcripts (NUTs) aresignificantly longer than CUTs, our data clearly shows that specific NNS-dependent transcripts rely on Rrp6 for proper termination while others do not. For instance, snr71 transcripts show a requirement for Rrp6, Nrd1, and Ssu72 for termination, while snR13 does not require Rrp6 (Figs. 3 and 5). The cryptic transcript CUT882 that is associated with FMP40/YPL22C-A also shows a requirement for Rrp6 for termination but it appears that the CUT882 transcript is significantly longer in nrd1∆151–214 temperature sensitive strains (Fig. 7). Previous studies have found other extended CUT transcripts (eCUTs; specifically CUT060 and CUT095) to be significantly longer in Nrd1 mutants than in rrp6Δ as well [48,58]. The mechanisms that underlie the specific requirement for Rrp6 at some transcripts for Nrd1 termination are unknown, but we would speculate that the requirement for Rrp6 in NNS-termination is mediated through its interaction with Nrd1 [7,8,49]. Future studies will also be needed to determine if the requirement for Rrp6 in NNS termination is direct or occurs through an indirect mechanism.
In this study, we have performed multiple biological replicates (n = 4) to provide highly accurate differential expression analysis for all classes of annotated transcripts. In addition, we have included differential expression analysis of 1215 antisense transcripts that were significantly changed in rrp6Δ, the majority of which (76%) were upregulated. These data will serve as a valuable resource to determine the diverse roles of the NNS pathway and Rrp6 in transcriptome-wide gene expression regulation. Our dataset also shows that the increased dynamic range and resolution provided by RNA-Seq and ChIP-exo gives a distinct advantage in precise mapping of the transcripts that accumulate following deletion of RRP6 [10,11,48]. Using RNA-Seq, we have determined that loss of Rrp6 leads to decreased expression of the majority of sn/snoRNA transcripts. Additionally, we have found that the mRNA transcripts from ribosomal protein coding genes show a strong dependence on Rrp6 for control of their steady state transcript levels. Previous ChIP studies on Nrd1 have shown that although Nrd1 localizes to many ribosomal protein-coding genes, it does not directly bind to the mRNA transcripts from those genes [39]. This class of mRNA transcripts may be regulated by Nrd1 and Rrp6 due to their short length and hence high levels of Ser5-phosphorylated RNAPII at their 3’ ends, which could facilitate interaction with Nrd1 and regulation of termination [8,37,74,75]. By comparison of our data to recent 4tU-Seq datasets following Nrd1-depletion from the nucleus, we have found that multiple ribosomal subunit mRNAs display transcript extension following loss of Nrd1 activity and have been annotated as NUTs [48]. These data strongly suggest that ribosomal protein coding mRNAs may require Rrp6 and the NNS pathway for proper 3’ processing and transcript stability.
It has previously been determined that the downstream region of snR13 contains both an Nrd1-dependent terminator and a polyA dependent terminator that is present only 50 nucleotides downstream of the Nrd1 terminator [63]. In rrp6Δ cells we observed proper termination for snR13 although termination was defective in other NNS-pathway components (Fig. 2). It is important to note, however, that overall levels of the mature processed snR13 transcript were not significantly changed in any of the mutant backgrounds, which is likely due to the presence of a second polyA-dependent terminator (Fig. 2D). It is likely that the differences in NNS-dependent termination efficiency at specific target genes observed in rrp6Δ is reliant on the context of both terminators and other downstream sequence elements that can contribute to proper RNAPII termination downstream. Additional failsafe mechanisms for transcription termination have also been described in yeast that involve the nuclear RNase III enzyme Rnt1 that could contribute to proper snRNA termination in the absence of NNS-termination pathway components including Rrp6 [76,77].
High-resolution RNAPII occupancy maps generated by ChIP-exo provide unique insights into the mechanisms of NNS-dependent termination. At NRD1, HRP1, and YPL222C-A, deletion of RRP6 lead to a 3’ shift in RNAPII localization indicating that NNS-dependent termination was delayed or less efficient (Figs. 6,7,and S4). This phenomena was also observed to a lesser extent at SRG1-SER3 (S5 Fig.). Interestingly, we observed distinct accumulation of RNAPII at these regions that occurred just 3’ to Nrd1 and/or Nab3 binding sites that have previously been mapped by RNA-protein crosslinking approaches (Fig. 6) [39,47]. These findings suggest that termination through the NNS-pathway may be an inefficient process requiring clusters of Nrd1/Nab3 binding sites to for facilitate higher order recruitment of multiple Nrd1-Nab3 heterodimers to carry out termination, which has support from other studies [27,59,78]. This hypothesis is further supported by studies that show that NNS-termination occurs in a ‘termination zone’ rather than a specific termination site as proposed for polyA-dependent termination pathways [40]. Through this mechanism, gene expression attenuation at genes such as NRD1 and SRG1-SER3 would remain leaky, allowing for transcription of a small percentage of full-length transcripts even in the presence of high Nrd1-Nab3 protein levels, which is what we observe in WT cells at these genes (Figs. 6, 7, S4, and S5). The requirement for the nuclear exosome and Rrp6 for efficient NNS-regulated attenuation adds a layer of complexity to this process and another path for regulation for these tightly controlled genes.
Methods
Yeast strains and RNA isolation
All yeast strains used are isogenic to BY4741. RRP6 deletion strain is from the yeast knockout collection (Open Biosystems) [79]. The Rpb3-FLAG WT strain was produced by yeast transformation with a tagging cassette containing sequences for a 3X-FLAG tag upstream of the URA3 gene from the plasmid pBS1539 (primer sequences available upon request) [80]. RRP6 deletion Rpb3–3XFLAG strains were produced by amplification of the RRP6 knockout cassette from the yeast knockout collection RRP6 deletion strain and transformation into the wild-type Rpb3-FLAG strain. RRP6 knockout was confirmed by PCR genotyping. RNA was extracted using the hot acid phenol method. Briefly, strains were grown in 100ml YPD medium to an OD600 of 0.8. Cells were collected by centrifugation, washed, and resuspended in 10ml AE buffer (50mM sodium acetate at pH 5.2, 10mM EDTA) in a Nalgene phenol-resistant 50ml tube. 800μl 20% SDS and 10ml cold acid phenol were added to each sample and mixed well by vortexing. Samples were incubated at 65°C for 10 minutes with vortexing every minute then cooled on ice for 5 minutes. Samples were centrifuged for 15 minutes at 10,000 rpm. The top phase was transferred to a pre-spun 50ml 5 PRIME Phase Lock Gel tube (Ref # 2302870). 13ml chloroform was added and well mixed before centrifuging for 10 minutes at 3000 rpm. The top phase was poured into a new phenol-resistant tube, and 1/10 volume sodium acetate at pH 5.2 and equal volume room temperature isopropanol was added. The precipitated RNA was collected by centrifuging for 45 minutes at 12,000 rpm. The pellet was washed with 70% ethanol, allowed to dry in a fume hood, and resuspended with molecular biology grade water. The Ambion DNase-turbo kit was used to degrade any contaminating DNA. The quality of the samples was determined with an Agilent Bioanalyzer before preparation of the sequencing libraries.
ChIP-exo analysis of Rpb3-FLAG localization
Chromatin IP followed by exonuclease treatment was performed using the protocol described by Rhee and Pugh with the following modifications [55]. Rpb3-FLAG WT and rrp6Δ strains were grown to an OD600 = 0.8–1 prior to crosslinking with formaldehyde (Sigma, catalog # F8775–25ML). Immunoprecipitation was performed with 50uL of anti-FLAG agarose (Sigma). Subsequent sample processing steps including exonuclease treatment and sequencing library preparation were performed as previously described [55].
Northern blot analysis
30ug of total RNA was loaded per lane on a 1.5% and separated by electrophoresis at 120 volts for 1 hour at 4°C. The RNA was transferred to Bio-Rad Zeta-Probe blotting membranes by capillary overnight. Transfer efficiency was determined by Methylene Blue staining. DNA oligonucleotide probes listed in S5 Table were 5’ end-labeled with gamma ATP-32P by T4 Polynucleotide Kinase. Probes were hybridized overnight to pre-blocked in Roche Life Science DIG Easy Hyb buffer at 37°C. Blots were washed in 6XSSC / 0.1%SDS once at room temperature and twice for 10 minutes at 50°C. Blots were exposed to a phosphorscreen overnight for snRNAs or 7 days for FMP40 and YPL22C-A followed by scanning using a phosphorimager (GE Healthcare).
SOLiD5500xl sequencing methods
Standard methods were used for RNA-seq library construction, EZBead preparation and Next-Gen sequencing, based on Life Technologies SOLiD5500xl system. Briefly, five microgram of total RNA per sample (RIN equal or higher than 6.0 by Agilent Bioanalyzer) was applied in library preparation. rRNA was first depleted using the standard protocol of RiboMinus Transcriptome Isolation Kit for yeast (Ambion, Cat# K1550–03), and rRNA-depleted RNA was concentrated with the PureLink RNA Micro Kit (Invitrogen, Cat# 12183–016) using 1 volume of Lysis Buffer and 2.5 volumes of 100% ethanol. Following the rRNA depletion, whole transcriptome library was prepared and barcoded per sample using the standard protocol of SOLiD Total RNA-seq Kit (Life Technologies, Cat# 4445374). Each barcoded library was quantified by quantitative PCR using SOLiD Library Taqman qPCR Module (Life Technologies, Cat#A12127), and pooled in equal molarity. Fifty microliters of 500 pM of pooled library was used in subsequent EZBead preparation, which involves bead emulsion, bead library amplification, and bead enrichment using Life Technologies EZ Bead E120 System (Cat# 4465571). Approximately six hundred million enriched beads then were deposited onto each lane of a 6-lane SOLiD5500xl flow chip. And finally sequencing by ligation was carried out using standard single-read, 5’-3’ strand-specific sequencing procedure (75b-read) on SOLiD5500xl Sequencer.
Sequencing data analysis
The libraries were sequenced on Solid 5500XL. The resulting 75 bp solid reads were mapped to Saccharomyces cerevisiae reference genome sacCer3 using in-house mapping pipelines that utilizes bfast-0.7.0a [81]. Briefly, using our RNA-seq pipeline, poor quality and rRNA/tRNAs reads were first discarded. The remaining reads were mapped to reference genome sacCer3 and a splice-junction library, respectively; the genomic and splice-junction library mapping were merged at the end. In a different pipeline, the rRNA/tRNAs were kept and the reads were mapped to the reference genome sacCer3 only based on the facts that in yeast there is some splicing but most genes do not have introns. Read counts were calculated using bamutils from NGSUtils [82]. Differential gene expression was analyzed using edgeR which calculated all normalized read counts, p-values, and FDR values listed in S1 Table [83]. All raw and processed files from the RNA sequencing performed for this study has been deposited to Gene Expression Omnibus [GEO] under the accession number GSE57155.
Manual annotation of snRNA extension and antisense transcripts
Following data alignment, snRNA transcripts were manually inspected individually using the Integrative Genomics Viewer [84,85]. For all snRNAs in a tail-to-tail orientation with a downstream gene, the snRNA ET annotation started just after the end of the snRNA annotation until continuous reads on the same strand were no longer detected. For all snRNAs in a tail-to-head orientation, the snRNA ET annotation started just after the end of the snRNA annotation and was ended just prior to the 5’ end of the annotation for the downstream gene. Annotations for ET for snRNAs that were encoded within introns were ended just prior to the 5’ end of the exon for the parent transcript. To identify antisense transcripts with significant changes in differential expression, the strand was reversed for all sense annotations for the coding region of each ORF-Ts and the text “AS_” was added in front of the ORF-T name. The annotations for the 5’ and 3’ UTR were not included. These annotations were then used for edgeR analysis and the annotations for antisense transcripts that showed significant changes in rrp6Δ were used for subsequent differential expression analysis to generate the final dataset in S1 Table.
GOStat analysis
GOStat analysis [86] was performed as previously described [87] for all significantly down-regulated ORF-T transcripts from our dataset. In brief, the list of 995 significantly upregulated ORF-Ts was entered into the GOStat web interface (http://gostat.wehi.edu.au/cgi-bin/goStat.pl) to search for the top 30 most over-represented GO terms and to obtain p-values to indicate the significance of enrichment (S3 Table). GO-term enrichment analysis was also performed using DAVID [88] and similar results were obtained.
Alignment of Nrd1-unterminated transcript data
4tU-seq reads of both wild type and Nrd1 depleted samples were downloaded from ftp.sra.ebi.ac.uk/vol1/ERA242/ERA242535/fastq/. The sequence data were processed according to the description in [48,89,90] with minor changes. Briefly, the reads were quality trimmed and mapped with bowtie-0.12.9 [91] to reference genome sacCer3 (mapping parameters:-q-p 4-S-n 0-e 70-l 28-y-k 1-m 2—best—strata—phred33-quals—norc/—nofw). The output SAM files were then converted to BAM files with SAMtools [92]. The BAM files were further processed to keep the uniquely and perfectly mapped reads using bamutils from NGSUtils [82].
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Bonneau F, Basquin J, Ebert J, Lorentzen E, Conti E (2009) The yeast exosome functions as a macromolecular cage to channel RNA substrates for degradation. Cell 139 : 547–559. doi: 10.1016/j.cell.2009.08.042 19879841
2. Schneider C, Tollervey D (2013) Threading the barrel of the RNA exosome. Trends in biochemical sciences 38 : 485–493. doi: 10.1016/j.tibs.2013.06.013 23910895
3. Makino DL, Baumgartner M, Conti E (2013) Crystal structure of an RNA-bound 11-subunit eukaryotic exosome complex. Nature 495 : 70–75. doi: 10.1038/nature11870 23376952
4. Makino DL, Halbach F, Conti E (2013) The RNA exosome and proteasome: common principles of degradation control. Nature reviews Molecular cell biology 14 : 654–660. doi: 10.1038/nrm3657 23989960
5. Mitchell P, Petfalski E., Shevchenko A., Mann M., Tollervey D. (1997) The exosome: a conserved eukaryotic RNA processing complex containing multiple 3′—>5′ exoribonucleases. Cell 91 : 457–466. 9390555
6. Lorentzen E, Basquin J, Tomecki R, Dziembowski A, Conti E (2008) Structure of the active subunit of the yeast exosome core, Rrp44: diverse modes of substrate recruitment in the RNase II nuclease family. Molecular cell 29 : 717–728. doi: 10.1016/j.molcel.2008.02.018 18374646
7. Vasiljeva L, Buratowski S (2006) Nrd1 interacts with the nuclear exosome for 3′ processing of RNA polymerase II transcripts. Molecular cell 21 : 239–248. 16427013
8. Heo DH, Yoo I, Kong J, Lidschreiber M, Mayer A, et al. (2013) The RNA Polymerase II C-terminal Domain-Interacting Domain of Yeast Nrd1 Contributes to the Choice of Termination Pathway and Couples to RNA Processing by the Nuclear Exosome. The Journal of biological chemistry. 288 : 36676–36690. doi: 10.1074/jbc.M113.508267 24196955
9. Wyers F, Rougemaille M, Badis G, Rousselle JC, Dufour ME, et al. (2005) Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell 121 : 725–737. 15935759
10. Neil H, Malabat C, d’Aubenton-Carafa Y, Xu Z, Steinmetz LM, et al. (2009) Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature 457 : 1038–1042. doi: 10.1038/nature07747 19169244
11. Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Munster S, et al. (2009) Bidirectional promoters generate pervasive transcription in yeast. Nature 457 : 1033–1037. doi: 10.1038/nature07728 19169243
12. Porrua O, Hobor F, Boulay J, Kubicek K, D’Aubenton-Carafa Y, et al. (2012) In vivo SELEX reveals novel sequence and structural determinants of Nrd1-Nab3-Sen1-dependent transcription termination. The EMBO Journal 31 : 3935–3948. doi: 10.1038/emboj.2012.237 23032188
13. Davis CA, Ares M Jr., (2006) Accumulation of unstable promoter-associated transcripts upon loss of the nuclear exosome subunit Rrp6p in Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences of the United States of America 103 : 3262–3267. 16484372
14. Dichtl B BD, Ohnacker M, Friedlein A, Roeder D, Langen H, Keller W. (2002) A Role for SSU72 in Balancing RNA Polymerase II Transcription Elongation and Termination. Molecular Cell 10 : 1139–1150. 12453421
15. Schmid M, Poulsen MB, Olszewski P, Pelechano V, Saguez C, et al. (2012) Rrp6p controls mRNA poly(A) tail length and its decoration with poly(A) binding proteins. Molecular cell 47 : 267–280. doi: 10.1016/j.molcel.2012.05.005 22683267
16. Grenier St-Sauveur V, Soucek S, Corbett AH, Bachand F (2013) Poly(A) tail-mediated gene regulation by opposing roles of Nab2 and Pab2 nuclear poly(A)-binding proteins in pre-mRNA decay. Molecular and cellular biology 33 : 4718–4731. doi: 10.1128/MCB.00887-13 24081329
17. Gudipati RK, Xu Z, Lebreton A, Seraphin B, Steinmetz LM, et al. (2012) Extensive degradation of RNA precursors by the exosome in wild-type cells. Molecular cell 48 : 409–421. doi: 10.1016/j.molcel.2012.08.018 23000176
18. Schneider C, Kudla G, Wlotzka W, Tuck A, Tollervey D (2012) Transcriptome-wide analysis of exosome targets. Molecular cell 48 : 422–433. doi: 10.1016/j.molcel.2012.08.013 23000172
19. Steinmetz EJC Nicholas K.; Brow David A.; Corden Jeffry L. (2001) RNA-binding protein Nrd1 directs poly(A)-independent 3′-end formation of RNA polymerase II transcripts. Nature 413 : 327–331. 11565036
20. Kim M, Vasiljeva L, Rando OJ, Zhelkovsky A, Moore C, et al. (2006) Distinct pathways for snoRNA and mRNA termination. Molecular cell 24 : 723–734. 17157255
21. Buratowski S (2009) Progression through the RNA polymerase II CTD cycle. Molecular cell 36 : 541–546. doi: 10.1016/j.molcel.2009.10.019 19941815
22. Kim MK Nevan J.; Vasiljeva Lidia; Rando Oliver J.; Nedea Eduard; Greenblatt Jack F.; Buratowski Stephen (2004) The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature 432 : 517–522. 15565157
23. Nedea E, He X, Kim M, Pootoolal J, Zhong G, et al. (2003) Organization and function of APT, a subcomplex of the yeast cleavage and polyadenylation factor involved in the formation of mRNA and small nucleolar RNA 3′-ends. The Journal of biological chemistry 278 : 33000–33010. 12819204
24. Zhao JK Marco; Helmling Steffen; O’Connor J. Patrick; Moore Claire (1999) Pta1, a component of yeast CF II, is required for both cleavage and poly(A) addition of mRNA precursor. Molecular and cellular biology 19 : 7733–7740. 10523662
25. Kyberz AS Martin; Dichtl Bernhard; Keller Walter (2003) The role of the yeast cleavage and polyadenylation factor subunit Ydh1p/Cft2p in pre-mRNA 3′-end formation. Nucleic acids research 31 : 3936–3945. 12853609
26. Lunde BM, Reichow SL, Kim M, Suh H, Leeper TC, et al. (2010) Cooperative interaction of transcription termination factors with the RNA polymerase II C-terminal domain. Nat Struct Mol Biol 17 : 1195–1201. doi: 10.1038/nsmb.1893 20818393
27. Carroll KL, Ghirlando R, Ames JM, Corden JL (2007) Interaction of yeast RNA-binding proteins Nrd1 and Nab3 with RNA polymerase II terminator elements. RNA 13 : 361–373. 17237360
28. Arigo JT, Eyler DE, Carroll KL, Corden JL (2006) Termination of cryptic unstable transcripts is directed by yeast RNA-binding proteins Nrd1 and Nab3. Mol Cell 23 : 841–851. 16973436
29. Carroll KL, Pradhan DA, Granek JA, Clarke ND, Corden JL (2004) Identification of cis elements directing termination of yeast nonpolyadenylated snoRNA transcripts. Molecular and cellular biology 24 : 6241–6252. 15226427
30. Steinmetz EJ, Conrad NK, Brow DA, Corden JL (2001) RNA-binding protein Nrd1 directs poly(A)-independent 3′-end formation of RNA polymerase II transcripts. Nature 413 : 327–331. 11565036
31. Thiebaut M, Kisseleva-Romanova E, Rougemaille M, Boulay J, Libri D (2006) Transcription termination and nuclear degradation of cryptic unstable transcripts: a role for the nrd1-nab3 pathway in genome surveillance. Molecular cell 23 : 853–864. 16973437
32. Honorine R, Mosrin-Huaman C, Hervouet-Coste N, Libri D, Rahmouni AR (2011) Nuclear mRNA quality control in yeast is mediated by Nrd1 co-transcriptional recruitment, as revealed by the targeting of Rho-induced aberrant transcripts. Nucleic acids research 39 : 2809–2820. doi: 10.1093/nar/gkq1192 21113025
33. Meinhart A CP (2004) Recognition of RNA polymerase II carboxy-terminal domain by 3′-RNA-processing factors. Nature 430 : 223–226. 15241417
34. Vasiljeva L, Kim M, Mutschler H, Buratowski S, Meinhart A (2008) The Nrd1-Nab3-Sen1 termination complex interacts with the Ser5-phosphorylated RNA polymerase II C-terminal domain. Nature structural & molecular biology 15 : 795–804. doi: 10.1111/nph.13282 25615841
35. Vasiljeva L, Kim M, Terzi N, Soares LM, Buratowski S (2008) Transcription termination and RNA degradation contribute to silencing of RNA polymerase II transcription within heterochromatin. Mol Cell 29 : 313–323. doi: 10.1016/j.molcel.2008.01.011 18280237
36. Conrad NK, Wilson SM, Steinmetz EJ, Patturajan M, Brow DA, et al. (2000) A yeast heterogeneous nuclear ribonucleoprotein complex associated with RNA polymerase II. Genetics 154 : 557–571. 10655211
37. Gudipati RK, Villa T, Boulay J, Libri D (2008) Phosphorylation of the RNA polymerase II C-terminal domain dictates transcription termination choice. Nat Struct Mol Biol 15 : 786–794. doi: 10.1038/nsmb.1460 18660821
38. Jamonnak N, Creamer TJ, Darby MM, Schaughency P, Wheelan SJ, et al. (2011) Yeast Nrd1, Nab3, and Sen1 transcriptome-wide binding maps suggest multiple roles in post-transcriptional RNA processing. RNA 17 : 2011–2025. doi: 10.1261/rna.2840711 21954178
39. Creamer TJ, Darby MM, Jamonnak N, Schaughency P, Hao H, et al. (2011) Transcriptome-wide binding sites for components of the Saccharomyces cerevisiae non-poly(A) termination pathway: Nrd1, Nab3, and Sen1. PLoS Genet 7: e1002329. doi: 10.1371/journal.pgen.1002329 22028667
40. Hazelbaker DZ, Marquardt S, Wlotzka W, Buratowski S (2013) Kinetic Competition between RNA Polymerase II and Sen1-Dependent Transcription Termination. Molecular cell 49 : 55–66. doi: 10.1016/j.molcel.2012.10.014 23177741
41. Brow DA (2011) Sen-sing RNA terminators. Mol Cell 42 : 717–718. doi: 10.1016/j.molcel.2011.06.002 21700218
42. Steinmetz EJ, Warren CL, Kuehner JN, Panbehi B, Ansari AZ, et al. (2006) Genome-wide distribution of yeast RNA polymerase II and its control by Sen1 helicase. Mol Cell 24 : 735–746. 17157256
43. Skourti-Stathaki K, Proudfoot NJ, Gromak N (2011) Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell 42 : 794–805. doi: 10.1016/j.molcel.2011.04.026 21700224
44. Lykke-Andersen S, Jensen TH (2007) Overlapping pathways dictate termination of RNA polymerase II transcription. Biochimie 89 : 1177–1182. 17629387
45. Grzechnik P, Kufel J (2008) Polyadenylation linked to transcription termination directs the processing of snoRNA precursors in yeast. Mol Cell 32 : 247–258. doi: 10.1016/j.molcel.2008.10.003 18951092
46. Stuparevic I, Mosrin-Huaman C, Hervouet-Coste N, Remenaric M, Rahmouni AR (2013) Cotranscriptional recruitment of RNA exosome cofactors Rrp47p and Mpp6p and two distinct Trf-Air-Mtr4 polyadenylation (TRAMP) complexes assists the exonuclease Rrp6p in the targeting and degradation of an aberrant messenger ribonucleoprotein particle (mRNP) in yeast. J Biol Chem 288 : 31816–31829. doi: 10.1074/jbc.M113.491290 24047896
47. Wlotzka W, Kudla G, Granneman S, Tollervey D (2011) The nuclear RNA polymerase II surveillance system targets polymerase III transcripts. EMBO J 30 : 1790–1803. doi: 10.1038/emboj.2011.97 21460797
48. Schulz D, Schwalb B, Kiesel A, Baejen C, Torkler P, et al. (2013) Transcriptome surveillance by selective termination of noncoding RNA synthesis. Cell 155 : 1075–1087. doi: 10.1016/j.cell.2013.10.024 24210918
49. Tudek A, Porrua O, Kabzinski T, Lidschreiber M, Kubicek K, et al. (2014) Molecular basis for coordinating transcription termination with noncoding RNA degradation. Mol Cell 55 : 467–481. doi: 10.1016/j.molcel.2014.05.031 25066235
50. Castelnuovo M, Rahman S, Guffanti E, Infantino V, Stutz F, et al. (2013) Bimodal expression of PHO84 is modulated by early termination of antisense transcription. Nat Struct Mol Biol 20 : 851–858. doi: 10.1038/nsmb.2598 23770821
51. Park D, Morris AR, Battenhouse A, Iyer VR (2014) Simultaneous mapping of transcript ends at single-nucleotide resolution and identification of widespread promoter-associated non-coding RNA governed by TATA elements. Nucleic Acids Res 42 : 3736–3749. doi: 10.1093/nar/gkt1366 24413663
52. Tan-Wong SM, Zaugg JB, Camblong J, Xu Z, Zhang DW, et al. (2012) Gene loops enhance transcriptional directionality. Science 338 : 671–675. doi: 10.1126/science.1224350 23019609
53. Castelnuovo M, Zaugg JB, Guffanti E, Maffioletti A, Camblong J, et al. (2014) Role of histone modifications and early termination in pervasive transcription and antisense-mediated gene silencing in yeast. Nucleic Acids Res 42 : 4348–4362. doi: 10.1093/nar/gku100 24497191
54. Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, et al. (2008) The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320 : 1344–1349. doi: 10.1126/science.1158441 18451266
55. Rhee HS, Pugh BF (2012) ChIP-exo method for identifying genomic location of DNA-binding proteins with near-single-nucleotide accuracy. Curr Protoc Mol Biol Chapter 21: Unit 21 24.
56. Ganem C, Devaux F, Torchet C, Jacq C, Quevillon-Cheruel S, et al. (2003) Ssu72 is a phosphatase essential for transcription termination of snoRNAs and specific mRNAs in yeast. EMBO J 22 : 1588–1598. 12660165
57. Steinmetz EJ, Brow DA (2003) Ssu72 Protein Mediates Both Poly(A)-Coupled and Poly(A)-Independent Termination of RNA Polymerase II Transcription. Molecular and cellular biology 23 : 6339–6349. 12944462
58. Marquardt S, Hazelbaker DZ, Buratowski S (2011) Distinct RNA degradation pathways and 3′ extensions of yeast non-coding RNA species. Transcription 2 : 145–154. 21826286
59. Loya TJ, O’Rourke TW, Degtyareva N, Reines D (2013) A network of interdependent molecular interactions describes a higher order Nrd1-Nab3 complex involved in yeast transcription termination. J Biol Chem 288 : 34158–34167. doi: 10.1074/jbc.M113.516765 24100036
60. Loya TJ, O’Rourke TW, Reines D (2012) A genetic screen for terminator function in yeast identifies a role for a new functional domain in termination factor Nab3. Nucleic Acids Res 40 : 7476–7491. doi: 10.1093/nar/gks377 22564898
61. Lowe TM, Eddy SR (1999) A computational screen for methylation guide snoRNAs in yeast. Science 283 : 1168–1171. 10024243
62. Samarsky DA, Fournier MJ (1999) A comprehensive database for the small nucleolar RNAs from Saccharomyces cerevisiae. Nucleic Acids Res 27 : 161–164. 9847166
63. Steinmetz EJ, Ng SB, Cloute JP, Brow DA (2006) cis - and trans-Acting determinants of transcription termination by yeast RNA polymerase II. Molecular and cellular biology 26 : 2688–2696. 16537912
64. Arigo JT, Carroll KL, Ames JM, Corden JL (2006) Regulation of yeast NRD1 expression by premature transcription termination. Mol Cell 21 : 641–651. 16507362
65. Martens JA, Laprade L, Winston F (2004) Intergenic transcription is required to repress the Saccharomyces cerevisiae SER3 gene. Nature 429 : 571–574. 15175754
66. Pelechano V, Steinmetz LM (2013) Gene regulation by antisense transcription. Nat Rev Genet 14 : 880–893. doi: 10.1038/nrg3594 24217315
67. Camblong J, Beyrouthy N, Guffanti E, Schlaepfer G, Steinmetz LM, et al. (2009) Trans-acting antisense RNAs mediate transcriptional gene cosuppression in S. cerevisiae. Genes Dev 23 : 1534–1545. doi: 10.1101/gad.522509 19571181
68. Houseley J, Rubbi L, Grunstein M, Tollervey D, Vogelauer M (2008) A ncRNA modulates histone modification and mRNA induction in the yeast GAL gene cluster. Mol Cell 32 : 685–695. doi: 10.1016/j.molcel.2008.09.027 19061643
69. Uhler JP, Hertel C, Svejstrup JQ (2007) A role for noncoding transcription in activation of the yeast PHO5 gene. Proceedings of the National Academy of Sciences of the United States of America 104 : 8011–8016. 17470801
70. Cloutier SC, Wang S, Ma WK, Petell CJ, Tran EJ (2013) Long noncoding RNAs promote transcriptional poising of inducible genes. PLoS Biol 11: e1001715. doi: 10.1371/journal.pbio.1001715 24260025
71. Terzi N, Churchman LS, Vasiljeva L, Weissman J, Buratowski S (2011) H3K4 trimethylation by Set1 promotes efficient termination by the Nrd1-Nab3-Sen1 pathway. Mol Cell Biol 31 : 3569–3583. doi: 10.1128/MCB.05590-11 21709022
72. Yassour M, Pfiffner J, Levin JZ, Adiconis X, Gnirke A, et al. (2010) Strand-specific RNA sequencing reveals extensive regulated long antisense transcripts that are conserved across yeast species. Genome Biol 11: R87. doi: 10.1186/gb-2010-11-8-r87 20796282
73. Lenstra TL, Tudek A, Clauder S, Xu Z, Pachis ST, et al. (2013) The role of Ctk1 kinase in termination of small non-coding RNAs. PLoS One 8: e80495. doi: 10.1371/journal.pone.0080495 24324601
74. Mayer A, Lidschreiber M, Siebert M, Leike K, Soding J, et al. (2010) Uniform transitions of the general RNA polymerase II transcription complex. Nat Struct Mol Biol 17 : 1272–1278. doi: 10.1038/nsmb.1903 20818391
75. Akhtar MS, Heidemann M, Tietjen JR, Zhang DW, Chapman RD, et al. (2009) TFIIH kinase places bivalent marks on the carboxy-terminal domain of RNA polymerase II. Mol Cell 34 : 387–393. doi: 10.1016/j.molcel.2009.04.016 19450536
76. Rondon AG, Mischo HE, Kawauchi J, Proudfoot NJ (2009) Fail-safe transcriptional termination for protein-coding genes in S. cerevisiae. Molecular cell 36 : 88–98. doi: 10.1016/j.molcel.2009.07.028 19818712
77. Ghazal G, Gagnon J, Jacques P-E, Landry J-R, Robert F, et al. (2009) Yeast RNase III Triggers Polyadenylation-dependent Transcription Termination. Molecular Cell 36 : 99–109. doi: 10.1016/j.molcel.2009.07.029 19818713
78. Loya TJ, O’Rourke TW, Reines D (2013) Yeast Nab3 protein contains a self-assembly domain found in human heterogeneous nuclear ribonucleoprotein-C (hnRNP-C) that is necessary for transcription termination. J Biol Chem 288 : 2111–2117. doi: 10.1074/jbc.M112.430678 23192344
79. Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, et al. (1999) Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285 : 901–906. 10436161
80. Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, et al. (2001) The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24 : 218–229. 11403571
81. Homer NM Barry; Nelson Stanley F. (2009) BFAST: An Alignment Tool for Large Scale Genome Resequencing. PLOS ONE 4: e7767. doi: 10.1371/journal.pone.0007767 19907642
82. Breese MR, Liu Y (2013) NGSUtils: a software suite for analyzing and manipulating next-generation sequencing datasets. Bioinformatics 29 : 494–496. doi: 10.1093/bioinformatics/bts731 23314324
83. Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26 : 139–140. doi: 10.1093/bioinformatics/btp616 19910308
84. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, et al. (2011) Integrative genomics viewer. Nat Biotechnol 29 : 24–26. doi: 10.1038/nbt.1754 21221095
85. Thorvaldsdottir H, Robinson JT, Mesirov JP (2013) Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14 : 178–192. doi: 10.1093/bib/bbs017 22517427
86. Beissbarth T, Speed TP (2004) GOstat: find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics 20 : 1464–1465. 14962934
87. Mosley AL, Sardiu ME, Pattenden SG, Workman JL, Florens L, et al. (2011) Highly reproducible label free quantitative proteomic analysis of RNA polymerase complexes. Mol Cell Proteomics 10: M110 000687. doi: 10.1074/mcp.M110.000687 21048197
88. Huang da W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4 : 44–57. doi: 10.1038/nprot.2008.211 19131956
89. Schwalb B, Schulz D, Sun M, Zacher B, Dumcke S, et al. (2012) Measurement of genome-wide RNA synthesis and decay rates with Dynamic Transcriptome Analysis (DTA). Bioinformatics 28 : 884–885. doi: 10.1093/bioinformatics/bts052 22285829
90. Sun M, Schwalb B, Schulz D, Pirkl N, Etzold S, et al. (2012) Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res 22 : 1350–1359. doi: 10.1101/gr.130161.111 22466169
91. Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. doi: 10.1186/gb-2009-10-3-r25 19261174
92. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25 : 2078–2079. doi: 10.1093/bioinformatics/btp352 19505943
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 2
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Genomic Selection and Association Mapping in Rice (): Effect of Trait Genetic Architecture, Training Population Composition, Marker Number and Statistical Model on Accuracy of Rice Genomic Selection in Elite, Tropical Rice Breeding Lines
- Discovery of Transcription Factors and Regulatory Regions Driving Tumor Development by ATAC-seq and FAIRE-seq Open Chromatin Profiling
- Evolutionary Signatures amongst Disease Genes Permit Novel Methods for Gene Prioritization and Construction of Informative Gene-Based Networks
- Proteotoxic Stress Induces Phosphorylation of p62/SQSTM1 by ULK1 to Regulate Selective Autophagic Clearance of Protein Aggregates
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy