
SPDEF Inhibits Prostate Carcinogenesis by Disrupting a Positive Feedback Loop in Regulation of the Foxm1 Oncogene
Development of prostate cancer is a multistep process that involves the loss of tumor suppressor functions and activation of oncogenes. SPDEF transcription factor is expressed in normal prostate epithelium and its expression changes during prostate carcinogenesis (PCa). Since the role of SPDEF in PCa remains controversial, we generated transgenic mice with loss - and gain-of-function of SPDEF to demonstrate that SPDEF functions as a tumor suppressor in PCa. In animal models, the loss of SPDEF promoted PCa and increased the levels of Foxm1, a well-known oncogenic protein. Overexpression of SPDEF in prostate epithelium decreased PCa and reduced Foxm1 levels. Proliferation defects in SPDEF-containing tumor cells were corrected by re-expression of Foxm1, providing direct evidence that SPDEF inhibits tumor cell proliferation through Foxm1. We further showed that SPDEF directly bound to Foxm1 promoter and prevented its auto-regulatory activation. In prostate cancer patients, the low SPDEF and high Foxm1 were found in most aggressive prostate tumors that were associated with poor prognosis. The combined two-gene signature of low SPDEF and high Foxm1 was a strong predictor of survival in prostate cancer patients. The present study identified novel molecular mechanism of prostate cancer progression, providing a crosstalk between SPDEF tumor suppressor and Foxm1 oncogene.
Published in the journal:
. PLoS Genet 10(9): e32767. doi:10.1371/journal.pgen.1004656
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004656
Summary
Development of prostate cancer is a multistep process that involves the loss of tumor suppressor functions and activation of oncogenes. SPDEF transcription factor is expressed in normal prostate epithelium and its expression changes during prostate carcinogenesis (PCa). Since the role of SPDEF in PCa remains controversial, we generated transgenic mice with loss - and gain-of-function of SPDEF to demonstrate that SPDEF functions as a tumor suppressor in PCa. In animal models, the loss of SPDEF promoted PCa and increased the levels of Foxm1, a well-known oncogenic protein. Overexpression of SPDEF in prostate epithelium decreased PCa and reduced Foxm1 levels. Proliferation defects in SPDEF-containing tumor cells were corrected by re-expression of Foxm1, providing direct evidence that SPDEF inhibits tumor cell proliferation through Foxm1. We further showed that SPDEF directly bound to Foxm1 promoter and prevented its auto-regulatory activation. In prostate cancer patients, the low SPDEF and high Foxm1 were found in most aggressive prostate tumors that were associated with poor prognosis. The combined two-gene signature of low SPDEF and high Foxm1 was a strong predictor of survival in prostate cancer patients. The present study identified novel molecular mechanism of prostate cancer progression, providing a crosstalk between SPDEF tumor suppressor and Foxm1 oncogene.
Introduction
Development of cancer is a multistep process that involves gain-of-function mutations in oncogenes and inactivation of tumor suppressor genes, leading to increased tumor cell proliferation, survival and resistance to cell cycle arrest [1]. In normal prostate epithelium, relatively low rates of cell proliferation are balanced by a low rate of apoptosis [2]. In contrast, prostatic intraepithelial neoplasia (PIN) and early invasive carcinomas are characterized by an increase in the proliferation rate. Advanced and/or metastatic prostate cancers also display a significant decrease in the rate of apoptosis. Altered cell-cycle control plays a key role in progression of prostate cancer. Published studies have demonstrated significant activation of the PI3K/Akt and Erk mitogen-activated protein kinase (MAPK) signaling pathways in prostate carcinomas [3], [4] and the loss of PTEN tumor suppressor [5].
The transgenic adenocarcinoma of the mouse prostate (TRAMP) model recapitulates multiple stages of human PCa by using the probasin promoter to drive the expression of the SV40 virus large and small T antigen (Tag) oncoprotein in prostate epithelial cells [6]. Tag inactivates the tumor suppressor proteins retinoblastoma (Rb), p53, and PP2A serine/threonine–specific phosphatase [7], inducing prostate tumors in adult mice. SV40 T antigens also induce expression of the Foxm1 oncogenic protein, a member of the Forkhead Box (Fox) family of transcription factors [8]. Foxm1 is activated by the Ras/Erk signaling pathway [9] and transcriptionally induces cell cycle-regulatory genes, including Cdc25b, Cyclin B1, Plk1, Aurora B. Recent studies demonstrated that Foxm1 is required for initiation and progression of various cancers, including prostate cancer [9]–[11].
SPDEF (SAM pointed domain containing ETS transcription factor) belongs to the family of ETS transcription factors containing a conserved DNA binding domain (ETS domain). The ETS domain binds to a conserved central “GGA” trinucleotide motif [12]. SPDEF expression is restricted to the epithelial layers of the prostate or other lumen-containing organs including lung, breast, ovary, stomach and colon [13]–[16]. SPDEF regulates mucus secretion, goblet cell differentiation, tumor progression and metastasis [13], [16]–[20]. In prostate, SPDEF directly interacts with the androgen receptor functioning as a co-activator to induce prostate-specific antigen (PSA) in LNCaP prostate tumor cells [21]. The prostate-specific Nkx3.1 nuclear protein directly inhibits SPDEF and prevents SPDEF-mediated PSA activation, indicating a potential role of SPDEF in prostate cancer [22].
Currently, the role of SPDEF in cancer pathogenesis remains controversial. Both, reduced expression of SPDEF in prostate, breast, ovarian and colon tumors [23]–[25], and increased SPDEF in breast, ovarian and prostate tumors [15], [25]–[27] has been reported. Expression of SPDEF in vitro in either PC3 prostate or MDA-MB231 breast carcinoma cells decreased cellular proliferation and increased apoptosis [24], [28]. On the other hand, transfection of MCF10A and MCF12A breast carcinoma cells with SPDEF increased cell growth, cell invasiveness and tumorigenicity [26]. It is unclear whether SPDEF functions as tumor suppressor or oncogene in prostate carcinogenesis. Given the apparent lack of in vivo data using animal models, we generated several distinct mouse models of prostate cancer with loss-of-function and gain-of-function of SPDEF to demonstrate that SPDEF inhibits prostate carcinogenesis by preventing a positive feedback mechanism regulating the Foxm1 oncogene.
Results
Prostate carcinogenesis is increased in SPDEF−/− mice
To determine the role of SPDEF during prostate carcinogenesis in vivo, we crossed SPDEF−/− mice [14] with transgenic TRAMP mice that express SV40 T large and small antigens under the control of probasin promoter to drive oncogenic transformation of prostate epithelial cells [7]. SPDEF−/−/TRAMP and control TRAMP mouse prostates were analyzed at 23 weeks of age. A significant increase in prostate weight and size was observed in SPDEF−/−/TRAMP mice compared to TRAMP mice (Figure 1A). SPDEF mRNA was undetectable in SPDEF−/−/TRAMP prostates, confirming the efficient knockout of SPDEF (Figure 1B). The numbers of PH3-positive and Ki67-positive cells were increased in SPDEF−/−/TRAMP tumors, indicating the increased cellular proliferation in SPDEF−/−/TRAMP prostates (Figure 1D). Moreover, the mRNA levels of several proliferation-specific genes, such as Cdc25b, Cyclin B1, Cyclin A2, Plk1, Cks1, Aurora B and Topo 2 alpha, were increased in the SPDEF−/−/TRAMP prostates, a finding consistent with the increased cellular proliferation (Figure 1C). In the absence of TRAMP transgene, SPDEF−/− mice did not develop prostate tumors or PINs, and expression of proliferation-specific genes in SPDEF−/− prostates was unchanged (Figure S1A–B). Thus, the loss of SPDEF was not sufficient to initiate prostate tumors; however SPDEF deficiency promoted prostate carcinogenesis in the TRAMP mouse model. These results suggest that SPDEF functions as tumor suppressor in SV40 T-antigen induced prostate cancer.

Expression of SPDEF in prostate adenocarcinoma cells decreased carcinogenesis in an orthotopic model
We next determined whether the transgenic expression of SPDEF was sufficient to inhibit prostate carcinogenesis. Since TRAMP C2 prostate adenocarcinoma cells do not express endogenous SPDEF, we used SPDEF lentivirus to generate TRAMP C2 cells with the stable over-expression of SPDEF (SPDEF OE, Figure 2A). Over-expression of SPDEF decreased cellular proliferation and decreased mRNA encoding several proliferation-specific genes in cultured TRAMP C2 cells (Figure 2A–B). Moreover, expression of SPDEF decreased cell migration and reduced anchorage-independent growth of TRAMP C2 cells on soft agar (Figure 2C–D). Similar effects were observed in MycCap prostate adenocarcinoma cells (Figure S2). Thus, SPDEF may function as a tumor suppressor in prostate adenocarcinoma cells.

To determine whether expression of SPDEF is sufficient to inhibit prostate carcinogenesis in vivo, SPDEF-overexpressing TRAMP C2 cells (SPDEF OE) were injected into prostates of syngeneic C57BL/6 mice and their tumorigenic potential was compared to parental TRAMP C2 cells. In this orthotopic model, high levels of SPDEF mRNA and protein were maintained in SPDEF OE tumors (Figure 3B and Figure 3C, left panels). Expression of SPDEF reduced the tumor burden (Figure 3A), decreased the number of Ki67 and PH3-positive cells (Figure 3C and 3D, left panels), and reduced mRNA levels of proliferation-specific genes Cdc25b, Cyclin B1, Cyclin A2, PLK1, CKS1, Aurora B and Topo2 alpha in the prostate tumors (Figure 3D, right panel). Altogether, our data indicate that overexpression of SPDEF decreased proliferation of TRAMP C2 and MycCap cancer cells and inhibited prostate carcinogenesis in the orthotopic model.

Transgenic expression of SPDEF in prostate epithelium decreased prostate carcinogenesis
We generated transgenic mice with prostate epithelial-specific expression of SPDEF under Doxycycline (Dox) control. These transgenic mice contained TRE-SPDEF [13], LoxP-stop-LoxP-rtTA(Rosa26) and the Probasin-Cre transgenes (Pb-Cretg/+/LoxP-stop-LoxP-rtTA(Rosa26)tg/tg/TRE-SPDEF mice, abbreviated as SPDEF OE). In SPDEF OE mice, Dox treatment induced the expression of SPDEF in prostate epithelial cells through excision of the LoxP-stop-LoxP cassette by the Probasin-driven Cre recombinase (Figure 4A). SPDEF OE mice were healthy and fertile, their prostates were normal. To induce prostate cancer, the SPDEF OE mice were bred with TRAMP transgenic mice to generate TRAMP/SPDEF OE mice (Figure 4A). SPDEF mRNA was increased in TRAMP/SPDEF OE mice after Dox treatment (Figure 4C). Expression of SPDEF in TRAMP mice was sufficient to inhibit prostate carcinogenesis as demonstrated by decreased tumor weight in TRAMP/SPDEF OE mice when compared to TRAMP mice (Figure 4B). The number of PH3-positive cells in TRAMP/SPDEF OE prostate tumors was reduced by 60% (Figure 4D). Moreover, the mRNA of several cell cycle regulatory genes, such as Cdc25b, Cyclin B1, Cyclin A2, Plk1, Cks1, Aurora B, and Topo 2 alpha were decreased in prostates of TRAMP/SPDEF OE mice (Figure 4C, right panel), a finding consistent with decreased cellular proliferation and decreased tumor sizes. These results indicate that increased expression of SPDEF in prostate epithelium is sufficient to decrease prostate carcinogenesis in the TRAMP mouse model.

SPDEF inhibits Foxm1 expression during prostate carcinogenesis
Our in vitro and in vivo studies demonstrated that SPDEF inhibits Cdc25b, Cyclin B1, Cyclin A2, Plk1, Cks1, Aurora B, and Topo 2 alpha mRNAs, all of which are known targets of Foxm1 transcription factor [29]. Since Foxm1 is up-regulated in mouse and human prostate cancers and is required for prostate carcinogenesis [8], [10], we tested whether SPDEF inhibits Foxm1. In transgenic TRAMP/SPDEF OE mice, overexpression of SPDEF in prostate decreased Foxm1 mRNA and protein (Figure 5A). Consistent with findings in the transgenic mice, expression of SPDEF in TRAMP C2 cells decreased Foxm1 mRNA and protein expression in orthotopic prostate tumors (Figure 5B). Deletion of SPDEF in TRAMP/SPDEF−/− mouse prostates caused a 30-fold increase in Foxm1 mRNA and increased Foxm1 staining (Figure 5C). Finally, an inverse correlation between SPDEF and Foxm1 was found in human prostate cancers using two independent human prostate cancer microarray datasets, GSE21034 [30] and GSE16560 [31]. Expression levels of SPDEF and Foxm1 were compared between indolent and lethal prostate cancers and high-risk and low-risk sample groups (Figure 6A–B). High-risk samples were derived from patients with surviving less than 12 months and low-risk samples from patients surviving more than 192 months [32]. In high-risk group of patients and in lethal prostate cancers, Foxm1 mRNA was significantly overexpressed while SPDEF mRNA was significantly decreased (Figure 6A). Inverse correlation between Foxm1 and SPDEF expression levels was also found when metastatic and primary tumor samples were compared (Figure S3). The Kaplan-Meier survival analysis demonstrated that, on its own, the low SPDEF expression or high Foxm1 expression are associated with worse overall survival (Figure 6C). However, combined two-gene signature of low SPDEF and high Foxm1 is a much stronger predictor of survival (Figure 6C). These results suggest the existence of inverse correlation between SPDEF and Foxm1 expression in mouse and human prostate cancers.


Re-expression of Foxm1 in the SPDEF-positive prostate adenocarcinoma cells restored tumor cell proliferation
We next examined Foxm1 levels in SPDEF OE TRAMP C2 cells in vitro. A 30-fold reduction in Foxm1 mRNA was observed in SPDEF OE cells compared to control cells expressing empty vector (Figure 7A, upper panel). Decreased Foxm1 levels in the SPDEF OE cells were associated with reduced cell proliferation (Figure 7A, middle and bottom panels). Thus, SPDEF inhibits cell proliferation and decreases Foxm1.

To determine whether SPDEF inhibits cell proliferation, at least in part, through Foxm1, we restored Foxm1 expression in SPDEF OE cells using a lentiviral vector. Increasing the levels of Foxm1 in cultured SPDEF OE cells restored their proliferation to the level of control TRAMP C2 cells (Figure 7A, middle and bottom panels). Flow cytometery demonstrated that increased expression of SPDEF delayed entry of TRAMP C2 cells into S-phase at 12 hours after serum stimulation (Figure 7B, upper and middle panels). Re-expression of Foxm1 in SPDEF OE cells restored the cell cycle progression (Figure 7B, bottom panels) in cultured cells. Furthermore, re-expression of Foxm1 in SPDEF-expressing cancer cells restored tumor weight in orthotopic mouse model of prostate cancer (Figure 7C, left panel), coinciding with increased number of Ki-67-positive (Figure 7D, upper panels) and PH3-positive (Figure 7D, bottom panels) tumor cells. Interestingly, re-expression of Foxm1 in SPDEF-deficient tumor cells restored cell migration (Figure S4A), coinciding with elevated levels of MMP2, MMP9 and MMP13 (Figure S4B). Since SPDEF inhibits cell migration, at least in part, through MMP9 and MM13 [18], increased expression of these MMP genes can contribute to Foxm1-mediated rescue of cell migration in SPDEF-deficient prostate tumor cells. Altogether, these results indicate that SPDEF decreases proliferation and migration of prostate cancer cells through inhibition of Foxm1.
SPDEF binds to and inhibits the Foxm1 promoter
Since SPDEF expression inversely correlated with Foxm1 expression in mouse and human prostate tumors (Figure 5), we examined the possibility that SPDEF directly represses the Foxm1 promoter. An evolutionary conserved Foxm1 binding site was identified in −745/−734 bp region of the mouse Foxm1 gene. We also identified three potential SPDEF binding sites in the −3.7 Kb Foxm1 promoter region (Figure 8A, schematic drawing). Interestingly, one of them, the −670/−660 SPDEF binding site is located near the Foxm1 binding site, suggesting that SPDEF may influence a positive feedback mechanism regulating the Foxm1 promoter. The −3.7 Kb mouse Foxm1 promoter and its deletion mutants were cloned into luciferase (LUC) reporter vectors and used in co-transfection experiments in TRAMP C2 prostate adenocarcinoma cells (Figure 8B). CMV-Foxm1 plasmid increased activity of the −3.7 Kb Foxm1 promoter (Figure 8B). Deletion of the −778/−500 bp region, containing −745/−734 bp Foxm1 binding site (Luc construct IV), completely abolished Foxm1 promoter activity; whereas deletion of −3758/−1167 bp or −1167/−778 bp regions had no effect (Figure 8B). Thus, the −745/−734 bp Foxm1 site is required for auto-regulation of the −3.7 Kb Foxm1 promoter by Foxm1. Co-transfection with CMV-SPDEF plasmid inhibited Foxm1 transcriptional activity in a dose-dependent manner (Figure 8B, right panel), indicating that SPDEF is a transcriptional repressor of Foxm1 gene. Interestingly, deletion of the −745/−660 bp SPDEF/Foxm1 site completely abolished the ability of SPDEF to inhibit the Foxm1 promoter, whereas deletion of −3663/−3653 and −1067/−1057 bp SPDEF sites had no effect (Figure 8B, middle panel). Furthermore, disruption of Foxm1 binding site (construct V, Figure 8B) inhibited Foxm1 promoter activity (Figure 8B and Figure S4C–D), confirming that the −745/−738 bp region is required for the auto-regulatory activation of the Foxm1 promoter. Likewise, disruption of the SPDEF binding site (construct VI, Figure 8B) prevented the inhibitory effect of SPDEF on Foxm1 promoter activity (Figure 8B and Figure S4C), indicating that SPDEF inhibits Foxm1 promoter through the −670/−660 bp region.
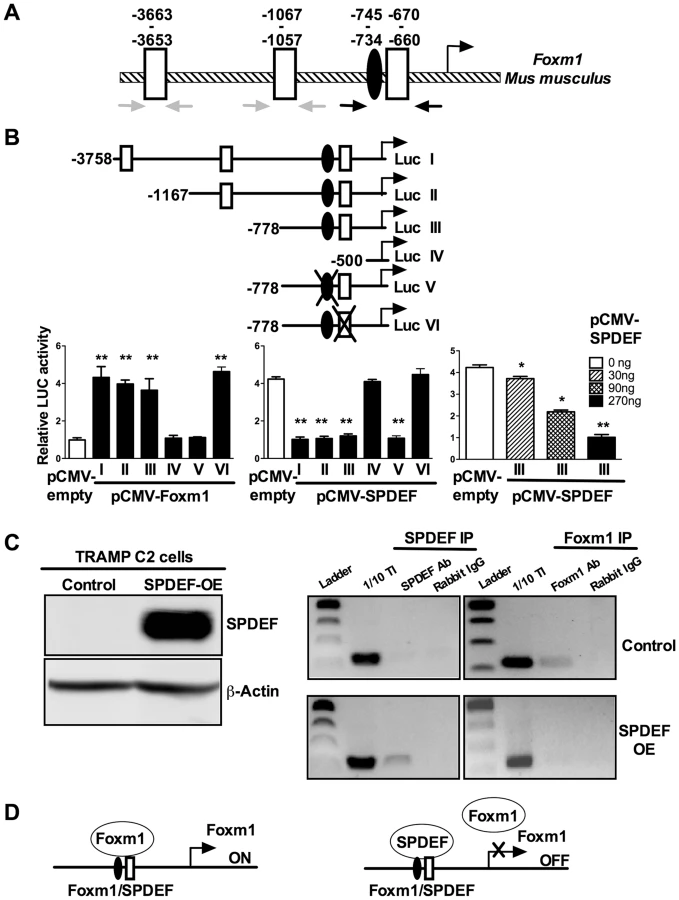
We next examined whether SPDEF physically binds to the −745/−660 bp Foxm1 promoter DNA and whether this binding inhibits the ability of Foxm1 to bind to its own binding site. Chromatin Immunoprecipitation (ChIP) assay was performed using TRAMP C2 cells. Since TRAMP C2 cells do not express endogenous SPDEF, we expressed SPDEF by lentiviral infection (Figure 8C, left panel SPDEF OE). In control cells lacking SPDEF, endogenous Foxm1 protein bound to the −745/−660 bp region, whereas there was no binding of SPDEF to this region (Figure 8C, right panels). In SPDEF OE cells, SPDEF bound to the −745/−660 bp Foxm1 promoter region, while the binding of Foxm1 to the same region was lost (Figure 8C, right panels). Neither Foxm1 nor SPDEF bound to the −3663/−3653 and to the −1067/−1057 bp Foxm1 sites. Altogether, SPDEF protein physically binds to the −745/−660 bp Foxm1 promoter region and directly inhibits the Foxm1 promoter activity by interfering with the ability of Foxm1 to activate its own promoter through an auto-regulatory element in the −745/−660 bp region (Figure 8D).
Discussion
Previous studies demonstrated that SPDEF is expressed in normal prostate epithelium and prostate tumors in mice and human patients [21], [27]; however, its role in prostate carcinogenesis is still controversial. SPDEF was originally discovered as a transcription factor that directly interacts with androgen receptor and functions as its co-activator to induce expression of prostate specific antigen (PSA) in LNCaP prostate tumor cells [21]. Several groups reported the increased expression of SPDEF during progression of prostate, breast and ovarian cancers, suggesting the oncogenic role of SPDEF [15], [27]. SPDEF was found to be required for tumorigenesis in ER-positive subset of breast cancers [33]. However, the loss of SPDEF during tumor progression was reported by other groups. In advanced prostate cancer and prostate cancer-derived cell lines, SPDEF was either decreased or lost [23], [34]. It was also shown that SPDEF expression is decreased during the transition from low-grade to high-grade prostate cancer [23], [34], [35]. Two studies had shown correlation between decreased expression of SPDEF and poor prognosis in prostate cancer [23], [35]. Likewise, high SPDEF levels were found in prostate cancer patients who had a prolong response to androgen deprivation therapy [36]. Expression and knock-down of SPDEF in different cell lines in vitro, provided controversial results [24], [26], [28], [37].
The diverse regulatory factors altering SPDEF expression may explain some of the discrepancies between published studies. Increased SPDEF mRNA, as reported in some studies, might not reflect protein levels of SPDEF, although it was suggested that the protein status of SPDEF strongly correlates with the transcript level [13]. Furthermore, differences in the specificity of antibodies and technical procedures used in each study may have contributed to contradicting results. Alternatively, the prostate cancer cell lines used in the studies have various genetic origins, and this heterogeneity may influence reported findings. While some studies examined SPDEF expression in tumor samples classified by Gleason scores [23], [34], [35], others used pooled tumor samples [27]. Nevertheless, the controversy regarding the role of SPDEF in prostate carcinogenesis remains unresolved. Our present findings provide both in vivo and in vitro support for tumor suppressive role of SPDEF in prostate cancer. We found inverse correlations between SPDEF and the Foxm1 oncogene in both mouse and human prostate tumors and demonstrated that SPDEF inhibits tumor cells proliferation through Foxm1 oncogene.
Our previous studies demonstrated that aberrant expression of Foxm1 in all cell types using ubiquitous Rosa26 promoter accelerated prostate carcinogenesis in TRAMP and LADY transgenic mice [9]. Prostate cancers contain heterogeneous populations of cells including epithelial, inflammatory and stromal cells that enhances Foxm1 expression during carcinogenesis [38]. Macrophage-specific inactivation of Foxm1 reduced cancer-associated inflammation and decreased tumor growth in chemically-induced lung cancer models [39], indicating that Foxm1 expression in macrophages is important for regulation of cancer-associated inflammation during tumor promotion. Recently, we established that prostate epithelial-specific expression of Foxm1 is required for prostate carcinogenesis. Deletion of Foxm1 from prostate epithelial cells in PB-Cre/Foxm1fl/fl/TRAMP mice prevented prostate carcinogenesis but did not change SPDEF levels [10]. Critical role of Foxm1 in proliferation of prostate tumor cells was confirmed in orthotopic model using Foxm1-deficient MycCap prostate adenocarcinoma cells [10]. Therefore, understanding the mechanisms regulating Foxm1 is particularly relevant to the pathogenesis of prostate cancer. In the present study, we established that SPDEF directly binds to an evolutionally-conserved region in the Foxm1 promoter and inhibits Foxm1 transcriptional activity via an auto-regulatory element of the Foxm1 promoter. Although, the ability of Foxm1 to activate its own promoter was previously shown, molecular mechanism underlying this regulation was not characterized [40]. Present studies establish, that Foxm1 activates its own promoter by binding to −745/−734 bp site, which is important for auto-regulatory loop. We also identified an evolutionally conserved SPDEF binding site in the close proximity to this Foxm1 binding site and demonstrated that SPDEF prevented Foxm1 binding to its own promoter, inhibits Foxm1 transcriptional activity and decreases expression of Foxm1 targets. Our data demonstrate that SPDEF functions as a tumor suppressor in SV40 T antigens and c-Myc-induced prostate cancers by inhibiting tumor cell proliferation via disruption of an auto-regulatory element in the Foxm1 promoter.
Increased expression of Foxm1 was found in human prostate adenocarcinomas and was correlated with the severity of the disease [8]. At the same time, the decrease in SPDEF expression was associated with transition from low-grade to high-grade human prostate cancer [23], [34], [35]. It is possible that the loss of SPDEF causes increased expression of oncogenic Foxm1, accelerating tumor cell proliferation and leading to poor outcome in prostate cancer patients. Our studies may serve as a foundation for the development of new therapeutic approaches in prostate cancer by targeting Foxm1 via SPDEF dependent pathways.
In summary, decreased expression of SPDEF in prostate epithelial cells was sufficient to increase prostate carcinogenesis, while increased SPDEF inhibited prostate carcinogenesis induced by SV40 T antigens. Decreased prostate carcinogenesis in SPDEF-deficient mice was associated with decreased proliferation of tumor cells and reduced expression of Cdc25b, Cyclin B1, Plk-1, AuroraB, and Topo2alpha, factors that are critical for tumor cell proliferation. SPDEF bound to −745/−734 bp Foxm1 promoter region and inhibited the ability of Foxm1 to bind to and activate its own promoter. Our results suggest that the loss of SPDEF during prostate carcinogenesis results in an increased activity of the oncogenic Foxm1.
Materials and Methods
Generation of TRAMP-C2R3 and MycCap cell lines expressing SPDEF and Foxm1
SPDEF expression plasmid pLenti-PGK-GFP-SPDEF [41], or Foxm1 expression plasmid pLenti-PGK-GFP-Foxm1 [42], and the control plasmid pLenti-PGK-GFP were used to generate lentiviruses at Cincinnati Children's Hospital Viral Vector Core. TRAMP-C2R3 (termed as TRAMP C2) prostate adenocarcinoma cells [10] and MycCap cells prostate adenocarcinoma cells [43] were transduced with lentiviruses. After two days, GFP expressing cells were sorted using flow cytometry. SPDEF expression was confirmed by qRT-PCR and Western blot.
Transgenic mice
Loss-of-function of SPDEF in prostate epithelium
SPDEF−/− mice [14] were bred with TRAMP transgenic mice containing Pb-driven SV40-T large and t small antigens [6] to generate SPDEF−/−/TRAMP mice. SPDEF−/−/TRAMP mice were fertile with no obvious abnormalities. SPDEF−/− and TRAMP littermates were used as controls. Mouse prostates were harvested 25 weeks of age.
Gain-of-function of SPDEF in prostate epithelium
Transgenic mice with doxycycline (Dox)-inducible Spdef expression (TRE-SPDEF) were generated [13]. The TRE-SPDEF mice were bred with Pb-Cretg/+/LoxP-stop-LoxP-rtTA(Rosa26)tg/tg mice that contained Cre recombinase expressed in prostate epithelial cells under control of rat probasin (PB) promoter and the reverse tetracycline activator (rtTA) inserted into Rosa26 locus [44]. In Pb-Cretg/+/LoxP-stop-LoxP-rtTA(Rosa26)tg/+/TRE-SPDEF mice (abbreviated as SPDEF OE), Dox treatment results in prostate epithelial-specific expression of SPDEF transgene due to excision of LoxP-stop-LoxP cassette by Cre recombinase. SPDEF OE mice were bred with TRAMP transgenic mice to generate TRAMP/SPDEF OE mice. To induce SPDEF, mice were given Dox in food chow beginning at 4 weeks of age and kept on Dox until the end of the experiment. Dox-treated TRE-SPDEF/TRAMP littermates lacking the Pb-Cre transgene were used as a control for Pb-Cretg/+/LoxP-stop-LoxP-rtTA(Rosa26)tg/+/TRE-SPDEF/TRAMP transgenic mice. Additional controls included Pb-Cretg/−/TRE-SPDEF tg/− or Pb-Cretg/+/LoxP-stop-LoxP-rtTA(Rosa26)tg/+/TRE-SPDEF/TRAMP mice without Dox.
Orthotopic model of prostate cancer
5×105 TRAMP C2R3 cells, expressing exogenous SPDEF, Foxm1 or GFP, were injected into the prostates of anesthetized C57BL/6 mice. Mouse prostates were harvested 4 weeks after the surgery. All animal studies were approved by the Animal Care and Use Committee of Cincinnati Children's Hospital.
Immunohistochemistry
The following antibodies were used for immunochistochemistry: anti-Foxm1 [42], [45], anti-SPDEF [46], anti-Ki67 (Thermo Scientific) and anti-phospho-Histone H3 (pH 3) antibody (Santa Cruz).
Cell growth assay
Control TRAMP C2 and SPDEF OE TRAMP C2 cells, or control MycCap and SPDEF OE MycCap cells were plated in triplicate. Alive cells were counted at 1, 2, 3, and 4 days using hemocytometer or WST-1 reagent (Roche) according to the manufacturers' recommendations. Optical density was quantified at 450 nm using spectrophotometer. Standard curves were made with increasing numbers of each of the prostate cancer cell lines to convert fluorescent readings into cell numbers. Experiments were performed in triplicates and presented as average numbers of cells ± S.D.
In vitro scratch wound healing assay
Control TRAMP C2 cells or SPDEF OE TRAMP C2 were plated onto 6-well plates and allowed to grow into confluent monolayers. Scrape wounds were generated using a 20 - µl pipette tip, and cell media were replaced. Phase-contrast images of the cells were taken at 0 h, 18 h and 24 h after wounding, and the average wound closure rate was measured. The width of the scratch was quantified using ImageJ (National Institutes of Health). The relative migration distance was calculated by dividing the width of scratch at each time point by the width of the scratch at time zero. The relative distance was then converted to a percentage by multiplying by 100. Experiments were done in triplicate.
Soft agar assay
For soft agar assay, control and SPDEF OE TRAMP C2 cells were plated on soft agar for two weeks to assay for anchorage-independent cell growth. The culture medium containing 10% fetal calf serum was replaced every 3 days. Colonies containing more than 50 cells were scored after 2 weeks. Triplicate plates were used to count colonies and determine the mean number of colonies ± SD.
Quantitative real-time RT-PCR (qRT-PCR)
Total RNA was prepared from mouse prostates and analyzed by qRT-PCR using the StepOnePlus Real-Time PCR system (Applied Biosystems, Foster City, CA) as described [46]. RNA was amplified with Taqman Gene Expression Master Mix (Applied Biosystems) combined with inventoried Taqman mouse gene expression assays: Foxm1, Mm00514924_m1; β-Actin, Mm00607939_g1; Cdc25b, Mm00499136_m1; Cyclin B1, Mm00838401_g1; Plk1, Mm00440924_g1; SPDEF, Mm01306245_m1; Topo2α, Mm00495703_m1; Cyclin A2, Mm00438063_m1; Aurora B, Mm01718146_g1; Cks1b, Mm01617993_gh. Reactions were analyzed in triplicates and expression levels were normalized to β-actin mRNA.
Analysis of SPDEF in human prostate cancer
Data from human prostate cancer microarray datasets, GSE21034 [30] and GSE16560 [31] were downloaded from the GEO archive [47]. For the GSE21034 dataset, expression measurements were first log2-tranformed and expression levels of SPDEF and FOXM1 genes were compared between metastatic and primary tumor samples. Three different probesets representing three different FOXM1 transcripts were available for the FOXM1 gene. Expression of each transcript was analyzed separately. Statistical analysis of differences in median expression levels for transcripts between the two sample groups were performed using the non-parametric Wilcoxon rank test. For the GSE16560, samples were split in two different ways. First, expression levels were compared between indolent and lethal prostate cancers. Second, high-risk and low-risk sample groups were formed as described in [32]. High-risk samples were derived from patients surviving less than 12 months and low-risk samples from patients surviving more than 192 months. Statistical analysis was again performed using the non-parametric Wilcoxon rank test. Survival analysis was performed by stratifying patients in GSE16560 dataset [31] by the expression level of FOXM1 and SPDEF genes into “low” (lowest third), “middle” (middle third) and “high” (highest third) expression groups. Statistical analysis of Kaplan-Meier survival curves for different strata was performed using the log-rank test [48]. All microarray data analysis was performed using R and Bioconductor packages [49]. In all comparisons, p-values less than or equal to 0.05 were considered to be statistically significant.
Cloning the mouse Foxm1 promoter region and luciferase assays
Mouse genomic DNA was used to amplify the −3758 bp to +1 bp region of the mouse Foxm1 promoter (Gene Bank Number NC_000072.6) using the primers 5′-GGT ACC TTT CTG GGA CTG TCT GCG-3′ and 5′-GAG CTC AGC GCC GCT TTC AGT TG-3′. To create deletion mutants of the −3758 bp Foxm1 promoter region, we used the following primers: −1167, 5′-GGT ACC GTG CTG GAA TTA AAG GTG TGC-3′ and 5′-GAG CTC AGC GCC GCT TTC AGT TG-3′; −778, 5′-GGT ACC GAA CGG CTT TAC TGT CCT AAG-3′ and 5′-GAG CTC AGC GCC GCT TTC AGT TG-3′; −500, 5′-GGT ACC CTT ATC GTA AAG TAC TTC GAG GG-3′ and 5′-GAG CTC AGC GCC GCT TTC AGT TG-3′. PCR products were cloned into a pGL3 firefly luciferase (LUC) reporter plasmid (Promega, Madison, WI) and verified by DNA sequencing. Site-directed mutagenesis was used to mutate several nucleotides in either Foxm1 or SPDEF site of the −778 bp Foxm1 promoter LUC construct (Suppl. Fig. 4D). FoxM1B-LUC constructs were co-transfected with CMV-FoxM1B [50], CMV-SPDEF or CMV-empty plasmids [41] in TRAMP-C2R3 cells using Lipofectamine (Invitrogen). CMV-Renilla was used as an internal control to normalize the transfection efficiency. A dual luciferase assay (Promega) was performed 24 hours post transfection as described previously [51].
ChIP assay
Control and SPDEF-overexpressing TRAMP-C2R3 cells were cross-linked by addition of formaldehyde, sonicated and used for immunoprecipitation with rabbit anti-SPDEF antibody (H-250, Santa Cruz) or rabbit anti-Foxm1 antibody [10] as described previously [52]. Rabbit polyclonal IgG (Vector Lab) was used as ChIP negative controls. DNA fragments were between 500 bp and 1000 bp in size. Reversed cross-linked ChIP DNA samples were subjected to PCR amplification with oligonucleotides specific to promoter regions of mouse Foxm1: −3758/−3551 (5′-TTT CTG GGA CTG TCT GCG-3′ and 5′-CCT TGT TAG CCT GAT GTC ATG-3′); −1169/−966 (5′-GTG CTG GAA TTA AAG GTG TGC-3′and 5′-AGG GTC TTC GCC TTT CTG-3′); −778/−562 (5′-GAA CGG CTT TAC TGT CCT AAG-3′and 5′-GAG GGA GCA CAG AAT GAG-3′).
Cell cycle analysis
Control and SPDEF expressing TRAMP-C2R3 cells were serum starved for 36 hr by using DMEM media supplemented with 0.1% FBS. A complete media with 10% FBS was added and cells were collected at the indicated time points, fixed in 70% ethanol, and stained with propidium iodide/RNase solution (Cell Signaling). Flow cytometry was used to count cells in G0/G1, S and G2/M phases of cell cycle.
Statistical analysis
Microsoft Excel was used to calculate SD and statistically significant differences between samples using the Student T-Test. P values<0.05 were considered statistically significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HanahanD, WeinbergRA (2011) Hallmarks of cancer: the next generation. Cell 144 : 646–674.
2. BergesRR, VukanovicJ, EpsteinJI, CarMichelM, CisekL, et al. (1995) Implication of cell kinetic changes during the progression of human prostatic cancer. Clin Cancer Res 1 : 473–480.
3. UzgareAR, KaplanPJ, GreenbergNM (2003) Differential expression and/or activation of P38MAPK, erk1/2, and jnk during the initiation and progression of prostate cancer. Prostate 55 : 128–139.
4. GaoH, OuyangX, Banach-PetroskyWA, GeraldWL, ShenMM, et al. (2006) Combinatorial activities of Akt and B-Raf/Erk signaling in a mouse model of androgen-independent prostate cancer. Proc Natl Acad Sci U S A 103 : 14477–14482.
5. CairnsP, OkamiK, HalachmiS, HalachmiN, EstellerM, et al. (1997) Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res 57 : 4997–5000.
6. GreenbergNM, DeMayoF, FinegoldMJ, MedinaD, TilleyWD, et al. (1995) Prostate cancer in a transgenic mouse. Proc Natl Acad Sci U S A 92 : 3439–3443.
7. KaplanPJ, MohanS, CohenP, FosterBA, GreenbergNM (1999) The insulin-like growth factor axis and prostate cancer: lessons from the transgenic adenocarcinoma of mouse prostate (TRAMP) model. Cancer Res 59 : 2203–2209.
8. KalinTV, WangIC, AckersonTJ, MajorML, DetrisacCJ, et al. (2006) Increased levels of the FoxM1 transcription factor accelerate development and progression of prostate carcinomas in both TRAMP and LADY transgenic mice. Cancer Res 66 : 1712–1720.
9. WangIC, MelitonL, TretiakovaM, CostaRH, KalinichenkoVV, et al. (2008) Transgenic expression of the forkhead box M1 transcription factor induces formation of lung tumors. Oncogene 27 : 4137–4149.
10. CaiY, BalliD, UstiyanV, FulfordL, HillerA, et al. (2013) Foxm1 expression in prostate epithelial cells is essential for prostate carcinogenesis. J Biol Chem 288 : 22527–22541.
11. WangIC, MelitonL, RenX, ZhangY, BalliD, et al. (2009) Deletion of Forkhead Box M1 transcription factor from respiratory epithelial cells inhibits pulmonary tumorigenesis. PLoS ONE 4: e6609.
12. WeiGH, BadisG, BergerMF, KiviojaT, PalinK, et al. (2010) Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J 29 : 2147–2160.
13. ParkKS, KorfhagenTR, BrunoMD, KitzmillerJA, WanH, et al. (2007) SPDEF regulates goblet cell hyperplasia in the airway epithelium. J Clin Invest 117 : 978–988.
14. GregorieffA, StangeDE, KujalaP, BegthelH, van den BornM, et al. (2009) The ets-domain transcription factor Spdef promotes maturation of goblet and paneth cells in the intestinal epithelium. Gastroenterology 137 : 1333–1345 e1331–1333.
15. RodabaughKJ, Mhawech-FaucegliaP, GrothJ, LeleS, SoodAK (2007) Prostate-derived Ets factor is overexpressed in serous epithelial ovarian tumors. Int J Gynecol Pathol 26 : 10–15.
16. HorstD, GuX, BhasinM, YangQ, VerziM, et al. (2010) Requirement of the epithelium-specific Ets transcription factor Spdef for mucous gland cell function in the gastric antrum. J Biol Chem 285 : 35047–35055.
17. CarverBS, TranJ, ChenZ, Carracedo-PerezA, AlimontiA, et al. (2009) ETS rearrangements and prostate cancer initiation. Nature 457: E1 discussion E2–3.
18. SteffanJJ, KoulS, MeachamRB, KoulHK (2012) The transcription factor SPDEF suppresses prostate tumor metastasis. J Biol Chem 287 : 29968–29978.
19. SchaeferJS, SabherwalY, ShiHY, SriramanV, RichardsJ, et al. (2010) Transcriptional regulation of p21/CIP1 cell cycle inhibitor by PDEF controls cell proliferation and mammary tumor progression. J Biol Chem 285 : 11258–11269.
20. NoahTK, LoYH, PriceA, ChenG, KingE, et al. (2013) SPDEF functions as a colorectal tumor suppressor by inhibiting beta-catenin activity. Gastroenterology 144 : 1012–1023 e1016.
21. OettgenP, FingerE, SunZ, AkbaraliY, ThamrongsakU, et al. (2000) PDEF, a novel prostate epithelium-specific ets transcription factor, interacts with the androgen receptor and activates prostate-specific antigen gene expression. J Biol Chem 275 : 1216–1225.
22. ChenH, NandiAK, LiX, BieberichCJ (2002) NKX-3.1 interacts with prostate-derived Ets factor and regulates the activity of the PSA promoter. Cancer Res 62 : 338–340.
23. JohnsonTR, KoulS, KumarB, KhandrikaL, VeneziaS, et al. (2010) Loss of PDEF, a prostate-derived Ets factor is associated with aggressive phenotype of prostate cancer: regulation of MMP 9 by PDEF. Mol Cancer 9 : 148.
24. FeldmanRJ, SementchenkoVI, GayedM, FraigMM, WatsonDK (2003) Pdef expression in human breast cancer is correlated with invasive potential and altered gene expression. Cancer Res 63 : 4626–4631.
25. GhadersohiA, OdunsiK, ZhangS, AzrakRG, BundyBN, et al. (2008) Prostate-derived Ets transcription factor as a favorable prognostic marker in ovarian cancer patients. Int J Cancer 123 : 1376–1384.
26. GunawardaneRN, SgroiDC, WrobelCN, KohE, DaleyGQ, et al. (2005) Novel role for PDEF in epithelial cell migration and invasion. Cancer Res 65 : 11572–11580.
27. SoodAK, SaxenaR, GrothJ, DesoukiMM, CheewakriangkraiC, et al. (2007) Expression characteristics of prostate-derived Ets factor support a role in breast and prostate cancer progression. Hum Pathol 38 : 1628–1638.
28. GuX, ZerbiniLF, OtuHH, BhasinM, YangQ, et al. (2007) Reduced PDEF expression increases invasion and expression of mesenchymal genes in prostate cancer cells. Cancer Res 67 : 4219–4226.
29. KalinTV, UstiyanV, KalinichenkoVV (2011) Multiple faces of FoxM1 transcription factor: Lessons from transgenic mouse models. Cell Cycle 10 : 396–405.
30. TaylorBS, SchultzN, HieronymusH, GopalanA, XiaoY, et al. (2010) Integrative genomic profiling of human prostate cancer. Cancer Cell 18 : 11–22.
31. SbonerA, DemichelisF, CalzaS, PawitanY, SetlurSR, et al. (2010) Molecular sampling of prostate cancer: a dilemma for predicting disease progression. BMC Med Genomics 3 : 8.
32. WangZA, MitrofanovaA, BergrenSK, Abate-ShenC, CardiffRD, et al. (2013) Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat Cell Biol 15 : 274–283.
33. BuchwalterG, HickeyMM, CromerA, SelforsLM, GunawardaneRN, et al. (2013) PDEF promotes luminal differentiation and acts as a survival factor for ER-positive breast cancer cells. Cancer Cell 23 : 753–767.
34. TurnerDP, FindlayVJ, MoussaO, SemenchenkoVI, WatsonPM, et al. (2011) Mechanisms and functional consequences of PDEF protein expression loss during prostate cancer progression. Prostate 71 : 1723–1735.
35. GhadersohiA, SharmaS, ZhangS, AzrakRG, WildingGE, et al. (2011) Prostate-derived Ets transcription factor (PDEF) is a potential prognostic marker in patients with prostate cancer. Prostate 71 : 1178–1188.
36. HallerAC, TanW, Payne-OndracekR, UnderwoodW, TianL, et al. (2014) High SPDEF may identify patients who will have a prolonged response to androgen deprivation therapy. Prostate 74 : 509–519.
37. SoodAK, WangJ, Mhawech-FaucegliaP, JanaB, LiangP, et al. (2009) Sam-pointed domain containing Ets transcription factor in luminal breast cancer pathogenesis. Cancer Epidemiol Biomarkers Prev 18 : 1899–1903.
38. de VisserKE, EichtenA, CoussensLM (2006) Paradoxical roles of the immune system during cancer development. Nat Rev Cancer 6 : 24–37.
39. BalliD, RenX, ChouFS, CrossE, ZhangY, et al. (2011) Foxm1 transcription factor is required for macrophage migration during lung inflammation and tumor formation. Oncogene 31 : 3875–3888.
40. HalasiM, GartelAL (2009) A novel mode of FoxM1 regulation: positive auto-regulatory loop. Cell Cycle 8 : 1966–1967.
41. ChenG, KorfhagenTR, XuY, KitzmillerJ, WertSE, et al. (2009) SPDEF is required for mouse pulmonary goblet cell differentiation and regulates a network of genes associated with mucus production. J Clin Invest 119 : 2914–2924.
42. BalliD, UstiyanV, ZhangY, WangIC, MasinoAJ, et al. (2013) Foxm1 transcription factor is required for lung fibrosis and epithelial-to-mesenchymal transition. EMBO J 32 : 231–244.
43. WatsonPA, Ellwood-YenK, KingJC, WongvipatJ, LebeauMM, et al. (2005) Context-dependent hormone-refractory progression revealed through characterization of a novel murine prostate cancer cell line. Cancer Res 65 : 11565–11571.
44. BeltekiG, HaighJ, KabacsN, HaighK, SisonK, et al. (2005) Conditional and inducible transgene expression in mice through the combinatorial use of Cre-mediated recombination and tetracycline induction. Nucleic Acids Res 33: e51.
45. UstiyanV, WangIC, RenX, ZhangY, SnyderJ, et al. (2009) Forkhead box M1 transcriptional factor is required for smooth muscle cells during embryonic development of blood vessels and esophagus. Dev Biol 336 : 266–279.
46. RenX, ShahTA, UstiyanV, ZhangY, ShinnJ, et al. (2013) FOXM1 promotes allergen-induced goblet cell metaplasia and pulmonary inflammation. Mol Cell Biol 33 : 371–386.
47. BarrettT, TroupDB, WilhiteSE, LedouxP, RudnevD, et al. (2009) NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res 37: D885–890.
48. MantelN (1966) Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother Rep 50 : 163–170.
49. GentlemanRC, CareyVJ, BatesDM, BolstadB, DettlingM, et al. (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5: R80.
50. MajorML, LepeR, CostaRH (2004) Forkhead Box M1B (FoxM1B) Transcriptional Activity Requires Binding of Cdk/Cyclin Complexes for Phosphorylation-Dependent Recruitment of p300/CBP Co-activators. Mol Cell Biol 24 : 2649–2661.
51. KalinTV, WangIC, MelitonL, ZhangY, WertSE, et al. (2008) Forkhead Box m1 transcription factor is required for perinatal lung function. Proc Natl Acad Sci U S A 105 : 19330–19335.
52. BalliD, ZhangY, SnyderJ, KalinichenkoVV, KalinTV (2011) Endothelial cell-specific deletion of transcription factor FoxM1 increases urethane-induced lung carcinogenesis. Cancer Res 71 : 40–50.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 9
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Admixture in Latin America: Geographic Structure, Phenotypic Diversity and Self-Perception of Ancestry Based on 7,342 Individuals
- Nipbl and Mediator Cooperatively Regulate Gene Expression to Control Limb Development
- Genome Wide Association Studies Using a New Nonparametric Model Reveal the Genetic Architecture of 17 Agronomic Traits in an Enlarged Maize Association Panel
- Histone Methyltransferase MMSET/NSD2 Alters EZH2 Binding and Reprograms the Myeloma Epigenome through Global and Focal Changes in H3K36 and H3K27 Methylation
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy