
Mitochondrial and Cytoplasmic ROS Have Opposing Effects on Lifespan
The accumulation of oxidative damage caused by reactive oxygen species (ROS) has been proposed to be one of the main causes of aging. However, recent work indicates that low levels of ROS can be beneficial and promote longevity. In this paper, we use a long-lived mitochondrial mutant C. elegans strain clk-1 to further examine the relationship between ROS and lifespan. While it was originally believed that clk-1 mutants had increased lifespan as a result of decreased ROS production, ROS levels have been shown to be increased in clk-1 worms. We show that this increase in ROS is required for the longevity of clk-1 worms as treatment with antioxidants decreases clk-1 lifespan. Further, by using a genetic approach to increase ROS in specific subcellular compartments, we show that the location of the ROS is crucial in determining its effect on lifespan. Increasing ROS in the mitochondria markedly increases clk-1 lifespan, while increasing ROS in the cytoplasm decreases it. Finally, we show that the effect of increased ROS on stress resistance and physiologic rates is also dependent on the location of ROS within the cell but that both of these factors can be experimentally dissociated from lifespan.
Published in the journal:
. PLoS Genet 11(2): e32767. doi:10.1371/journal.pgen.1004972
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004972
Summary
The accumulation of oxidative damage caused by reactive oxygen species (ROS) has been proposed to be one of the main causes of aging. However, recent work indicates that low levels of ROS can be beneficial and promote longevity. In this paper, we use a long-lived mitochondrial mutant C. elegans strain clk-1 to further examine the relationship between ROS and lifespan. While it was originally believed that clk-1 mutants had increased lifespan as a result of decreased ROS production, ROS levels have been shown to be increased in clk-1 worms. We show that this increase in ROS is required for the longevity of clk-1 worms as treatment with antioxidants decreases clk-1 lifespan. Further, by using a genetic approach to increase ROS in specific subcellular compartments, we show that the location of the ROS is crucial in determining its effect on lifespan. Increasing ROS in the mitochondria markedly increases clk-1 lifespan, while increasing ROS in the cytoplasm decreases it. Finally, we show that the effect of increased ROS on stress resistance and physiologic rates is also dependent on the location of ROS within the cell but that both of these factors can be experimentally dissociated from lifespan.
Introduction
Reactive oxygen species (ROS) are toxic oxygen-containing molecules that can cause molecular damage within the cell. While it has been proposed that ROS-mediated damage contributes to aging [1], ROS have also been shown to act in signaling pathways [2]. In fact, recent work has demonstrated that mildly increasing ROS levels through treatment with the superoxide-generator paraquat can promote longevity [3–5]. In addition, elevated ROS have been proposed to alter physiologic rates [4,6,7] and contribute to the longevity of multiple long-lived mutants including clk-1 worms [3,7,8].
The clk-1 gene, which was identified in the worm Caenorhabditis elegans, was one of the first genes shown to increase lifespan in any organism [9,10]). clk-1 encodes a hydroxylase involved in the synthesis of ubiquinone (coenzyme Q) [11], a redox-active lipid which acts as an electron carrier in the electron transport chain of the mitochondria [12]. Worms with a mutation in clk-1 exhibit an overall slowing of numerous physiological rates including slow development, slow defecation cycle length, decreased brood size and slow rate of thrashing [9]. Interestingly, clk-1 worms also exhibit a slow rate of aging leading to a significant extension of mean and maximum lifespan [10].
As clk-1 worms have been shown to have a decreased rate of oxidative phosphorylation compared to wild-type worms [13–15], as well as decreased electron transport in the mitochondria [16,17], it was originally believed that the increased longevity of clk-1 worms resulted from decreased production of ROS as a result of less electron transport chain activity. However, altered mitochondrial function in clk-1 worms could also lead to increased leakage of electrons thereby increasing ROS generation. Distinguishing between these two possibilities has been made difficult by an inability to measure ROS directly in live worms [18].
Measurement of oxidative damage in clk-1 worms has demonstrated that clk-1 worms have decreased levels of protein carbonylation [14,19], decreased lipofuscin accumulation [20], and decreased 4-HNE [21,22]. In contrast, clk-1 worms have increased levels of F3-isoprostanes [23]. Attempts to measure ROS levels using redox dyes have suggested that ROS levels are increased in clk-1 worms. clk-1 worms have increased 2’,7’-dichlorofluorescein diacetate (DCF) fluorescence in whole worm extracts and increased dihydroethidium (DHE) fluorescence in the heads of whole worms [5], as well as increased DCF fluorescence (overall ROS) but normal MitoSox staining (superoxide levels) in isolated mitochondria [3]. In addition, ROS production, as measured by Amplex Red, which detects hydrogen peroxide, was found to be normal in whole mitochondria but increased in sub-mitochondrial particles from clk-1 worms [22]. Superoxide production potential has also been shown to be increased in clk-1 worms using a lucigenin-mediated light production assay [20]. Combined, these results indicate that clk-1 worms have increased levels of ROS but decreased levels of at least some types of oxidative damage. As oxidative damage is a balance between ROS production, ROS detoxification and repair, this suggests that antioxidant defense or damage repair may be increased in clk-1 worms. While the level of catalase activity has been shown to be increased in clk-1 worms [24,25], differing results have been obtained as to whether or not SOD protein or mRNA are increased [14,15,26,27].
In this paper, we determine the contribution of ROS to clk-1 lifespan and physiologic rates. We find that elevated ROS in clk-1 worms results in the upregulation of multiple classes of antioxidant genes during adulthood. We show that increased levels of ROS in the mitochondria further increase clk-1 lifespan, while cytoplasmic ROS decrease it. We also observe compartment specific effects of ROS on stress resistance and physiologic rates that do not account for the effects on lifespan. Finally, our data suggest that both ROS-dependent and ROS-independent mechanisms contribute to the longevity of clk-1 worms.
Results
clk-1 worms have increased antioxidant defenses
As clk-1 worms have increased levels of ROS [3,5] but decreased oxidative damage [14,21], we measured the expression levels of antioxidant enzymes to determine the extent to which increased antioxidant defenses could account for the observed levels of oxidative damage in clk-1 worms. We used quantitative real-time RT-PCR to measure the levels of four different antioxidant enzymes: superoxide dismutase (SOD), catalase (CTL), peroxiredoxin (PRDX) and thioredoxin (TRX). SOD detoxifies superoxide, CTL and PRDX detoxify hydrogen peroxide, and TRX reduces various substrates in the cell including PRDX. By examining all of the SODs, CTLs, PRDXs and TRXs, we were able to also determine which specific subcellular compartments had increased antioxidant defense.
We found that all five sod genes are upregulated in clk-1 worms compared to WT (Fig. 1A). We also observed increased expression of SOD-3 protein in clk-1 worms expressing SOD-3:GFP under a sod-3 promoter (Fig. 1B). Similarly, we observed increased expression of all three prdx genes (prdx-2, prdx-3, prdx-6), all three ctl genes (ctl-1, ctl-2, ctl-3) and trx-2 in clk-1 worms (Fig. 1C-E). In contrast, the levels of trx-3, which is expressed in the cytoplasm of the intestine [28], were significantly decreased in clk-1 worms. Thus, clk-1 worms exhibit significantly increased expression of antioxidant genes in multiple sub-cellular compartments of the cell including the cytoplasm (sod-1, ctl-1, prdx-2, trx-1), mitochondria (sod-2, sod-3, trx-2), peroxisome (ctl-2) and extra-cellularly (sod-4).

clk-1 worms are sensitive to acute oxidative stress but resistant to chronic oxidative stress in adulthood
Having shown that clk-1 worms have increased antioxidant defenses, we next wanted to determine whether this increase in antioxidant defense was sufficient to protect clk-1 worms from their elevated ROS production. To do this, we measured sensitivity to oxidative stress during development and adulthood using two different superoxide-generating compounds, paraquat and juglone. At the L2 stage of development, we found that clk-1 worms have decreased survival compared to WT on plates containing either 200 mM paraquat or 180 μM juglone (Fig. 2A,B). Similarly, we found that clk-1 worms fail to develop to adulthood under conditions of oxidative stress that are insufficient to prevent the development of WT worms (S1A Fig.). Combined, this indicates that clk-1 worms have increased sensitivity to oxidative stress during development.

To assess sensitivity to an acute exposure to oxidative stress during adulthood, we exposed clk-1 and WT worms to plates containing 200 mM paraquat or 180–300 μM juglone. In both cases, clk-1 worms had decreased survival compared to WT worms, indicating increased sensitivity to acute oxidative stress on day 1 of adulthood (Fig. 2C,D). In contrast, we found that in a chronic oxidative stress assay where worms were exposed to 4 mM paraquat beginning at day 1 of adulthood, clk-1 worms show increased survival compared to WT worms (Fig. 2E). This suggests that either clk-1 worms become resistant to oxidative stress during adulthood or that clk-1 worms respond differently to acute versus chronic stresses. To distinguish between these two possibilities, we examined sensitivity to an acute exposure to oxidative stress at various time points throughout adulthood by treating worms with 180 μM juglone. We found that at each time point, clk-1 worms exhibited increased sensitivity to oxidative stress compared to wild-type worms (Fig. 2F, S2 Fig.). Thus, clk-1 worms are sensitive to acute exposure to oxidative stress but resistant to chronic exposure. The increased resistance to chronic oxidative stress is consistent with clk-1 worms having decreased levels of oxidative damage. Note that testing sensitivity to chronic oxidative stress using juglone is unfeasible as its toxicity decreases rapidly within 8 hours of pouring plates (S1B Fig.).
Stress response pathways are upregulated in clk-1 worms and decline with age
As clk-1 worms have been shown to have elevated levels of ROS, it is plausible that the elevated levels of ROS trigger a protective response that results in their increased resistance to chronic oxidative stress and decreased oxidative damage. To investigate the mechanism underlying the increased resistance to chronic oxidative stress in clk-1 worms, we used reporter strains to monitor different stress response pathways that have been implicated in clk-1 lifespan. We used a gst-4 reporter to monitor upregulation of phase II detoxification pathways (skn-1 target)[29], an hsp-6 reporter to monitor the upregulation of the mitochondrial unfolded protein response (UPR)[30] and an nhr-57 reporter to monitor the upregulation of a hypoxic response (hif-1 target)[31].
On a wild-type background, Pgst-4 reporter activity decreased mildly with increasing age (Fig. 3A). In clk-1 worms, we found that activity of the Pgst-4 reporter was already markedly increased on day 1 of adulthood (Fig. 3A). As in WT worms, Pgst-4 reporter activity declined with age and was similar to WT worms at day 5 and beyond. Using the Phsp-6 reporter, we found that the mitoUPR was upregulated in clk-1 worms at day 1 of adulthood (Fig. 3B). In contrast to the Pgst-4 reporter, the maximum increase was observed at days 3 and 4 of adulthood and then declined with age. Finally, with the Pnhr-57 reporter, we observed a mild activation of a hypoxic response that was relatively constant as the worms aged (Fig. 3C).
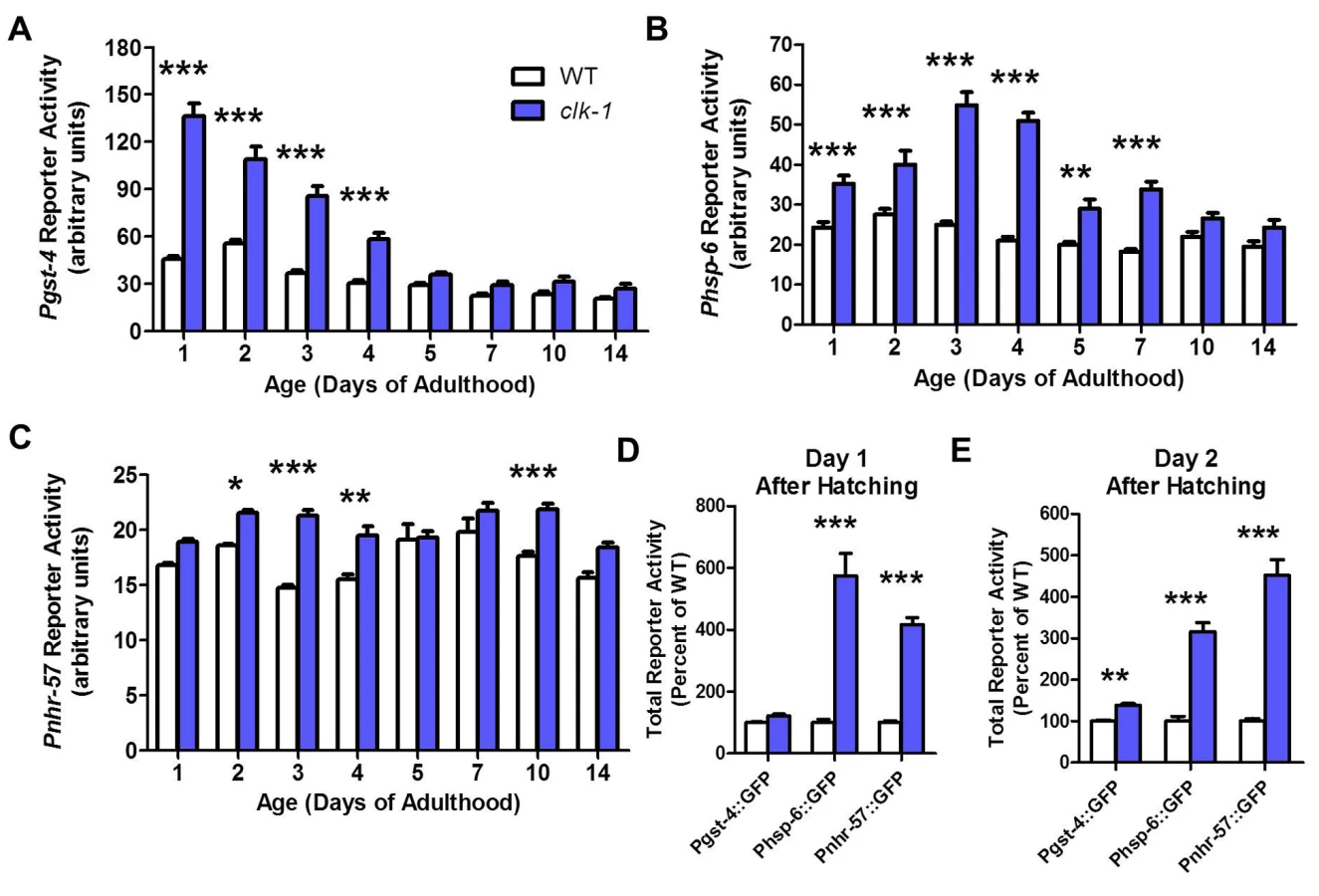
To determine whether the decline in Pgst-4 reporter activity resulted from a decrease in oxidative stress or a decrease in the ability to activate skn-1 in response to oxidative stress, we treated day 1 adult worms and day 10 adult worms with 2 mM paraquat. We found that aged worms failed to activate the gst-4 promoter (S3 Fig.). This suggests that the decline in Pgst-4 reporter activity in clk-1 worms results from a decrease in the ability to respond to oxidative stress with increasing age and does not indicate that the levels of oxidative stress decline to wild-type levels in clk-1 worms. Interestingly, treatment with paraquat does not further increase Pgst-4 reporter activity in clk-1 worms suggesting that these worms already have a maximal response (S3 Fig.).
Since all of the reporters were already upregulated in young adult clk-1 worms, we next sought to determine if there is a critical time period during development when these reporters become activated. Examination of clk-1 and WT worms revealed that the Phsp-6 and Pnhr-57 reporters were already upregulated on day 1 after hatching (Fig. 3D), while the Pgst-4 reporter was not significantly activated until day 2 (Fig. 3E). Combined this indicates that all three stress response pathways are engaged during embryonic or very early development and continue to be upregulated into adulthood.
Antioxidant genes become upregulated during adulthood in clk-1 worms
To further investigate the time course of gene expression changes in clk-1 worms that contribute to stress resistance and longevity, we next used quantitative real-time RT-PCR to examine the expression of select antioxidant genes at three time points: the L2 stage of larval development, day 1 of adulthood and day 4 of adulthood. We found that for all three antioxidant genes tested (sod-3, prdx-2 and ctl-1), mRNA levels were not increased in L2 worms but were upregulated during adulthood (Fig. 4A-C). We also found that the skn-1 target gcs-1 (gamma glutamylcysteine synthetase), which performs the rate limiting step in glutathione synthesis, was upregulated in adult clk-1 worms (Fig. 4D). The upregulation of antioxidant genes during adulthood likely contributes to the increased resistance to chronic oxidative stress observed in clk-1 worms.

Elevated superoxide levels exhibit a compartment specific effect on clk-1 lifespan
We have previously shown that clk-1 lifespan is dramatically increased by the deletion of sod-2 [7]. As SOD-2 encodes the primary mitochondrial sod gene, this indicates that increasing mitochondrial superoxide levels through decreased detoxification increases clk-1 lifespan. To determine whether this increase in lifespan is specific to mitochondrial superoxide, or whether increased superoxide in other subcellular compartments would also increase clk-1 lifespan, we used a genetic approach to specifically decrease superoxide detoxification in different subcellular compartments. As C. elegans have five sod genes [32], we generated clk-1; sod double mutants for the remaining four sod genes and measured lifespan. sod-1, sod-2 and sod-4 encode the primary cytoplasmic, mitochondrial and extracellular SODs, respectively, while sod-3 and sod-5 encode inducible mitochondrial and cytoplasmic SODs, which are normally expressed at low levels [33–38].
In contrast to the increased lifespan observed in clk-1;sod-2 double mutants, we found that deletions in either of the cytoplasmic sod genes, sod-1 or sod-5, resulted in a significant decrease in lifespan (Fig. 5A,B; S4A–S4B Fig.). Although the magnitude of the decrease in lifespan between clk-1 and clk-1;sod-5 worms is small, the difference is highly significant (p < 0.001 for mean lifespan, maximum lifespan and Log-rank test comparing survival curves). As we have previously reported, the deletion of sod-2 dramatically increased the lifespan of clk-1 worms (Fig. 5C; S4C Fig.). Finally, the deletion of sod-3 or sod-4 had no effect on the lifespan of clk-1 worms (Fig. 5D,E; S4D–S4E Fig.).

We also examined the lifespan of clk-1 worms with deletions in both cytoplasmic sod genes (clk-1;sod-1;sod-5 worms) and both mitochondrial matrix sod genes (clk-1;sod-2;sod-3 worms). We found that clk-1;sod-1;sod-5 worms had decreased lifespan compared to clk-1 worms, while clk-1;sod-2;sod-3 worms have increased lifespan compared to clk-1 worms, though the increase was less than in clk-1;sod-2 worms (Fig. 5F). Combined these results demonstrate a compartment specific effect of ROS on lifespan: elevated superoxide levels in the mitochondria increases clk-1 lifespan, while elevated superoxide in the cytoplasm decreases it (Fig. 5G,H).
Compartment specific effect of ROS on stress resistance does not account for the effect of ROS on lifespan
Having shown that clk-1 lifespan is modulated by levels of ROS, we next sought to determine the extent to which the different effects on longevity could be attributed to changes in resistance to stress. If differences in stress resistance contributed to the changes in longevity observed in clk-1;sod double mutants, we would expect clk-1;sod-2 mutants to have increased resistance to stress compared to clk-1 worms and clk-1;sod-1 and clk-1;sod-5 worms to have decreased resistance. We measured sensitivity to oxidative stress during development, acutely during adulthood and in a chronic assay during adulthood. We found that deletion of sod-1 or sod-2 further increased sensitivity to oxidative stress during development in clk-1 worms (Fig. 6A), while deletion of other sod genes had no effect. At this concentration of 0.2 mM paraquat, we previously showed that all of the individual sod deletion mutants except for sod-2 were able to develop to adulthood [7]. Of note, clk-1;sod-1 double mutants are more sensitive to oxidative stress during development than either single mutant.

To examine sensitivity to oxidative stress acutely on day 1 of adulthood, we measured the survival of the clk-1;sod double mutants on plates containing 240 μM juglone. We found that deletion of either cytoplasmic sod gene (sod-1, sod-5) or the extracellular sod gene (sod-4) resulted in increased sensitivity to oxidative stress in clk-1 worms (Fig. 6B). We previously showed that deletion of sod-1 or sod-2 increases sensitivity to juglone on a WT background, with the greatest deficit being observed in the sod-1 deletion mutants [7].
Finally, we examined sensitivity to oxidative stress throughout adulthood by treating worms with 4 mM paraquat beginning on day 1 of adulthood. In this assay, we found that deletion of sod-1, sod-2 or sod-5 all increased sensitivity to oxidative stress in clk-1 worms (Fig. 6C). We have previously shown that deletion of sod-1, sod-2 or sod-3 decreases survival in this assay on a wild-type background [7].
Overall, the effect of deleting individual sod genes on sensitivity to oxidative stress in clk-1 worms does not account for the observed effects on lifespan. While both sod-1 and sod-2 deletions markedly increase the sensitivity of clk-1 worms to paraquat-induced oxidative stress, these sod genes have opposite effects on clk-1 lifespan.
Compartment specific effect of ROS on physiologic rates does not account for the effect of ROS on lifespan
In addition to increased lifespan, clk-1 and other mitochondrial mutants have been shown to exhibit slow physiologic rates, which have been proposed to contribute to their longevity. To determine whether alterations in physiologic rates could account for the effect of deleting individual sod genes on clk-1 lifespan, we compared the physiologic rates of clk1;sod double mutants to clk-1 worms. clk-1 worms develop significantly slower than wild-type worms. Deletion of sod-1 or sod-2 further lengthened the development time in clk-1 worms while deletion of sod-4 restored development time towards that of wild-type (S5A Fig.). Defecation cycle length is also significantly longer in clk-1 worms compared to wild-type worms. Deletion of sod-2 further exacerbated this phenotype while deletion of sod-1 or sod-4 resulted in near wild-type rates of defecation (S5B Fig.). The self-brood size of clk-1 worms is decreased compared to wild-type worms and is further decreased by the deletion of sod-1 or sod-2 (S5C Fig.). In contrast, deletion of sod-4 partially rescues the decreased brood size of clk-1 worms (S5C Fig.). Finally, thrashing rate is decreased in clk-1 worms compared to wild-type worms but is restored towards that of wild-type by deletion of sod-1, sod-2 or sod-4 (S5D Fig.). Thus, aside from defecation cycle length, the deletion of sod-1 and sod-2 has similar effects on clk-1 physiologic rates, despite having opposite effects on lifespan. This indicates that changes in clk-1 physiologic rates induced by deletion of individual sod genes cannot account for the observed differences in lifespan. Interestingly, the deletion of sod-4, the extracellular sod gene, rescues the slow development, slow defecation, decreased brood size and slow thrashing rate of clk-1 worms, yet has no impact on clk-1 lifespan. This provides a clear demonstration that clk-1 physiologic rates can be experimentally dissociated from lifespan.
Modulating ROS levels in clk-1 worms suggests multiple mechanisms of lifespan extension
As ROS have been implicated in the longevity of clk-1 worms [3], we next examined the effect of pharmacologic alterations of ROS levels on clk-1 lifespan. We have previously shown that treating wild-type worms with the superoxide generator paraquat at concentrations between 0.05 and 0.5 mM increased lifespan, while paraquat concentrations of 2 mM and above resulted in decreased lifespan [4]. If elevated ROS levels in clk-1 worms contribute to their longevity, we would predict that (1) treating clk-1 worms with antioxidants would result in decreased lifespan and (2) clk-1 worms would exhibit a decrease in their optimum paraquat concentration compared to wild-type worms, since ROS levels are already increased in clk-1 worms [3,5].
To test the effect of antioxidants on clk-1 lifespan, we treated clk-1 worms with three different antioxidants: 10 mM vitamin C, 25 μM α-lipoic acid and 25 μM epigallocatechin gallate (EGCG). In each case, we found that antioxidant treatment significantly reduced clk-1 lifespan (Fig. 7A). However, antioxidant-treated clk-1 worms were still long-lived compared to wild-type worms, suggesting the possibility that there are ROS-dependent and ROS-independent mechanisms involved in clk-1 longevity. Treatment of clk-1 worms with 10 mM vitamin C did not affect development time, brood size or thrashing rate, thereby providing further confirmation that clk-1 longevity can be experimentally dissociated from their slow physiologic rates (S6 Fig.).

To determine the optimum level of ROS for clk-1 lifespan, we treated clk-1 and wild-type worms with a concentration dilution series of paraquat ranging from 0.05 mM to 4 mM. As we have previously shown, wild-type worms exhibit a maximum lifespan at 0.1 mM paraquat (Fig. 7B) [4]. Surprisingly, we found that clk-1 worms do not exhibit a decrease in their optimum paraquat concentration with respect to lifespan. In fact, we observed the opposite. Despite having elevated levels of ROS compared to wild-type worms, we found that clk-1 worms exhibit an increase in their optimum paraquat concentration with respect to lifespan of 0.2 mM (Fig. 7B). Comparing the pattern observed for wild-type and clk-1 worms reveals an upward shift in the pattern for clk-1 worms compared to wild-type.
For comparison, we examined the lifespan of sod-2 worms exposed to the same concentration dilution series of paraquat. sod-2 worms have increased lifespan that is due to elevated levels of mitochondrial superoxide [7]. In contrast to clk-1 worms, treatment with paraquat does not increase the lifespan of sod-2 worms at any concentration (Fig. 7C). As would be predicted for worms with elevated ROS, the optimum paraquat concentration with respect to lifespan is decreased compared to wild-type. It is also noteworthy that the peak lifespans for sod-2 worms and wild-type worms treated with paraquat is similar but occur at different concentrations of paraquat, 0 mM and 0.1 mM, respectively. The fact that clk-1 worms exhibit a distinct pattern from what is observed in sod-2 mutants suggests that clk-1 longevity is at least partially mediated by a ROS-independent mechanism (S7 Fig.).
As deletion of sod-2 markedly increases the lifespan of clk-1 worms, we next sought to determine the optimum paraquat concentration for clk-1;sod-2 worm lifespan. As with sod-2 worms, we found that there was no concentration of paraquat that could increase clk-1;sod-2 lifespan (Fig. 7D). Interestingly, at very low concentrations of paraquat (0–0.05 mM paraquat), clk-1;sod-2 worms live longer than wild-type worms, at low concentrations of paraquat they are equally long-lived while at high concentrations of paraquat (0.5 mM paraquat) wild-type worms have increased survival compared to clk-1;sod-2 worms.
Increasing mitochondrial ROS levels can still extend lifespan in worms with a detrimental level of cytoplasmic ROS
If mitochondrial and cytoplasmic superoxide have different effects on lifespan then increasing mitochondrial superoxide should still be able to increase the lifespan of clk-1;sod-1 and clk-1;sod-5 worms, which have decreased lifespan compared to clk-1 worms resulting from elevated levels of cytoplasmic superoxide. To test this, we examined whether treatment with paraquat could increase the lifespan of the clk-1;sod double mutants. We found that, in contrast to clk-1;sod-2 double mutants, increasing ROS levels through treatment with paraquat could increase the lifespan of all of the other clk-1;sod double mutants, including clk-1;sod-1 double mutants (Fig. 8A-E, S8 Fig.). This clearly indicates that the decreased lifespan in clk-1;sod-1 worms does not result from too much ROS. Instead, it suggests that it is high levels of ROS specifically in the cytoplasm that act to decrease clk-1 lifespan. This result also demonstrates that increasing mitochondrial ROS can increase lifespan despite detrimental levels of cytoplasmic ROS. Conversely, the fact that clk-1;sod-1 worms treated with paraquat have a shorter lifespan than clk-1 worms treated with paraquat indicates that increasing cytoplasmic ROS through deletion of sod-1 shortens the lifespan of worms that have extended longevity resulting from increased mitochondrial ROS.

To provide further support for the compartment specific effects of ROS on lifespan, we generated clk-1;sod-1;sod-2 triple mutants, which are predicted to have increased cytoplasmic ROS compared to clk-1;sod-2 worms and increased mitochondrial ROS compared to clk-1;sod-1 worms. We found that clk-1;sod-1;sod-2 worms have increased lifespan compared to clk-1;sod-1 worms but decreased lifespan compared to clk-1;sod-2 worms (Fig. 8F). Thus, increasing cytoplasmic ROS through the deletion of sod-1 decreases clk-1;sod-2 lifespan, while increasing mitochondrial ROS through the deletion of sod-2 increases clk-1;sod-1 lifespan. This indicates that mitochondrial and cytoplasmic ROS have opposing effects on lifespan.
Discussion
While ROS have been proposed to be one of the main causes of aging, accumulating evidence suggests that ROS can also be beneficial. Deletion of the mitochondrial sod gene, sod-2, results in increased lifespan despite increased levels of oxidative damage[7]. In addition, treating wild-type worms with low concentrations of paraquat (paraquat), a compound that increases levels of superoxide, also results in increased lifespan[4,5]. There are now multiple examples of interventions that cause elevated ROS that also result in an increase in lifespan that is reduced by antioxidants, including treatment of worms with 2-deoxy-D-glucose[39], lonidamine[40], arsenic[41], complex I inhibitors[42], or D-glucosamine[43]. Similarly, mutations in the insulin-IGF1 receptor gene daf-2[8]; overexpression of sirtuin (sir-2.1)[44]; and mutations in genes that affect electron transport chain function (clk-1, isp-1, and nuo-6)[9,45,46] have been shown to increase both ROS levels and lifespan. Importantly, the ability of ROS to increase lifespan is conserved across species. Yeast treated with menadione to elevate mitochondrial ROS have increased lifespan[47]. As in worms, mice with a mutation in the clk-1 ortholog Mclk1[48] and mice treated with D-glucosamine[43] exhibit elevated ROS levels and increased lifespan. In order to elucidate the mechanism by which ROS act to increase lifespan, it will be important to define the levels and localization of ROS that are required to promote longevity.
Compartment specific effect of ROS on lifespan
To gain insight into the sub-cellular localization requirements for ROS to increase lifespan, we used a genetic approach to specifically increase ROS in different sub-cellular compartments in clk-1 worms, which have increased sensitivity to ROS. As it is currently not possible to detect levels of superoxide in a specific subcellular compartment in a living worm, we were unable to show directly that deletion of the sod genes results in increased superoxide levels in the compartment in which the gene was expressed. However, our previous work showing increased sensitivity to oxidative stress and increased oxidative damage in the sod deletion mutants suggests that superoxide levels are increased. Since superoxide is not able to cross biological membranes[49], any increase in superoxide levels should be limited to the subcellular compartment in which the sod gene was normally expressed.
While deletion of the primary mitochondrial sod gene (sod-2) results in a dramatic increase in clk-1 lifespan, loss of either cytoplasmic sod gene (sod-1 or sod-5) significantly decreased clk-1 lifespan. This is particularly surprising for sod-5, which is normally expressed at very low levels[50], and is only mildly upregulated in clk-1 worms. Deletion of sod-1 has previously been shown to have either no impact or a small negative impact on lifespan on a wild-type background, while sod-5 has not been shown to affect longevity [7,50,51]. The deletion of sod-3, the inducible mitochondrial sod gene, or sod-4, the extracellular sod gene, had no impact on lifespan in clk-1 or wild-type worms. While sod-3 is expressed in the same subcellular compartment as sod-2, it is likely that deletion of sod-3 does not increase mitochondrial superoxide levels enough to increase clk-1 lifespan as sod-3 normally accounts for only 1% of the total sod mRNA in a cell [50]. Overall these results indicate that elevated superoxide levels act specifically in the mitochondria, and not other sub-cellular compartments, to increase lifespan. Interestingly, mice that are heterozygous for the targeted inactivation of the mouse ortholog of clk-1 (Mclk1), which also exhibit extended longevity [52], have increased oxidative damage in the mitochondria and decreased damage in the cytoplasm in young mice [53]. Based on our present results, both the increased mitochondrial ROS and decreased cytoplasmic ROS could contribute to the extended lifespan of Mclk1+/- mice.
Multiple mechanisms of lifespan extension in clk-1 worms
To specifically test the role of elevated ROS in the longevity of clk-1 worms, we examined the effect of pharmacologically increasing and decreasing ROS levels on clk-1 lifespan. In contrast to the predictions of the free radical theory of aging, we found that decreasing ROS decreased clk-1 lifespan, while increasing ROS increased clk-1 lifespan. Treating clk-1 worms with three different antioxidants (vitamin C, α-lipoic acid and epigallocatechin gallate) all resulted in a similar partial reduction of clk-1 lifespan. This suggests that the increased ROS levels in clk-1 worms are required for their full longevity. The fact that the anti-oxidant treated lifespan was still longer than wild-type suggests the possibility that there are also ROS-independent mechanisms contributing to clk-1 longevity. A previous study examined the effect of a different antioxidant, N-acetyl cysteine (NAC), on clk-1 lifespan and observed no decrease [3]. While it is unclear why treatment with NAC did not decrease clk-1 lifespan, it may have been due to differences in the mechanism of action between the different antioxidants, due to the fact that NAC affects bacterial growth, due to differences in experimental conditions or due to non-antioxidant effects of NAC on lifespan.
Since clk-1 worms have elevated levels of ROS at baseline, we predicted that their optimum superoxide concentration would be decreased compared to wild-type worms. However, we observed that the maximum lifespan for clk-1 worms occurred at a concentration of paraquat that was higher than the optimum concentration for wild-type worms with respect to longevity. This was in stark contrast to what we observed in sod-2 deletion mutants where even small increases in superoxide levels resulted in a decrease in lifespan. The differing response of clk-1 and sod-2 worms to paraquat treatment suggests that different mechanisms are responsible for their longevity. Combined with our observation that antioxidant treatment decreases clk-1 lifespan, this suggests that clk-1 has both ROS-dependent and ROS-independent contributions to their longevity.
Compartment specific effect of ROS on stress resistance and physiologic rates
In addition to having a compartment specific effect on lifespan, we also found that ROS have a compartment specific effect on both sensitivity to oxidative stress and physiologic rates (summarized in S1 Table). However, the effect of sod gene deletions on physiologic rates or sensitivity to oxidative stress was in no case predictive of the effect on lifespan. Relative to clk-1 worms, both clk-1;sod-1 and clk-1;sod-2 worms have slow development, decreased brood size, arrestment of larval development under conditions of oxidative stress and increased sensitivity to chronic oxidative stress during adulthood. Thus, deletion of either sod-1 or sod-2 in clk-1 worms results in similar changes in physiologic rates and stress sensitivity despite the fact that they have opposite effects on lifespan. This indicates that stress resistance and physiologic rates can be experimentally dissociated from longevity.
In examining the effect of increased ROS on the already slow physiologic rates of clk-1 worms, we found that the deletion of sod-4 partially restores all of the clk-1 phenotypes towards wild-type except for lifespan. This finding is particularly interesting given the fact that deletion of sod-4 does not alter physiologic rates on a wild-type background [4]. The fact that sod-4 deletion rescues development rate, defecation rate, brood size and thrashing rate but does not affect clk-1 lifespan indicates that the slowing of other physiologic rates is not necessary for the extended lifespan of clk-1 worms. Conversely, we observed that treating clk-1 worms with vitamin C had no effect on development time, brood size or thrashing rate, but significantly decreased clk-1 lifespan towards wild-type. Combined, these findings indicate that independent mechanisms are responsible for the increased longevity and slow physiologic rates in clk-1 worms.
Upregulation of antioxidant genes during adulthood causes increased resistance to chronic oxidative stress
In examining the sensitivity of clk-1 worms to oxidative stress, we found that sensitivity to oxidative stress was dependent on the age of the worms and whether it was an acute or chronic exposure to stress. During larval development and with acute exposure to oxidative stress during adulthood, clk-1 worms have increased sensitivity to oxidative stress. However, in a chronic assay throughout adulthood, clk-1 worms exhibit increased resistance to oxidative stress. This is consistent with observations of decreased oxidative damage in clk-1 worms [14,19,21,22,54]. We found that the change in resistance to oxidative stress corresponds to the upregulation of antioxidant genes. During larval development, we found that the expression of representative antioxidant genes (sod-3, prdx-2, ctl-1) were equally expressed in clk-1 and wild-type worms. In contrast, adult clk-1 worms have increased expression of sod, prdx, ctl and some trx genes, demonstrating a broad upregulation of antioxidant defense. Thus, it is plausible that altered electron transport chain function in clk-1 worms leads to elevated levels of ROS, which result in increased sensitivity to oxidative stress early in life but also trigger the upregulation of antioxidant defense genes that compensate for the increased level of ROS production leading to an increased resistance to chronic exposure to oxidative stress during adulthood. We also show that clk-1 worms have increased activity of a gst-4 reporter construct and increased gcs-1 mRNA levels. As both gst-4 and gcs-1 are targets of skn-1, this indicates that phase II detoxification pathways are also upregulated in clk-1 worms and likely also contribute to their resistance to oxidative stress during adulthood. Our results are in agreement with a previous microarray experiment showing increased expression of antioxidant genes in clk-1 worms including sod-3, gst-4 and gst-8[27].
Conclusions
Overall, we show that mitochondrial superoxide increases clk-1 lifespan, while cytoplasmic superoxide decreases it. Combined with our previous results showing that mild elevation of mitochondrial superoxide levels can increase lifespan, while high levels of superoxide are toxic [4], this clearly indicates that the effect of superoxide on lifespan is dependent on where and how much superoxide is present. Our data demonstrates that clk-1 worms have increased expression of antioxidant defenses during adulthood that results in resistance to chronic exposure to oxidative stress, and suggest that there are ROS-dependent and ROS-independent mechanisms contributing to clk-1 longevity (S9 Fig.).
Materials and Methods
Strains
The following strains were used in these experiments: N2 (wild-type), sod-1(tm776), sod-1(tm783), sod-2(gk257), sod-2(ok1030), sod-3(tm760), sod-4(gk101), sod-5(tm1146), sod-5(tm1246), clk-1(qm30), isp-1(qm150). Strains obtained from external sources were outcrossed with our N2 worms for a minimum of 6 generations. For these experiments the following double and triple mutant strains were generated: clk-1(qm30);sod-1(tm776), clk-1(qm30);sod-1(tm783), clk-1(qm30);sod-2(ok1030), clk-1(qm30);sod-3(tm760), clk-1(qm30);sod-4(gk101), clk-1(qm30);sod-5(tm1146), clk-1(qm30);sod-5(tm1246), clk-1(qm30);sod-1(tm783);sod-2(ok1030), clk-1(qm30);sod-2(ok1030);sod-3(tm760). We did not observe any phenotypic differences between clk-1(qm30);sod-1(tm776) and clk-1(qm30);sod-1(tm783) or clk-1(qm30);sod-5(tm1146) and clk-1(qm30);sod-5(tm1246) worms. All of the sod deletions and the clk-1 deletion were confirmed by PCR. All strains were maintained at 20°C.
Lifespan analysis
Lifespan studies were completed at 20°C with a minimum of 3 independent trials and an initial number of 80 worms per strain per trial on plates containing 100 μM FUdR (Sigma). Survival plots shown represent pooled data from multiple trials. For lifespan assays involving 10 mM vitamin C, 25 μM α-lipoic acid, 25 μM epigallocatechin gallate or 0.05–4 mM paraquat, concentrated solutions of the chemical to be added were prepared fresh on the day that the plates were made. The chemical was added to the plates just prior to pouring. Plates were made fresh weekly. In assessing the effect of antioxidants on lifespan, we planned to include 10 mM N-acetyl cysteine (NAC). However, we observed abnormal bacterial growth on these plates, which had the potential to confound the lifespan results. Accordingly, lifespan was tested only on the three antioxidants listed above.
Paraquat and juglone sensitivity assays
Juglone sensitivity assays were completed in triplicate with 30–40 worms per strain per trial at 20°C on plates containing 180–300 μM juglone (Sigma). For this assay, plates were made fresh on the day of the assay as the toxicity of juglone decreases rapidly over time (S1B Fig.). Worms that developed on NGM plates were transferred to juglone plates at either the L2 or young adult stage and survival was monitored for 6 hours. To assess the ability of worms to develop under oxidative stress, a minimum of 40 eggs were placed on plates containing 0.1–0.25 mM paraquat and seeded with OP50 bacteria. The furthest developmental stage reached was recorded. As a chronic assay of oxidative stress, 20 worms per strain per trial were grown to adulthood on NGM plates and then transferred to plates containing 4 mM paraquat at the young adult stage (day 1 of adulthood). The plates contained 100 μM FUdR to prevent paraquat-induced internal hatching of progeny. Survival was monitored daily. To assess sensitivity to acute oxidative stress using paraquat, worms that were grown on NGM plates were transferred to plates containing 200 mM paraquat at either the L2 or young adult stage. Survival was monitored hourly for a period of 15 hours. For each assay a minimum of 3 biological replicates was completed.
Quantitative real-time RT-PCR
RNA was isolated from young adult worms using TRIZOL reagent (Invitrogen). Subsequently, 1 μg of RNA was converted to cDNA using the Quantitect Reverse Transcription kit (Qiagen). 1 μl of the resulting cDNA preparation was used for quantitative real-time PCR using the Quantitect SYBR Green PCR kit and a Biorad iCycler RT-PCR machine. Results represent the average of at least three independent biological samples.
Measurement of fluorescent reporter activity
A Cellomics arrayscan high-content imager was used to measure reporter activity in larval worms. Worms were suspended in M9 buffer, immobilized with levamisole to a final concentration of 2 mM and transferred to a 96-well dish at a density such that worms were not touching. Brightfield and fluorescent images were captured by the imager and used to quantify whole worm fluorescence. Reporter activity in adult worms was assessed through measurement of whole worm florescence. For each of 3 biological replicates, approximately 10 worms were paralyzed with levamisole (2mM), mounted on an NGM plate and florescent images were captured using an AVT Stingray F145B camera and VimbaViewer 1.1.2 software. ImageJ was used to measure the average pixel intensity.
Post-embryonic development
Eggs were collected and allowed to hatch over a period of 3 hours. After 3 hours, L1 worms were transferred to a new plate and monitored for development to an adult worm. Results are the average of at least three independent trials with 20 worms per trial.
Defecation
Defecation cycle length in young adult worms was measured as the average time between consecutive pBoc contractions. Results represent a minimum of 3 trials with 10 worms per trial.
Self-brood size
To determine the average number of progeny produced by each strain, L4 worms were placed on individual NGM plates. Worms were transferred daily until egg laying ceased and the total number of live progeny produced was counted.
Statistical analysis
Survival plots were compared using the log-rank test. The maximum lifespan of a given strain was measured as the average of the lifespan of the longest-living 5% of worms of that strain. A one way ANOVA was used to compare mean and maximum lifespan between strains. For analyses involving multiple groups and time points, a two way ANOVA was used to assess significance followed by Bonferroni post-hoc tests for detecting specific differences between groups.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Harman D (1956) Aging: a theory based on free radical and radiation chemistry. JGerontol 11 : 298–300. 13332224
2. D'Autreaux B, Toledano MB (2007) ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. NatRevMolCell Biol 8 : 813–824.
3. Yang W, Hekimi S (2010) A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol 8: e1000556. doi: 10.1371/journal.pbio.1000556 21151885
4. Van Raamsdonk JM, Hekimi S (2012) Superoxide dismutase is dispensable for normal animal lifespan. Proc Natl Acad Sci U S A 109 : 5785–5790. doi: 10.1073/pnas.1116158109 22451939
5. Lee SJ, Hwang AB, Kenyon C (2010) Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol 20 : 2131–2136. doi: 10.1016/j.cub.2010.10.057 21093262
6. Shibata Y, Branicky R, Landaverde IO, Hekimi S (2003) Redox regulation of germline and vulval development in Caenorhabditis elegans. Science 302 : 1779–1782. 14657502
7. Van Raamsdonk JM, Hekimi S (2009) Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet 5: e1000361. doi: 10.1371/journal.pgen.1000361 19197346
8. Zarse K, Schmeisser S, Groth M, Priebe S, Beuster G, et al. (2012) Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell metabolism 15 : 451–465. doi: 10.1016/j.cmet.2012.02.013 22482728
9. Wong A, Boutis P, Hekimi S (1995) Mutations in the clk-1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics 139 : 1247–1259. 7768437
10. Lakowski B, Hekimi S (1996) Determination of life-span in Caenorhabditis elegans by four clock genes. Science 272 : 1010–1013. 8638122
11. Ewbank JJ, Barnes TM, Lakowski B, Lussier M, Bussey H, et al. (1997) Structural and functional conservation of the Caenorhabditis elegans timing gene clk-1. Science 275 : 980–983. 9020081
12. Stepanyan Z, Hughes B, Cliche DO, Camp D, Hekimi S (2006) Genetic and molecular characterization of CLK-1/mCLK1, a conserved determinant of the rate of aging. ExpGerontol.
13. Braeckman BP, Houthoofd K, De Vreese A, Vanfleteren JR (1999) Apparent uncoupling of energy production and consumption in long-lived Clk mutants of Caenorhabditis elegans. Curr Biol 9 : 493–496. 10330373
14. Yang W, Li J, Hekimi S (2007) A Measurable increase in oxidative damage due to reduction in superoxide detoxification fails to shorten the life span of long-lived mitochondrial mutants of Caenorhabditis elegans. Genetics 177 : 2063–2074. 18073424
15. Van Raamsdonk JM, Meng Y, Camp D, Yang W, Jia X, et al. (2010) Decreased energy metabolism extends life span in Caenorhabditis elegans without reducing oxidative damage. Genetics 185 : 559–571. doi: 10.1534/genetics.110.115378 20382831
16. Felkai S, Ewbank JJ, Lemieux J, Labbe JC, Brown GG, et al. (1999) CLK-1 controls respiration, behavior and aging in the nematode Caenorhabditis elegans. Embo J 18 : 1783–1792. 10202142
17. Kayser EB, Sedensky MM, Morgan PG, Hoppel CL (2004) Mitochondrial oxidative phosphorylation is defective in the long-lived mutant clk-1. JBiolChem 279 : 54479–54486. 15269213
18. Van Raamsdonk JM, Hekimi S (2010) Reactive Oxygen Species and Aging in Caenorhabditis elegans: Causal or Casual Relationship? Antioxid Redox Signal 13 : 1911–1953. doi: 10.1089/ars.2010.3215 20568954
19. Baker BM, Nargund AM, Sun T, Haynes CM (2012) Protective coupling of mitochondrial function and protein synthesis via the eIF2alpha kinase GCN-2. PLoS Genet 8: e1002760. doi: 10.1371/journal.pgen.1002760 22719267
20. Braeckman BP, Houthoofd K, Brys K, Lenaerts I, De Vreese A, et al. (2002) No reduction of energy metabolism in Clk mutants. Mech Ageing Dev 123 : 1447–1456. 12425951
21. Kayser EB, Sedensky MM, Morgan PG (2004) The effects of complex I function and oxidative damage on lifespan and anesthetic sensitivity in Caenorhabditis elegans. Mech Ageing Dev 125 : 455–464. 15178135
22. Yang YY, Gangoiti JA, Sedensky MM, Morgan PG (2009) The effect of different ubiquinones on lifespan in Caenorhabditis elegans. Mech Ageing Dev 130 : 370–376. doi: 10.1016/j.mad.2009.03.003 19428456
23. Labuschagne CF, Stigter EC, Hendriks MM, Berger R, Rokach J, et al. (2013) Quantification of in vivo oxidative damage in Caenorhabditis elegans during aging by endogenous F3-isoprostane measurement. Aging Cell 12 : 214–223. doi: 10.1111/acel.12043 23279719
24. Taub J, Lau JF, Ma C, Hahn JH, Hoque R, et al. (1999) A cytosolic catalase is needed to extend adult lifespan in C. elegans daf-C and clk-1 mutants. Nature 399 : 162–166. 10335847
25. Petriv OI, Rachubinski RA (2004) Lack of peroxisomal catalase causes a progeric phenotype in Caenorhabditis elegans. J Biol Chem 279 : 19996–20001. 14996832
26. Honda Y, Honda S (1999) The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. Faseb J 13 : 1385–1393. 10428762
27. Cristina D, Cary M, Lunceford A, Clarke C, Kenyon C (2009) A regulated response to impaired respiration slows behavioral rates and increases lifespan in Caenorhabditis elegans. PLoS Genet 5: e1000450. doi: 10.1371/journal.pgen.1000450 19360127
28. Jimenez-Hidalgo M, Kurz CL, Pedrajas JR, Naranjo-Galindo FJ, Gonzalez-Barrios M, et al. (2014) Functional characterization of thioredoxin 3 (TRX-3), a Caenorhabditis elegans intestine-specific thioredoxin. Free radical biology & medicine 68 : 205–219. doi: 10.1016/j.ejpn.2014.12.007 25553845
29. Leiers B, Kampkotter A, Grevelding CG, Link CD, Johnson TE, et al. (2003) A stress-responsive glutathione S-transferase confers resistance to oxidative stress in Caenorhabditis elegans. Free radical biology & medicine 34 : 1405–1415. doi: 10.1016/j.ejpn.2014.12.007 25553845
30. Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, et al. (2004) Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. Journal of cell science 117 : 4055–4066. 15280428
31. Zhang Y, Shao Z, Zhai Z, Shen C, Powell-Coffman JA (2009) The HIF-1 hypoxia-inducible factor modulates lifespan in C. elegans. PloS one 4: e6348. doi: 10.1371/journal.pone.0006348 19633713
32. Landis GN, Tower J (2005) Superoxide dismutase evolution and life span regulation. MechAgeing Dev 126 : 365–379. 15664623
33. Giglio AM, Hunter T, Bannister JV, Bannister WH, Hunter GJ (1994) The copper/zinc superoxide dismutase gene of Caenorhabditis elegans. BiochemMolBiolInt 33 : 41–44.
34. Giglio MP, Hunter T, Bannister JV, Bannister WH, Hunter GJ (1994) The manganese superoxide dismutase gene of Caenorhabditis elegans. BiochemMolBiolInt 33 : 37–40.
35. Suzuki N, Inokuma K, Yasuda K, Ishii N (1996) Cloning, sequencing and mapping of a manganese superoxide dismutase gene of the nematode Caenorhabditis elegans. DNA Res 3 : 171–174. 8905235
36. Hunter T, Bannister WH, Hunter GJ (1997) Cloning, expression, and characterization of two manganese superoxide dismutases from Caenorhabditis elegans. JBiolChem 272 : 28652–28659. 9353332
37. Fujii M, Ishii N, Joguchi A, Yasuda K, Ayusawa D (1998) A novel superoxide dismutase gene encoding membrane-bound and extracellular isoforms by alternative splicing in Caenorhabditis elegans. DNA Res 5 : 25–30. 9628580
38. Jensen LT, Culotta VC (2005) Activation of CuZn superoxide dismutases from Caenorhabditis elegans does not require the copper chaperone CCS. JBiolChem 280 : 41373–41379. 16234242
39. Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, et al. (2007) Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell metabolism 6 : 280–293. 17908557
40. Schmeisser S, Zarse K, Ristow M (2011) Lonidamine extends lifespan of adult Caenorhabditis elegans by increasing the formation of mitochondrial reactive oxygen species. Hormone and metabolic research = Hormon - und Stoffwechselforschung = Hormones et metabolisme 43 : 687–692. doi: 10.1055/s-0031-1286308 21932172
41. Schmeisser S, Schmeisser K, Weimer S, Groth M, Priebe S, et al. (2013) Mitochondrial Hormesis Links Low-Dose Arsenite Exposure to Lifespan Extension. Aging cell doi: 10.1111/acel.12076 23534459
42. Schmeisser S, Priebe S, Groth M, Monajembashi S, Hemmerich P, et al. (2013) Neuronal ROS signaling rather than AMPK/sirtuin-mediated energy sensing links dietary restriction to lifespan extension. Mol Metab 2 : 92–102. doi: 10.1016/j.molmet.2013.02.002 24199155
43. Weimer S, Priebs J, Kuhlow D, Groth M, Priebe S, et al. (2014) D-Glucosamine supplementation extends life span of nematodes and of ageing mice. Nature communications 5 : 3563. doi: 10.1038/ncomms4563 24714520
44. Schmeisser K, Mansfeld J, Kuhlow D, Weimer S, Priebe S, et al. (2013) Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nature chemical biology doi: 10.1038/nchembio.1352 24077178
45. Feng J, Bussiere F, Hekimi S (2001) Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Developmental cell 1 : 633–644. 11709184
46. Yang W, Hekimi S (2010) Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging cell 9 : 433–447. doi: 10.1111/j.1474-9726.2010.00571.x 20346072
47. Pan Y, Schroeder EA, Ocampo A, Barrientos A, Shadel GS (2011) Regulation of yeast chronological life span by TORC1 via adaptive mitochondrial ROS signaling. Cell metabolism 13 : 668–678. doi: 10.1016/j.cmet.2011.03.018 21641548
48. Liu X, Jiang N, Hughes B, Bigras E, Shoubridge E, et al. (2005) Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes & development 19 : 2424–2434. doi: 10.1016/j.etap.2014.11.031 25555259
49. Gus'kova RA, Ivanov II, Kol'tover VK, Akhobadze VV, Rubin AB (1984) Permeability of bilayer lipid membranes for superoxide (O2-.) radicals. Biochim Biophys Acta 778 : 579–585. 6095912
50. Doonan R, McElwee JJ, Matthijssens F, Walker GA, Houthoofd K, et al. (2008) Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev 22 : 3236–3241. doi: 10.1101/gad.504808 19056880
51. Yen K, Mobbs CV (2010) Evidence for only two independent pathways for decreasing senescence in Caenorhabditis elegans. Age (Dordr) 32 : 39–49. doi: 10.1007/s11357-009-9110-7 19662517
52. Liu X, Jiang N, Hughes B, Bigras E, Shoubridge E, et al. (2005) Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev 19 : 2424–2434. 16195414
53. Lapointe J, Hekimi S (2008) Early Mitochondrial Dysfunction in Long-lived Mclk1+/ - Mice. JBiolChem 283 : 26217–26227. doi: 10.1074/jbc.M803287200 18635541
54. Braeckman BP, Houthoofd K, Brys K, Lenaerts I, De Vreese A, et al. (2002) No reduction of energy metabolism in Clk mutants. Mech Ageing Dev 123 : 1447–1456. 12425951
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 2
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Genomic Selection and Association Mapping in Rice (): Effect of Trait Genetic Architecture, Training Population Composition, Marker Number and Statistical Model on Accuracy of Rice Genomic Selection in Elite, Tropical Rice Breeding Lines
- Discovery of Transcription Factors and Regulatory Regions Driving Tumor Development by ATAC-seq and FAIRE-seq Open Chromatin Profiling
- Evolutionary Signatures amongst Disease Genes Permit Novel Methods for Gene Prioritization and Construction of Informative Gene-Based Networks
- Proteotoxic Stress Induces Phosphorylation of p62/SQSTM1 by ULK1 to Regulate Selective Autophagic Clearance of Protein Aggregates
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy