
Contribution of the Two Genes Encoding Histone Variant H3.3 to Viability and Fertility in Mice
Histones package DNA and regulate chromosome activity. Histone H3 is particularly important in this regard. The H3.3 isoform is unique, among H3 histones, in being able to incorporate into chromosomes independent of DNA replication. Thus, in slowly - or non-dividing somatic cells, H3.3 histone can take on the bulk of H3 functions. The developing germ line is very slowly dividing, and undergoes large-scale changes in chromosome activity. H3.3 is therefore likely to be very important in regulating these processes. Here, we have studied the effects of null mutations in each of the two genes encoding H3.3. We demonstrate that H3.3 is very important in germ cell development, regulating oogenesis, spermatogenesis, and fertilization. Also, we reveal H3.3 to be important in somatic growth. Each of the two genes is required to varying extents in regulating these processes.
Published in the journal:
. PLoS Genet 11(2): e32767. doi:10.1371/journal.pgen.1004964
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004964
Summary
Histones package DNA and regulate chromosome activity. Histone H3 is particularly important in this regard. The H3.3 isoform is unique, among H3 histones, in being able to incorporate into chromosomes independent of DNA replication. Thus, in slowly - or non-dividing somatic cells, H3.3 histone can take on the bulk of H3 functions. The developing germ line is very slowly dividing, and undergoes large-scale changes in chromosome activity. H3.3 is therefore likely to be very important in regulating these processes. Here, we have studied the effects of null mutations in each of the two genes encoding H3.3. We demonstrate that H3.3 is very important in germ cell development, regulating oogenesis, spermatogenesis, and fertilization. Also, we reveal H3.3 to be important in somatic growth. Each of the two genes is required to varying extents in regulating these processes.
Introduction
The core histones H2A, H2B, H3 and H4, package DNA into nucleosomes and modify higher-order chromatin structure. H3 in mammals is represented by three near-identical 135 amino acid isoforms: canonical H3.1 and H3.2, and the variant H3.3. Chromatin incorporation is replication-coupled for the canonical forms, and is replication-independent for H3.3 [1–4]. H3.3 plays fundamental roles in regulating genome function and stability. It can accumulate in terminally differentiated or non-dividing cells, and can become the predominant H3 isoform [3, 5]. Through the chaperone HIRA (histone cell cycle regulation defective homolog A (S. cerevisiae)), H3.3 is deposited at active genes and regulatory regions, forming more accessible nucleosomes [3, 4, 6–8], and underlies the formation of epigenetic bivalent domains at the promoter regions of poised genes in embryonic stem (ES) cells [8, 9]. Through the chaperones ATRX (alpha thalassemia/mental retardation syndrome X-linked) and DAXX (Fas death domain-associated protein), H3.3 is deposited at repetitive regions such as telomeres and centromeres, serving genome stabilization functions [10–12]. H3.3 is required for heterochromatin formation in mammalian preimplantation-stage embryos [13, 14], and has been reported to preferentially localize to the heterochromatic XY-body in meiosis-I, where it could serve functions related to meiotic sex chromosome inactivation (MSCI) [15]. Finally, specific amino acid substitutions in H3.3 drive cancer [16, 17], underscoring the fundamental importance of H3.3 in regulating epigenetic states.
Canonical histones are encoded by numerous clustered intronless genes, while histone variants are typically encoded by one intron-containing gene. H3.3 presents a unique genetic paradigm in that the identical protein is encoded by two intron-containing unlinked genes, H3f3a, and H3f3b (histone 3, family 3A and -B). We will refer to the encoded proteins as H3.3A and H3.3B, respectively. The two genes have divergent regulatory and intervening regions, suggesting some qualitatively different functional roles. The presence of two H3.3-encoding genes, and their genomic arrangement, is highly conserved in mammals and birds [18].
Here, we have investigated the developmental effects of null mutations of H3f3a and H3f3b in the mouse (Mus musculus), each mutated by the same experimental strategy, and in the identical mouse strain—129S1/SvImJ (129S1). H3f3a mutants were viable to adulthood. By contrast, H3f3b mutants were growth deficient, and inviable at birth. Failures in gametogenesis and fertilization were seen, with the defects severer on ablation of H3f3b. Our results demonstrate important functions of H3.3 in growth, viability, and fertility, and reveal stages of gametogenesis and fertilization that are adversely affected by H3.3 deficiency in the mouse.
Results
H3f3a mutants are viable, with males slightly growth deficient
H3f3a+/- heterozygous young were produced by mating ES cell chimaeras to strain 129S1 Cre-deleter females [19] (see Materials and Methods). H3f3a+/- females and males were overtly normal and fertile. Timed matings were obtained from H3f3a+/- ♀ × H3f3a+/- ♂ intercrosses so that newborns could be monitored. In 14 litters in which 65 pups were born, only four dead pups were seen: two of these dead pups were H3f3a-/-. All remaining young survived to adulthood, except two H3f3a-/- runts that were culled. From all timed pregnancies, and other intercrosses, 29, 84 and 46 animals surviving to adulthood were H3f3a-/- mutant, H3f3a+/- and H3f3a+/+ (WT), respectively—not significantly different from a 1 : 2:1 ratio (P>0.05, chi-squared test). H3f3a-/- females were of normal size. By contrast, H3f3a-/- males were slightly smaller than H3f3a+/- and WT males at 3 wk and 6 wk (Fig. 1A). H3f3a-/- mutants of both sexes were overtly normal in behaviour and appearance.
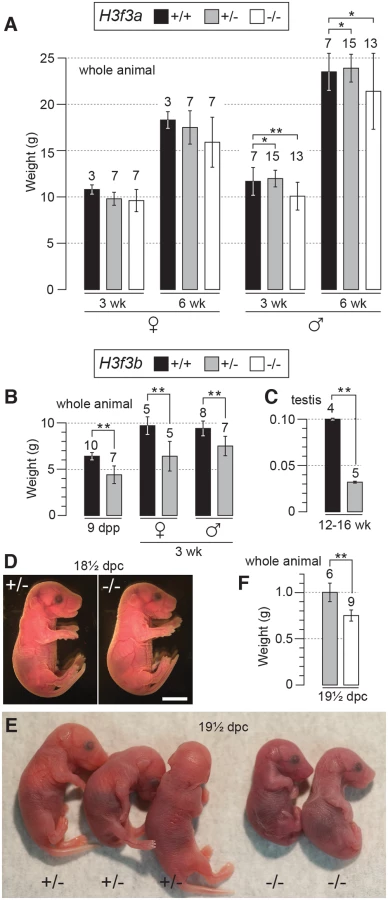
H3f3b heterozygotes are growth deficient, while H3f3b mutants die at birth
H3f3b+/- young were bred by mating ES cell chimaeras to strain 129S1 Cre-deleter females (see Materials and Methods). These young often appeared smaller than WT sibs at weaning, but developed into robust adults. H3f3b+/- females were fertile on mating with WT 129S1 males. From these matings, altricial and weaned H3f3b-/+ young were significantly smaller than WT littermates, revealing haploinsufficiency for H3f3b (Fig. 1B).
H3f3b-/- mutants could not be bred in intercrosses because H3f3b-/+ males were sterile, with small testes (Fig. 1C). To produce H3f3b-/- zygotes, we again made use of the 129S1 Cre-deleter line. The cre cds in this line is X-linked (Xcre). Floxed alleles are always mutated on sperm entry into Cre-containing oocytes when XXcre females are used [19]. Two sequential matings were performed. First, H3f3b-/+, XXcre females were bred in H3f3bc/c, XX ♀ × WT, XcreY ♂ matings—these females were converted to the H3f3b-/+ genotype early in development because of the presence of Xcre. Second, H3f3b-/+, XXcre ♀ × H3f3bc/c, XY ♂ matings were performed to obtain timed pregnancies. The paternal H3f3bc allele was converted to H3f3b- in the zygote in these matings. Therefore, a 1 : 1 ratio of H3f3b-/- to H3f3b+/- zygotes was expected. Results of fetal recovery are shown (Table 1). Overall, 37 H3f3b-/- and 69 H3f3b+/- fetuses were obtained, different from a 1 : 1 ratio. Thirty-six deciduae were counted in these pregnancies, these being implantation sites in which development did not proceed beyond the early implantation stage. It is possible that most of these deciduae represented failures in H3f3b-/- development, accounting for the fetal deficit. This failure was not associated with sex chromosome constitution or to the presence of Xcre (Table 1).

Twenty four percent (9/37) of H3f3b-/- fetuses at 13½-18½ dpc were dead or severely abnormal (Table 1). Live 18½ dpc H3f3b-/- fetuses, two days before birth, had no gross abnormalities (Fig. 1D). However, no live H3f3b-/- pups were seen in newborn litters. Thirty two H3f3b+/- animals were born, 29 of which survived to weaning. A single H3f3b-/- dead pup was found on the day of birth. These results strongly suggest that all H3f3b-/- pups died at birth, then were consumed by the mothers. The immediate death of pups at birth is usually associated with respiratory failure [20]. Examining 18½ dpc H3f3b-/- fetuses histologically revealed no gross pathology in any tissues, including the lungs (S1 Fig.). To confirm that H3f3b-/- fetuses could not survive parturition, fetuses were delivered by Caesarean at 19½ dpc. From 5 pregnancies, of 11 H3f3b+/- fetuses delivered, all initiated normal respiration within a few minutes, remaining oxygenated or ‘pink’, and mobile. By contrast, all 6 H3f3b-/- fetuses delivered failed to initiate respiration after initially gulping. Within a few minutes they were immobile, and ‘blue’ (Fig. 1E). They were also smaller than H3f3b+/- littermates (Fig. 1F).
H3f3a and H3f3b mutants contain similar amounts of residual H3.3 in somatic tissues
H3f3a-/- mutants contain residual H3.3B, while H3f3b-/- mutants contain residual H3.3A. These amounts of residual H3.3 were determined in immunoblots in the brain, kidney, liver and lung of 18½ dpc fetuses, and in the trunk of 13½ dpc fetuses (Fig. 2). In both mutants, H3.3 was detected in all organs, consistent with H3.3A and H3.3B being essentially ubiquitous in the developing fetus. Given that H3f3b-/- mutants had the severer phenotype, we expected to see relatively less residual H3.3 in these animals. However, the only clear difference between the two mutants was in the brain, where H3f3b-/- animals had relatively higher residual H3.3A (Fig. 2).

H3f3a mutant males are subfertile
Three H3f3a-/- males were fertility-tested alongside three WT littermates within the first three months after reaching adulthood. Each male was caged for one week with two nulliparous 8–10 wk outbred Swiss females, this being repeated over several weeks. For H3f3a-/- males, 7 pregnancies from 34 female exposures resulted, less than the frequency for WT males—16 pregnancies from 28 exposures (P<0.001, Fisher’s exact test). Also, litters sired by H3f3a-/- males were small: WT, 10.63 ± 2.55 (16); H3f3a-/-, 6.14 ± 4.85 (7) [mean ± s.d. (n)] (P<0.01, t-test). These three littermate pairs, and two additional pairs, were then fertility-tested at 6–10 months of age. Over this period, each male was caged with two Swiss females. All WT males consistently sired large litters, while three H3f3a-/- males sired no litters, and two H3f3a-/- males sired the occasional small litter. At the end of this period, the ability of H3f3a-/- males to copulate was tested. Vaginal plugs, at ½ days post coitum (dpc), were readily obtained when H3f3a-/- males were mated with nulliparous Swiss females. Oviducts were flushed at 1½ dpc. H3f3a-/- males fertilized substantially fewer ova than WT littermates, indicating that the subfertility was caused by a defect in spermatogenesis (Table 2). The reproductive tracts were overtly normal, except the vasa deferentia appeared to be devoid of sperm. Representative histological sections of the testes and caudae epididymides are shown (Fig. 3 A–E). A substantial production of sperm was seen in H3f3a-/- males, although some sloughed germ cells were present in the caudae, indicating developmental failure of some spermatids (Fig. 3D, E). Sperm isolated from the caudae showed reduced motility, failing to efficiently swim out in media. The large majority of spermatozoa possessed tail defects, with some possessing head defects (Figs. 3F, 4). These data are consistent with a requirement for H3.3A in spermiogenesis.



H3f3b heterozygous males are sterile, with developmental arrest of round spermatids
H3f3b heterozygous males derived from chimaeras mated to Cre-deleter females, or from the H3f3b+/- ♀ × WT ♂ backcross, were sterile. Their testes were less than one third of normal weight, indicative of failed spermatogenesis (Fig. 1C). Histological analysis revealed a reduction of round spermatids and very few elongating spermatids in the lumen of seminiferous tubules (Fig. 3G, H). The caudae epididymides were devoid of sperm, and often contained sloughed germ cells (Fig. 3I).
H3.3 has been reported to preferentially localize to the XY-body, where it may play a role in MSCI [15]. Therefore, we thought there may a level of MSCI failure in H3f3b-/+ males. MSCI acts as a checkpoint. Its failure is associated with autosomal asynapsis, leads to pachytene apoptosis with no progression of spermatocytes beyond meiosis-I [21, 22]. The presence of pachytene spermatocytes with XY-bodies, and round spermatids in H3f3b-/+ sterile males (Fig. 3H), indicates that there is no serious failure of MSCI. Yet, we decided to investigate autosomal synapsis in H3f3b-/+ males more directly. First, we examined the localization of γH2A.X (H2A histone family, member X) in mid - to late-pachytene spermatocytes of H3f3b-/+ males, when autosomes should be fully synapsed. These stages were marked by positive staining for H1FNT (H1 histone family, member N, testis-specific) [23]. All H3f3b-/+ H1FNT-positive cells examined (>50) displayed discrete localization of γH2A.X to the presumptive XY-body, with no labelling evident outside this region, indicating that autosomal synapsis and MSCI proceeded normally (Fig. 5A–D). Complete autosomal synapsis in H3f3b-/+ males was confirmed in double-staining for SYCP3 and SYCP1 (synaptonemal complex proteins 3 and 1). SYCP3 marks chromosomal axial elements that have assembled in meiosis-I, and is present before and during synapsis, while SYCP1 marks synapsed chromatin. All spreads with 20 discrete SYCP3-positive axial elements examined (>30) were equivalently positive for SYCP1, demonstrating complete synapsis. The unsynapsed sex chromosomes remained SYCP1-negative (Fig. 5E–H). Finally, no increase in apoptosis was seen in spermatocytes or round spermatids by TUNEL staining (S2 Fig.). Overall, these data strongly suggest that meiosis-I is essentially normal in H3f3b-/+ spermatocytes. Thus, the failure in spermatogenesis is localized to round spermatids, which appear to arrest through mechanisms that do not involve apoptosis.

These data suggest a crucial role for H3.3B in early spermiogenesis, one that is relatively more important than for H3.3A. In H3f3a mutants, spermiogenesis was also abnormal, but did lead to the formation of sperm. RNA in situ hybridization analysis has shown that the expression of H3f3b is restricted to primary spermatocytes, while the expression of H3f3a is ubiquitous across spermatogenic cell types [24]. It therefore appears that the requirement for H3.3B in spermiogenesis is fulfilled by translation in meiosis-I. The difference in severity of phenotype was not reflected in the relative amount of RNA, as we measured more H3f3a than H3f3b RNA in pachytene spermatocytes (Fig. 6A). Also, the amounts of H3.3 detected in pachytene spermatocytes and round spermatids of H3f3a-/- and H3f3b+/- males were not discernibly different to WT (Fig. 6B).

H3f3a mutant females are fertile
H3f3a-/- females were of normal fertility. Of eight 6 wk females caged with WT 129S1 males, seven gave birth within 10 wk. Litter size was 4.86 ± 1.86 (7) [mean ± s.d. (n)], not significantly different to that obtained with H3f3a+/- females in intercrosses; 5.54 ± 2.54 (14) (P>0.05, t-test). H3f3a-/- females raised their young normally. Three H3f3a-/- second-generation female young, bred in H3f3a-/- ♀ × H3f3a-/- ♂ matings, were also fertile: when mated to a WT outbred Swiss male, each had three sequential litters no more that one month apart, and with no fewer than five pups in any litter. All pups were raised normally. These results show that H3.3A is dispensable in the female germ line, and for fertilization and development.
H3f3b mutagenesis at the beginning of folliculogenesis results in severe female subfertility
To determine the requirement for H3.3B in folliculogenesis, we used the Zp3 (zona pellucida protein 3) promoter-cre cds transgenic mouse line, Tg(Zp3-cre), which excises floxed sequences at primordial follicle activation [25, 26]. We bred a Tg(Zp3-cre) congenic line in strain 129S1 to perform all the experiments described. H3f3bc/c, 0/0 ♀ × H3f3bc/c, 0/Tg(Zp3-cre) ♂ matings produced control H3f3bc/c, 0/0, and experimental H3f3bc/c, 0/Tg(Zp3-cre) females. These females were mated with WT males to obtain timed pregnancies. Less H3f3bc/c, 0/Tg(Zp3-cre) or experimental females became pregnant, and in these, fetal recovery was very low—only 6% in successful matings, relative to control females (Table 3). A few females were allowed to give birth, and pups were viable to adulthood. No deciduae were observed in any of the pregnant or non-pregnant plugged experimental females, indicating that there was a failure in ovulation, fertilization, or cleavage. Ova were scored at ½ dpc. The number obtained in control and experimental female matings was equivalent. However, fewer ova in experimental females were fertilized (Table 3). Zygotes, with visible pronuclei, were cultured overnight to 1½ dpc: only 10% of experimental pronuclear zygotes could cleave. Cleavage failure was also observed in vivo, when oviducts of experimental females were flushed at 1½ dpc (Table 3). Experimental zygotes that failed to cleave are shown (Fig. 7A).


To gain insight into the cause of cleavage failure in H3f3b-/- zygotes, we studied aspects of chromatin structure using immunofluorescence. A hallmark of late prophase and mitosis is the serine phosphorylation of histone H3 (H3Sph). The deposition dynamics of this mark are highly conserved, suggestive of important roles in mitosis. Indeed, it has been recently shown that H3S10ph is required for proper chromosome condensation in budding yeast (Saccharomyces cerevisiae) [27]. H3f3b-/- zygotes did not accumulate this mark, even after overnight culture, indicating that they failed to progress to late prophase (Fig. 8). Staining for H3K9me3, being specific for the maternal pronucleus [28, 29], revealed that the paternal pronucleus was often similar in size or smaller than the maternal, instead of being consistently larger, as in controls (Fig. 8). We also typically observed small and dispersed nucleolar precursor bodies (NPBs) in the maternal pronuclei of H3f3b-/- zygotes—evident under DAPI staining. Control H3f3bc/c zygotes typically contained one large NPB in each pronucleus (Fig. 8). Similarly, the central regions of intense DAPI staining in the paternal pronuclei of H3f3b-/- zygotes could be related to a reduction in the development of NPBs: normally, intense DAPI staining is concentrated at the periphery of NPBs, corresponding to constitutive heterochromatin [29, 30]. DNA replication was examined in H3f3b-/- zygotes using Edu incorporation, concomitant with H3K9me3 immunofluorescence so that the maternal and paternal pronuclei could be definitively discerned. Three classes of H3f3b-/- zygote were seen in respect to relative pronuclear size and DNA synthesis: (1) paternal larger than the maternal, robust replication in both—3 of 21 zygotes, (2) paternal similar to the maternal, reduced replication in at least the paternal—8 of 21 zygotes, and (3) paternal smaller than the maternal, replication in only the maternal—10 of 21 zygotes (Fig. 8). Thus, all mutant zygotes appeared to have smaller NPBs, while most showed reductions in DNA replication.

H3f3a, H3f3b double-mutagenesis in folliculogenesis results in primary oocyte failure
The developmental potential of H3f3a and H3f3b mutant oocytes as described above, indicates that H3f3b is either the predominant source, or the only source, of H3.3 during folliculogenesis. To distinguish between these possibilities, we first examined the relative amounts of H3f3a and H3f3b RNA in ovulated unfertilized oocytes by quantitative real-time PCR. Both RNAs are present, with the amount of H3f3b RNA being double that of H3f3a (Fig. 6A). Next, we assessed the developmental potential of double-conditional experimental H3f3ac/c, H3f3bc/c, 0/Tg(Zp3-cre) oocytes, in which H3.3A and H3.3B should be eliminated soon after primary oocyte activation. Presumptive double-mutant primary oocytes were inviable. Experimental females at 4 wk had very small ovaries, with no apparent oocyte growth. A similar phenotype was seen in 10 wk experimental females (Fig. 7B). Primordial follicles, of ~11 μm in diameter, and very early-stage primary oocytes, were of normal appearance. By contrast, nearly all primary oocytes developing a second layer of follicle cells were degenerating, reaching a maximum of ~50 μm in diameter (Fig. 7C). Very few larger primordial oocytes were seen, except for a rare early-stage antral follicle. This result indicates that H3f3a and H3f3b are both sources of H3.3 during folliculogenesis, and clearly shows that H3.3 is absolutely required. Despite the inviability of double-mutant primary oocytes, they had undergone a significant development of the zona pellucida, indicating that the genes encoding the zona glycoproteins were being actively transcribed up to the stage of oocyte death (Fig. 7C).
Discussion
We have assessed the contribution to viability and fertility of the two unlinked genes encoding the histone variant H3.3 in mice. The findings provide implications for the relative importance of each gene. H3f3a and H3f3b mutants could develop to late gestation, demonstrating no absolute requirement for either gene up to this stage. H3f3a is strongly ubiquitously expressed during development [31, 32], and the present study indicates this is also true for H3f3b. Both genes have CpG-island promoters, consistent with housekeeper expression. Given the wide expression profile of each gene, a level of functional redundancy is likely. The co-ablation of H3f3a and H3f3b RNA in zygotes by morpholinos resulted in morula-stage arrest, revealing functional importance of H3.3 at this early stage [33]. It remains of interest to determine the developmental potential of double-mutant animals in our experimental system. Double-mutants may survive the preimplantation period because of the presence of maternal H3f3a and H3f3b RNAs. Also, mouse ES cells in which H3.3 is depleted are viable, and are able to differentiate into all three germ layers in teratomas [9].
The presence of two H3.3-encoding genes appears to be important, given its conservation in at least mammals and birds [18]. This gene doubling may be necessary to satisfy the high demand for this protein in DNA packaging and other downstream functions, akin to the multiplicity of canonical H3.1 - and H3.2-encoding genes. Sequences outside the cds of H3f3a and H3f3b are nevertheless highly divergent. From the present results, H3f3b is generally functionally more important than H3f3a, given H3f3b mutants were more severely affected than H3f3a mutants. This was seen in fetal and postnatal growth, perinatal survivability, spermiogenesis, and zygote development. However, this conclusion assumes that there is no compensatory regulation between the two genes. This cannot be examined at the protein level in WT animals, as H3.3A and H3.3B cannot be distinguished. Surprisingly, H3f3a and H3f3b mutants contained similar amounts of residual H3.3 in a range of tissues examined, except brain, where the residual H3.3A was higher. These findings indicate that differences in expression of the two genes at the sub-organ tissue-specific level are the most important determinants of the relative functional importance of H3.3A and H3.3B. Differences in tissue-specificity of expression is suggested in the recent findings that paediatric brain tumors, and giant cell tumors of bone, contain driver amino acid substitution mutations in H3F3A, while chondroblastomas contain driver mutations in H3F3B [16, 17]. A detailed investigation of the tissue-specific expression of each of these genes in development would be of interest.
Approximately one half of H3f3b mutants appeared to die at the implantation or early postimplantation stage. The other half developed to fetal stages, with the majority reaching birth. One possible explanation for this developmental bipotentiality is chromosomal instability. A higher level of aneuploidy was reported in H3f3b mutant primary embryo fibroblasts [34], therefore there could exist a potential for aneuploidy at the preimplantation stage. Most aneuploid blastocysts die at implantation, or shortly thereafter [35]. The failure of H3f3b mutants to initiate respiration at birth was not accompanied by any lung histopathology, but there could be lung surfactant or neural deficits that lead to immediate respiratory failure [20].
The defects induced by H3.3B deficiency in the germ line were striking. The constitutive loss of one H3f3b allele led to the arrest of round spermatids, and sterility. By contrast, the loss of both H3f3a alleles resulted in dysmorphic spermatozoa, and subfertility. This difference in severity occurred despite our lack of evidence that H3f3b is expressed at a higher level than H3f3a, or is the major source of H3.3, in pachytene spermatocytes and round spermatids. This raises the possibility that there could be qualitative differences in the distribution of H3.3A and H3.3B in spermatogenic cells. Differential expression of the two genes during the prolonged meiotic cycle could lead to regional and functional differences in the incorporation of each protein into chromatin, depending on what sites are vacant for incorporation, and when.
The phenotypes described here in H3f3a and H3f3b mutants differ substantially from phenotypes described in other studies. A hypomorphic gene-trap mutation in H3f3a led to very high mortality by weaning [36]. In explaining our milder phenotype, genetic background is an unlikely explanation, as both H3f3a mutations were propagated in a ‘Steel’ substrain of strain 129 [37]. Environmental influences are a possible explanation: the survivability of H3f3a mutants to weaning was increased when litter size was reduced [36]. Finally, the H3f3a retroviral gene-trap mutation was reported to produce up to ~20% normal levels of full length H3f3a RNA [36]. Therefore, retroviral sequences could have resulted in a deregulation of residual H3.3A that was detrimental to viability.
In contrast to the situation for H3f3a, our H3f3b mutants were considerably more affected than those previously described [34, 38]. In this earlier study, a normal frequency of H3f3b mutants was obtained at midgestation, although most were substantially growth retarded. Despite this prominent defect, many mutants survived parturition and developed to adulthood. No growth deficit was seen in postnatal H3f3b heterozygous or mutant animals [34]. By marked contrast, our H3f3b mutants could not survive beyond birth, and a significant number died even before this stage. Further, we saw marked reductions in growth in perinatal and postnatal heterozygous and mutant animals.
The greater phenotypic severity associated with our H3f3b mutation was also seen in the male germ line. We saw a near-uniform arrest of round spermatids in heterozygous males. By contrast, previously reported heterozygous males were fertile [34]. Indeed, the phenotypic severity in our heterozygous animals exceeded even that seen in recently described H3f3b mutants, where appreciable progression through spermiogenesis was observed. These mutants were sterile due to the production of sperm with motility and head defects [38]. Other differences were apparent. We saw no increase in apoptosis by TUNEL staining in seminiferous tubules of heterozygotes, while an increase was seen in mutants [38]. Reduced protamine loading was reported in mutants, and this could be a major contributor to the defective head formation in mature sperm [38]. This phenotype was similar to that seen in our H3f3a mutant males, therefore there could be a similar mechanistic basis. However, in our H3f3b heterozygous males, other explanations are required, as the stage of arrest precedes the phase of histone displacement. In round spermatids, there is a large increase in transcriptional activity [39,40], and defects in this transcriptional activation could lead to their arrest. The simplest explanation for the differences in effect of the two H3f3b mutations is genetic background. Ours was kept in strain 129S1, while the other was kept in strain C57BL/6 [38]. A caveat in interpreting the present, and previous, results is that the H3f3b mutations were constitutive. H3.3B deficiency in Sertoli cells, which support spermatogenesis, could exacerbate the germ cell defects observed.
H3.3B deficiency in folliculogenesis led to some fertilization failure, and zygotic arrest, demonstrating that H3f3b is a maternal effect gene. Loss of maternal Hira, a H3.3 chaperone, leads to zygote failure in flies (Drosophila melanogaster). After sperm entry, H3.3 fails to incorporate into paternal chromatin, leading to failed decondensation of sperm chromatin [41]. A similar effect has recently been described in mouse, where depletion of HIRA in folliculogenesis using the Tg(Zp3.cre) transgene resulted in zygote arrest. The loss of HIRA led to a failure of paternal pronucleus formation. Also, the loss of DNA replication and ribosomal RNA transcription in paternal and maternal chromatin was observed, and either one of these deficiencies can lead to zygote arrest [42]. The phenotype in our H3.3B-deficient mutant oocytes was similar to, but less severe than, the HIRA-deficient phenotype, and could be explained by the presence of residual H3.3A. Our phenotype was variable, ranging from reduced, to overtly normal, paternal pronuclear expansion. Only a small number of mutant zygotes showed robust DNA replication in both pronuclei. If these represent the few zygotes able to cleave, then incomplete DNA replication could be the predominant cause of failed entry into first mitosis. Another possible explanation is a deficiency of H3Sph, given the importance of H3.3 in DNA packaging in the zygote, and the probable requirement for H3Sph in chromosome condensation at the end of prophase [27]. This could be investigated by determining the developmental potential of H3.3B-deficient zygotes supplemented with mutant H3.3 lacking relevant serines. H3.3 incorporation has also been recently shown to be required for pronuclear pore formation [43] and it is possible that a defect in this assembly could explain the mitosis failure.
An important requirement for H3.3 during the early phase of oocyte growth was seen in the developmental failure of H3f3a, H3f3b double-mutant primary oocytes. Given that HIRA-deficient oogonia can develop and be ovulated, this result clearly demonstrates a requirement for other H3.3 chaperones, possibly DAXX and ATRX, in regulating folliculogenesis. Conditional mutagenesis of these chaperones during folliculogenesis could be informative. The death of H3.3-deficient primary oocytes could be related to the lack of incorporation by a chaperone aside from HIRA, or to the combined lack of incorporation by two or more chaperones.
This study provides insight into the relative importance of the two genes encoding H3.3 for development and fertility in the mouse, and provides a basis for further investigations into the various biological roles of this basic building block of chromatin.
Materials and Methods
Mouse lines
The mouse lines carrying Cre/loxP conditional mutant alleles of H3f3a and H3f3b, termed H3f3ac and H3f3bc, have been described [32]. The Cre-deleter line used to derive mice carrying constitutive mutant alleles, termed H3f3a- and H3f3b-, was 129S1/Sv-Hprttm1(cre)Mnn/J (Jackson Laboratory, stock no. 004302).
Histology
Testes and ovaries from adult mice were fixed in Bouin’s fixative. Sections were embedded in paraffin and stained by Periodic-Acid Schiff (PAS). Fetuses at 18½ dpc were decapitated, transferred to 20 mL of Bouin’s fixative, rocked slowly for 3 days, then washed in four changes of 70% (v/v) ethanol over 2 days. Sections were stained with haematoxylin and eosin. Spreads of mature sperm were made by squeezing the cauda epididymis with the back of curved forceps for release into 0.3 mL of medium M2 [44]. Most of the droplet was transferred to a tube with a wide-bore yellow tip, then an equal volume of 10% phosphate buffered formalin added, and gently mixed. After fixation (15 min, RT), a small drop of the sperm suspension was smeared onto a slide, air-dried, and stained with haematoxylin and eosin.
Preparation of spermatocyte and sperm spreads, and immunofluorescence
Spermatocyte spreads from 18–22 days post partum mice and immunofluorescence (IF) were performed as described [45], with the following modifications: both testes of an 18–22 days post partum (dpp) male were transferred to 2 mL of Dulbecco’s phosphate buffered saline (without calcium and magnesium, with 5.6 mM glucose and 0.4 mg/mL bovine serum albumin), in a petri dish, the tunicae removed, and cut into small pieces with scissors. The pieces were transferred to 3 mL of 0.25% trypsin-EDTA (Gibco, 25200) in a polystyrene tube (Falcon, 352054), incubated (37°C, 20 min), and triturated vigorously with a transfer pipette (30 sec). The crude suspension was passed through a cell strainer (Falcon, 352350) into 1 mL of fetal bovine serum in a 15 mL centrifuge tube, and mixed. The spermatocytes were pelleted (250 × g, 4 min), the pellet resuspended in 3 mL of medium M2, and the tube placed on ice. Protease inhibitors (Sigma Aldrich, P8340) were added to all of the solutions.
To prepare the spreads, 50 μL of the sperm suspension was combined with 0.1 mL of medium M2, and pelleted (500 × g, 2 min). The supernatant was removed, and the pellet resuspended in 40 μL of alkaline sucrose solution [45]. Two drops of this suspension were then dispensed from a yellow tip into ~0.1 mL of the 1% (w/v) paraformaldehyde fixative solution, this being previously dispensed within a rectangle of ~1.5 cm2 drawn on a SuperFrost slide with a PAP pen. Spermatocytes were allowed to settle onto the slides in a humidified chamber (2 h, RT). Protease inhibitors were added to all solutions, except the fixative solution. After removing from the chamber, slides were rinsed in water, then in 0.004% (v/v) Photo-Flo 200 (Kodak, 14634510) in water, and air-dried. Slides were stored at -20°C until hybridization with Abs. Imaging was performed with a Imager.M1 microscope and AxioCam MRm camera (Carl Zeiss).
Primary Abs used, dilution factor: anti-γH2A.X, rabbit mcAb, 400× (Cell Signaling, 9718); anti-H1FNT, guinea pig pcAb, 250× (kindly provided by Mary Ann Handel); anti-SYCP3, mouse mcAb, 100× (Abcam, 97672); anti-SYCP1, rabbit pcAb, 200× (Abcam, 15090). Secondary Abs used, dilution factor: anti-rabbit IgG, goat, 1000×, AlexaFluor 488; anti-guinea pig IgG, goat, 1000×, AlexaFluor 594; anti-mouse IgG, goat, 1000×, AlexaFluor 488; anti-rabbit IgG, goat, 1000×, AlexaFluor 594 (Life Technologies, A-11008, A-11076, A-11001 and A-11012, respectively).
Spermatocyte and round spermatid purification
Pachytene spermatocytes (larger 4c cells) and round spermatids (smaller 1c cells) were purified by flow cytometry as described [46]: testis suspensions were obtained as described above, except that DNAseI (Sigma Aldrich, D4527) was added to the trypsin digest at 5 U/mL, and the cell pellet resuspended in 1.5 mL of medium M2 containing 1 U/mL of DNAseI. The final suspension was incubated with Hoechst 33342 (Life Technologies, H3570) for gating on larger pachytene spermatocytes (4c) and smaller round spermatids (1c), and propidium iodide (Life Technologies, P3566) for gating on live cells (S3 Fig.). Cells were sorted into 0.3 mL of medium M2 in 1.5 mL centrifuge tubes using an Influx cell sorter (Becton Dickinson), then pelleted (2,500 rpm, 3 min), the supernatant removed, and stored at -80°C.
Quantitative real-time PCR
RNA was isolated using TRI Reagent (Life Technologies, AM9738) according to the instructions. Pachytene spermatocytes (200,000) were purified from WT prepubertal 129S1 males by flow cytometry as described above. Oocytes (200) were obtained from WT superovulated prepubertal 129S1 females. The zona pellucida was removed using acid tyrode’s solution [47] before lysis in TRI Reagent. Two random-primed reverse transcription (RT) reactions were performed with SuperScript III Reverse Transcriptase (Life Technologies, 18080). For each RT, triplicate PCR reactions (20 μL, 10 ng cDNA) were performed using GoTaq qPCR Master Mix (Promega, A6001) to yield six Ct values. Equipment used was the 7500 Real-Time PCR System (Applied Biosystems). These values were converted to the relative amount of RNA by multiplying the fold increase in product per cycle—equivalent to amplification efficiency, to the power of ∆Ct. Amplification efficiencies for each transcript were pre-determined in standard curves. Each value was normalized to the mean obtained for the Rsp7 (ribosomal protein S7) housekeeper RNA. Primers, 5′-3′: H3f3a, GCAG CTAT TGGT GCTT TGC (JRM-1227), ATGT TTCC CCTC ATAG TGGA CTC (JRM-1228), amplicon 173 bp; H3f3b, GGAG ATCG CCCA GGAT TTC (JRM-1231), ATGC CATA AAAA CCGC TTCA AC (JRM-1232), amplicon 215 bp; Rps7, TGGT CTTC ATTG CTCA GAGG AGG (JRM-609), TGCC ATCC AGTT TCAC ACGG (JRM-610), amplicon 177 bp.
Immunoblotting
Immunoblotting was performed as described [48]. Tissues were homogenized by trituration with a 19 G needle and syringe in RIPA buffer. Primary Abs used, dilution factor: anti-H3.3, rabbit pcAb, 1000× (Millipore, 09–838); anti-H3 (panH3), rabbit pcAb, 2000× (Abcam, 1791); anti-TUBB (β-tubulin), rabbit mcAb, 1000× (Cell Signaling, 2128); anti-GAPD (glyceraldehyde-3-phosphate dehydrogenase), rabbit mcAb, 1000× (Cell Signaling, 5174). Secondary Abs used, dilution factor: anti-rabbit IgG, goat, horseradish peroxidase conjugate, 15000× (Millipore, 12–348).
Immunofluorescence in zygotes
The light cycle in the mouse colony was 0630–1830 h. Zygotes were collected at ~1200 h on the day of obtaining a vaginal plug, when usually they were at the mid-pronuclear stage. They were cultured for ~2 h for pronuclei to become more prominent before fixation. For H3S10ph immunofluorescence, WT zygotes were not fixed until at least two in the batch had undergone pronuclear envelope breakdown, indicating entry into mitosis. Immunofluorescence was performed as described [30], except goat serum was used in blocking, and fixation and permeabilization solutions contained 0.2% (w/v) and 0.1% (w/v) BSA, respectively, to prevent zygote sticking. Fixation, permeabilization, washing and secondary Ab incubation steps were performed in volumes of 0.1–0.15 mL in 12-well staining dishes (ProSciTech, H421–12), while blocking and primary Ab incubations were carried out in 10 μL drops under paraffin oil. In mounting, zygotes were transferred to a 1 μL drop of Fluorescence Mounting Medium (Dako, S3023) on a SuperFrost slide, coverslipped, and sealed with nail polish. 5-ethynyl-2′-deoxyuridine (Edu) incorporation and detection was carried out using a Click-iT Edu Imaging Kit (Life Technologies, C10339): freshly isolated zygotes with pronuclei were cultured in 20 μM Edu in medium KSOM-GAA [49] (2 h) prior to fixation. H3K9me3 primary and secondary Ab incubations were carried out immediately before Edu detection. Primary Abs, dilution factor: anti-H3K9me3, rabbit pcAb, 1000× (Abcam, 8898); anti-H3S10ph, rabbit mcAb, 1000× (Cell Signaling, 3377). Secondary Ab, dilution factor: anti-rabbit IgG, goat, 1500×, AlexaFluor 488 (Life Technologies, A-11008). The third wash after secondary Ab staining (10 min, RT) contained DAPI at 1 μg/mL, then zygotes were rinsed in DAPI-free wash solution (1 min) before mounting.
Genotyping
Tail tips were digested in 0.1 mL of 50 mM Tris-HCl pH 8.0, 0.5% (v/v) Triton X-100, 20 mM EDTA, 400 μg/mL proteinase K, (55°C, overnight). An aliquot was diluted 20× in water, then heat inactivated (70°C, 10 min). This template solution was diluted 10× in the final PCR reaction, performed using MyTaq Red DNA polymerase (Bioline, 21108). Primers: H3f3a, CTGG TTTT GGCT GTTT TATC GCTC GG (WT primer, JRM-1272), GCTA TTGC TTTA TTTG TAAC CATT ATAA GCTG C (mutant/conditional, JRM-1461), and AGGG CGCA CTCT TGCG AGC (common, JRM-1276); triplex reaction, amplicons WT—362, and mutant/conditional—205 bp. H3f3b, CTCA CCGC TACA GGTA GGC (WT primer, JRM-1465), GCTA TTGC TTTA TTTG TAAC CATT ATAA GCTG C (mutant/conditional, JRM-1461), TCTC CCTC ACCA ATCT CTGG (common, JRM-1466); triplex reaction, amplicons WT—211, mutant/conditional—357. Xcre, specific for the cre cds, TGCT GTTT CACT GGTT ATGC GGCG (JRM-391), TGCC TTCT CTAC ACCT GCGG TGCT (JRM-392); amplicon 304 bp. Y chromosome, nos. 211 and 212 [50].
Ethics statement
This work was approved by the Animal Ethics Committee of the Murdoch Children’s Research Institute, approval no. A692.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. Peters AH, O’Carroll D, Scherthan H, Mechtler K, Sauer S, et al (2001) Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107 : 323–337. 11701123
2. Waterborg JH, Robertson AJ (1996) Common features of analogous replacement histone H3 genes in animals and plants. J Mol Evol 43 : 194–206. 8703085
3. Urban MK, Zweidler A (1983) Changes in nucleosomal core histone variants during chicken development and maturation. Dev Biol 95 : 421–428. 6825941
4. Ahmad K, Henikoff S (2002) Histone H3 variants specify modes of chromatin assembly. Proc Natl Acad Sci USA 99 : 16477–16484. 12177448
5. Castiglia D, Cestelli A, Scaturro M, Nastasi T, Di Liegro I (1994) H1(0) and H3.3B mRNA levels in developing rat brain. Neurochem Res 19 : 1531–1537. 7877725
6. Mito Y, Henikoff JG, Henikoff S (2005) Genome-scale profiling of histone H3.3 replacement patterns. Nat Genet 37 : 1090–1097. 16155569
7. Jin C, Felsenfeld G (2007) Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev 21 : 1519–1529. 17575053
8. Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, et al (2010) Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140 : 678–691. doi: 10.1016/j.cell.2010.01.003 20211137
9. Banaszynski LA, Wen D, Dewell S, Whitcomb SJ, Lin M, et al (2013) Hira-dependent histone H3.3 deposition facilitates PRC2 recruitment at developmental loci in ES cells. Cell 155 : 107–120. doi: 10.1016/j.cell.2013.08.061 24074864
10. Drane P, Ouararhni K, Depaux A, Shuaib M, Hamiche A (2010) The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev 24 : 1253–1265. doi: 10.1101/gad.566910 20504901
11. Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, Allis CD (2010) Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci USA 107 : 14075–14080. doi: 10.1073/pnas.1008850107 20651253
12. Wong LH, McGhie JD, Sim M, Anderson MA, Ahn S, et al (2010) ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res 20 : 351–360. doi: 10.1101/gr.101477.109 20110566
13. Santenard A, Ziegler-Birling C, Koch M, Tora L, Bannister AJ, Torres-Padilla ME (2010) Heterochromatin formation in the mouse embryo requires critical residues of the histone variant H3.3. Nat Cell Biol 12 : 853–862. doi: 10.1038/ncb2089 20676102
14. Akiyama T, Suzuki O, Matsuda J, Aoki F (2011) Dynamic replacement of histone H3 variants reprograms epigenetic marks in early mouse embryos. PLoS Genet 7: e1002279. doi: 10.1371/journal.pgen.1002279 21998593
15. van der Heijden GW, Derijck AA, Posfai E, Giele M, Pelczar P, et al (2007) Chromosome-wide nucleosome replacement and H3.3 incorporation during mammalian meiotic sex chromosome inactivation. Nat Genet 39 : 251–258. 17237782
16. Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, et al (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22 : 425–437. doi: 10.1016/j.ccr.2012.08.024 23079654
17. Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, et al (2013) Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet 45 : 1479–1482. doi: 10.1038/ng.2814 24162739
18. Brush D, Dodgson JB, Choi OR, Stevens PW, Engel JD (1985) Replacement variant histone genes contain intervening sequences. Mol Cell Biol 5 : 1307–1317. 2863747
19. Tang SH, Silva FJ, Tsark WM, Mann JR (2002) A Cre/loxP-deleter transgenic line in mouse strain 129S1/SvImJ. Genesis 32 : 199–202. 11892008
20. Turgeon B, Meloche S (2009) Interpreting neonatal lethal phenotypes in mouse mutants: insights into gene function and human diseases. Physiol Rev 89 : 1–26. doi: 10.1152/physrev.00040.2007 19126753
21. Burgoyne PS, Mahadevaiah SK, Turner JM (2009) The consequences of asynapsis for mammalian meiosis. Nat Rev Genet 10 : 207–216. doi: 10.1038/nrg2505 19188923
22. Ichijima Y, Ichijima M, Lou Z, Nussenzweig A, Camerini-Otero RD, et al (2011) MDC1 directs chromosome-wide silencing of the sex chromosomes in male germ cells. Genes Dev 25 : 959–971. doi: 10.1101/gad.2030811 21536735
23. Inselman A, Eaker S, Handel MA (2003) Temporal expression of cell cycle-related proteins during spermatogenesis: establishing a timeline for onset of the meiotic divisions. Cytogenet Genome Res 103 : 277–284. 15051948
24. Bramlage B, Kosciessa U, Doenecke D (1997) Differential expression of the murine histone genes H3.3A and H3.3B. Differentiation 62 : 13–20. 9373943
25. de Vries WN, Binns LT, Fancher KS, Dean J, Moore R, et al (2000) Expression of Cre recombinase in mouse oocytes: a means to study maternal effect genes. Genesis 26 : 110–112. 10686600
26. Lan ZJ, Xu X, Cooney AJ (2004) Differential oocyte-specific expression of Cre recombinase activity in GDF-9-iCre, Zp3cre, and Msx2Cre transgenic mice. Biol Reprod 71 : 1469–1474. 15215191
27. Wilkins BJ, Rall NA, Ostwal Y, Kruitwagen T, Hiragami-Hamada K, et al (2014) A cascade of histone modifications induces chromatin condensation in mitosis. Science 343 : 77–80. doi: 10.1126/science.1244508 24385627
28. Arney KL, Bao S, Bannister AJ, Kouzarides T, Surani MA (2002) Histone methylation defines epigenetic asymmetry in the mouse zygote. Int J Dev Biol 46 : 317–320. 12068953
29. Santos F, Peters AH, Otte AP, Reik W, Dean W (2005) Dynamic chromatin modifications characterise the first cell cycle in mouse embryos. Dev Biol 280 : 225–236. 15766761
30. Puschendorf M, Terranova R, Boutsma E, Mao X, Isono K, et al (2008) PRC1 and Suv39h specify parental asymmetry at constitutive heterochromatin in early mouse embryos. Nat Genet 40 : 411–420. doi: 10.1038/ng.99 18311137
31. Diez-Roux G, Banfi S, Sultan M, Geffers L, Anand S, et al (2011) A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol 9: e1000582. doi: 10.1371/journal.pbio.1000582 21267068
32. Tang MC, Jacobs SA, Wong LH, Mann JR (2013) Conditional allelic replacement applied to genes encoding the histone variant H3.3 in the mouse. Genesis 51 : 142–146. doi: 10.1002/dvg.22366 23315948
33. Lin CJ, Conti M, Ramalho-Santos M (2013) Histone variant H3.3 maintains a decondensed chromatin state essential for mouse preimplantation development. Development 140 : 3624–3634. doi: 10.1242/dev.095513 23903189
34. Bush KM, Yuen BT, Barrilleaux BL, Riggs JW, O’Geen H, et al (2013) Endogenous mammalian histone H3.3 exhibits chromatin-related functions during development. Epigenetics Chromatin 6 : 7. doi: 10.1186/1756-8935-6-7 23570311
35. Epstein CJ (1986) Developmental genetics. Experientia 42 : 1117–1128. 3021509
36. Couldrey C, Carlton MB, Nolan PM, Colledge WH, Evans MJ (1999) A retroviral gene trap insertion into the histone 3.3A gene causes partial neonatal lethality, stunted growth, neuromuscular deficits and male sub-fertility in transgenic mice. Hum Mol Genet 8 : 2489–2495. 10556297
37. Simpson EM, Linder CC, Sargent EE, Davisson MT, Mobraaten LE, Sharp JJ (1997) Genetic variation among 129 substrains and its importance for targeted mutagenesis in mice. Nat Genet 16 : 19–27. 9140391
38. Yuen BT, Bush KM, Barrilleaux BL, Cotterman R, Knoepfler PS (2014) Histone H3.3 regulates dynamic chromatin states during spermatogenesis. Development 141 : 3483–3494. doi: 10.1242/dev.106450 25142466
39. Schmidt EE, Schibler U (1995) High accumulation of components of the RNA polymerase II transcription machinery in rodent spermatids. Development 121 : 2373–2383. 7671803
40. Soumillon M, Necsulea A, Weier M, Brawand D, Zhang X, et al (2013) Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell Rep 3 : 2179–2190. doi: 10.1016/j.celrep.2013.05.031 23791531
41. Bonnefoy E, Orsi GA, Couble P, Loppin B (2007) The essential role of Drosophila HIRA for de novo assembly of paternal chromatin at fertilization. PLoS Genet 3 : 1991–2006. 17967064
42. Lin CJ, Koh FM, Wong P, Conti M, Ramalho-Santos M (2014) Hira-mediated H3.3 incorporation is required for DNA replication and ribosomal RNA transcription in the mouse zygote. Dev Cell 30 : 268–279. doi: 10.1016/j.devcel.2014.06.022 25087892
43. Inoue A, Zhang Y (2014) Nucleosome assembly is required for nuclear pore complex assembly in mouse zygotes. Nat Struct Mol Biol 21 : 609–616. doi: 10.1038/nsmb.2839 24908396
44. Wood MJ, Whittingham DG, Rall WF (1987) The low temperature preservation of mouse oocytes and embryos. In Monk M, editor. Mammalian development: A practical approach. Oxford, Washington DC: IRL Press. pp. 255–280.
45. Peters AH, Plug AW, van Vugt MJ, de Boer P (1997) A drying-down technique for the spreading of mammalian meiocytes from the male and female germline. Chromosome Res 5 : 66–68. 9088645
46. Lefevre C, Mann JR (2008) RNA expression microarray analysis in mouse prospermatogonia: identification of candidate epigenetic modifiers. Dev Dyn 237 : 1082–1089. doi: 10.1002/dvdy.21482 18330932
47. Behringer R, Gertsenstein M, Nagy K, Nagy A (2014) Manipulating the mouse embryo: A laboratory manual. New York: Cold Spring Harbor Laboratory Press. 814 p.
48. Tang MC, Binos S, Ong EK, Wong LH, Mann JR (2014) High histone variant H3.3 content in mouse prospermatogonia suggests a role in epigenetic reformatting. Chromosoma 123 : 587–595. doi: 10.1007/s00412-014-0475-8 25007861
49. Biggers JD, McGinnis LK, Lawitts JA (2005) One-step versus two-step culture of mouse preimplantation embryos: is there a difference? Hum Reprod 20 : 3376–3384. 16123096
50. Navin A, Prekeris R, Lisitsyn NA, Sonti MM, Grieco DA, et al (1996) Mouse Y-specific repeats isolated by whole chromosome representational difference analysis. Genomics 36 : 349–353. 8812464
51. Theiler K (1989) The house mouse: Atlas of embryonic development. New York: Springer-Verlag. 185p.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 2
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Genomic Selection and Association Mapping in Rice (): Effect of Trait Genetic Architecture, Training Population Composition, Marker Number and Statistical Model on Accuracy of Rice Genomic Selection in Elite, Tropical Rice Breeding Lines
- Discovery of Transcription Factors and Regulatory Regions Driving Tumor Development by ATAC-seq and FAIRE-seq Open Chromatin Profiling
- Evolutionary Signatures amongst Disease Genes Permit Novel Methods for Gene Prioritization and Construction of Informative Gene-Based Networks
- Proteotoxic Stress Induces Phosphorylation of p62/SQSTM1 by ULK1 to Regulate Selective Autophagic Clearance of Protein Aggregates
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy