
Modeling of the Human Alveolar Rhabdomyosarcoma Chromosome Translocation in Mouse Myoblasts Using CRISPR-Cas9 Nuclease
Many cancers carry recurrent chromosome translocations, which often result in the formation of fusion genes that are directly involved in the tumorigenic process. Alveolar rhabdomyosarcoma, a muscle tumor in children, is typified by a translocation that fuses the PAX3 gene on chromosome 2 to the FOXO1 gene on chromosome 13. For translocation to occur both genes need to break and the disparate ends need to fuse via a process called non-homologous end joining. We determined that physical proximity of Pax3 and Foxo1 in mouse muscle progenitor cells (myoblasts) facilitates fusion gene formation. Because Pax3 and Foxo1 in the mouse are in an opposite orientation, we used a chromosome engineering strategy to invert the orientation of Foxo1 so that upon translocation a productive Pax3-Foxo1 fusion gene is created. Co-localization of the Pax3 and Foxo1 loci is higher in fore limb than in hind limb myoblasts. Simultaneous induction of a targeted double strand DNA break in each gene by CRISPR-Cas9 nuclease generated more fusion genes in fore limb than in hind limb myoblasts. Thus, gene proximity facilitates fusion gene formation. We propose that CRISPR-Cas9 nuclease can be used for the precise modeling of chromosome translocations of human cancer in mice.
Published in the journal:
. PLoS Genet 11(2): e32767. doi:10.1371/journal.pgen.1004951
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004951
Summary
Many cancers carry recurrent chromosome translocations, which often result in the formation of fusion genes that are directly involved in the tumorigenic process. Alveolar rhabdomyosarcoma, a muscle tumor in children, is typified by a translocation that fuses the PAX3 gene on chromosome 2 to the FOXO1 gene on chromosome 13. For translocation to occur both genes need to break and the disparate ends need to fuse via a process called non-homologous end joining. We determined that physical proximity of Pax3 and Foxo1 in mouse muscle progenitor cells (myoblasts) facilitates fusion gene formation. Because Pax3 and Foxo1 in the mouse are in an opposite orientation, we used a chromosome engineering strategy to invert the orientation of Foxo1 so that upon translocation a productive Pax3-Foxo1 fusion gene is created. Co-localization of the Pax3 and Foxo1 loci is higher in fore limb than in hind limb myoblasts. Simultaneous induction of a targeted double strand DNA break in each gene by CRISPR-Cas9 nuclease generated more fusion genes in fore limb than in hind limb myoblasts. Thus, gene proximity facilitates fusion gene formation. We propose that CRISPR-Cas9 nuclease can be used for the precise modeling of chromosome translocations of human cancer in mice.
Introduction
Rhabdomyosarcoma (RMS) is the third most common soft-tissue sarcoma in children with an annual incidence of five new cases per million. It accounts for 5–8% of all pediatric cancer. RMS belongs to the family of small round blue cell tumors of childhood and exhibits histological features of skeletal muscle. Two major histological subtypes of RMS can be distinguished, embryonal (E-RMS) and alveolar (A-RMS). E-RMS has its highest incidence in infants and young children whereas A-RMS is more frequent in older children and adolescents. A-RMS has a more aggressive clinical behavior with early dissemination, a poor response to chemotherapy, frequent relapses, and a 5-year failure-free survival of 65% after treatment [1]. A-RMS is found predominantly in the extremities (42%), parameningeal (17%), head and neck (11%) and other locations (21%) [1] including the trunk, perirectal and perianal areas [2, 3]. Cytogenetically A-RMS is distinguished from E-RMS by one of two recurrent chromosome translocations: t(2;13) or t(1;13), which result in fusion of PAX3 or PAX7 to FOXO1, respectively [4].
In spite of multiple attempts to identify the cell of origin in which the t(2;13) occurs the question remains unanswered. It was shown previously that transcription occurs at a few hundred discrete nuclear sites called transcription factories [5]. Some genes frequently involved in a recurrent chromosome translocation (MYC and IGH in B lymphoid progenitors, TMPRSS2 and ERG or ETV1 in prostate cancer, RET and H4 in in radiation-associated papillary thyroid cancer) co-localize to the same transcription factory [6–9]. Initial chromosome conformation capture experiments in activated mouse B cells suggested that physical proximity of the IGH and MYC loci is a minor contributor to the frequency of chromosome translocation [10]. However, combined high resolution Hi-C mapping and genome-wide translocation sequencing in transformed mouse pre-B cells found good coincidence between chromosomal translocation and spatial proximity [11]. A possible driver of double strand DNA breaks might be the co-localization of replication stress-induced early replication fragile sites (ERFSs) with highly expressed gene clusters [12]. Though it was demonstrated that ectopic expression of PAX3-FOXO1/Pax3-Foxo1 can transform mouse mesenchymal stem cells in vitro [13] as well as Myf6+ myofibers in vivo [14] in view of the above these cell types seem unlikely hosts for the chromosome translocation given that they do not express Pax3. In fact, the suggestion that Myf6+ myofibers might be the host of the PAX3-FOXO translocation was recently rectified [15].
In contrast, Pax3 is expressed in activated myoblasts upon muscle injury or in growing muscles during normal development [16]. Moreover, PAX3-FKHR, in cooperation with loss of p16INK4A expression, transforms both fetal and postnatal primary human skeletal muscle cell precursors [17]. Together these observations suggest that translocation might occur in a population of activated myoblasts that express PAX3 (PAX3+). It has been shown that Pax3 expression differs among different muscles in the mouse [18, 19]. There are many more Pax3+ cells in fore limb than in hind limb muscles [19]. Muscle satellite cells from the masseter and soleus did not express Pax3 while only 7% of those from the extensor digitorum longus (EDL) did. In contrast 49% of satellite cells from the biceps were Pax3+. In addition, most ventral trunk muscles were Pax3-positive and 64% of satellite cells from the diaphragm expressed Pax3. Importantly, primary myoblast cultures of Pax3+ satellite cells remain Pax3+, while Pax3- satellite cells from hind limb remain negative [19].
Studies addressing the relation between spatial chromosome proximity and translocation have been performed in cells of the B-lymphoid lineage or of hormone-responsive lineages mostly using transformed cell lines [6, 7, 9]. Recently CRISPR-Cas9 nuclease (Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-associated systems) [10] was used to engineer human tumor-associated translocations [20]. To answer the question if locus proximity of Pax3 and Foxo1 in low-passage primary mouse myoblasts contributes to the frequency of Pax3-Foxo1 fusion gene formation we used the CRISPR-Cas9 system to induce double strand DNA breaks (DSBs), which spurred by non-homologous end joining repair (NHEJ) produce chromosome translocations between these two loci. We used synthetic single-guide RNAs (sgRNA) to program Cas9 to induce DNA double-strand breaks (DSBs) in Pax3 and Foxo1 [21–23]. Unlike the human PAX3/7 and FOXO1 genes, mouse Pax3/7 and Foxo1 are in opposite orientation on their respective chromosomes (1, 4, and 3). Compared with human chromosome 13, Foxo1 is part of an inverted 4.9Mb syntenic region on mouse chromosome 3. Although a recurrent complex inversion/translocation event involving the oppositely oriented ETV6 and c-ABL genes in humans gives rise to the ETV6-ABL fusion gene in some myeloid and lymphoid malignancies, the frequency of this event is extremely low [24]. Therefore, to successfully generate a CRISPR-Cas9-mediated Pax3-Foxo1 fusion gene we used chromosomal engineering via Cre recombinase-mediated genetic alterations to create a mouse in which the Foxo1 containing 4.9 Mb syntenic region is inverted (Foxo1-inv+/+ mice). Previously, Cre recombinase-mediated inversions of large fragments of chromosomes have been used to create balanced chromosomes [25–29].
We show that myoblasts isolated from fore and hind limb keep their Pax3-expressing identity and co-localization of Pax3 and Foxo1 loci strongly correlates with the level of Pax3 expression and generation of a CRISPR-Cas9 induced t(1;3), which is more frequent in fore limb myoblasts. Our Foxo1inv+/+ mice will be a valuable tool for studying mechanisms underlying the initial stages of the A-RMS implicated chromosome translocations resulting in development of better animal models for this pediatric cancer and other human diseases caused by chromosome translocations.
Results
Expression of Pax3 in primary myoblasts correlates with co-localization of the Pax3 and Foxo1 loci
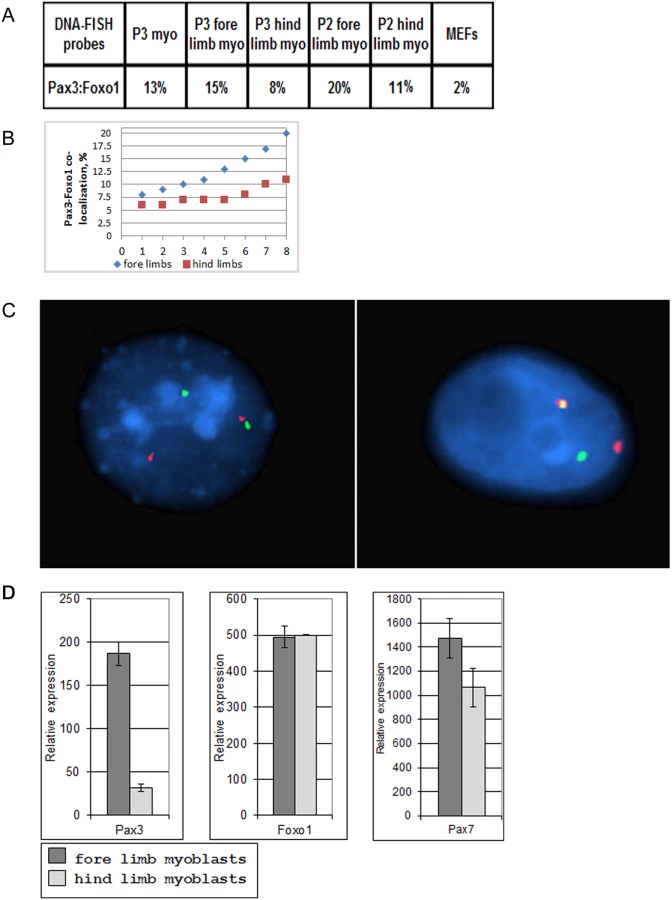
Since close physical proximity of translocation partners might facilitate chromosome translocation, we determined if Pax3 and Foxo1—the translocation partners in A-RMS—are co-localized in actively proliferating low-passage primary mouse myoblasts. DNA-FISH analyses of the Pax3 and Foxo1 loci in interphase nuclei of primary limb myoblasts of newborn pups, after one week in culture showed 13% co-localization, which was significantly higher than in similarly cultured MEFs (2%, the background of this method; Fig. 1A). We hypothesized that co-localization of Pax3 and Foxo1 loci in myoblasts reflects the percentage of Pax3+ cells in the original newborn muscles. To test this hypothesis we isolated myoblasts from hind and fore limbs of newborn pups and compared the frequency of co-localization of Pax3 and Foxo1 loci in proliferating myoblasts from these two sources. It was shown previously with a Pax3 knock-in reporter gene that many more satellite cells in fore limb muscle express Pax3 than in hind limb muscle [19]. In accordance, the percentage of co-localized Pax3 and Foxo1 loci was notably higher in fore limb than in hind limb myoblasts in 8 independent experiments (Fig. 1A–C). In addition, Q-RT-PCR of RNA from these myoblasts confirmed that expression of Pax3 was six-fold higher in fore limb myoblasts (Fig. 1D). These results are in agreement with the published observation that satellite cells maintain their Pax3+ identity upon activation in vitro. Expression of other genes such as Foxo1 and Pax7 was similar in the two types of myoblasts (Fig. 1D). The results for Pax3 expression were reproducible given that a number of independent experiments produced similar data (S1 Fig.). Because diaphragm was shown to contain the highest number of Pax3+ myoblasts [19], we compared by FISH the co-localization of the Pax3 and Foxo1 loci in myoblasts isolated from fore limb, hind limb, and diaphragm of the same adult mouse. Indeed, diaphragm myoblasts showed a higher co-localization of the two loci (20%) than fore limb (11%) or hind limb (9%) myoblasts.
Chromosome engineered Foxo1-inv+/+ mice
The mouse Foxo1 gene is located on chromosome 3 in a 4.9 Mb DNA fragment (ch3 : 52,059,615–56,995,963) that is syntenic with human chromosome 13 (ch13 : 41,254,213–34,463,185) but positioned in the opposite orientation (Fig. 2A). This places Foxo1 in the mouse in a reverse transcriptional direction with respect to that of the Pax3 or Pax7 genes. To engineer a mouse capable of acquiring productive Pax3/7-Foxo1 fusion genes via a simple balanced t(1;3) or t(4;3), we performed two consecutive rounds of ES cell targeting in which we introduced two pairs of non-compatible LoxP sites at either border of this syntenic region with the goal to create a Cre-recombinase mediated permanent inversion of the 4.9Mb DNA fragment (S2 Fig.). Without inversion there would only be two ways to produce a productive fusion: 1) Via a translocation in which the resulting chromosomes would carry a double centromere and no centromere, respectively, an option likely to be non-viable in primary myoblasts and 2) Via a complex inversion/translocation event as described for the human ETV6-ABL fusion gene [24], a rare event, which likely would reduce the frequency of fusion gene formation below detectable levels.

The centromeric border of the mouse/human syntenic region is located 15 kb upstream of the Foxo1 start codon (Fig. 2B). To precisely target this border in ES cells we used recombineering in E. coli [30] to modify the RP24–391O12 BAC (bacterial artificial chromosome) clone, so that it carries non-compatible mutant 511-ILoxP and wtLoxP sites [31] flanking the hph (hygromycin B resistance) and tk (HSV1-thymidine kinase) selectable marker genes (Figs. 2B, S2). The precise targeting of the border of the syntenic region minimizes the chance of disturbing any potentially important regulatory sequences that might affect Foxo1 expression (Fig. 2B, top). We targeted ES cells with linearized RP24–391O12-LoxP-hygro-TK BAC DNA and counter selected hygromycin B resistant clones carrying random integrations by screening for the presence of vector sequences remaining on either side of the insert. Colonies containing such vector segments were discarded [32]. The remaining clones were subjected to the ‘loss-of-native-allele’ assay using real-time quantitative PCR [33]. For copy number control of stably integrated target DNA we used the telomeric border of the syntenic region as a reference. In total 273 clones were analyzed, two of which contained a single copy of the wild type locus (Fig. 2C). These clones were submitted to FISH analysis and karyotyping which confirmed the presence of only two native signals on chromosome 3 when hybridized with a wild type BAC RP24–391O12 probe (Fig. 2D). For consecutive targeting of the telomeric border of the syntenic region we selected clone XIIB3, which had a 100% normal diploid karyotype.
For targeting of the telomeric border of the syntenic region we followed the same strategy and engineered a BAC clone carrying the 511-ILoxp-Neo-TK-wtLoxP cassette inserted at the precise syntenic border (Fig. 2B, bottom). The recombinant RP23–422I13-LoxP-Neo-TK BAC was linearized in such a way that only very short vector fragments remained at either side of the insert. After targeting in ES cells, analysis with the ‘loss-of-native-allele’ assay of 48 clones proved sufficient to obtain the desired recombinant. Two clones carrying a single copy of the wild type telomeric locus (Fig. 2E) were analyzed by FISH using the RP24–391O12 and RP23–422I13 BAC probes. One of them (13D3) showed two native signals on chromosome 3 with either BAC probe (Fig. 2F). This clone had a 90% normal diploid karyotype and we next determined if it carried cis - or trans-targeted borders of the 4.9 Mb syntenic region.
To discriminate between these two possibilities we transiently transfected a Cre recombinase plasmid into the double-targeted 13D3 ES cells and DNA isolated from the pool of electroporated cells was analyzed by PCR using only forward or reverse primers from both targeted borders. Both PCRs produced bands indicating that the pool contained cells carrying the 4.9Mb inversion. The same pool of cells was counter selected with FIAU and 23 resistant clones were analyzed by PCR (Fig. 3A). Sixteen clones harbored the 4.9Mb inversion and two of these were selected, A6 and C5, which had a 100% and 93% normal karyotype, respectively. Inversion of the 4.9Mb region in these clones was subsequently confirmed by FISH analysis (Fig. 3B) using the RP24–391O12 and RP23–422I13 BAC probes. The chromosome containing the inversion showed split hybridization signals while the wild type chromosome produced contiguous signals with these probes. These ES cell clones were used to generate chimeric mice that transmitted the inversion of the Foxo1 syntenic region to heterozygous Foxo1-inv+/- offspring.

Foxo1-inv+/- mice were fertile and produced Foxo1-inv+/+ offspring at the expected Mendelian frequency. Foxo1-inv+/+ animals did not exhibit any obvious phenotypic abnormalities and showed normal fecundity and life span. Moreover, western blot analysis confirmed that Foxo1-inv+/+ primary myoblasts and wild type myoblasts expressed equal amounts of Foxo1 protein (Fig. 3C) and co-localization of the Pax3 and Foxo1 loci was equal in Foxo1-inv+/+ and wild type myoblasts (8%, S5 Fig.). Finally, DNA-FISH analysis of Foxo1-inv+/+ fibroblasts with RP24–391O12 and RP23–422I13 BAC probes confirmed that both chromosomes 3 carried the 4.9Mb inversion (Fig. 3D).
CRISPR-Cas9 induced t(1;3) reciprocal translocation
Nuclear receptor-induced chromosomal proximity of TMPRSS2 and ERG or TMPRSS2 and ETV1 promotes the occurrence of nonrandom ligation sites upon translocation between these partner genes, thereby generating unique breakpoint “hot spots” [6]. It is possible that translocations in A-RMS are non-random and occur predominantly at sites, coming in close proximity during co-regulated expression. We hypothesized that directing DSBs to sites in mouse Pax3 and Foxo1 homologous to those in PAX3 and FOXO1 in an ARMS cell line carrying a t(2;13) might increase the chance of generating a t(1;3) in proliferating Foxo1-inv+/+ myoblasts after Cas9 induced DSBs. We chose to mimic the breakpoints of the widely used ARMS cell line RH30 (S3 and S4 Figs.). Alignment of human and mouse Pax3 and Foxo1 sequences mapped the RH30-like breakpoints at positions 78105273 on mouse chromosome 1 and 52300558 on mouse chromosome 3 (Fig. 4A). We chose unique protospacer sequences followed by a 5’-GGT PAM as close as possible to the RH30-like breakpoints in both Pax3 and Foxo1 (Fig. 4B). Cas9 introduces DSB three nucleotides downstream of the two PAM sequences, which would result in DSBs between nucleotides 78105248 and 78105247 on chromosome 1 in intron 7 of Pax3 and between nucleotides 52300541 and 52300542 (coordinates in the non-inverted sequence) on chromosome 3 in intron 1 of Foxo1 (Fig. 4B).

For gene delivery to the myoblasts we cloned the human codon optimized Cas9 (hCas9) into the pCL20C [34] lentiviral vector downstream of the MSCV promoter and upstream of an IRES-YFP fluorescent marker (Fig. 4C). In order to express two different sgRNAs form a single vector we constructed a second pCL20C dual sgRNA vector in which the Pax3-specific sgRNA was driven by the human U6 promoter and the Foxo1-specific sgRNA by the mouse U6 promoter (Fig. 4C,D).
We first determined that with our current batch of serum maximum co-localization of Pax3 and Foxo1 occurred at 7–8 days of culture after myoblast isolation. This time point synchronized with Cas9 and sgRNA expression should therefore maximize the probability of introducing DSB in closely positioned Pax3 and Foxo1 loci. Hence 24 hours after isolation we transduced primary fore and hind limb myoblasts of Foxo1-inv+/+ pups, Foxo1-inv+/+ MEFs and fore limb myoblasts from wild type mice with Cas9 lentivirus (Fig. 4C). After FACS sorting for YFP, cells were expanded and transduced with lentivirus expressing the RH30-like sgRNAs at day 7 after isolation and with an SV40 large T antigen expressing lentivirus at day 8. The latter was done to prevent senescence of the myoblasts during puromycin selection and allows subsequent expansion of the culture. To detect the Pax3-Foxo1 fusion DNA fragments from Cas9/sgRNAs expressing myoblasts and MEFs we used the Pax3-RH30F (forward) and Foxo1-RH30R (reverse) primers for PCR analysis, which are positioned downstream and upstream of the putative Cas9-induced Pax3 and Foxo1 DSBs (Fig. 5A). PCR amplification of DNA from 104 cells produced bands of 250 bp or shorter in Cas9/sgRNAs expressing hind limb and fore limb Foxo1-inv+/+ myoblast (Fig 5B, lanes 2 and 4). However, no product was detected upon PCR amplification of DNA from 104 hind and fore limb Foxo1-inv+/+ myoblasts not treated with Cas9/sgRNAs (Fig. 5B, lanes 1 and 3) or from 104 Cas9/sgRNAs expressing Foxo1-inv+/+ MEFs or wild type fore limb myoblasts (Fig. 5B, lanes 5 and 6). As a control we verified that the difference in translocation frequency between myoblasts and MEFs was not caused by differences in CRISPR-Cas9’s accessibility to chromatin, given that Pax3 is not expressed in MEFs. The CRISPR-Cas9 breakpoint in Pax3 falls within a MaeIII restriction endonuclease site and that in Foxo1 within a DdeI site. Therefore we PCR amplified the Pax3 and Foxo1 fragments spanning the breakpoints and digested them with MaeIII or DdeI. This showed that 96% (Pax3) and 97% (Foxo1) of the PCR products of CRISPR-Cas9 treated myoblasts were resistant to MaeIII or DdeI digestion, whereas in CRISPR-Cas9 treated MEFs these numbers were 72% for both enzymes (Fig. 5C,D). Thus, there was no great difference in chromatin accessibility. Moreover, the Pax3 and Foxo1 chromatin in MEFs was equally accessible to CRISPR-Cas9, despite the fact that Foxo1 is and Pax3 is not expressed in these cells.

Cloning of the CRISPR-Cas9 induced fusion DNAs, followed by sequencing of 45 individual clones of each of the PCR products, produced 39 and 34 translocation breakpoint sequences from fore and hind limb myoblasts, respectively. This identified 6 different breakpoint sequences from fore limb and 3 different breakpoint sequences from the hind limb myoblasts. This represents the minimal number of translocation events per 104 cells (Fig. 5E, top and bottom). Taking into account the percentage of locus co-localization (Fig. 5F) these numbers translate to a minimal translocation frequency of 1 in 150 in fore limb and 1 in 200 in hind limb myoblasts, respectively. The only sequence in common between the fusion fragments from these two types of myoblasts was the cleanly re-ligated fusion, without missing or added base pairs. The other 7 (5 from fore limb myoblast and 2 from hind limb myoblast) were all unique and carried NHEJ-mediated deletions varying from 6 to 71 bp. Superimposed on the deletion, two of the clones also contained randomly added base pairs. Notably, three additional breakpoint sequences obtained from an independent experiment (S5 Fig.) were different from the 7 shown in Fig. 5E and underline the mutation-prone repair of the NHEJ DNA-repair machinery during the translocation event. Together these results show excellent correlation between the frequency of translocation, co-localization, and expression of the Pax3 and Foxo1 loci in primary myoblasts. It was highest in fore limb myoblasts, lower in hind limb myoblasts and undetectable in MEFs. Although wild type myoblasts show the same frequency of locus co-localization as Foxo-inv+/+ myoblasts (S6 Fig.), the opposite orientation of Foxo1 prevented the formation of a productive Pax3-Foxo1 fusion gene. Next we performed RT-PCR on equal amounts of total RNA from fore limb and hind limb myoblasts to detect the Pax3-Foxo1 fusion mRNA. In support of the higher frequency of chromosome translocation in fore limb myoblasts, we were able to RT-PCR amplify the Pax3-Foxo1 cDNA from these myoblasts (Fig. 5G) but not from the hind limb myoblasts using an equal amount of input RNA (not shown). Sequence analysis of the cDNA confirmed the correctly spliced Pax3 exon 7-Foxo1 exon 2 fusion (Fig. 5G).
FISH detection of the t(1;3) reciprocal translocation
To further characterize the t(1;3) we repeated the experiment in Foxo1-inv+/+/Ink4a-ARF-/ - myoblasts. Due to loss of a functional p53 pathway Ink4a-ARF-/ - myoblasts do not senesce during further experimental manipulation. Based on the Pax3 and Foxo1 co-localization data at the time of induction of the t(1;3) (11% in fore limb myoblasts and 7% in hind limb myoblasts) we assumed that the frequency of translocation events in these myoblasts should not be lower than in the myoblasts used in Fig. 5, i.e. at least 6 independent translocation events per 104 fore limb myoblasts. This frequency is too low for further molecular and functional analyses. To enrich the cell pool for the t(1;3) carrying cells, we evenly distributed 104 cells between the wells of three 96-well plates (on average 30 cells per well). PCR analyses of the DNA of 95 wells from the first plate identified 3 potentially t(1;3)-enriched cell pools (S7 Fig.). Pool 1E10 was lost during the freeze-thawing cycle but FISH analyses detected the reciprocal t(1;3) in 64% of pool 1G3 metaphase cells (Fig. 6A–C) and in 4% of pool 1D10 metaphase cells. Both the derivative chromosomes 1 and 3 were detected in all t(1;3) positive cells, confirming that the translocation was reciprocal.

Functional analyses of cells harboring the reciprocal t(1;3)
To determine if the t(1;3) resulted in expression of Pax3-Foxo1 protein we immunoprecipitated three cell lysates each of the t(1;3)-negative (1H3) and t(1;3)-positive (1G3) pools with either an anti-Pax3 or an anti-Foxo1 antibody. The Pax3 IPs were then immunoblotted with the anti-Pax3 antibody, showing the Pax3 and Pax3-Foxo1 bands (Fig. 6D, Pax3/Pax3 panel), or with anti-Foxo1 antibody showing only the Pax3-Foxo1 band (Fig. 6D, Pax3/Foxo1 panel). Similarly, immunoblotting the Foxo1 IPs with anti-Pax3 antibody again showed the Pax3-Foxo1 fusion protein (Fig. 6D, Foxo1/Pax3 panel) while immunoblotting with the Foxo1 antibody showed both Foxo1 and the fusion protein (Fig. 6D, Foxo1/Foxo1 panel). This confirmed that the engineered t(1;3) expressed the fusion protein, which allowed us to assess if it affected the expression of Pax3-Foxo1’s transcriptional targets. We performed RNA-seq analysis comparing the mapped sequence reads of presumed PAX3-FOXO1 target genes [2] in the 1G3 pool (64% Pax3-Foxo1 positive) with those in the 1H3 pool (Pax3-Foxo1 negative) (S1 Table). This showed that roughly half the targets of PAX3-FOXO1 were correctly up or down regulated in the 1G3 pool. The same comparison with a PAX3-FOXO1 expression signature obtained with the ectopic PAX3-FOXO1 expressing ERMS cell line RD [35], also showed coincident regulation of half the targets (S2 Table), suggesting that the t(1;3) generated fusion protein is active.
Discussion
For the precise modeling of human recurrent chromosome translocations and their impact on disease development in mice, reenactment of the actual translocation would be the closest possible recapitulation of the sequence of events in humans. Until now such reenactment was a daunting task as the translocation would require introduction of LoxP [36, 37] or Frt recombination sites into both translocation partners via homologous recombination in ES cells, followed by expression of Cre or Flp recombinase to create DSBs that would mediate the translocation. As shown by others [20] and here, the availability of the CRISPR-Cas9 system has paved the way to implementing this approach without such major technical or time investment. Given the high homology between mouse and human genes and their regulatory sequences, this approach is likely to include all sequences that are important for the precise regulation of the mouse fusion gene as it occurs in humans. The first and only published model for ARMS [38] in which expression of a conditional Pax3-Foxo1 knock-in fusion oncogene is induced by a Myf6 driven Cre had a low incidence and long latency of tumor development, requiring the presence of two Pax3-Foxo1 alleles on a Trp53-null or Ink4a/Arf-null background. One reason for this might be that the level of expression of the fusion oncogene in this KI model is inadequate for shorter latency tumor development. An argument against this possibility is that a high level of PAX3-FOXO1 expression induces cell death [39], most likely due to transcriptional activation of the Pax3-Foxo1 pro-apoptotic target gene Noxa1 [40]. Unlike other studies [41, 42], the KI Pax3-Foxo1 gene contained some Foxo1 genomic sequences that allowed expression of the fusion gene in adult mice, but despite their presence the construct might lack sequences that mediate human-like regulation of fusion gene expression, which in turn might be crucial for efficient tumor development.
In agreement with published data [19] we established that co-localization of Pax3 and Foxo1 in our culture system was higher in forelimb than in hind limb myoblasts, which coincided with higher Pax3 expression in forelimbs. Due to experimental variability the percentage of co-localization of the two loci varied in 8 independent experiments, but co-localization in the fore limbs was always higher than in the hind limbs. Therefore our myoblast model represents a graded system to determine if these features contributed to the frequency of chromosome translocation in low passage primary myoblasts upon introduction of targeted DSBs. To perform these experiments and to eventually develop a precise mouse model of ARMS, the transcriptional orientation of Foxo1 on chromosome 3 needed to be inverted. We followed the Cre-dependent one-way inversion of a DNA fragment in mice as was previously demonstrated by Schnütgen and colleagues [43]. To avoid disturbing the transcriptional regulation of the inverted Foxo1, we decided to invert the mouse/human 4.9 Mb syntenic region encompassing Foxo1, rather than the gene itself. Although the centromeric border of this region is only 15 kb upstream of Foxo1, we reasoned that all important Foxo1 regulatory sequences should be contained within this region otherwise it would not be syntenic with human FOXO1 on chromosome 13q14.1. Although we did not analyze the detailed expression of Foxo1 in Foxo1-inv+/+ mice during pre - and postnatal life, the animals did not show any obvious phenotypic abnormalities. In addition, they had a normal lifespan, normal fecundity, and the level of Foxo1 protein expression and co-localization of the Pax3 and Foxo1 loci in myoblasts were identical to those of wild type mice. Together these observations made the Foxo1-inv+/+ myoblasts suitable for our translocation experiments.
To determine if the level of co-localization of Pax3 and Foxo1 in primary myoblasts affected the frequency of chromosome translocation between these loci upon induction of targeted DSB, we transduced the cells with Cas9 and dual sgRNA expressing lentiviruses. Combining the three genes into a single lentiviral vector failed to produce viral particles. We targeted the CRISPR-Cas9 DSBs to sequences in Pax3 and Foxo1 that mediated the t(2;13) in the A-RMS cell line Rh30. Both breakpoints are present in sequences conserved between the mouse and human genes, suggesting that they occurred in non-redundant sequences that might bind factors with a role in expression regulation of both genes. Currently we do not know if this affects the frequency of translocation, which is a possibility that can be tested in future by choosing sgRNAs targeting non-conserved sequences within the target Pax3 and Foxo1 introns. We found excellent positive correlation between the frequency of the t(1;3) and the percentage of locus co-localization using FISH analysis. This also correlated with the level of Pax3 expression, which is much higher in fore limb than hind limb myoblasts and absent in MEFs, while Foxo1 expression is ubiquitous. Given that the frequency of CRISPR-Cas9 induced DSBs in Pax3 and Foxo1 is comparable in myoblasts and MEFs, it is the proximity of the loci in these primary cells that facilitates trans-chromosomal ligation producing the two expected derivative chromosomes during NHEJ DNA repair. The derivative chromosome 3 produced correctly spliced Pax3-Foxo1 mRNA, encoding active Pax3-Foxo1 protein that up/down-regulated expression of approximately half the presumed PAX3-FOXO1 targets in the 64% Pax3-Foxo1-positive cell pool (S1 Table). The genes compiled in this table are differentially expressed in ARMS versus ERMS tumors or have been identified by forced expression of PAX3-FOXO1 in different cell lines, including NIH3T3 cells, MEFs, SAOS2 cells and C2C12 cells ([2] and references therein). Because the cell background affects the range of PAX3-FOXO1 target gene expression [44], none of the published scenarios reflect expression of Pax3-Foxo1 in primary p16/Arf-/- mouse myoblasts. Possibly this is the reason for the 45% match of reported PAX3-FOXO1 up or down regulated genes. Comparison with genes up or down regulated in the ERMS cell line RD transduced with PAX3-FOXO1 retrovirus [35] showed 52% coincident regulation (S2 Table). Clearly, the t(1;3) generated Pax3-Foxo1 protein in mouse myoblasts is active and changes the expression of target genes in an ARMS-like manner.
One would expect that the frequency of translocation in myoblast that show co-localization of the two translocation partners would be the same irrespective of the source of myoblasts. We found a frequency of 1/150 and 1/200 in fore and hind limb myoblasts, respectively, which we believe does not represent a difference given the uncertainty of how many translocation events actually took place (we can only count those that give distinguishable fusion products). Our results in mouse myoblasts suggest that human myoblasts can be a cell of origin for the PAX3-FOXO1 translocation as they would provide a favorable environment for the translocation to occur, i.e. expression of both genes and spatial co-localization. It is curious that A-RMS is more frequent in the lower than in the higher extremities in humans, as reported by Neville and co-workers [45]. This apparent inconsistency with our mouse data might be explained by the possibility that humans may not have a difference in the distribution of PAX3 expression in the upper and lower extremities. In addition, the muscle mass and presumably the number of satellite cells in the lower extremities in humans is much higher than in the upper extremities, hence increasing the number of translocation-competent cells and frequency of translocation.
By using CRISPR-Cas9 nuclease we showed that targeted chromosome translocations could be induced with high efficiency. Unlike other approaches that have relied on induction of chromosome translocation using γ−irradiation, DSB-inducing chemicals, or the lymphoid cell-specific gene rearrangement machinery, CRISPR-Cas9 can be employed in any cell type. Due to its specificity the system is suitable for use in vivo in cell culture or in mice. Application of this system will greatly facilitate the development of mouse models that precisely recapitulate chromosome translocation-induced human diseases.
Materials and Methods
Strains of E. coli, BAC clones, PCR Primers and oligonucleotides
A complete list of E. coli strains used for this work can be found in S1 Protocol.
BAC clones RP24–391O12 (centromeric border of the 4.9 Mb syntenic region) and RP23–422I13 (telomeric border of the 4.9 Mb syntenic region) were purchased from the BACPAC Resource Center (BPRC), Children’s Hospital Oakland Research Institute in Oakland, California, USA (http://bacpac.chori.org).
The complete list of PCR Primers and oligonucleotides can be found in S1 Protocol.
Construction of plasmid and BAC-based targeting vectors
A modified pNeoTKLoxP was recombineered into BAC RP23–422I13 (telomeric border of the syntenic region). In pNeoTKLoxP we replaced the wild type (wt) LoxP site downstream of the TK gene with the 511-ILoxP sequence (annealed oligonucleotides TK-511-ILoxP and TK-511-ILoxP-C). Then, via recombineering, we introduced the EM7 promoter upstream of the Neo gene. We therefore transformed electrocompetent DY380 Ecoli cells, containing the wtLoxPNeoTK-511-IloxP plasmid with the TK-EM7-Neo fragment (ends of the annealed oligonucleotides TK-EM7 and EM7-NeoC had been filled-in with Klenow DNA polymerase (Invitrogen) following the manufacturer’s protocol). For recombineering we followed the protocol posted on the Frederick National Laboratory for Cancer Research web site: http://ncifrederick.cancer.gov/research/brb/protocol/Protocol1_DY380.pdf. A short 5’-arm (annealed phosphorylated oligonucleotides 5-tel-s and 5-tel-s-C) was cloned downstream of 511-IloxP and a short 3’-arm was cloned upstream of wtLoxP (annealed phosphorylated oligonucleotides 3-tel and 3-tel-C).
A modified pBSLoxPTKhygro plasmid (kind gift from Drs. M. Roussel and F. Zindy, SJCRH) was recombineered into BAC RP24–391O12 (centromeric border of the syntenic region). In this construct we inserted a 511-ILoxP sequence upstream of the TK-promoter-Neo sequence. Since the activity of TK promoter in prokaryotic cells was sufficient to ensure Hygromycin B resistance, we did not introduce the bacterial EM7 promoter in this construct. A short 5’-arm (annealed phosphorylated oligonucleotides 5-cent-s and 5-cent-s-C) was cloned upstream of the 511-IloxP site and a short 3’-arm was cloned downstream of wtLoxP (annealed phosphorylated oligonucleotides 3-cent and 3-cent-C).
The pCL20c-MSCV-IRES-YFP vector backbone was generated by replacing GFP of pCL20c-MSCV-GFP [46] with I-YFP from MSCV-I-YFP [38]. hCas9 [23] was then cloned downstream of MSCV into pCL20c-MSCV-IRES-YFP. The mU6 fragment was generated by PCR using pSicoR-GFP (Addgene, Cambridge, MA, USA) and cloned downstream of hU6 in pLKO.1 (Addgene, Cambridge, MA, USA). The cassette containing the human and mouse U6 promoters (hU6 and mU6) followed by AgeI and EcoNI cloning sites was cloned upstream of the β-actin promoter of the modified pCL20c vector, containing the β-actin-puro cassette from pJ6.OMEGA.puro [47]. The spacer sequence of hU6 driven sgRNA starts with GG followed by 18 specific nucleotides from the target sequence, and mU6 driven sgRNA starts with GT followed by 18 specific nucleotides from the second target sequence (Fig. 4C). Synthetic ds-DNA fragments, coding Pax3_RH30 sgRNA and Foxo1_RH30 sgRNA were cloned into AgeI and EcoNI sites under control of hU6 and mU6 promoters of pCL20C-hU6-mU6-βact-puro, respectively (Fig. 4C,D).
pCL20C-MSCV-Luc2–2A-LgT was constructed by replacing IRES-YFP with a Luc2–2A-LgT cassette.
Lentivirus production
Lentivirus was produced as described in [46].
ES cell targeting, Cre-mediated inversion, and screening of ES cell clones
F12 (129SvJ-derived) embryonic stem (ES) cells were electroporated and selected for hygromycin B or G418 resistance using standard procedures. In short, 25–45 μg of linearized BAC DNA was electroporated into 2*107 ES cells followed by selection with 100 μg/ml Hygromycin B or 200 μg/ml G418. RP24–391O12-LoxP-hygro-TK was linearized with PI-SceI (NEB) and RP23–422I13-LoxP-Neo-TK was linearized with NotI (NEB). Drug resistant clones were picked after 7–9 days of selection. DNA from these clones was used for PCR analysis.
Screening of homologously recombined ES cell clones was done by PCR and qPCR. The presence of vector arms remaining on either side of the insert was detected by PCR with primers pTARBAC1–3F and pTARBAC1–3R for 3’-located sequences and pTARBAC1–5F and RP24–5R for 5’-located sequences. The “loss-of-native allele” assay was performed as described in [33] with some minor modification. For quantitative (q)PCR we used SYBR®Green PCR Master Mix (Applied Biosystems). qPCR was performed with the RP24-F and RP24-R primers to determine the copy number of the centromeric locus and the RP23-F and RP23-R primers to determine the copy number of the telomeric locus. Ratios between the copy numbers of the two loci were determined either by a standard dilution curve or by the Δct method.
A double targeted ES cell clone was electroporated with a Cre-expressing plasmid (pMC-CRE) using the Amaxa™ Mouse ES Cell Nucleofector™ Kit (Lonza, Germany) according to the manufacturer’s protocol. After 5 days of selection with 0.2μM of fialuridine (FIAU) (a kind gift of Bristol Myers) ES cells were collected for DNA isolation as a pool or as single clones. Cre-mediated inversion was detected by standard PCR using the RP24-F/RP23-F2, and RP24-R/RP23-R2 primer pairs.
Fluorescence in situ hybridization (FISH)
For FISH analyses of Pax3 and Foxo1 co-localization and detection of t(1;3) reciprocal translocation we used BAC probes RP23–260F1 and RP24–391O12. For FISH analyses of targeted ES cells and Foxo1-inv+/+ fibroblasts we used BAC probes RP24–391O12 (centromeric border of the syntenic region) and RP23–422I13 (telomeric border of the syntenic region). BAC probes were labeled with nick translation using either Green (RP23–260F1and RP24–391O12) or Red (RP23–422I13) dUTP (Abbott Molecular). Probes were hybridized to metaphase and/or interphase cells either separately or as a 1 : 1 mixture in hybridization solution (50% formamide, 10% dextran sulfate, and 2X SSC). Slides were washed in 2X Saline-Sodium Citrate (SSC) buffer containing 50% formamide at 37°C for 5 minutes. Cells were counterstained with DAPI and analyzed using a Nikon E80i fluorescence microscope (Nikon) with a 100× oil immersion objective. Successfully targeted clones showed 2 native signals for the centromeric or telomeric targeted regions. Inverted chromosomes 3 appeared as two linked pairs of red and green signals on interphase cells, each pair representing one end of the inverted chromosome segment. Normal chromosomes 3 appeared as a single loosely paired red and green signal. One hundred interphase nuclei were scored for the presence of co-localization of Pax3/Pax7 and Foxo1 signals. Only nuclei with discernible red and green signals were scored. Fifty metaphase cells from CRISPR-Cas nuclease treated myoblasts were scored for the presence of cells containing the reciprocal translocation between Pax3 and Foxo1a.
Identification of the translocation breakpoints in A-RMS
Experimental details are provided in S1 Protocol. Position of primers, used for LD-PCR, gel electrophoresis of LD-PCR products and sequences flanking the breakpoint in the Rh30 cell line are shown in S3 and S4 Figs.
Detection of RH30-like translocation in primary myoblasts
Cas9 induced translocation was detected by PCR of chromosomal DNA from 104 cells using Pax3-RH30F and Foxo1-RH30R primers.
qRT-PCR and RT-PCR of myoblasts
For each qRT-PCR reaction we used RNA isolated from either 2.5×103 (data in Fig. 1) or 1.6×103 (data in S1 Fig.) cells. For each RT-PCR we used RNA isolated from 6.7×103 cells. For RT we used SuperScript III First-Strand Synthesis SuperMix (Invitrogen) with an equimolar mix of Pax3R primer and random hexonucleotides and performed the reaction following the manufacturer’s protocol. For the qPCR step we used TaqMan Gene Expression Master Mix (Applied Biosystems). Ratios between gene expression in different cell lines were determined by a standard dilution curve.
Immunoprecipitation and western-blot analyses
Myoblasts (5×106) were lysed in 0.5 ml CHAPS lysis buffer (40 mM HEPES [pH 7.4], 1 mM EDTA 120 mM NaCl, 10 mM sodium pyrophosphate, 10 mM β-glycerophosphate, 0.3% CHAPS, 50 mM NaF, 1.5 mM NaVO, 1 mM PMSF, and 1 tablet of EDTA-free protease inhibitors [Roche] per 10 mL solution) and freeze-thawed 3 times, followed by centrifugation at 20,000 ×g for 10 min at 4°C. After adding 2 μg anti-Pax3 antibody [48] or anti-Foxo1 (C29H4) Rabbit mAb (Cell Signaling) Pax3, Foxo1 and Pax3-Foxo1 were immunoprecipitated overnight at 4°C. Immunoprecipitated material was bound onto 10 μl protein G-coated Dynabeads (Invitrogen) for 90 minutes at 4°C, which were captured using a DYNA-Mag-2 magnet (Invitrogen), washed 4 times with CHAPS buffer, and removed from the beads by heating to 70°C in 1.25xLDS loading buffer (Invitrogen) in CHAPS and separated on pre-cast 4%–12% bis-tris polyacrylamide gels. Western-blotting was performed using the same anti-Foxo1 and anti-Pax3 antibodies.
Enrichment of t(1;3) harboring cells
To enrich for cells harboring the t(1;3), 104 of the Cas9/sgRNAs expressing fore limb myoblasts were evenly distributed over three 96-well plates (on average 30 cells per well). DNA from each cell pool was isolated and analyzed for the presence of t(1;3) translocation using PCR.
RNA sequence analysis
Libraries were generated from ~ 500 ng total RNA of the 1H3 (no Pax3-Foxo1) and 1G3 (64% Pax3-FOXO1) cell pools using the Illumina TruSeq Stranded mRNA Sample Preparation Kit. Libraries were sequenced on an Illumina HiSeq 2500 using paired-end 100 bp sequencing chemistry. Paired-end reads from RNA-seq were aligned to the following 4 database files using BWA (0.5.10) aligner: (1) the human GRCh37-lite reference sequence, (2) RefSeq, (3) a sequence file representing all possible combinations of non-sequential pairs in RefSeq exons, (4) AceView database flat file downloaded from UCSC representing transcripts constructed from human ESTs. The mapping results from (2) to (4) were aligned to human reference genome coordinates. In addition, they were aligned using STAR 2.3.0 to the human GRCh37-lite reference sequence without annotations. The final BAM file was constructed by selecting the best alignment among the five map events. We used HTSeq [49] to count the number of fragments that mapped to each gene (Gencode v 15), where each gene is considered as the union of all its exons. Then we normalized the count to FPKM (fragments per kilobase of exons per million fragments mapped) as the expression value of the gene. RNA-seq of both samples produced 55M reads each, with a 20X coverage of 43.561% of the exons in 1H3 and 43.992% of the exons in 1G3.
Ethics statement
This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The Institutional Animal Care and Use Committee (IACUC) of St. Jude Children’s Research Hospital (Animal protocol number 209–100171) approved the protocol.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Meza JL, Anderson J, Pappo AS, Meyer WH, Children’s Oncology G (2006) Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: the Children’s Oncology Group. J Clin Oncol 24 : 3844–3851. doi: 10.1200/JCO.2005.05.3801 16921036
2. Marshall AD, Grosveld GC (2012) Alveolar rhabdomyosarcoma—The molecular drivers of PAX3/7-FOXO1-induced tumorigenesis. Skelet Muscle 2 : 25. doi: 10.1186/2044-5040-2-25 23206814
3. Wexler LH, Ladanyi M (2010) Diagnosing alveolar rhabdomyosarcoma: morphology must be coupled with fusion confirmation. J Clin Oncol 28 : 2126–2128. doi: 10.1200/JCO.2009.27.5339 20351321
4. Douglass EC, Valentine M, Etcubanas E, Parham D, Webber BL, et al. (1987) A specific chromosomal abnormality in rhabdomyosarcoma. Cytogenet Cell Genet 45 : 148–155. doi: 10.1159/000132446 3691179
5. Osborne CS, Chakalova L, Brown KE, Carter D, Horton A, et al. (2004) Active genes dynamically colocalize to shared sites of ongoing transcription. Nat Genet 36 : 1065–1071. doi: 10.1038/ng1423 15361872
6. Lin C, Yang L, Tanasa B, Hutt K, Ju BG, et al. (2009) Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 139 : 1069–1083. doi: 10.1016/j.cell.2009.11.030 19962179
7. Mani RS, Tomlins SA, Callahan K, Ghosh A, Nyati MK, et al. (2009) Induced chromosomal proximity and gene fusions in prostate cancer. Science 326 : 1230. doi: 10.1126/science.1178124 19933109
8. Nikiforova MN, Stringer JR, Blough R, Medvedovic M, Fagin JA, et al. (2000) Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science 290 : 138–141. doi: 10.1126/science.290.5489.138 11021799
9. Osborne CS, Chakalova L, Mitchell JA, Horton A, Wood AL, et al. (2007) Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol 5: e192. doi: 10.1371/journal.pbio.0050192 17622196
10. Hakim O, Resch W, Yamane A, Klein I, Kieffer-Kwon KR, et al. (2012) DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature 484 : 69–74. doi: 10.1038/nature10909 22314321
11. Zhang Y, McCord RP, Ho YJ, Lajoie BR, Hildebrand DG, et al. (2012) Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell 148 : 908–921. doi: 10.1016/j.cell.2012.02.002 22341456
12. Barlow JH, Faryabi RB, Callen E, Wong N, Malhowski A, et al. (2013) Identification of early replicating fragile sites that contribute to genome instability. Cell 152 : 620–632. doi: 10.1016/j.cell.2013.01.006 23352430
13. Ren YX, Finckenstein FG, Abdueva DA, Shahbazian V, Chung B, et al. (2008) Mouse mesenchymal stem cells expressing PAX-FKHR form alveolar rhabdomyosarcomas by cooperating with secondary mutations. Cancer Res 68 : 6587–6597. doi: 10.1158/0008-5472.CAN-08-0859 18701482
14. Keller C, Hansen MS, Coffin CM, Capecchi MR (2004) Pax3:Fkhr interferes with embryonic Pax3 and Pax7 function: implications for alveolar rhabdomyosarcoma cell of origin. Genes Dev 18 : 2608–2613. doi: 10.1101/gad.1243904 15520281
15. Abraham J, Nunez-Alvarez Y, Hettmer S, Carrio E, Chen HI, et al. (2014) Lineage of origin in rhabdomyosarcoma informs pharmacological response. Genes Dev 28 : 1578–1591. doi: 10.1101/gad.238733.114 25030697
16. Lagha M, Sato T, Bajard L, Daubas P, Esner M, et al. (2008) Regulation of skeletal muscle stem cell behavior by Pax3 and Pax7. Cold Spring Harb Symp Quant Biol 73 : 307–315. doi: 10.1101/sqb.2008.73.006 19022756
17. Linardic CM, Naini S, Herndon JE, 2nd, Kesserwan C, Qualman SJ, et al. (2007) The PAX3-FKHR fusion gene of rhabdomyosarcoma cooperates with loss of p16INK4A to promote bypass of cellular senescence. Cancer Res 67 : 6691–6699. doi: 10.1158/0008-5472.CAN-06-3210 17638879
18. Calhabeu F, Hayashi S, Morgan JE, Relaix F, Zammit PS (2013) Alveolar rhabdomyosarcoma-associated proteins PAX3/FOXO1A and PAX7/FOXO1A suppress the transcriptional activity of MyoD-target genes in muscle stem cells. Oncogene 32 : 651–662. doi: 10.1038/onc.2012.73 22710712
19. Relaix F, Montarras D, Zaffran S, Gayraud-Morel B, Rocancourt D, et al. (2006) Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. J Cell Biol 172 : 91–102. doi: 10.1083/jcb.200508044 16380438
20. Torres R, Martin MC, Garcia A, Cigudosa JC, Ramirez JC, et al. (2014) Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system. Nat Commun 5 : 3964. doi: 10.1038/ncomms4964 24888982
21. Cong L, Ran FA, Cox D, Lin S, Barretto R, et al. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339 : 819–823. doi: 10.1126/science.1231143 23287718
22. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, et al. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337 : 816–821. doi: 10.1126/science.1225829 22745249
23. Mali P, Yang L, Esvelt KM, Aach J, Guell M, et al. (2013) RNA-guided human genome engineering via Cas9. Science 339 : 823–826. doi: 10.1126/science.1232033 23287722
24. Zuna J, Zaliova M, Muzikova K, Meyer C, Lizcova L, et al. (2010) Acute leukemias with ETV6/ABL1 (TEL/ABL) fusion: poor prognosis and prenatal origin. Genes Chromosomes Cancer 49 : 873–884. doi: 10.1002/gcc.20796 20589932
25. Chick WS, Mentzer SE, Carpenter DA, Rinchik EM, You Y (2004) Modification of an existing chromosomal inversion to engineer a balancer for mouse chromosome 15. Genetics 167 : 889–895. doi: 10.1534/genetics.104.026468 15238537
26. Klysik J, Dinh C, Bradley A (2004) Two new mouse chromosome 11 balancers. Genomics 83 : 303–310. doi: 10.1016/j.ygeno.2003.08.011 14706459
27. Nishijima I, Mills A, Qi Y, Mills M, Bradley A (2003) Two new balancer chromosomes on mouse chromosome 4 to facilitate functional annotation of human chromosome 1p. Genesis 36 : 142–148. doi: 10.1002/gene.10207 12872245
28. Zheng B, Mills AA, Bradley A (1999) A system for rapid generation of coat color-tagged knockouts and defined chromosomal rearrangements in mice. Nucleic Acids Res 27 : 2354–2360. doi: 10.1093/nar/27.11.2354 10325425
29. Zheng B, Sage M, Cai WW, Thompson DM, Tavsanli BC, et al. (1999) Engineering a mouse balancer chromosome. Nat Genet 22 : 375–378. doi: 10.1038/11949 10431243
30. Lee EC (2003) Clinical manifestations of sarin nerve gas exposure. JAMA 290 : 659–662. doi: 10.1001/jama.290.5.659 12902371
31. Siegel RW, Jain R, Bradbury A (2001) Using an in vivo phagemid system to identify non-compatible loxP sequences. FEBS Lett 505 : 467–473. doi: 10.1016/S0014-5793(01)02806-X 11576551
32. Yang Y, Seed B (2003) Site-specific gene targeting in mouse embryonic stem cells with intact bacterial artificial chromosomes. Nat Biotechnol 21 : 447–451. doi: 10.1038/nbt803 12627171
33. Valenzuela DM, Murphy AJ, Frendewey D, Gale NW, Economides AN, et al. (2003) High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat Biotechnol 21 : 652–659. doi: 10.1038/nbt822 12730667
34. Hanawa H, Hematti P, Keyvanfar K, Metzger ME, Krouse A, et al. (2004) Efficient gene transfer into rhesus repopulating hematopoietic stem cells using a simian immunodeficiency virus-based lentiviral vector system. Blood 103 : 4062–4069. doi: 10.1182/blood-2004-01-0045 14976042
35. Davicioni E, Finckenstein FG, Shahbazian V, Buckley JD, Triche TJ, et al. (2006) Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res 66 : 6936–6946. doi: 10.1158/0008-5472.CAN-05-4578 16849537
36. Marshall AD, van der Ent MA, Grosveld GC (2012) PAX3-FOXO1 and FGFR4 in alveolar rhabdomyosarcoma. Mol Carcinog 51 : 807–815. doi: 10.1002/mc.20848 21882254
37. Smith AJ, De Sousa MA, Kwabi-Addo B, Heppell-Parton A, Impey H, et al. (1995) A site-directed chromosomal translocation induced in embryonic stem cells by Cre-loxP recombination. Nat Genet 9 : 376–385. doi: 10.1038/ng0495-376 7795643
38. Keller C, Arenkiel BR, Coffin CM, El-Bardeesy N, DePinho RA, et al. (2004) Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev 18 : 2614–2626. doi: 10.1101/gad.1244004 15489287
39. Xia SJ, Barr FG (2004) Analysis of the transforming and growth suppressive activities of the PAX3-FKHR oncoprotein. Oncogene 23 : 6864–6871. doi: 10.1038/sj.onc.1207850 15286710
40. Marshall AD, Picchione F, Geltink RI, Grosveld GC (2013) PAX3-FOXO1 induces up-regulation of Noxa sensitizing alveolar rhabdomyosarcoma cells to apoptosis. Neoplasia 15 : 738–748. 23814486
41. Lagutina I, Conway SJ, Sublett J, Grosveld GC (2002) Pax3-FKHR knock-in mice show developmental aberrations but do not develop tumors. Mol Cell Biol 22 : 7204–7216. doi: 10.1128/MCB.22.20.7204-7216.2002 12242297
42. Relaix F, Polimeni M, Rocancourt D, Ponzetto C, Schafer BW, et al. (2003) The transcriptional activator PAX3-FKHR rescues the defects of Pax3 mutant mice but induces a myogenic gain-of-function phenotype with ligand-independent activation of Met signaling in vivo. Genes Dev 17 : 2950–2965. doi: 10.1101/gad.281203 14665670
43. Schnutgen F, Doerflinger N, Calleja C, Wendling O, Chambon P, et al. (2003) A directional strategy for monitoring Cre-mediated recombination at the cellular level in the mouse. Nat Biotechnol 21 : 562–565. doi: 10.1038/nbt811 12665802
44. Begum S, Emami N, Cheung A, Wilkins O, Der S, et al. (2005) Cell-type-specific regulation of distinct sets of gene targets by Pax3 and Pax3/FKHR. Oncogene 24 : 1860–1872. doi: 10.1038/sj.onc.1208315 15688035
45. Neville HL, Andrassy RJ, Lobe TE, Bagwell CE, Anderson JR, et al. (2000) Preoperative staging, prognostic factors, and outcome for extremity rhabdomyosarcoma: a preliminary report from the Intergroup Rhabdomyosarcoma Study IV (1991–1997). J Pediatr Surg 35 : 317–321. doi: 10.1016/S0022-3468(00)90031-9 10693687
46. Hanawa H, Persons DA, Nienhuis AW (2005) Mobilization and mechanism of transcription of integrated self-inactivating lentiviral vectors. J Virol 79 : 8410–8421. doi: 10.1128/JVI.79.13.8410-8421.2005 15956585
47. Morgenstern JP, Land H (1990) Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res 18 : 3587–3596. doi: 10.1093/nar/18.12.3587 2194165
48. Lam PY, Sublett JE, Hollenbach AD, Roussel MF (1999) The oncogenic potential of the Pax3-FKHR fusion protein requires the Pax3 homeodomain recognition helix but not the Pax3 paired-box DNA binding domain. Mol Cell Biol 19 : 594–601. 9858583
49. Anders S, Pyl PT, Huber W (2014) HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics.
50. Davicioni E (2006) Molecular classification, diagnosis and prognosis of pediatric rhabdomyosarcoma by oligonucleotide microarray analysis, PhD Thesis [PhD Thesis]. USC digital library, University of Southern California.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 2
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Genomic Selection and Association Mapping in Rice (): Effect of Trait Genetic Architecture, Training Population Composition, Marker Number and Statistical Model on Accuracy of Rice Genomic Selection in Elite, Tropical Rice Breeding Lines
- Discovery of Transcription Factors and Regulatory Regions Driving Tumor Development by ATAC-seq and FAIRE-seq Open Chromatin Profiling
- Evolutionary Signatures amongst Disease Genes Permit Novel Methods for Gene Prioritization and Construction of Informative Gene-Based Networks
- Proteotoxic Stress Induces Phosphorylation of p62/SQSTM1 by ULK1 to Regulate Selective Autophagic Clearance of Protein Aggregates
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy