
A Nitric Oxide Regulated Small RNA Controls Expression of Genes Involved in Redox Homeostasis in
Bacteria have evolved various strategies to continually monitor the redox state of the internal and external environments to prevent cell damage and/or to protect them from host defense mechanisms. These signals modify the expression of genes, allowing bacteria to adapt to altered redox environments and to maintain homeostasis. Studies in Enterobacteriaceae have shown that sRNAs play central roles in adaptation to oxidative stress. We show here that the conserved sRNA, RoxS is induced by the presence of nitric oxide (NO) in the medium, through the ResDE and SrrAB two-component systems of Bacillus subtilis and Staphylococcus aureus, respectively. B. subtilis RoxS regulates functions related to oxidation-reduction reactions and acts as an antisense RNA to control translation initiation and the degradation of ppnKB mRNA, encoding an NAD+/NADH kinase. Interestingly, RNase Y processes the 5′ end of the RoxS sRNA leading to a truncated sRNA that in turn interacts more efficiently with a second target, the sucCD mRNA, encoding succinyl-CoA synthase. Taken together this work shows that RoxS is part of a complex regulatory network that allows the cell to sense and respond to redox perturbations, and revealed a novel process that allows an expansion of the repertoire of sRNA targets.
Published in the journal:
. PLoS Genet 11(2): e32767. doi:10.1371/journal.pgen.1004957
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004957
Summary
Bacteria have evolved various strategies to continually monitor the redox state of the internal and external environments to prevent cell damage and/or to protect them from host defense mechanisms. These signals modify the expression of genes, allowing bacteria to adapt to altered redox environments and to maintain homeostasis. Studies in Enterobacteriaceae have shown that sRNAs play central roles in adaptation to oxidative stress. We show here that the conserved sRNA, RoxS is induced by the presence of nitric oxide (NO) in the medium, through the ResDE and SrrAB two-component systems of Bacillus subtilis and Staphylococcus aureus, respectively. B. subtilis RoxS regulates functions related to oxidation-reduction reactions and acts as an antisense RNA to control translation initiation and the degradation of ppnKB mRNA, encoding an NAD+/NADH kinase. Interestingly, RNase Y processes the 5′ end of the RoxS sRNA leading to a truncated sRNA that in turn interacts more efficiently with a second target, the sucCD mRNA, encoding succinyl-CoA synthase. Taken together this work shows that RoxS is part of a complex regulatory network that allows the cell to sense and respond to redox perturbations, and revealed a novel process that allows an expansion of the repertoire of sRNA targets.
Introduction
Small regulatory RNAs (sRNA) have been shown to play key roles in the regulation of a wide variety of cellular processes in bacteria, including stress responses, environmental signaling and virulence [1,2]. They generally regulate at the post-transcriptional level by altering mRNA translation or stability. Most sRNAs identified to date base pair with the 5’ untranslated region (5’-UTR) and alter ribosome binding to the mRNA. Changes in translation rates often have indirect consequences for mRNA stability as ribosomes can shield mRNA from attack by ribonucleases. A number of sRNAs have also been shown to directly affect mRNA stability without altering translation initiation rates through interactions with the 5’-UTR, the 3’-UTR or the coding sequence [3,4,5,6].
Although bacterial sRNAs have been studied most extensively in Escherichia coli and closely related organisms, the link to virulence has led to the identification and characterization of sRNAs in a wide range of both Gram-negative and Gram-positive bacterial pathogens. The Gram-positive model organism Bacillus subtilis trails conspicuously behind in these efforts, where only two trans-acting sRNAs, SR1 and FsrA, have been studied in detail [7–11]. The RNA chaperone Hfq has been shown to play a key role in sRNA association with its mRNA target in Proteobacteria. However, its role in Firmicutes seems to be less evident [7,8,12–14], suggesting that alternative RNA chaperones remain to be discovered in these organisms. Furthermore, there are important differences in the mRNA degradation machineries and pathways of these two bacterial clades, most notably the widespread occurrence of a 5’-3’ exoribonuclease activity provided by RNase J in the Firmicutes and the ability of stalled ribosomes to protect long stretches of downstream mRNA from ribonucleolytic attack [15,16]. The RNases involved in the regulation of mRNA stability by sRNAs in the Firmicutes have not been identified in many cases.
RsaE was first discovered in Staphylococcus aureus as a member of a family of sRNAs that contain multiple C-rich regions (CRR) that can potentially pair with the G-rich Shine Dalgarno (SD) sequences of ribosome binding sites to inhibit translation [17]. RsaE shows some strain-dependence in its expression patterns [17,18], but in all tested clinical isolates expression of RsaE was maximal during mid-exponential growth and declined in late-exponential/pre-stationary phase [19]. Expression of RsaE in S. aureus strain RN6390 was activated by the agr quorum sensing system that plays a key role in S. aureus virulence [17] and was further shown to be induced by both oxidative stress and high salt conditions [17,18]. Transcriptome and proteome analysis of RsaE deletion strains or overexpressing strains pointed to a role for S. aureus RsaE in governing the expression of genes involved in central metabolism, notably folate metabolism and the TCA cycle [17,18].
RsaE is highly conserved between Bacillus and Staphylococcus species at both the primary sequence and predicted secondary structure level [17] (Fig. 1A). The two best-studied representatives of these groups, B. subtilis and S. aureus, occupy very different ecological niches, the soil and the mammalian skin and respiratory tract, respectively. In these environments, both organisms frequently encounter nitric oxide (NO), a key signaling molecule in both bacteria and eukaryotes (for review, see [20]). Indeed NO, which is toxic at high doses through the production of reactive nitrogen species (RNS), is produced primarily by denitrifying bacteria in the soil and by macrophages in the mammalian host, but some species, notably Bacilli, Staphylococci and Streptomyces, can also synthesize NO via bacterial NO synthases (bNOS) [21]. NO has been shown in a number of bacteria to provide protection from oxidative stress, provoked either by peroxide [22,23,24] or antibiotics [25,26]. The beneficial effects of NO can also be shared between bacteria and their hosts; NO produced by B. subtilis in the intestine of C. elegans has been shown to increase the lifespan of the nematode [27]. Despite its importance as both a signaling and potentially stress-inducing molecule, no bacterial sRNA that responds to NO levels has been identified to date. Given that B. subtilis is a non-pathogenic organism that occupies a very different niche to S. aureus, we were curious as to the physiological role and the targets of this sRNA in B. subtilis. We found that expression of RsaE, which we have renamed RoxS in B. subtilis for related to oxidative stress, is induced by NO in both B. subtilis and S. aureus. Despite their similarity in sequence and regulation in the two organisms, the genes affected by deletion of this sRNA are mostly different. Our data illustrate how the functions of a highly conserved sRNA have evolved in distantly related bacteria.

Results
RoxS expression is induced by the response regulator ResD in response to increased NO levels in B. subtilis
The chromosomal context of the S. aureus rsaE and B. subtilis roxS genes is very similar and, interestingly, many of the genes have functions related to redox homeostasis or show increased expression under conditions of diamide or peroxide-induced oxidative stress in B. subtilis (S1 Fig.) [28]. An alignment of the homologous roxS/rsaE genes from several Bacilli and Staphylococci showed significant sequence conservation in the promoter region (S2 Fig.). An examination of a conserved 8-nucleotide (nt) sequence around position −65 suggested that ResD, the response regulator of the two-component system (TCS) ResDE, that is sensitive to both O2 and NO levels [29,30], might recognize this promoter region. Indeed, the sequence upstream of the roxS promoter is highly similar to the validated ResD binding site found upstream of the yclJ gene [31]. We therefore tested whether the expression of RoxS was altered in a mutant lacking the ResDE TCS. In mid-log phase, a ResDE deletion strain showed a three to four-fold decrease in RoxS expression and this effect was complemented by a plasmid expressing ResDE under IPTG control (Fig. 1B), indicating that ResD is an activator of RoxS transcription. The effect of the ResDE deletion was even stronger as the cells progressed towards stationary phase, confirming its importance as a regulator of RoxS expression (Fig. 1C). In agreement with our data, a recent chromatin immunoprecipitation study has shown a ResD binding at this location of the B. subtilis chromosome [32]. In contrast, the thiol specific oxidative stress regulator Spx, also shown to bind in this region [33], had little effect on RoxS expression under the conditions tested.
The membrane-bound ResE sensor kinase responds to either decreased dissolved O2 or increased NO levels [34] by a mechanism that is still not completely understood. It then activates the ResD response regulator through phosphorylation. We tested the effect of NO on RoxS expression by adding spermine NONOate to growing cultures. Spermine NONOate dissolves at neutral pH with a half-life of about 39 mins to produce NO. Expression of RoxS decreased slightly before increasing to a peak 30 mins to 1 h after addition of spermine NONOate (Fig. 2A). Although RoxS expression also decreased slightly upon addition of spermine NONOate to the ΔresDE mutant strain, no significant increase was observed after 1 h of incubation. Thus, the NO-dependent induction of RoxS expression depends on the ResDE TCS.

RsaE expression is also NO and ResDE/SrrAB-dependent in S. aureus
Given the strong conservation of the predicted ResD binding site upstream of RsaE in Staphylococci, we asked whether expression of RsaE was subjected to a similar regulation in S. aureus. The S. aureus homolog of the ResDE is called SrrAB and this TCS is also known to respond to low O2 levels and NO [30]. The effect of NO on RsaE expression was also tested by adding spermine NONOate under identical conditions to those described for B. subtilis. As for B. subtilis RoxS, we observed a weak but significant increase in RsaE expression about 1 h after the addition of spermine-NONOate to the growth medium in the wild-type (WT) strain (Fig. 2B). As anticipated, expression of RsaE was significantly lower in steady state conditions in the srrAB mutant, suggesting that SrrA is an activator of RsaE transcription. Furthermore, expression of RsaE was no longer induced by the addition of spermine-NONOate in this strain, clearly showing that the induction of RsaE by NO is dependent on the SrrAB TCS. Therefore, the signaling pathway and the expression of S. aureus RsaE and B. subtilis RoxS have been maintained during evolution.
B. subtilis strains lacking RoxS show increased expression of genes with redox-related functions
To get insight into the regulatory role(s) of RoxS in B. subtilis, we performed both proteome and transcriptome analysis in strains lacking RoxS. We detected 1092 proteins in whole cell extracts by LC-MS/MS and identified 63 proteins with significantly increased levels in the ΔroxS strain compared to the WT strain (≥1.5-fold increase by two independent methods of analysis: Spectral Counting and MS1 Filtering; S1 Table). No proteins showed significantly decreased expression in the ΔroxS strain relative to WT. Nineteen of the up-regulated candidates (30%) had functions related to oxidation-reduction processes (Fig. 3), a significant enrichment from the 8.5% of B. subtilis genes associated with this Gene Ontology (GO) term (GO:0055114) genome-wide.

A number of the candidates showing increased expression in the ΔroxS strain (and therefore down-regulated by RoxS) are predicted to be involved in oxidative stress protection. These include the putative peroxiredoxins Tpx, AhpC and YgaF, and the thioredoxin-like protein YdbP. We also observed increased expression of the peptide methionine sulfoxide reductase MsrA, the DNA-protecting ferritin Dps and two heme-degrading mono-oxygenases HmoA and HmoB, all of which have been shown involved in resistance to oxidative stress in different Bacilli [35–37]. The increased expression of these proteins suggests that cells lacking RoxS are either experiencing, or behave as if they are experiencing oxidative stress, in conditions that are not normally stressful for WT cells.
Eleven of the proteins showing increased expression in the ΔroxS strain use prosthetic groups (NAD, FAD, FMN, heme, iron-sulfur clusters) for their oxido-reduction/electron transfer reactions (Fig. 3 and S1 Table). These include the short-chain flavodoxins YkuN and YkuP. YkuN has been shown to be capable of transporting electrons to B. subtilis nitric oxide synthase (bsNOS) to generate NO from arginine [38]. Interestingly, five of the proteins showing increased levels in the RoxS deletion mutant were members of the ferric uptake regulator (Fur) regulon, YkuN, YkuP, HmoA, YcgT and FeuA (iron hydroxamate binding lipoprotein), and four were members of the general stress sigma B (SigB) regulon, YdbP, Dps, YtkL (a predicted metal hydrolase) and SigB itself. One of the proteins that showed the greatest increase in expression levels was PpnKB, an inorganic polyphosphate/ATP-NAD kinase that converts NAD+ to NADP+. Although this enzyme is not directly involved in a redox reaction, it does have an influence on the cell’s levels of reducing power through the production of NADPH. The ppnkB mRNA is predicted to be a direct target for RoxS repression (see below).
The transcriptome analysis was performed using tiling arrays with 22 nt resolution as described previously [39]. A comparison of the RoxS deletion strain to the WT parental strain showed 46 mRNAs with increased expression levels and 48 with decreased synthesis (≥2-fold; q-value <0.05; S2 Table). Most (28/48) of the genes with decreased expression levels in the deletion strain were from the PBSX prophage, including all 12 members of the sigma factor Xpf regulon. Nine of the genes with augmented expression in the ΔroxS strain were members of the Fur regulon, consistent with the proteome data, although some of the genes concerned were different (Fig. 3). They include the yxeB and yusV genes, involved in the acquisition of iron, the dhbABCE operon involved in siderophore biosynthesis and the flavodoxin-encoding ykuNOP operon. YkuN and YkuP were among six candidates also identified in the proteome analysis, the others being Tpx, LplJ (lipoate protein ligase), YhfE (putative endogluconase) and YktB (unknown). We confirmed the increase in ykuNOP expression in the ΔroxS strain by Northern blot (S3A Fig.) although we suspect it may be an indirect consequence of the roxS deletion on Fur activity (see below). Furthermore, genes encoding a thioredoxin (resA) and the putative peroxiredoxin (tpx) also showed increased expression in the absence of RoxS. The resA gene encodes an extracytoplasmic thioredoxin involved in the maturation of cytochrome C, while the function of Tpx is still unknown. Two genes involved in the pentose phosphate pathway, tkt, encoding transketolase and gndA, encoding 6-phospho-gluconate dehydrogenase, also showed increased transcript levels. The pentose phosphate pathway is a major source of NADPH production in the cell for use as a reducing agent in anabolic reactions such as lipid and nucleic acid synthesis. Overall, fourteen of the up-regulated genes in the tiling array (30%) had annotated functions related to oxidation-reduction reactions (GO term 0055114), consistent with the functional enrichment seen in the proteome study (Fig. 3 and S2 Table). These data are in good agreement with a general role for RoxS in the redox state/oxidative stress response.
RoxS inhibits initiation of ppnKB mRNA translation
Because most sRNAs base-pair to mRNA targets, we used several programs (TargetRNA2 [40], CopraRNA [41], RNApredator [42]) to predict potential direct mRNA targets of RoxS, with a particular focus on the translation initiation region. The targets suggested by CopraRNA were highly enriched for mRNAs involved in a relatively small number of cellular processes including electron transport, respiration, lipid metabolism and metal binding (S4 Fig.). The best target proposed by TargetRNA2 and RNA predator, the ppnKB mRNA (Fig. 4A), was consistent with this functional enrichment. Furthermore, the synthesis of the PpnKB protein was 4.5 to 9-fold increased in the ΔroxS strain by Spectral Counting and MS1 Filtering, respectively (Fig. 3). We therefore chose to study the RoxS-dependent regulation of ppnkB in more detail.

To determine whether RoxS could directly bind to the ppnKB mRNA to inhibit translation initiation, we tested the effect of RoxS on the formation of the ribosomal initiation complex on the ppnKB mRNA by toeprinting assays. Addition of 30S ribosomal subunits and initiator tRNA to the ppnKB transcript, showed a clear toeprint at position +16 relative to the ppnKB start codon (Fig. 4B; lane 5). Incubation of the ppnKB transcript with equimolar and higher concentrations of RoxS resulted in complete inhibition of the 30S ribosome toeprint, while a band specific to the binding of RoxS appeared at position −9/10 (Fig. 4B; lane 6 and 7). In contrast, RoxS had a much weaker effect on the formation of the initiation complex on the ykuN transcript (S3B Fig.) and did not show evidence for a stable interaction around the SD sequence, consistent with the fact that, despite the presence of four consecutive G-residues in the SD, it was not predicted as a target by any of the three algorithms (including the ORFs, for TargetRNA2). This experiment shows that RoxS specifically binds to the ppnKB mRNA and forms a stable complex that is sufficient to prevent the formation of the ternary translation initiation complex. The toeprinting assays, coupled with the fact that RoxS is not predicted to make significant interactions with any portion of the ykuNOP mRNA, suggest that RoxS-dependent effect on the expression this operon, observed in both the transcriptome and proteome analysis, most likely results from an indirect effect.
C-rich region 3 is key for RoxS binding to the ppnKB mRNA and inhibiting translation initiation complex formation
The base-pairing interaction between RoxS and ppnKB predicted by TargetRNA is extensive (Fig. 4A) and includes the first three C-rich regions (CRR1-3). However, the strong reverse transcriptase (RT) stop at nt −9/10 provoked by duplex formation is close to the SD sequence, suggesting the most stable interaction is between CCR3 and the ppnKB ribosome binding site. However, the six nts downstream of CRR1 are identical to those downstream of CRR3, creating a 10 nt duplication (CCCCUUUGUU) in RoxS and leaving open the possibility that the two sequences were functionally redundant. We therefore performed toeprinting assays with RoxS variants where the four consecutive C-residues of CRR1 or CRR3, or both, were changed to A. These mutations are not predicted to alter the secondary structure of RoxS.
The data clearly showed that mutation of CRR3 alone abolished the ability of RoxS to bind the mRNA and to inhibit 30S ribosome binding to ppnKB (Fig. 4B, lanes 14–15). Conversely, mutation of CRR1 alone had no effect (Fig. 4B, lanes 10–11). RoxS mutants lacking both CRR1 and CRR3, or the three CRR’s 1, 2 and 3, behaved similarly to the CRR3 mutant in failing to interact with the ppnKB mRNA or to inhibit translation initiation complex formation (Fig. 4B, lanes 18–19 and 22–23). Hence, these data show that CRR3 plays the most important role in inhibition of translation initiation and that the two repeat motifs of RoxS are not functionally equivalent for the regulation of ppnKB.
RoxS overexpression leads to degradation of the ppnKB mRNA
Binding of sRNAs can have direct effects on target mRNA stability, by creating new sites for endoribonuclease cleavage, or indirect effects through the increased exposure of existing cleavage sites following translational repression. We therefore asked whether overexpression of RoxS would lead to degradation of the ppnKB mRNA. For these studies, we used the ΔroxS mutant strain transformed with a plasmid expressing RoxS from a tetracycline-dependent promoter (strain CCB498: ΔroxS + pDG-Ptet-roxS) or with a control plasmid (strain CCB505: ΔroxS + pDG-Ptet). Induction of RoxS expression with increasing concentrations of anhydrotetracylcine (aTc) in strain CCB498 caused a gradual reduction (about two-fold) in ppnKB mRNA levels compared to the empty vector control strain (Fig. 5), showing that RoxS affects the amount of ppnKB mRNA in the cell, in addition to controlling its translation.

Expression of RoxS from the pDG-Ptet vector is transient, reaching a peak about 5 mins after addition of aTc before decreasing rapidly (Fig. 6A, D), presumably due to an accumulation of the TetR repressor driven by the same promoter. We exploited this property of the plasmid to analyze whether ppnKB mRNA levels would recover upon shut-down of RoxS expression. Indeed, ppnKB mRNA levels fell to a minimum about 5 mins after induction of RoxS and were rapidly restored as RoxS levels decreased (Fig. 6A, D). The RoxS-dependent reduction in ppnKB levels was only slightly less efficient in a strain lacking the double-strand specific endonuclease RNase III, encoded by the rnc gene (Fig. 6B, D). However, it was significantly reduced in a strain lacking the single-strand specific nuclease RNase Y, encoded by rny (Figs. 6C, D). These results suggest that RNase Y is a key enzyme for RoxS-mediated ppnKB mRNA turnover, while RNase III plays a secondary role under these experimental conditions. It should be noted that RoxS is slow to shut-off in the rny mutant (Fig. 6C, D); we will see later that this is due to an effect of RNase Y on RoxS RNA stability.

To further show that RoxS controls ppnKB expression at the level of mRNA stability, we measured the half-life of the ppnKB mRNA in WT strains and mutant strains lacking either RNase III or RNase Y under steady state conditions (Fig. 7). The ppnKB mRNA was stabilized about 1.8-fold in cells lacking RoxS (8.4 vs. 15 mins half life, respectively in WT and ΔroxS strains), consistent with a role for RoxS in controlling ppnKB mRNA stability (Fig. 7A). In the absence of RNase III, a similar increase in ppnKB stability was seen, but was not further amplified by the additional deletion of roxS (Fig. 7B). The simplest explanation is that RNase III and RoxS collaborate to degrade a portion of ppnKB transcripts; the lack of either component, or both, leading to a similar increase in ppnKB mRNA stability.

The ppnKB mRNA was also significantly stabilized (8.4 vs. 24 mins) in the Δrny mutant compared to the WT strain (Fig. 7C), consistent with a role for RNase Y initiating the degradation of the ppnKB mRNA. In this case, however, further deletion of roxS had an additional stabilizing effect (24 vs. >40 mins half life, respectively). This suggests the existence of a RoxS-mediated ppnKB turnover pathway that is independent of RNase Y and that inactivation of both pathways are required for maximal stabilization of ppnKB. We propose that the second pathway is the RoxS/RNase III dependent pathway described above. The data also indicate an effect of RNase Y that is independent of RoxS (15 mins vs. >40 mins half-life, respectively, in ΔroxS vs. Δrny ΔroxS strains), consistent with a role for RNase Y in the non-regulated turnover of the ppnKB mRNA.
Interestingly, the ppnKB mRNA was highly unstable in a strain lacking the 5′-3′ exoribonuclease RNase J1 (Fig. 7D) and this destabilization was attenuated upon deleting RoxS (3.1 vs. 9.4 mins half-life, respectively, in ΔrnjA vs. ΔrnjA ΔroxS strains). Data presented in the next section will shed light on this phenomenon. Globally, our data provide an illustration of the complex interplay between ribonucleases involved in the turnover of the ppnKB mRNA, both dependent and independent of RoxS-mediated repression.
Evidence for two pathways of RoxS turnover
We also analyzed the importance of the three main ribonucleases in the degradation of the RoxS sRNA. The half-life of the chromosomal copy of RoxS was first measured in WT cells and in cells lacking either RNase III, RNase Y or RNase J1. In WT cells and in cells lacking RNase III, RoxS showed bi-phasic RNA degradation upon transcription arrest with rifampicin (Fig. 8A, C), suggesting that two populations of this sRNA exist in vivo. The simplest interpretation is that these populations represent free RoxS or RoxS bound to its targets. While the half-life of the rapidly decaying population was similar in both strains, the slowly decaying population was strongly stabilized in the Δrnc mutant (Fig. 8C). Because of its specificity for double-stranded RNA, it is most likely that the stabilisation of the slowly decaying phase represents stabilisation of RoxS molecules that are hybridized to its mRNA targets. In cells lacking RNase J1 (ΔrnjA), full length RoxS was stabilized compared to the WT strain, but in addition a very long-lived degradation/processing intermediate was detected (Fig. 8B, C). This intermediate was not detected in the absence of RNase Y and the full-length RoxS had a much longer half-life in the Δrny mutant strain (Fig. 8B, C). Using primer extension, we mapped the 5’ end of the short RoxS fragment to nt +20 of RoxS (S5 Fig.). Together these results suggest that RNase Y initiates RoxS turnover by cleaving around nt +20 (Fig. 1) and RNase J1 degrades the downstream cleavage product, in addition to having some activity on the full-length RNA.

Because a small amount of RNase Y-cleaved RoxS was visible in WT cells (Fig. 8A), we asked whether this truncated form was functional and might contribute to regulation. We cloned a 5′ truncated version of RoxS beginning at nt 20, called RoxS(Y), into the plasmid vector pDG-Ptet. In a manner similar to full-length RoxS, aTc induction of RoxS(Y) resulted in a rapid and efficient reduction in ppnKB levels, which then recovered as RoxS(Y) levels fell (S6A-S6B Fig.). Thus the truncated form of RoxS that accumulates in an RNase J1 mutant is fully functional and may explain the RoxS-dependent destabilization of ppnKB in the absence of RNase J1 (Fig. 7D). When tested in the toeprinting assay, the truncated RoxS species formed a more extensive hybrid than the full-length sRNA with the ppnKB mRNA, indicated by additional reverse transcriptase stops around nt −2 and nt +23 (Fig. 4B, lane 24). The short form was equally efficient as the full-length RoxS in inhibiting ppnKB translation initiation complex formation at the concentrations tested.
RoxS forms a duplex with the ribosome binding site of ppnKB mRNA and creates RNase III cleavage site
To characterize the interactions between ppnKB and RoxS and its various mutant forms, we performed structure probing experiments on the ppnKB mRNA using the double-strand-specific enzymes RNase V1 and RNase III, and RNase T1, which cleaves principally 3′ to unpaired guanines (Fig. 9 and S7 Fig.). The data suggested that in the absence of RoxS, the ppnKB mRNA folds into a long, but relatively unstable hairpin structure that extends from nt −37 to nt +34 relative to the translation initiation site (Fig. 9A). Indeed, two major RNase T1 cleavages occur 3′ to G-3 and G-4 and two lesser cleavages 3′ to nts +2/+3 in the apical loop containing the AUG initiation codon while a number of RNase V1 cleavages are located in the irregular helix (Fig. 9A and S7A-S7B Fig.; lane 2). Consistent with this model, RNase III cleaves the large irregular helix of ppnKB at four sites (nts −24, +10, +22 and +32), with the cleavages at −24 and +22 producing the two-nt 3’ overhang characteristic of RNase III processing (Fig. 9A and 9D; lane 3).
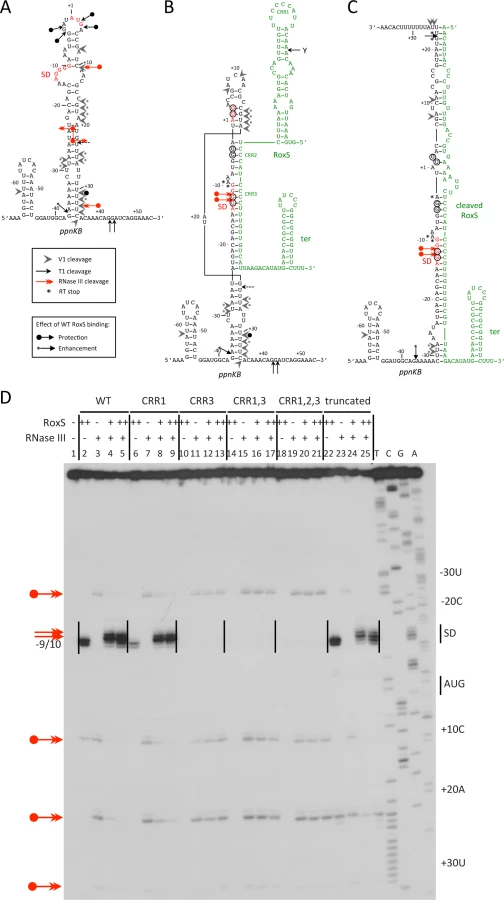
Binding of RoxS induces strong protection of the RNase T1 cleavages in the apical loop (G+3, G-3, G-4) and at G-12 and G-13 of the SD sequence while the cleavage at G-37 is slightly enhanced (Fig. 9B and S7A Fig.; lanes 3, 4). Concomitantly, RNase V1 cleavages are slightly enhanced at nts −29/30, +16 to +18, +24/25 and +31 (Fig. 9B and S7B Fig.; lanes 3, 4). Remarkably, all four RNase III cleavage sites are significantly reduced upon binding to RoxS while two strong adjacent cleavages appear in the ppnKB SD sequence at nts −12/13 (Fig. 9D; lanes 4, 5). These data suggest that the large hairpin loop of ppnkB undergoes a partial melting to promote basepairing interactions with RoxS, leading to the sequestration of the SD sequence. Identical changes in the RNase III cleavage patterns were observed if complex formation was performed with the truncated RoxS(Y) (Fig. 9C and 9D, lanes 22–25) or with the CRR1 mutant (Fig. 9D; lanes 6–9). However, RoxS derivatives with a mutation in CRR3 had no effect on the RNase III cleavages, showing that the mutated RNAs fail to interact with ppnKB (Fig. 9D). Identical conclusions were reached in the probing experiments with RNases T1 and V1 (S7 Fig.).
The proposed models for the interaction of ppnkB with full-length or truncated RoxS (Fig. 9B and C) take into account most of the data although we cannot completely distinguish between RoxS-dependent changes that are due to the formation of an extended RoxS/ppnKB duplex or due to a stabilization of existing ppnKB helices upon RoxS binding. However, the data unambiguously show that the CCR3 motif is responsible for the interaction with the SD sequence to prevent the formation of the translation initiation complex and to create a novel site for RNase III binding and cleavage.
The sucCD operon mRNA is a direct target of the cleaved form of RoxS
TargetRNA2 and CopraRNA both predicted the sucC gene, the first cistron of the sucCD operon encoding the two subunits of succinyl-coA synthase, as another potential target of RoxS (Fig. 10A). Differential proteomic analysis showed a 2-fold increased expression of SucD in the mutant ΔroxS strain, while SucC narrowly missed the dual 1.5-fold cut-off (1.4 fold increase by spectral counting; 1.8 fold increase by MS filtering) (Fig. 3, S1 Table). Interestingly, the sucCD mRNA was also shown to be a target of RsaE in S. aureus [17,18]. We therefore probed the membranes shown in Fig. 6 for the sucCD mRNA to see whether its mRNA levels were affected by RoxS expression. Transient expression of RoxS by aTc addition led to a similar decrease in sucCD expression as was observed for ppnKB (Fig. 10B, E). This decrease in expression was slightly attenuated in the absence of both RNase III (Fig. 10C, E) and RNase Y (Fig. 10D, E), suggesting roles for both of these enzymes in the turnover of the sucCD mRNA in response to RoxS expression.

We then analyzed the effect of RoxS and its variants on the formation of the translation initiation complex formed with the sucC mRNA using toeprinting assays. In contrast to the ppnKB mRNA, RoxS binding did not prevent the formation of the initiation complex on sucC, even at the highest RoxS concentration (Fig. 11, lanes 6 and 7). Only a weak RT pause characteristic of RoxS binding was observed at nt +4/5 relative to the first nt of the open reading frame, indicating that RoxS did not form a stable complex with sucC (Fig. 11, lane 4). To our surprise, the truncated form of RoxS(Y) bound far more efficiently than RoxS to the sucCD mRNA, causing a very strong RT pause at position −2/−3 and an increased signal at +4/5 (Fig. 11, lane 24). As a consequence, this led to a strong and efficient inhibition of the toeprint at +16 by the truncated form of RoxS (Fig. 11, lanes 26 and 27) comparable to that seen with ppnKB (Fig. 4, lanes 6 and 7). This experiment suggests that processing of RoxS is necessary for regulation of sucC and that truncation at the 5’ end of RoxS expands the repertoire of effective targets for this sRNA.

Discussion
The RoxS sRNA belongs to the ResD regulon and regulates the response to NO
In this paper, we have shown that expression of the regulatory RNA RoxS/RsaE is induced by nitric oxide in both B. subtilis and S. aureus, in a mechanism that is dependent on their respective orthologous two-component systems, ResDE and SrrAB. The membrane-bound sensor protein ResE/SrrB is autophosphorylated in response to both NO and limiting O2 levels, and in turn phosphorylates the response regulator ResD (SrrA in S. aureus) [31,43]. NO and hypoxia inhibit terminal oxidases and limit the flow of electrons through the electron transport chain. It was recently suggested that the resulting accumulation of reduced menaquinones in the membrane is likely to be the trigger that activates the ResDE/SrrAB TCS [30], similar to the quinone-sensitive ArcAB TCS in E. coli [44]. In S. aureus, SrrAB is important for cell survival in the host environment and in biofilms. This system senses and responds to both NO and hypoxia, and regulates genes required for cytochrome biosynthesis and assembly, anaerobic metabolism, iron-cluster repair, and NO detoxification [43]. In B. subtilis, ResD is known to activate the expression of about 30 genes involved in the anaerobic respiration of nitrate, the production of cytochromes, the fermentation of pyruvate and in NO detoxification [45].
Here, we have studied in more detail the regulatory functions of B. subtilis RoxS, which further expands the regulatory impact of ResD. Interestingly, regulation by ResD is significantly more efficient as growth begins to slow down and RoxS expression is essentially completely ResD-dependent in early stationary phase (Fig. 1C). Despite the fact that a number of the surrounding genes show increased expression in the presence of diamide, the thiol stress regulator Spx had little effect on RoxS expression. This is consistent with a recent study by Rochat et al. that showed an effect of an spx deletion on the neighboring genes but not on roxS itself [33]. However, a significant number of genes with functions related to oxidation-reduction reactions or oxidative stress resistance showed increased expression in B. subtilis cells lacking RoxS. This surprising result suggests that ΔroxS cells are suffering from a deficit of reducing power. The derepression of many members of the Fur regulon, including the ykuNOP operon, in the ΔroxS deletion strain is consistent with this observation. Since Fur uses reduced iron (Fe2+) as a co-repressor, a deficit in reducing power would be predicted to lead to decreased Fur repressor activity and increased expression of these genes.
We validated the direct role of RoxS in regulation of expression of ppnKB at both the level of translation initiation and mRNA turnover (Fig. 12). By converting NAD+ to NADP+ using ATP or inorganic polyphosphate as a phosphate donor, PpnKB shifts the metabolic balance from the production of energy from respiration, which uses primarily NADH, towards more anabolic reactions that use NADPH as a reducing agent. Expression of RoxS would therefore be expected to limit NADPH production. The increased expression of the gndA and tkt genes observed in the ΔroxS strain in the transcriptome experiment is consistent with this response. Tkt (transketolase) and GndA (6-phospho-gluconate dehydrogenase) are involved in the pentose phosphate pathway, a major source of NADPH synthesis in the cell. Furthermore, a recent paper has suggested that up to 40% of cellular levels NADPH come from folate metabolism in higher eukaryotes [46]. Interestingly, folate metabolism was a key target of RsaE in S. aureus [17,18]. We therefore suggest that one of the physiological roles of RoxS in both B. subtilis and S. aureus is to turn down expression of genes no longer required under conditions where electron transport is limited (e.g. NO stress or hypoxia), the intracellular environment is reduced and less NADPH is required. Conversely, the increased expression of ppnKB, gndA and tkt genes observed in the RoxS deletion strain is a consistent response for a cell in need of reducing power.

A direct comparison of the genes affected by the RoxS/RsaE deletion in B. subtilis and S. aureus showed only four genes in common, namely sucD, pgk, citZ and yjcG/SA0873 [17,18]. Thus, although the TCA cycle is a common target for this sRNA in both species, the majority of the genes affected are different. This may reflect fundamental differences in the regulation of the expression of RoxS or its targets in relation to their respective ecological niches and circumstances in which they encounter NO. Nevertheless, it provides an interesting example of how a similarly regulated sRNA has evolved to control different targets in two distantly related bacteria.
B. subtilis RoxS acts in concert with ribonucleases to regulate mRNA targets.
The toeprinting analysis showed that RoxS forms a complex with the ppnKB mRNA sufficiently stable to prevent ribosome initiation complex formation. Structure probing and reverse transcription stops at nts −9/10 of ppnKB suggested that the primary ppnKB/RoxS interaction occurs via CRR3, although we expected more extensive intermolecular pairings. However, it is likely that the 5′ stem does not easily unwind in full-length RoxS. In this regard, it is of interest that additional RT stops were seen with the truncated form of RoxS around nts −2 and +23, coherent with interactions that extend all the way to the 5′ end of the cleaved sRNA. This can be explained by the additional nucleotides in the 5′ stem that become available for base-pairing with ppnKB upon cleavage of the RoxS sRNA. Intriguingly, this cleavage event is also the first step in RoxS turnover. Although this processed/partially degraded form of RoxS is just as functional as the full-length species in promoting ppnKB mRNA turnover in vivo, cleavage is not necessary for inhibition of translation initiation complex formation on the ppnKB transcript in vitro. In contrast, we showed that processing of RoxS is necessary to prevent ribosome binding to the sucCD mRNA in vitro. Prediction of basepairing interactions suggest that CCR3 binds to the SD sequence in agreement with the toeprinting assays while a second stretch of nts located in the early coding region of sucC is complementary to CCR1 of RoxS. We propose that the key to the stabilization of the interaction between RoxS(Y) and sucCD is the opening of the 5’ stem of RoxS to favor basepairing with the early coding region of sucC (Fig. 10A). Although full-length RoxS is unable to bind efficiently to the sucCD mRNA in vitro, a specific RoxS-dependent degradation of sucCD mRNA was observed in vivo (Fig. 10) suggesting that a trans-acting factor, such as an RNA chaperone, might assist RoxS in the regulatory mechanism. Alternatively, sufficient quantities of the RNase Y cleaved form of RoxS may be generated in vivo for regulation of sucCD. If it were rapidly co-degraded with its target, this might explain why large quantities of the truncated form do not accumulate in vivo. The predicted RNase Y cleavage site in RoxS is in a region of secondary structure, unusual for this endonuclease with a preference for single-stranded RNA [47]. It is therefore possible that RNase Y benefits from its proposed association with the DEAD box helicase CshA [48] to cleave at this site. A number of other sRNAs have been shown to be processed from larger transcripts, e.g. DicF and MicL in E. coli, ArcZ in E. coli and Salmonella enterica, MicX in Vibrio cholerae [49–53]. In most of these cases, the processed form is the major form found in the cell and is considered to be the active form of the molecule. In the case of ArcZ, it was shown that while the long form was a better binder of Hfq, only the RNase E-processed species could form a Hfq-dependent complex with its target rpoS [54]. In the case studied here, the unprocessed form of RoxS is the major form found in cells and likely to be the principal regulatory species. Cleavage of the sRNA potentially allows it to diversify and bind to other mRNA targets, essentially creating two functional sRNAs for the price of one.
Hfq has been shown to play an important role in sRNA regulation in the Proteobacteria, but only very few sRNAs have Hfq-dependent regulation in the Firmicutes. We nonetheless addressed the question of whether deletion of the hfq gene had an effect on RoxS-dependent down-regulation of the ppnKB mRNA, but did not observe any major impact of the hfq mutation (S8 Fig.). This result is tempered by the observation that, in the Proteobacteria, over-expression of sRNA has sometimes been shown to by-pass the need for Hfq [54].
Because RoxS binds to the SD sequence and inhibits the initiation complex formation in vitro, it clearly points to a role for RoxS in inhibiting ppnKB translation. In addition to this regulation, ppnKB is controlled at the level of mRNA turnover. First, RNase III can cleave the duplex between ppnKB and RoxS (Fig. 12). Cleavage in the SD sequence would immediately and irreversibly render the ppnKB mRNA translationally non-functional. A similar mechanism has been previously proposed in the regulation of the ompA and lamB mRNAs by the MicA sRNA in Salmonella [55] and the sodB mRNA by RyhB in E. coli [56]. In vitro, RNase III cleaved the large and irregular helix in the ppnKB mRNA in the absence of RoxS binding, albeit weakly. We therefore cannot rule out the involvement of RNase III in the turnover of ppnKB in the absence of RoxS in vivo; however, this relatively weak structure may not be able to compete efficiently with ribosome initiation in vivo. Our in vivo data suggest that RNase Y is also an important player in turnover of the ppnKB mRNA in both a RoxS-dependent (Fig. 6) and RoxS-independent (Fig. 7) manner. We propose that in the absence of RoxS, RNase Y can cleave the ppnKB mRNA occasionally between passing ribosomes (Fig. 12A). Upon inhibition of translation by RoxS, cleavage by RNase Y would be more efficient. Although the role of RNase E in sRNA regulation is now well-established in the Enterobacteria, the equivalent enzyme in Firmicutes, which in most cases do not have RNase E, was not known. Here we show for the first time that RNase Y plays an analogous role in Gram-positive sRNA regulation.
As two endonucleases cannot generally be rate limiting for the initiation of degradation on the same RNA, the fact that the ppnKB mRNA half-life increases in both an Δrnc and Δrny mutant suggests that at least two different populations of the ppnKB mRNA exist. The simplest interpretation of the data measured under equilibrium conditions in RoxS and RNase deletion strains (Fig. 7) is that RNase III cleaves the population of ppnKB bound to RoxS, while RNase Y cleaves the free ppnKB population. However, the transient high-level RoxS induction experiment gives a different picture. Indeed, this experiment suggests that RNase Y plays the major role in RoxS-mediated ppnKB turnover, while the RoxS-dependent role of RNase III is relatively minor (Fig. 6). The fundamental difference between these two experiments is the amount of RoxS in the cell. We therefore hypothesize that both RNase Y and RNase III can turnover the ppnKB mRNA in a RoxS-dependent manner, but that the equilibrium between the two pathways depends on the amount of RoxS in the cell in ways that we do not yet fully understand.
CRR1 and CCR3 are each contained within a 10-nucleotide (nt) sequence duplication CCCCUUUGUU that can potentially base pair with the ppnKB SD sequence. Surprisingly, mutation of CRR1 had little effect on ribosome binding, while mutation of CRR3 essentially abolished it. Furthermore, the mutation of CRR1 and CRR3 together behaved similarly to the CRR3 mutant alone in both toeprinting assays and in the structure probing assays of the ppnKB/RoxS duplex, clearly showing the predominant role of CRR3 in ppnKB binding (Fig. 4, 9 and S7 Fig.). The structure of RoxS is most likely an important factor that explains why the two homologous sequences are not functionally redundant for ppnKB regulation. However, our data do not rule out a role for CRR1 in the regulation of other targets.
In addition to a whole family of sRNAs in S. aureus that contain CRRs [17], at least two other B. subtilis sRNAs, CsfG and FsrA [7,57], have CRRs in predicted single stranded regions. FsrA is a member of the Fur regulon. It contains three CRRs, two of which are separated by two nucleotides similar to the CRR2/CRR3 configuration in RoxS. CsfG also has three CRRs and, although it was characterized as an sRNA specific to sporulation in B. subtilis, recent evidence has suggested it may also be induced under conditions of oxidative stress [28]. These intriguing overlaps in both structure and regulation are suggestive of a functional redundancy and/or shared mechanisms of action between this class of sRNAs and their targets in Firmicutes.
Materials and Methods
Strains and constructs
Strains and oligonucleotides used are given in S3–S4 Tables, respectively. Strain CCB485 (roxS::kan) was constructed using a PCR fragment generated by re-amplifying (oligo pair CC1154/1159) three overlapping PCR fragments corresponding to upstream (oligo pair CC1154/CC1231), downstream (oligo pair CC1233/CC1159) regions of the roxS gene and the kanamycin resistance cassette (oligo pair CC1230/CC1232) from plasmid pDG780 [58]. The resulting PCR fragment was used to transform B. subtilis W168 (SSB1002)and the correct insertion verified by PCR and sequenced. Strain CCB282 (roxS::spc) was constructed by first constructing a plasmid where fragments corresponding to regions upstream (oligo pair RsaE-up-BamHI/RsaE-low-HindIII) and downstream (oligo pair RsaE-up-XhoI/RsaE-low-ApaI) of the roxS gene were amplified by PCR, cut with BamHI/HindIII and XhoI/ApaI, respectively and cloned on either side of the spectinomycin resistance cassette in plasmid pBS-Spc [59] cut with the same enzymes. The resulting plasmid was used to transform B. subtilis 168 (CCB281) and the correct insertion verified by PCR.
Plasmid pDG-ResDE was made by amplifying the resDE gene by PCR using oligo pair CC1222/1223, cutting with SphI and SalI and inserting between the same sites of pDG148 [58].
Plasmid pDG-Ptet-roxS was made from a previously made plasmid (pDG-Ptet-txpA/ratA-Plac) containing a XbaI-Pxyl/tet promoter-AatII-txpA/ratA-AatII-Plac-BstB1 construct cloned between the BstB1 and XbaI sites of pDG148 [58]. The roxS gene was amplified with oligos CC1216/1217 and inserted in the above plasmid cut with AatII and BstB1. The resulting plasmid pDG-Ptet-roxS permits the expression of roxS from its native +1 under the control of aTc. The empty vector control pDG-Ptet was made by inserting the hybridized oligos CC1328/1329 between the AatII and BstB1 sites of pDG-Ptet-txpA/ratA-Plac (above), creating a HindIII site.
Mutant derivatives of RoxS were created by PCR or overlapping PCR and cloning between the AatII and HindIII sites of pDG-Ptet. The four C’s of CRR1 were mutated to four A residues using oligo pair CC1323/1399. The four C’s of CRR3 were mutated to four A residues using overlapping oligo pairs CC1160/1327 and CC1326/1399 and reamplified with CC1160/1399. The CRR1 and CRR3 mutants were combined using oligos CC1323/1327 and CC1326/1399 and reamplified with oligos CC1323/1399. RoxS(Y) was made using oligo pair CC1398/1399. The CRR1,2,3 mutant was made by overlapping PCR using oligo pairs CC1323/1324 and CC1325/1399 and reamplified using oligos CC1323/1399. These plasmids were used for PCR amplification of templates for in vitro transcription of mutant RoxS RNAs.
RNA isolation and northern blots
RNA was isolated from mid-log phase B. subtilis cells growing 2×YT medium either by the glass beads/phenol method described in [60] or by the RNAsnap method described in [61]. Northern blots were performed as described previously [39]. S. aureus strains (HG001, HG001-ΔsrrAB) were grown to post-exponential phase by inoculating 50 ml of BHI medium with an overnight culture (1 : 100) at 37°C for 7 h. S. aureus total RNA was prepared according to the FastRNA Pro protocol (Qbiogene). After separation on agarose gels (1–2%) containing 20 mM guanidine thiocyanate, RNA was transferred onto Hybond-N+ membranes (Amersham). Detection of transcripts was done with DIG-labeled RNA according to the protocol provided by Roche.
Tiling array and data analysis
Strains CCB281 (wild-type) and CCB282 ΔroxS (S3 Table) were used for transcriptome analysis. Cells were grown in 2×YT medium to OD600 ∼0.6. 20 mL aliquots were centrifuged and RNA was prepared by the glass beads method [60], with an additional RQ DNase (Promega) step (0.01 units/μL, 37°C for 30 mins) after the second phenol extraction. RNA concentrations were measured and duplicate samples sent to Roche/Nimblegen for labeling and analysis on second-generation (T2) tiling arrays according to the BaSysBio protocol described in [28]. The data were quantile normalized and moderated t-test p-values were computed for the difference between the ΔroxS and WT genetic backgrounds (R package limma [62]). The p-values were converted to q-values to account for multiple testing via control of the False Discovery Rate (FDR) (R package fdrtool [63]). The cut-off value chosen for significant effects was ≥ 2, with a Q-value of ≤ 0.05.
Mass spectrometry based quantification: Spectral counting and MS1 label-free approaches
Strains CCB281 (WT) and CCB282 (ΔroxS) were used for comparative proteomic analysis. Crude extracts were obtained by trizol (Sigma) extraction, followed by acetone precipitation. Proteins (5 μg) were precipitated with 0.1 M ammonium acetate in 100% methanol, and then resuspended in 50 mM ammonium bicarbonate. After a reduction-alkylation step (5 mM DTT—10 mM iodoacetamide), proteins were digested overnight with 1/25 (W/W) of trypsin and only 3 μg of proteins was vacuum dried. The dried peptides were resuspended in 20 μL of water containing 0.1% formic acid (solvent A). The peptide mixtures were analyzed using a NanoLC-2DPlus system (with nanoFlex ChiP module; Eksigent, ABSciex, Concord, Ontario, Canada) coupled to a TripleTOF 5600 mass spectrometer (ABSciex) operating in positive mode. Each sample (5 μl; 750 ng) was loaded on a ChIP C-18 precolumn (300 μm ID × 5 mm ChromXP; Eksigent) at 2 μL/min in solvent A. After 10 min of desalting and concentration in the trap, the pre-column was switched online with the analytical ChIP C-18 analytical column (75 μm ID × 15 cm ChromXP; Eksigent) equilibrated in 95% solvent A and 5% solvent B (0.1% formic acid in acetonitrile). Peptides were eluted by using a 5%–40% gradient of solvent B for 120 min at a flow rate of 300 nL/min. The TripleTOF 5600 was operated in data-dependent acquisition mode (DDA) with Analyst software (v1.6, ABSciex). Survey MS scans were acquired during 250 ms in the 350–1250 m/z range. Up to 20 of the most intense multiply charged ions (2+ to 4+) were selected for Collision-Induced Dissociation (CID) fragmentation, if they exceeded a 150 counts per second intensity threshold. Ions were fragmented using a rolling collision energy script within a 60 ms accumulation time and an exclusion time of 15s. This so-called “Top20” method, with a constant cycle time of 1.5s, was set to high-sensitivity mode.
Data were searched against the B. subtilis strain 168 database from SwissProt (release 2013-11-20). The first algorithm used was Mascot (version 2.2, Matrix Science, London, UK) through the ProteinScape 3.1 package (Bruker). Variable peptide modifications allowed during the search were: N-acetyl (protein), carbamidomethylation (C) and oxidation (M). Mass tolerances in MS and MS/MS were set to 20 ppm and 0.5 Da, respectively. Two missed trypsin cleavages sites were allowed. Peptide identifications obtained from Mascot were validated with a peptide false discovery rate (FDR) of 1%. A Spectral Counting quantitative strategy was first carried on using the Mascot identification results and Proteinscape 3.1 package [64,65]. To normalize the data, a correction factor was applied for each condition according to the average total spectra number for all samples.
The Paragon algorithm (ProteinPilot package, AB Sciex) was then used to perform a second database search on the same nanoLC-MS/MS dataset and with the same decoy B. subtilis database. Proteins validated by Paragon at FDR 1% were submitted to a MS1 label-free quantification. For that purpose, only non-modified and unshared peptides were considered, as well as the Paragon identification confidence threshold set at 99%. Precursor ions fulfilling these criteria were transferred into PeakView package (v 2.0 with Protein Quantitation plug-in, AB Sciex) and their corresponding eXtracted Ion Chromatograms (XIC) were automatically integrated, using the following parameters: RT window ± 2 min, MS tolerance ± 0.05Da. To normalize and further process the MS1 label-free data, MarkerView software (v 1.2, ABSciex) was used: a correction factor was first applied for each condition according to the total area sum function. The statistical module from MarkerView finally allowed us to perform a Principle Component Analysis (PCA) and a Student t-test on the repeated triplicate experiments.
Primer extension assays
Primer extension assays were performed as described previously on total RNA [59].
Toeprinting assays
The RNA fragments were transcribed in vitro from PCR fragments containing the leader regions and coding sequences of ppnkB (oligos ppnKB T7 fw/ppnKB rev2), ykuN (oligos ykuN T7 fw/ykuN rev 2) and sucC (oligos sucC T7 fw/sucC rev2). RoxS and its mutant derivatives were made similarly using PCR fragments (oligos RsaE T7 fw/RsaE rev) amplified from their respective plasmids. The transcribed RNAs were purified by 8% polyacrylamide-8 M urea gel electrophoresis. After elution in 0.5 M ammonium acetate/1 mM EDTA buffer, the RNAs were precipitated twice with ethanol. The 5′ end-labeling of dephosphorylated DNA oligonucleotides was performed with T4 polynucleotide kinase and [γ-32P]ATP. Before use, RNAs were renatured by incubation at 90°C for 2 min in the absence of magnesium and salt, 1 min on ice, followed by an incubation step at 37°C for 15 mins in TMN buffer (20 mM Tris-acetate pH 7.5, 10 mM magnesium-acetate, 150 mM Na-acetate).
Toeprints were performed with E. coli 30S subunits. The preparation ribosomal subunits, the formation of a simplified translational initiation complex with mRNA, and the extension inhibition conditions were described according to Fechter et al. [66]. Standard conditions contained 15 nM RNA transcript annealed to a 5’ end-labeled oligonucleotide (ppnKB rev1, ykuN rev1: complementary to codons 16 to 22; and sucC rev1: complementary to codons 24 to 31) 250 nM E. coli 30S ribosomal subunits (250 nM), and 40 to 80 nM of RoxS in 10 μl of buffer containing 20 mM Tris-acetate, pH 7.5, 60 mM NH4Cl, 10 mM magnesium acetate, and 3 mM ß-mercaptoethanol. After 10 mins at 37°C, the initiator tRNA (1 μM) was added and the reaction was incubated for a further 5 mins at 37°C. Reverse transcription was conducted with one unit of AMV reverse transcriptase for 15 mins at 37°C.
Structure probing experiments
RoxS-ppnKB mRNA formation was carried out at 37°C for 15 mins in TMN buffer containing 50 mM Hepes-NaOH pH 7.5, MgCl2 5 mM, KCl 50 mM. The concentrations of RoxS or its derivatives were 40 nM and 80 nM. Enzymatic hydrolysis was performed with unlabeled ppnKB (0.5 pmol) in 10 μl of TMN, in the presence of 1 μg carrier tRNA at 37°C for 5 mins: RNase T1 (0.002 units, Thermo scientific), RNase V1 (1 unit, Ambion), purified B. subtilis RNase III (165 nM, 330 nM, 660 nM). The reactions were stopped by phenol extraction followed by RNA precipitation. The enzymatic cleavages were detected by primer extension with reverse transcriptase according to [67].
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Storz G, Vogel J, Wassarman KM (2011) Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 43 : 880–891. doi: 10.1016/j.molcel.2011.08.022 21925377
2. Caldelari I, Chao Y, Romby P, Vogel J (2013) RNA-mediated regulation in pathogenic bacteria. Cold Spring Harb Perspect Med 3: a010298. doi: 10.1101/cshperspect.a010298 24003243
3. Ramirez-Pena E, Trevino J, Liu Z, Perez N, Sumby P (2010) The group A Streptococcus small regulatory RNA FasX enhances streptokinase activity by increasing the stability of the ska mRNA transcript. Mol Microbiol 78 : 1332–1347. doi: 10.1111/j.1365-2958.2010.07427.x 21143309
4. Obana N, Shirahama Y, Abe K, Nakamura K (2010) Stabilization of Clostridium perfringens collagenase mRNA by VR-RNA-dependent cleavage in 5′ leader sequence. Mol Microbiol 77 : 1416–1428. doi: 10.1111/j.1365-2958.2010.07258.x 20572941
5. Opdyke JA, Fozo EM, Hemm MR, Storz G (2011) RNase III participates in GadY-dependent cleavage of the gadX-gadW mRNA. J Mol Biol 406 : 29–43. doi: 10.1016/j.jmb.2010.12.009 21147125
6. Pfeiffer V, Papenfort K, Lucchini S, Hinton JC, Vogel J (2009) Coding sequence targeting by MicC RNA reveals bacterial mRNA silencing downstream of translational initiation. Nat Struct Mol Biol 16 : 840–846. doi: 10.1038/nsmb.1631 19620966
7. Gaballa A, Antelmann H, Aguilar C, Khakh SK, Song KB, et al. (2008) The Bacillus subtilis iron-sparing response is mediated by a Fur-regulated small RNA and three small, basic proteins. Proc Natl Acad Sci U S A 105 : 11927–11932. doi: 10.1073/pnas.0711752105 18697947
8. Heidrich N, Chinali A, Gerth U, Brantl S (2006) The small untranslated RNA SR1 from the Bacillus subtilis genome is involved in the regulation of arginine catabolism. Mol Microbiol 62 : 520–536. 17020585
9. Gimpel M, Preis H, Barth E, Gramzow L, Brantl S (2012) SR1 - a small RNA with two remarkably conserved functions. Nucleic Acids Res 40 : 11659–11672. doi: 10.1093/nar/gks895 23034808
10. Smaldone GT, Antelmann H, Gaballa A, Helmann JD (2012) The FsrA sRNA and FbpB protein mediate the iron-dependent induction of the Bacillus subtilis lutABC iron-sulfur-containing oxidases. J Bacteriol 194 : 2586–2593. doi: 10.1128/JB.05567-11 22427629
11. Smaldone GT, Revelles O, Gaballa A, Sauer U, Antelmann H, et al. (2012) A global investigation of the Bacillus subtilis iron-sparing response identifies major changes in metabolism. J Bacteriol 194 : 2594–2605. doi: 10.1128/JB.05990-11 22389480
12. Huntzinger E, Boisset S, Saveanu C, Benito Y, Geissmann T, et al. (2005) Staphylococcus aureus RNAIII and the endoribonuclease III coordinately regulate spa gene expression. EMBO J 24 : 824–835. doi: 10.1038/sj.emboj.7600572 15678100
13. Hammerle H, Amman F, Vecerek B, Stulke J, Hofacker I, et al. (2014) Impact of Hfq on the Bacillus subtilis transcriptome. PLoS One 9: e98661. doi: 10.1371/journal.pone.0098661 24932523
14. Dambach M, Irnov I, Winkler WC (2013) Association of RNAs with Bacillus subtilis Hfq. PLoS One 8: e55156. doi: 10.1371/journal.pone.0055156 23457461
15. Even S, Pellegrini O, Zig L, Labas V, Vinh J, et al. (2005) Ribonucleases J1 and J2: two novel endoribonucleases in B. subtilis with functional homology to E. coli RNase E. Nucleic Acids Res 33 : 2141–2152. doi: 10.1093/nar/gki505 15831787
16. Mathy N, Benard L, Pellegrini O, Daou R, Wen T, et al. (2007) 5′-to-3′ exoribonuclease activity in bacteria: role of RNase J1 in rRNA maturation and 5′ stability of mRNA. Cell 129 : 681–692. 17512403
17. Geissmann T, Chevalier C, Cros MJ, Boisset S, Fechter P, et al. (2009) A search for small noncoding RNAs in Staphylococcus aureus reveals a conserved sequence motif for regulation. Nucleic Acids Res 37 : 7239–7257. doi: 10.1093/nar/gkp668 19786493
18. Bohn C, Rigoulay C, Chabelskaya S, Sharma CM, Marchais A, et al. (2010) Experimental discovery of small RNAs in Staphylococcus aureus reveals a riboregulator of central metabolism. Nucleic Acids Res 38 : 6620–6636. doi: 10.1093/nar/gkq462 20511587
19. Song J, Lays C, Vandenesch F, Benito Y, Bes M, et al. (2012) The expression of small regulatory RNAs in clinical samples reflects the different life styles of Staphylococcus aureus in colonization vs. infection. PLoS One 7: e37294. doi: 10.1371/journal.pone.0037294 22629378
20. Demple B (1999) Genetic responses against nitric oxide toxicity. Braz J Med Biol Res 32 : 1417–1427. 10559844
21. Crane BR, Sudhamsu J, Patel BA (2010) Bacterial nitric oxide synthases. Annu Rev Biochem 79 : 445–470. doi: 10.1146/annurev-biochem-062608-103436 20370423
22. Gusarov I, Nudler E (2005) NO-mediated cytoprotection: instant adaptation to oxidative stress in bacteria. Proc Natl Acad Sci U S A 102 : 13855–13860. doi: 10.1073/pnas.0504307102 16172391
23. Shatalin K, Gusarov I, Avetissova E, Shatalina Y, McQuade LE, et al. (2008) Bacillus anthracis-derived nitric oxide is essential for pathogen virulence and survival in macrophages. Proc Natl Acad Sci U S A 105 : 1009–1013. doi: 10.1073/pnas.0710950105 18215992
24. Hochgrafe F, Wolf C, Fuchs S, Liebeke M, Lalk M, et al. (2008) Nitric oxide stress induces different responses but mediates comparable protein thiol protection in Bacillus subtilis and Staphylococcus aureus. J Bacteriol 190 : 4997–5008. doi: 10.1128/JB.01846-07 18487332
25. Gusarov I, Shatalin K, Starodubtseva M, Nudler E (2009) Endogenous nitric oxide protects bacteria against a wide spectrum of antibiotics. Science 325 : 1380–1384. doi: 10.1126/science.1175439 19745150
26. van Sorge NM, Beasley FC, Gusarov I, Gonzalez DJ, von Kockritz-Blickwede M, et al. (2013) Methicillin-resistant Staphylococcus aureus bacterial nitric-oxide synthase affects antibiotic sensitivity and skin abscess development. J Biol Chem 288 : 6417–6426. doi: 10.1074/jbc.M112.448738 23322784
27. Gusarov I, Gautier L, Smolentseva O, Shamovsky I, Eremina S, et al. (2013) Bacterial nitric oxide extends the lifespan of C. elegans. Cell 152 : 818–830. doi: 10.1016/j.cell.2012.12.043 23415229
28. Nicolas P, Mader U, Dervyn E, Rochat T, Leduc A, et al. (2012) Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science 335 : 1103–1106. doi: 10.1126/science.1206848 22383849
29. Baruah A, Lindsey B, Zhu Y, Nakano MM (2004) Mutational analysis of the signal-sensing domain of ResE histidine kinase from Bacillus subtilis. J Bacteriol 186 : 1694–1704. doi: 10.1128/JB.186.6.1694-1704.2004 14996800
30. Kinkel TL, Roux CM, Dunman PM, Fang FC (2013) The Staphylococcus aureus SrrAB two-component system promotes resistance to nitrosative stress and hypoxia. MBio 4: e00696–00613. doi: 10.1128/mBio.00696-13 24222487
31. Hartig E, Geng H, Hartmann A, Hubacek A, Munch R, et al. (2004) Bacillus subtilis ResD induces expression of the potential regulatory genes yclJK upon oxygen limitation. J Bacteriol 186 : 6477–6484. doi: 10.1128/JB.186.19.6477-6484.2004 15375128
32. Henares B, Kommineni S, Chumsakul O, Ogasawara N, Ishikawa S, et al. (2014) The ResD Response Regulator, through Functional Interaction with NsrR and Fur, Plays Three Distinct Roles in Bacillus subtilis Transcriptional Control. J Bacteriol 196 : 493–503. doi: 10.1128/JB.01166-13 24214949
33. Rochat T, Nicolas P, Delumeau O, Rabatinova A, Korelusova J, et al. (2012) Genome-wide identification of genes directly regulated by the pleiotropic transcription factor Spx in Bacillus subtilis. Nucleic Acids Res 40 : 9571–9583. doi: 10.1093/nar/gks755 22904090
34. Nakano MM (2002) Induction of ResDE-dependent gene expression in Bacillus subtilis in response to nitric oxide and nitrosative stress. J Bacteriol 184 : 1783–1787. doi: 10.1128/JB.184.6.1783-1787.2002 11872732
35. Antelmann H, Engelmann S, Schmid R, Sorokin A, Lapidus A, et al. (1997) Expression of a stress - and starvation-induced dps/pexB-homologous gene is controlled by the alternative sigma factor sigmaB in Bacillus subtilis. J Bacteriol 179 : 7251–7256. 9393687
36. You C, Sekowska A, Francetic O, Martin-Verstraete I, Wang Y, et al. (2008) Spx mediates oxidative stress regulation of the methionine sulfoxide reductases operon in Bacillus subtilis. BMC Microbiol 8 : 128. doi: 10.1186/1471-2180-8-128 18662407
37. Kiran MD, Bala S, Hirshberg M, Balaban N (2010) YhgC protects Bacillus anthracis from oxidative stress. Int J Artif Organs.
38. Wang ZQ, Lawson RJ, Buddha MR, Wei CC, Crane BR, et al. (2007) Bacterial flavodoxins support nitric oxide production by Bacillus subtilis nitric-oxide synthase. J Biol Chem 282 : 2196–2202. 17127770
39. Durand S, Gilet L, Bessieres P, Nicolas P, Condon C (2012) Three essential ribonucleases-RNase Y, J1, and III-control the abundance of a majority of Bacillus subtilis mRNAs. PLoS Genet 8: e1002520. doi: 10.1371/journal.pgen.1002520 22412379
40. Tjaden B (2012) Computational identification of sRNA targets. Methods Mol Biol 905 : 227–234. doi: 10.1007/978-1-61779-949-5_14 22736007
41. Wright PR, Richter AS, Papenfort K, Mann M, Vogel J, et al. (2013) Comparative genomics boosts target prediction for bacterial small RNAs. Proc Natl Acad Sci U S A 110: E3487–3496. doi: 10.1073/pnas.1303248110 23980183
42. Eggenhofer F, Tafer H, Stadler PF, Hofacker IL (2011) RNApredator: fast accessibility-based prediction of sRNA targets. Nucleic Acids Res 39: W149–154. doi: 10.1093/nar/gkr467 21672960
43. Geng H, Nakano S, Nakano MM (2004) Transcriptional activation by Bacillus subtilis ResD: tandem binding to target elements and phosphorylation-dependent and -independent transcriptional activation. J Bacteriol 186 : 2028–2037. doi: 10.1128/JB.186.7.2028-2037.2004 15028686
44. Georgellis D, Kwon O, Lin EC (2001) Quinones as the redox signal for the arc two-component system of bacteria. Science 292 : 2314–2316. 11423658
45. Ye RW, Tao W, Bedzyk L, Young T, Chen M, et al. (2000) Global gene expression profiles of Bacillus subtilis grown under anaerobic conditions. J Bacteriol 182 : 4458–4465. 10913079
46. Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, et al. (2014) Quantitative flux analysis reveals folate-dependent NADPH production. Nature 510 : 298–302. doi: 10.1038/nature13236 24805240
47. Shahbabian K, Jamalli A, Zig L, Putzer H (2009) RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J 28 : 3523–3533. doi: 10.1038/emboj.2009.283 19779461
48. Lehnik-Habrink M, Rempeters L, Kovacs AT, Wrede C, Baierlein C, et al. (2013) DEAD-Box RNA helicases in Bacillus subtilis have multiple functions and act independently from each other. J Bacteriol 195 : 534–544. doi: 10.1128/JB.01475-12 23175651
49. Faubladier M, Cam K, Bouche JP (1990) Escherichia coli cell division inhibitor DicF-RNA of the dicB operon. Evidence for its generation in vivo by transcription termination and by RNase III and RNase E-dependent processing. J Mol Biol 212 : 461–471. 1691299
50. Guo MS, Updegrove TB, Gogol EB, Shabalina SA, Gross CA, et al. (2014) MicL, a new sigmaE-dependent sRNA, combats envelope stress by repressing synthesis of Lpp, the major outer membrane lipoprotein. Genes Dev 28 : 1620–1634. doi: 10.1101/gad.243485.114 25030700
51. Papenfort K, Said N, Welsink T, Lucchini S, Hinton JC, et al. (2009) Specific and pleiotropic patterns of mRNA regulation by ArcZ, a conserved, Hfq-dependent small RNA. Mol Microbiol 74 : 139–158. doi: 10.1111/j.1365-2958.2009.06857.x 19732340
52. Mandin P, Gottesman S (2010) Integrating anaerobic/aerobic sensing and the general stress response through the ArcZ small RNA. EMBO J 29 : 3094–3107. doi: 10.1038/emboj.2010.179 20683441
53. Davis BM, Waldor MK (2007) RNase E-dependent processing stabilizes MicX, a Vibrio cholerae sRNA. Mol Microbiol 65 : 373–385. doi: 10.1111/j.1365-2958.2007.05796.x 17590231
54. Soper T, Mandin P, Majdalani N, Gottesman S, Woodson SA (2010) Positive regulation by small RNAs and the role of Hfq. Proc Natl Acad Sci U S A 107 : 9602–9607. doi: 10.1073/pnas.1004435107 20457943
55. Viegas SC, Silva IJ, Saramago M, Domingues S, Arraiano CM (2011) Regulation of the small regulatory RNA MicA by ribonuclease III: a target-dependent pathway. Nucleic Acids Res 39 : 2918–2930. doi: 10.1093/nar/gkq1239 21138960
56. Afonyushkin T, Vecerek B, Moll I, Blasi U, Kaberdin VR (2005) Both RNase E and RNase III control the stability of sodB mRNA upon translational inhibition by the small regulatory RNA RyhB. Nucleic Acids Res 33 : 1678–1689. doi: 10.1093/nar/gki313 15781494
57. Marchais A, Duperrier S, Durand S, Gautheret D, Stragier P (2011) CsfG, a sporulation-specific, small non-coding RNA highly conserved in endospore formers. RNA Biol 8 : 358–364. doi: 10.4161/rna.8.3.14998 21532344
58. Guerout-Fleury AM, Shazand K, Frandsen N, Stragier P (1995) Antibiotic-resistance cassettes for Bacillus subtilis. Gene 167 : 335–336. 8566804
59. Britton RA, Wen T, Schaefer L, Pellegrini O, Uicker WC, et al. (2007) Maturation of the 5′ end of Bacillus subtilis 16S rRNA by the essential ribonuclease YkqC/RNase J1. Mol Microbiol 63 : 127–138. 17229210
60. Bechhofer DH, Oussenko IA, Deikus G, Yao S, Mathy N, et al. (2008) Analysis of mRNA decay in Bacillus subtilis. Methods Enzymol 447 : 259–276. doi: 10.1016/S0076-6879(08)02214-3 19161848
61. Stead MB, Agrawal A, Bowden KE, Nasir R, Mohanty BK, et al. (2012) RNAsnap: a rapid, quantitative and inexpensive, method for isolating total RNA from bacteria. Nucleic Acids Res 40: e156. doi: 10.1093/nar/gks680 22821568
62. Smyth GK (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article3. 16646809
63. Strimmer K (2008) A unified approach to false discovery rate estimation. BMC Bioinformatics 9 : 303. doi: 10.1186/1471-2105-9-303 18613966
64. Zybailov B, Coleman MK, Florens L, Washburn MP (2005) Correlation of relative abundance ratios derived from peptide ion chromatograms and spectrum counting for quantitative proteomic analysis using stable isotope labeling. Anal Chem 77 : 6218–6224. 16194081
65. Bauer KM, Lambert PA, Hummon AB (2012) Comparative label-free LC-MS/MS analysis of colorectal adenocarcinoma and metastatic cells treated with 5-fluorouracil. Proteomics 12 : 1928–1937. doi: 10.1002/pmic.201200041 22623418
66. Fechter P, Chevalier C, Yusupova G, Yusupov M, Romby P, et al. (2009) Ribosomal initiation complexes probed by toeprinting and effect of trans-acting translational regulators in bacteria. Methods Mol Biol 540 : 247–263. doi: 10.1007/978-1-59745-558-9_18 19381565
67. Chevalier C, Geissmann T, Helfer AC, Romby P (2009) Probing mRNA structure and sRNA-mRNA interactions in bacteria using enzymes and lead(II). Methods Mol Biol 540 : 215–232. doi: 10.1007/978-1-59745-558-9_16 19381563
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 2
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Genomic Selection and Association Mapping in Rice (): Effect of Trait Genetic Architecture, Training Population Composition, Marker Number and Statistical Model on Accuracy of Rice Genomic Selection in Elite, Tropical Rice Breeding Lines
- Discovery of Transcription Factors and Regulatory Regions Driving Tumor Development by ATAC-seq and FAIRE-seq Open Chromatin Profiling
- Evolutionary Signatures amongst Disease Genes Permit Novel Methods for Gene Prioritization and Construction of Informative Gene-Based Networks
- Proteotoxic Stress Induces Phosphorylation of p62/SQSTM1 by ULK1 to Regulate Selective Autophagic Clearance of Protein Aggregates
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy