
Low Levels of p53 Protein and Chromatin Silencing of p53 Target Genes Repress Apoptosis in Endocycling Cells
In order to maintain genome integrity, eukaryotic cells have evolved multiple ways to respond to DNA damage stress. One of the major cellular responses is apoptosis, during which the cell undergoes programmed cell death in order to prevent the propagation of the damaged genome to daughter cells. Although clinical observations and other studies have shown that tissues can differ in their apoptotic response, the molecular mechanisms underlying these differences are largely unknown. We have shown in our model system, Drosophila, that endocycling cells do not initiate cell death in response to DNA damage. The endocycle is a cell cycle variation that is widely found in nature and conserved from plant to animals. During the endocycle, cells duplicate their genomic DNA but do not enter mitosis to segregate chromosomes, resulting in a polyploid genome content. In this study, we investigate how the apoptotic response to DNA damage is repressed in endocycling cells. We find that the Drosophila ortholog of the human p53 tumor suppressor protein is expressed at very low levels in endocycling cells. Moreover, the downstream pro-apoptotic genes that are regulated by p53 are epigenetically silenced in endocycling cells. Our results provide important insights into tissue-specific apoptotic responses in development, with possible broader impact on understanding radiation therapy response and cancer of different tissues.
Published in the journal:
. PLoS Genet 10(9): e32767. doi:10.1371/journal.pgen.1004581
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004581
Summary
In order to maintain genome integrity, eukaryotic cells have evolved multiple ways to respond to DNA damage stress. One of the major cellular responses is apoptosis, during which the cell undergoes programmed cell death in order to prevent the propagation of the damaged genome to daughter cells. Although clinical observations and other studies have shown that tissues can differ in their apoptotic response, the molecular mechanisms underlying these differences are largely unknown. We have shown in our model system, Drosophila, that endocycling cells do not initiate cell death in response to DNA damage. The endocycle is a cell cycle variation that is widely found in nature and conserved from plant to animals. During the endocycle, cells duplicate their genomic DNA but do not enter mitosis to segregate chromosomes, resulting in a polyploid genome content. In this study, we investigate how the apoptotic response to DNA damage is repressed in endocycling cells. We find that the Drosophila ortholog of the human p53 tumor suppressor protein is expressed at very low levels in endocycling cells. Moreover, the downstream pro-apoptotic genes that are regulated by p53 are epigenetically silenced in endocycling cells. Our results provide important insights into tissue-specific apoptotic responses in development, with possible broader impact on understanding radiation therapy response and cancer of different tissues.
Introduction
Eukaryotic cells respond to DNA damage via multiple pathways. Checkpoints arrest the cell cycle and mobilize the DNA repair machinery to fix the damage [1], [2]. If this genotoxic stress is severe, however, the cells can enter a quiescent state known as senescence, or initiate programmed cell death (PCD), with one important type called apoptosis [3], [4]. Mutations in genes that compromise these pathways result in genome instability and cancer [5]. Cells from different tissues respond to DNA damage in different ways, but the mechanism(s) underlying this difference among tissues remains poorly characterized [6], [7]. Here, we use Drosophila as a model to define the mechanisms by which cells in development differ in their apoptotic response to genotoxic stress.
Many investigations have focused on how cells in culture respond to genotoxic stress, but much less is known about the different stress responses of tissues in vivo [7]–[9]. It is known that some tissues in mice and humans have higher levels of apoptosis in response to the genotoxic stress of ionizing radiation, which is largely dependent on the p53 tumor suppressor [9]–[11]. Other studies have described the transcriptional outputs downstream of p53 in different tissues in response to radiation [12]–[17]. Very little is known, however, about the molecular mechanisms that determine these tissue-specific activities of p53 and the apoptotic response to ionizing radiation [7], [18]. Defining the mechanisms of the tissue-specific apoptotic response is important to fully understand the sensitivity of different tumors to radiation therapy and the deleterious effects of this therapy on healthy tissues.
We had previously shown that Drosophila tissues that are composed of cells in the endocycle repress the apoptotic response to DNA damage [19]. The endocycle is a cell cycle variation with alternating gap (G) and DNA synthesis (S) phases without mitosis (M), and results in large, polyploid cells [20]–[22]. Cells switch into polyploid cell cycles as a part of normal development or regeneration in a wide variety of organisms including humans [20]. In addition, emerging evidence suggests that cancer cells polyploidize by inappropriately switching into an endocycle, which may contribute to genome instability and oncogenesis [23]–[25].
Our previous evidence suggested that Drosophila endocycling cells do not apoptose in response to DNA damage because both p53-dependent and independent pathways are repressed [19]. During the normal response to DNA damage, the ATM checkpoint kinase phosphorylates the Chk2 kinase, which in turn phosphorylates p53 [26]–[31]. Phosphorylation of p53 induces transcription of a number of downstream genes, including pro-apoptotic genes at one large complex locus called H99 [32]–[37]. In most tissues, the expression of the H99 genes, reaper (rpr), Head involution defective (Hid), and sickle (skl) are dependent on p53 after DNA damage, and evidence suggests that at least rpr is a direct target of p53 [3], [29], [38]. In endocycling cells, however, we found that the expression of the H99 genes was 10–100's of fold lower relative to mitotic cells, despite activation of the upstream ATM kinase by DNA damage [19], [39]. Endocycling cells also did not have a delayed apoptotic response to DNA damage, which in other tissues is mediated by p53-independent pathways that induce Hid expression [19], [40]–[43]. Together, this evidence suggested that the block to apoptosis in the endocycling cells is at or upstream of H99 gene expression, but the mechanism remained unknown.
In this study, we investigate the molecular mechanisms underlying the different apoptotic responses of mitotic cycling and endocycling cells. Our evidence suggests that p53-dependent and p53-independent apoptotic pathways are repressed in endocycling cells through epigenetic silencing of pro-apoptotic genes at the H99 locus. In addition, we find that although p53 mRNA levels are comparable between mitotic cycling and endocycling cycling cells, p53 protein is undetectable in endocycling cells. Thus, similar to humans, Drosophila p53 protein levels are regulated. In mitotic cycling cells, p53 protein and a paused RNA polymerase complex at the H99 gene promoters may prepare these cells to rapidly respond to genotoxic stress. We discuss the significance of these results in the context of tissue-specific apoptotic responses in development and cancer.
Results
H99 genes rpr and hid are transcriptionally repressed in endocycling cells
Our previous RT-qPCR data showed that mRNA levels for the pro-apoptotic H99 genes rpr, hid, sickle, and grim are much lower in endocycling larval salivary gland (SG) and fat body (FB) tissues than in mitotic cycling larval brain and imaginal discs (B–D) [19]. To test whether this is a result of transcriptional regulation, we examined the promoter activity for two of the genes at the H99 locus, rpr and hid. The hid 5′F-WT (hereafter hid-GFP) and rpr-11-lacZ reporters contain only the promoter and part of the enhancer regions of the rpr and hid gene, but lack the transcription units, and therefore are insensitive to post-transcriptional regulation [29], [44]. In untreated animals, the hid-GFP reporter was expressed at very low levels in both the mitotic cycling B–D and endocycling SG cells (Figures 1A and 1C). Within four hours after irradiation with 4000 rads of γ-rays, hid-GFP was induced to high levels in mitotic cycling B–D cells, consistent with previous reports (Figure 1B) [41], [44]. In contrast, hid-GFP expression was not induced by irradiation in the endocycling SG cells (Figure 1D). The hid-GFP reporter was also not induced in SG cells 24 hours after radiation, suggesting that both p53-dependent and the delayed p53-independent activation of this reporter is repressed in endocycling cells. Similar results were obtained with the rpr-11-lacZ promoter reporter, which was induced by IR in mitotic cycling but not endocycling cells (Figures S1A–S1D). These reporters were also not induced by radiation in other endocycling cells of larval and adult tissues, for example the larval fat body and follicle cells and nurse cells of the adult ovary (Figures S1E–S1H). These results suggest that the block to apoptosis in the endocycling cells acts in part through reduced transcription of the pro-apoptotic H99 genes.

We had previously shown that over-expression of p53 from a UAS:6xMyc:p53 transgene induces apoptosis in mitotic cycling cells, but it does not in endocycling cells. One possibility is that over-expressed p53 cannot induce apoptosis in endocycling cells because the checkpoint pathway upstream is uncoupled, and thus p53 may not be activated by Chk2. To address this, we determined whether Chk2 is required for apoptosis in mitotic cycling B–D cells when p53 is over-expressed. Over-expression of UAS:6xMyc:p53 in B–D cells using an hsp70:GAL4 driver resulted in comparable levels of apoptosis in Chk2 null and Chk2 wild type animals (Figures S1I–S1L). This result indicates that over-expressed p53 induces apoptosis in diploid cells in the absence of activation by Chk2. This result further suggests that uncoupling of checkpoint signaling upstream of p53 is inadequate to explain the absence of apoptosis in endocycling cells after p53 over-expression.
Epigenetic silencing at H99 genes in endocycling cells
The results with the rpr and hid reporters prompted us to ask whether chromatin silencing represses their transcription in endocycling cells. To test this, we performed Chromatin Immuno-Precipitation (ChIP) using antibodies against chromatin silencing and activating marks in mitotic cycling B–D and endocycling SG tissues. Both the rpr and hid enhancer-promoter regions had higher levels of the silencing mark H3K27Me3 in SG than in B–D cells (SG/B–D ∼10 fold) (Figures 1E and 1F). Within the enhancer-promoter region of both rpr and hid, H3K27Me3 peaked around the predicted p53 binding sites (p53 response elements, hereafter p53REs) (Figures 1E and 1F). ChIP for another silent chromatin mark, H3K9Me3, showed that it was also higher at rpr and hid in SG cells (Figure 1G). ChIP for the activating marks poly-acetylated H3 (H3Ac) and H4 (H4Ac) revealed that they were lower at rpr and hid in SG compared to B–D, although the difference between tissues was not as extreme as for silencing marks (Figure 1H, S2A). Analysis of extant ChIP-array data for H3K27Me3 in salivary glands from the Orr-Weaver lab showed that this silencing mark is heavily enriched across a >400 kb domain spanning the H99 locus (Figure S2B) [45]. Together with the promoter reporter results, these ChIP results suggest that the regulatory regions of the p53 target genes at the H99 locus are epigenetically silenced in endocycling cells.
To further address whether chromatin silences H99 genes, we tested whether RNAi knockdown of epigenetic regulators would sensitize salivary gland cells to p53 over-expression. We created a fly strain that expressed both UAS:6xMyc:p53 and UAS:GFP transgenes under control of the salivary gland driver, Fkh:GAL4 (Figures S3A-A″, S3C-C″) [46]. We crossed this strain to gene-specific UAS:RNAi strains (200 total, with ∼50% against epigenetic regulators), and screened the living G1 larvae for the appearance of the fluorescent salivary glands. The expression of hairpin RNA corresponding to three different H3K9 histone methyl-transferases (HMT), Su(var)3-9, Eggless and G9a, all resulted in salivary gland cell death [47]–[50]. Knockdown of Su(var)3-9 was the most severe, with three different RNAi constructs resulting in extremely small salivary glands and high levels of apoptotic cell death as evidenced by pycnotic nuclei, Caspase-3 labeling, and TUNEL (Table 1, Figures S3B-B″). We also observed a more mild, variably-expressive salivary gland cell death after knockdown of Enhancer of Polycomb (E(Pc)) (Figures S3D-D″). E(Pc) is known to modify the silencing of genes by Polycomb complexes, which are writers and readers of the H3K27me3 mark, and is also a suppressor of heterochromatic variegation of genes with the H3K9me3 mark [51]–[53]. Knockdown of E(Pc) or the H3K9 HMTs also induced some salivary gland apoptosis in the absence of p53 over-expression, which may be triggered by the constitutive heterochromatic DNA damage that is known to occur in endocycling cells (Figures S3E-E″) [19], [54]. The E(Pc) phenotype, however, was clearly more severe when p53 was over-expressed (Figures S3D–E″). These genetic results, combined with the H99 gene expression and ChIP data, are consistent with the idea that chromatin silencing contributes to the repression of apoptosis in endocycling cells.

Over-expression of p53B, but not p53A, induces apoptosis in endocycling cells
The UAS:6XMyc:p53 transgene that we used encodes the 385 amino acid (AA) p53A isoform (hereafter UAS:6XMyc:p53A), which has been the most widely studied isoform over the last decade. The current Drosophila genome annotation predicts four p53 mRNA transcripts (A,B,C,E), which potentially encode three different protein isoforms (Figure 2A) [55]. The shortest mRNA isoform, p53E, is predicted to encode 334 AA of the p53 C-terminus. The physiological relevance of this protein prediction is in question because RNA-Seq data indicates this mRNA is extremely rare throughout development [55]. The longest 495 AA protein isoform, p53B, differs from p53A by having a 110 AA N-terminal extension that is somewhat conserved with the N-terminal transactivation domain of full-length human p53 (Figure 2A) [56]. RNA-seq data suggests that the p53B mRNA is expressed throughout Drosophila development, although at lower levels than p53A mRNA [55].

To more fully understand the role of p53 in the tissue-specific regulation of apoptosis, we transformed fly strains with P-elements carrying the epitope-tagged and GAL4-inducible UAS:6XMyc:p53A or UAS:6XMyc:p53B isoforms [19]. Within six hours of heat-induced over-expression with hsp70:GAL4, both p53A and p53B induced apoptosis in mitotic cycling B–D cells (Figures 2B–E, J). This result is consistent with a recent report which also showed that p53B can induce apoptosis in imaginal discs when over-expressed [57]. We found, however, that p53B over-expression resulted in a significantly larger fraction of apoptotic cells in discs than did p53A (Figures 2C, 2E and 2J). Surprisingly, unlike p53A, the over-expressed p53B isoform was also a potent inducer of cell death in salivary glands (Figures 2F–I). Salivary gland cell death was also observed in hsp70:GAL4; UAS:6XMyc:p53B controls without heat induction, which is a result of leaky expression of hsp70:GAL4 in salivary glands (Figure 2H). Therefore, to compare the relative effect of acute expression of p53A versus p53B in salivary glands, we induced expression of these isoforms using Sgs3:GAL4, which first becomes active in mid-3rd instar larval salivary glands. Within hours after this induction, acute p53B expression strongly induced apoptosis in salivary glands while p53A did not (Figure S4 A–B′). Analysis of multiple transformed lines by immunofluorescence and Western blotting showed that while transgene expression was subject to genomic position effect, it did not explain the stronger apoptotic activity of p53B (Figure 2K, Figure S5A–H). In fact, some UAS:6XMyc:p53A transgenes were expressed at higher levels than UAS:6XMyc:p53B, yet still did not induce apoptosis in endocycling cells (Figure 2K, Figure S5). Transformation of UAS:6XMyc:p53A or UAS:6XMyc:p53B into the same genomic docking site resulted in similar induced levels of p53A and p53B protein [58], and again showed that over-expressed p53B was a more potent inducer of apoptosis in mitotic cycling and endocycling cells (Figure 2K, Fig. S5I–L).
We next investigated whether p53B over-expression caused salivary gland cell death by inducing H99 gene expression. We first used the rpr-11-lacZ and hid-GFP reporters. p53B over-expression induced these reporters in both mitotic cycling B-D and SG cells, whereas p53A over-expression induced them only in B–D cells (Figures 3A–3D) [19]. RT-qPCR also indicated that over-expression of p53B, but not p53A, induced transcription of the endogenous rpr and hid genes in endocycling SG cells (Figure 3E). These data indicate that, when over-expressed, the longer p53B isoform is intrinsically a stronger inducer of H99 gene transcription and apoptosis than p53A in both mitotic cycling and endocycling cells.

Over-expression of p53B, but not p53A, may activate a paused RNA pol II complex in endocycling cells
We further explored the different abilities of p53A and p53B over-expression to induce apoptosis in endocycling cells as an inroad to define the mechanism of apoptotic repression. We first determined if Myc:p53A and Myc:p53B differed in binding to the rpr and hid genes by using anti-Myc antibody for ChIP experiments. The results indicated that both over-expressed p53A and p53B were bound to the p53REs upstream of the rpr and hid genes in both the mitotic cycling B–D and endocycling SG cells (Figures 4A and 4B, Figures S6A, B). The level of p53 occupancy at the hid promoter, however, was much lower in SG cells than in B–D cells, even though the p53 isoforms were highly over-expressed (Figures 4A, B). This is consistent with the idea that chromatin silencing may act to partly restrict p53 promoter binding in endocycling cells. At the hid locus we saw two peaks of binding, one at the previously predicted p53RE at −2,000 and another minor peak around −200 upstream of transcription (Figures 4A, B) [41]. This is the first molecular evidence, to our knowledge, that p53 directly regulates the hid gene by binding to this predicted p53RE [38], [41]. Importantly, the relative binding of p53A and p53B was comparable. This data suggested, therefore, that the greater transcriptional potency of p53B relative to p53A in SG cells is not because of differential promoter binding.

We therefore tested whether p53A and p53B differed in their ability to change local chromatin environment in the promoter-enhancer regions of rpr and hid genes. Chromatin ChIP indicated that over-expression of p53A or p53B in B–D cells increased poly-acetylated H4 levels within the vicinity of the p53RE in the hid promoter-enhancer, with p53B inducing a higher level of H4Ac than p53A (Figure 4C). At the rpr locus, only p53B increased AcH4 around the p53RE in B–D cells (Figure S6C). These higher levels of acetylation after p53B over-expression correlate with the greater strength of p53B to induce transcription and apoptosis in B–D cells. In the endocycling SG cells, however, p53A and p53B increased histone acetylation to comparable levels at the p53REs in the rpr and hid genes (Figure 4D, Figure S6D). This suggests that both p53A and p53B may be capable of recruiting HAT co-activators that partially reverse the relatively low level of histone acetylation in SG cells. Therefore, this evidence does not explain the unique ability of p53B to activate transcription in SG cells.
We therefore examined downstream recruitment of RNA pol II to the rpr and hid promoters. ChIP with an antibody against the initiation form of RNA pol II phosphorylated on serine 5 indicated that in untreated, wild type, control animals there was a high level of RNA Pol II occupancy around the TSS of the rpr and hid gene in both mitotic cycling B–D and endocycling SG cells, although occupancy was lower in SG cells (Figures 4E, F, Figures S6E, F). These data suggest that there is a paused RNA Pol II complex at the rpr and hid genes even in the absence of genotoxic stress. Activation of this paused RNA Pol II may mediate an immediate response to genotoxic stress in B–D cells, consistent with the previously observed rapid transcriptional induction of these genes after IR [59]. ChIP with antibodies against the elongating form of RNA pol II phosphorylated on serine 2 indicated that over-expression of p53B was stronger than p53A in stimulating movement of RNA polymerase through the body of the hid and rpr genes, and also increased occupancy of RNA Pol II near the promoter (Figure 4G, and Figure S6G). All together, the mRNA expression and ChIP data suggest that p53B has the unique ability to induce H99 transcription in endocycling cells by promoting the transition from a paused to elongating RNA Pol II.
Endocycling cells have very low levels of p53 protein
Our data was consistent with the idea that apoptosis is repressed in endocycling cells through chromatin silencing of H99 genes, which could be overridden by high-level over-expression of p53B. To further explore this idea, we examined the endogenous expression of p53. RT-qPCR indicated that total p53 mRNA levels are similar between mitotic cycling B–D and endocycling SG and FB cells, consistent with our previous RT-qPCR and microarray results (Figure 5A) [19], [39]. Isoform-specific primers showed, however, that p53B mRNA is actually expressed at three fold higher levels in SG and FB than in B–D cells. Therefore, reduced transcription of p53 does not contribute to the repression of apoptosis in endocycling cells.

We next evaluated the levels of p53 protein in different tissues. To potentially detect all the isoforms, we transformed flies with a 24 kb BAC with mCherry fused to the common C-terminus of all the isoforms (p53-Ch). Within this genomic BAC clone, expression of p53 is under control of its normal regulatory sequences (Figure 5B) [60]. Fly strains transformed with the p53-Ch BAC, or an untagged version of the same BAC, rescued the apoptotic response of two different p53 null mutants (p535A-1-4 and p5311-1B-1) in imaginal discs, suggesting that these BACs recapitulate normal p53 function (Figure S7A–D).
We then used a DsRed/mCherry antibody for ChIP to evaluate promoter binding by p53-Ch, which showed that it binds to the p53RE in the enhancer-promoter of both rpr and hid genes in the mitotic cycling B–D cells, even in the absence of genotoxic stress (Figures 5C and 5D). In contrast, binding of p53-Ch to the rpr and hid promoters was not detected in endocycling SG cells, even though p53A and p53B can bind to these promoters in SG cells when over-expressed (Figures 5C and 5D).
To gain insight into why p53-Ch promoter binding is not detected in SG cells, we analyzed mCherry fluorescence by microscopy in the p53-Ch; p535A-1-4 strain. This indicated that p53-Ch was concentrated in 1–2 distinct nuclear foci within B–D cells, which were often in close proximity to the DAPI-bright heterochromatic chromocenter (Figures 5E and 5E′). In larval SG and FB cells, however, p53-Ch fluorescence was not detectible except for very rare, small cytoplasmic puncta (Figures 5F and 5F′). Immunolabeling of fixed cells with the DsRed/mCherry antibody gave similar results. These results suggest that p53 protein levels are low in endocycling cells, explaining why promoter binding is not detected.
To evaluate the tissue-specific abundance of endogenous p53 protein isoforms, we examined their protein levels by Western Blotting. Antibody raised against the conserved C-terminus of human p53 recognized Drosophila p53, and indicated that the most abundant isoform in B–D cells is ∼48 kDa, close to the predicted 44 kDa size of the p53A isoform (Figure 5G, lanes 1, 2, 3, 4) [61]. The predicted 56 kDa p53B and 38 kDa p53E isoforms were not detected in B–D cells. Western blotting of p53-Ch confirmed that there is one major isoform in B–D cells corresponding to the predicted p53A, which is shifted to higher molecular weight by the mCherry tag (Figure 6A). Importantly, none of these isoforms were detected in either SG or FB endocycling cells (Figure 5G, lanes 5, 6, 7, 8). These results showed that p53A is the major isoform in B–D cells, and confirmed that all p53 protein isoforms are expressed at extremely low levels in endocycling SG and FB cells.
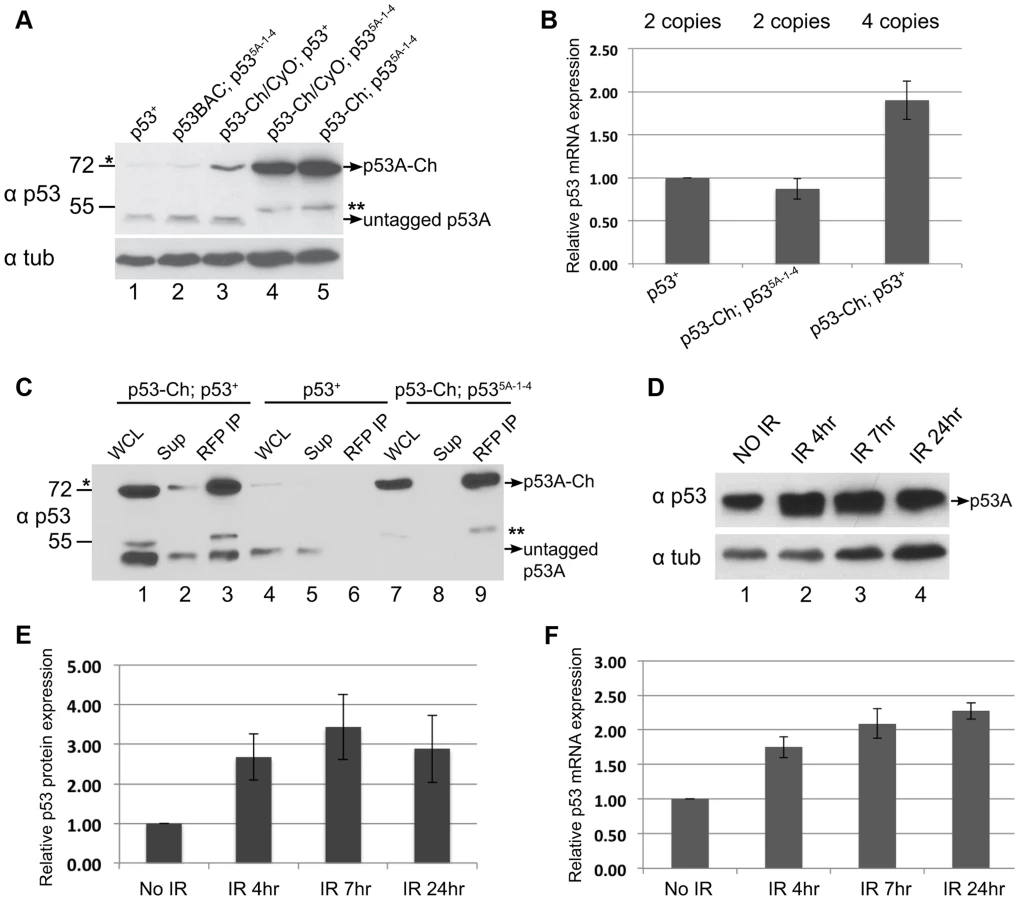
The difference between the p53 mRNA and protein levels raised the possibility that p53 protein may be targeted for degradation in endocycling cells. To test this, we inhibited proteasome function in larval salivary glands by using Fkh:GAL4 to drive expression of two temperature-sensitive, dominant-negative subunits of the proteasome, P{UAS-Pros261} and P{UAS-Prosbeta21}, created by the Belote lab [62]. We then compared the levels of p53 protein in B–D and SG cells from the same animals at the permissive (25°C) and non-permissive (29°C) temperature for proteasome function in SG cells. Although alpha-Tubulin was used as a loading control, it is only appropriate for comparison within the same tissue type and not between different tissues, and, therefore, Bradford quantification and Ponceau S staining were also used as normalization controls. The results indicated that p53A protein increased to detectable levels in SG cells when proteasome function was compromised at 29°C, but p53B and the other isoforms remained undetectable (Figure 5H). It is important to stress that proteasome function was only inhibited in SG cells, and that expression in B–D cells from the same animals is shown for comparison only (Figure 5H). Immunofluorescence and Western blotting indicated that levels of the tagged p53A-Ch isoform protein also increased in SG cells when proteasome function was compromised (Figure 5I, J, K lane 1,2). This also showed that inhibition of proteasome function had a pleiotropic effect on SG cell morphology (Figure 5J). Immunoprecipitation with an antibody against ubiquitin followed by Western blotting for p53 indicated that the p53A-Ch isoform is ubiquitinated, including higher molecular weight ubiquitinated forms, which increased when proteasome function was compromised at 29°C (Figure 5K, lane 3,4). Taken together, these data indicate that p53A protein is ubiquitinated and degraded by the proteasome in endocycling cells.
p53 complexes are turned over in mitotic cycling cells
The results clearly indicated that the steady-state level of p53A protein was higher in B–D cells than SG cells, but the p53B and other predicted isoforms were undetectable in both tissue types. Upon further analysis of p53 in B–D cells, however, we noticed that the level of the tagged p53A-Ch isoform was elevated compared to untagged p53A that was encoded either by the endogenous locus or untagged BAC transgenes (Figure 6A, compare lanes 1, 2 with 4, 5). RT-qPCR analysis of B–D cells indicated that mRNA levels for the tagged and untagged p53 was similar (Figure 6B). One interpretation is that p53A protein is also degraded in mitotic cycling B–D cells, although not to the extent it is in endocycling cells, and that the epitope tag at least partially impairs this degradation. In support of this interpretation, we observed that the tagged p53A-Ch isoform was stabilized in B–D cells only in the p53 null mutant background, but not when the endogenous p53 locus was wild type, a difference that is not due to mRNA levels (Figures 6A compare lane 3 with 4, 5, Figure 6B). This suggests that in the p53 mutant background p53-Ch homomeric complexes are stabilized, whereas in the p53 wild type background complexes comprised of p53-Ch and untagged p53 are turned over similar to wild type complexes. Consistent with this, IP with a highly-efficient, single-chain nanobody against RFP/mCherry followed by Western blotting with p53 antibodies indicated that tagged and untagged p53A isoforms are present in the same complex in vivo (Figure 6C). These results imply that the wild type untagged p53A exerts a trans-degradation effect on p53A-Ch, and that p53 tetramers are subject to proteolytic turnover in mitotic cycling cells.
In human cells, p53 protein is constantly degraded, but this degradation is inhibited in response to genotoxic stress. We did observe ∼3 fold increase in p53A protein levels in B–D cells at different time points after IR (Figures 6D, E). This increase, however, was proportional to an increase in p53A mRNA after irradiation, consistent with previous reports (Figure 6F) [38], [63]. Thus, there is no evidence that Drosophila p53 protein is stabilized in response to genotoxic stress.
Discussion
We have used Drosophila as a model system to define the molecular mechanisms for tissue-specific apoptotic responses to genotoxic stress. Our data suggest that Drosophila endocycling cells repress the apoptotic response in two ways: low level expression of the p53 transcription factor and epigenetic silencing of the p53 target genes at the H99 locus (Figure 7). In mitotic cycling B–D cells, the major p53 protein isoform is p53A, and we did not detect expression of the other predicted p53 protein isoforms. In endocycling SG and FB cells, all of the p53 protein isoforms, including p53A, were below the level of detection. Our data suggest that, similar to human p53, Drosophila p53A is ubiquitinated and degraded by the proteasome in endocycling cells. Over-riding this proteolysis by forced expression of p53A did not activate H99 gene transcription or apoptosis in endocycling cells. Together with our other data, these results suggest that downstream chromatin silencing of the H99 locus represses apoptosis in endocycling cells even when p53A protein is abundant. In contrast, we found that over-expression of the longer p53B isoform induced H99 gene expression and apoptosis in endocycling cells. However, the normal physiological expression of p53B protein and binding to the H99 locus was undetectable in endocycling cells, suggesting that the low level of expression of this isoform also contributes to the repression of apoptosis. In the absence of genotoxic stress, we found a paused RNA Pol II at the H99 gene promoters in both mitotic cycling and endocycling cells. In endocycling cells, this paused RNA Pol II complex is activated only when the longer p53B isoform is highly over-expressed. This result implicates polymerase activation as one step that is blocked after DNA damage or p53A over-expression. In mitotic cycling cells, both paused RNA pol II and p53A protein are bound to H99 promoters in the absence of stress, which may prepare cells for a rapid apoptotic response to DNA damage. In addition, our data suggest that p53A protein levels are regulated in mitotic cycling cells, which likely ensures that apoptosis occurs only in response to stress. Together, our results have revealed new mechanisms by which different cells in development modulate their apoptotic response.

Human and Drosophila p53 proteins are targeted for degradation at the proteasome
Previous evidence suggested that Drosophila p53 is regulated primarily by Chk2 phosphorylation and not protein stability [26], [59], [64]. Consistent with this, we found that in mitotic cycling cells p53A protein levels do not increase during the early response to radiation, a time when H99 genes are highly induced. At later times after irradiation, p53A protein levels increased only 2–3 fold, a magnitude that is proportional to the increase in p53 mRNA levels, as has been previously reported [38], [43], [63]. Therefore, there is no evidence that the protein stability of p53A or other p53 isoforms changes in response to genotoxic stress. Both with and without genotoxic stress, the cellular levels of p53A protein were relatively low in mitotic cycling cells, and we observed that the epitope tag on p53-Ch increased the abundance of p53A protein in p53 mutant but not p53 wild type cells [59]. A cogent model is that the epitope-tag on p53-Ch partially interferes with p53A proteolysis in mitotic cycling cells, and that untagged p53 can promote the degradation of tagged p53-Ch in the same tetramer. Dampening of p53 protein levels may be critically important to prevent inappropriate apoptosis in the absence of stress. Consistent with this idea, we found that elevated levels of p53A or p53B protein were sufficient to induce apoptosis in mitotic cycling cells even in Chk2 null animals. We propose that regulation of p53 protein levels in mitotic cycling cells tunes a threshold level of p53 protein that is poised to rapidly activate H99 gene expression when phosphorylated by activated Chk2 in response to DNA damage.
In endocycling cells, however, we were unable to detect any of the p53 protein isoforms using a variety of methods. This tissue-specific regulation of p53 protein abundance is post-transcriptional because mRNA levels were similar between mitotic cycling and endocycling cells. This low level of p53 protein suggests that either its translation is repressed and/or that it is more efficiently proteolyzed in endocycling cells. We favor a model wherein it is p53 proteolysis that is regulated in endocycling cells (Figure 7). In support of this model, compromising proteasome function elevated p53A protein levels in salivary glands. Moreover, p53A is ubiquitinated in endocycling cells, and these modified forms increase when proteasome function is compromised, which is consistent with previous data that p53 turnover is regulated by ubiquitination in Drosophila S2 cells [65]. In contrast, the longer p53B isoform remained undetectable when the proteasome function was reduced. Given that proteasome function was only partially compromised, our inability to detect p53B may reflect a more efficient degradation of this longer isoform. This idea is consistent with the known correlation between transactivation domains and ubiquitin-mediated proteolysis for mammalian p53 and other proteins [66].
Although our results suggest that at least the p53A isoform is modified and targeted for degradation by a ubiquitin ligase, the identity of this ligase is unknown. The Drosophila genome does not have an obvious ortholog of the ubiquitin ligase MDM2, which targets p53 for degradation in mammalian cells [67]–[70]. It remains possible that another family of ubiquitin ligases mediate p53 degradation in endocycling cells [71], [72]. Nonetheless, our results indicate that regulation of p53 is more similar between flies and humans than previously suspected, a finding that is interesting in the context of growing evidence for conserved p53 functions in flies and humans, including the response to hyperplasia [73].
p53 target genes at the H99 locus are repressed in endocycling cells
Our data suggest that apoptosis in endocycling cells is repressed in part through chromatin silencing of the pro-apoptotic genes at the H99 locus (Figure 7). Our evidence for silent chromatin marks H3K9me3 and H3K27me3 at H99 are consistent with cytogenetic observations that the H99 chromosome region (75C) is a highly-condensed constriction on salivary gland polytene chromosomes, and genome-wide studies that showed that H3K27me3 is enriched at H99 relative to other loci in salivary glands [45], [74], [75]. Although our genetic data indicate that knockdown of the writers and readers of H3K9me3 and H3K27me3 results in salivary gland apoptosis, it remains possible that knockdown of these regulators causes other types of stress that triggers apoptosis. It is important to note, however, that our results in endocycling cells are also consistent with a previous analysis that indicated that chromatin silencing at H99 dampens the apoptotic response during late embryogenesis [76].
It was previously shown that the chromatin organization at the H99 locus impedes its DNA replication in endocycling cells [45], [74], [77], [78]. As a result, DNA at this locus is not duplicated every endocycle S phase, resulting in a final lower DNA copy number relative to euchromatic loci. This “under-replication” is not the cause of apoptotic repression because we found that in Suppressor of Underreplication (Su(UR)) mutants, in which the H99 locus is almost fully replicated, endocycling SG cells still did not apoptose in response to DNA damage [45], [77], [79].
Our data suggest that the apoptotic response to genotoxic stress is repressed in endocycling cells because paused RNA Pol II is not activated at rpr and hid genes (Figure 7). One possibility is that chromatin silencing in endocycling cells restricts recruitment of transcription elongation factors to H99 promoters. We found that over-expressed p53A and p53B were similar in binding and recruitment of acetylation to rpr and hid promoters, but only p53B activated transcription and apoptosis in endocycling cells. This difference between p53A and p53B isoform activity is attributable to an additional 110 AA amino - terminal transactivation domain in p53B that is somewhat conserved with human p53 [56]. The N-terminus of over-expressed p53B, therefore, may bypass silencing of the H99 genes in endocycling cells by activating this paused RNA polymerase to promote transcriptional elongation. The normal biological function of these paused RNA pol II complexes may be to coordinate a rapid response to developmental signals that trigger apoptosis and autophagy of endocycling larval tissues during metamorphosis [80]–[82].
We propose that low levels of p53 protein and downstream silencing of its target genes both prevent endocycling cell apoptosis (Figure 7). We previously proposed that the apoptotic response to genotoxic stress must be tightly repressed in polyploid endocycling cells because they have constitutive genotoxic stress caused by under-replication of heterochromatic DNA [19], [83], [84]. Consistent with a possible linkage between the endocycle program and apoptotic repression, we recently found that experimentally-induced endocycling cells (iECs) repress apoptosis independent of cell differentiation [25]. It is clear that low levels of p53 protein is not the only mechanism of repression because over-expression of p53A resulted in abundant protein in endocycling cells, but failed to induce H99 transcription or apoptosis. Notably, over-expressed p53 had lower occupancy at H99 promoters in SG than B–D cells, another possible mechanism by which chromatin organization represses apoptosis downstream of p53. Moreover, the complete absence of endocycling cell apoptosis in response to IR suggests that both p53-dependent and p53-independent apoptotic pathways are repressed through silencing of the H99 locus, a point where these pathways intersect. Our data, however, do not rule out the possibility that endocycling cells may use other mechanisms to repress the apoptotic response to DNA damage to ensure their survival despite the continuous genotoxic stress caused by under-replication.
Mitotic cycling cells are poised to respond to genotoxic stress
In mitotic cycling cells, the p53 protein and paused RNA Pol II were bound to rpr and hid gene promoters in the absence of stress. This suggests that Chk2 phosphorylation of p53 pre-bound to these promoters activates the paused RNA Pol II to elicit a coordinated and rapid transcriptional response to genotoxic stress [85]. This is consistent with previous evidence that p53-dependent activation of rpr and hid transcription is readily detectable within 15 minutes of ionizing radiation [59]. This strategy to rapidly respond to stress appears to be conserved to humans where it has been shown that p53 activates paused RNA Pol II at some of its target genes, by indirect or direct physical interaction of p53 with elongation factors [8], [86]. Together, our results suggest that mitotic cycling cells in Drosophila are poised to respond to stress by tuning a threshold level of p53 protein that is bound to H99 promoters with a stalled RNA Pol II (Figure 7).
Cell cycle and tissue-specific regulation of apoptosis
Our data raise the question as to whether similar mechanisms repress apoptosis in mammalian polyploid cells. The transcriptome signatures of fly endocycles is very similar to that of polyploid cycles of mouse liver, megakaryocytes, and placental Trophoblast Giant Cells (TGCs), suggesting a conservation of cell cycle regulation [21], [39], [87]–[91]. It is also known that mouse TGCs do not apoptose in response to UV [92]. Moreover, evidence suggests that p53 protein levels decline when trophoblast stem cells switch into the endocycle and differentiate into TGCs, suggesting that the endocycle repression of apoptosis may be a theme conserved to mammals [93]. The ubiquitin ligase that targets p53 for degradation in TGCs has not been identified, and it is possible that in both Drosophila and mouse the same family of ubiquitin ligases targets p53 for degradation in endocycling cells. In addition to developmentally-programmed endocycles, recent evidence suggests that cells can inappropriately switch from mitotic cycles into endocycles, and that this cell cycle switch contributes to genome instability and oncogenesis [23]–[25]. Similar to developmental endocycles, apoptosis may be repressed in these endocycling cancer cells. In support of this idea, recent evidence showed that pro-apoptotic p53 target genes are epigenetically silenced in polyploid cancer cells [94]. Therefore, the mechanisms that repress apoptosis in Drosophila endocycling cells may be conserved to humans and relevant to tissue-specific radiation therapy response and oncogenesis [7], [9].
Materials and Methods
Drosophila genetics
Fly strains were raised at 25°C prior to and during experimental procedures. Fly strains were obtained from the Bloomington Drosophila Stock Center (BDSC, Bloomington, IN, USA) unless otherwise noted. hid-GFP fly strain was kindly provided by W. Du. A y w strain was used as wild type. For the proteasome dominant negative experiments, crosses were performed at 25°C with 24 hr egg lay. The vials were then transferred to 29°C until 3rd instar larvae.
Chromatin Immunoprecipitation
The ChIP protocol was modified from previous methods (ChIP-chip protocol for the mod-ENCODE project by Kevin P. White lab and 17–295; Millipore) and entailed at least two biological replicates from separate isolations of 3rd larval instar brains and imaginal discs (B–D) and salivary glands (SG).
RNA isolation and real-time qPCR
Total RNA was isolated from hand-dissected tissues using TRIzol (15596-026, Invitrogen). 1 µg of RNA from each sample was reverse-transcribed using the QuantiTect Reverse Transcription Kit (Qiagen) according to manufacturer's instructions. qPCR analysis was done on a Stratagene (Santa Clara, CA) Mx3005P machine with SYBR Green Master Mix (600843; Agilent, Santa Clara, CA). For mRNA quantification, Act 5C was used as a reference gene to calculate the relative expression (fold difference). For ChIP-qPCR experiments, the amount of DNA in the pellet was expressed as percentage of input DNA estimated by a standard curve generated from a serial dilution of the input. The values were then normalized to a control. Antibodies used for ChIP are described in Text S1 and PCR primer sequences are listed in Table S1.
Construction of p53 transgenes
construction was described previously [19]. construction was performed using the same strategy. For phiC 31 mediated integration, p53A and B cDNA fragments were cloned into pUAST-w+-attB vector and then transformed into the attP docking site at 65B2 (strain 24871) [58]. Unless otherwise stated, the analyses of p53 isoform over-expression were performed using staining corresponding to UAS:6xMyc:p53A (P #44) and UAS:6xMyc:p53B (P#20).
BAC recombineering
BAC CH322-178C12 from the P[acman] library was tagged with mCherry at the end of the common last exon of the p53 gene [60], [95]. The resulting construct and untagged BAC were transformed into attP docking sites at 22A3 (strain 24872) using phiC31 integrase.
Immunoblotting and nanobody immunoprecipitation
Protein extracts were prepared from hand-dissected tissues of mid-late 3rd instar larvae by standard methods using RIPA buffer [96]. For Western blots that compared p53 protein abundance between tissues, we loaded equal amounts of total protein as determined by standard Bradford assay in triplicate using BSA for a standard curve (BSA), and Ponceau S staining, as we have previously reported [39]. For comparison within the same tissue type, mouse anti-α-tubulin was used as loading control. Western blotting was performed as previously described [25], [39]. Antibody dilutions are: mouse anti Myc (9E10, Developmental Studies Hybridoma Bank, University of Iowa) 1∶500, mouse anti p53 (C11, Santa Cruz) 1∶500, mouse anti-α-tubulin (clone DM1A, Sigma) 1∶5,000, and anti-mouse secondary antibody, peroxidase labeled (KPL) at 1∶5,000. The average intensity of the bands for each sample was quantified using ImageJ software.
For RFP nanobody IP [97], 10–15 µl of Chromotek-RFP-Trap beads were added to the protein extracts and incubated for 2 hours at 4°C and precipitated by brief centrifugation. SDS-PAGE sample buffer was added to the washed beads. Mouse monoclonal antibody FK2 (Enzo BML-PW8810) were used to IP for ubiquitin conjugated proteins. 30 µl of Protein A agarose beads were used to pull down the antibody.
Immunofluorescent microscopy
Mid-late 3rd instar larvae were dissected in either 1× PBS or Grace's solution, and fixed in 6% formaldehyde as previously described [98], and immunolabeled using anti-cleaved-Caspase-3, 1∶50 (Cell Signaling). Secondary antibody was anti-rabbit 488 at 1∶500 dilutions, and DNA was counterstained with DAPI. X-gal staining was performed 6 hours after heat shock treatment as previously described [19]. TUNEL staining (In Situ cell death detection kit, TMR red, Roche) was performed according to manufacture's instructions. Wide-field micrographs were taken on a Leica DMRA2 and analyzed using OpenLab (Improvision) software. Confocal micrographs were captured on a Leica SP5 confocal.
Gamma irradiation
Larvae were irradiated with a total of 4,000 rad (40 Gy) from a Cesium source, and 4, 6 and 24 hours later labeled with anti-activated-Caspase-3.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. WeinertTA, HartwellLH (1993) Cell cycle arrest of cdc mutants and specificity of the RAD9 checkpoint. Genetics 134 : 63–80.
2. CicciaA, ElledgeSJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40 : 179–204.
3. FuchsY, StellerH (2011) Programmed cell death in animal development and disease. Cell 147 : 742–758.
4. Di MiccoR, FumagalliM, CicaleseA, PiccininS, GaspariniP, et al. (2006) Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444 : 638–642.
5. HanahanD, WeinbergRA (2011) Hallmarks of cancer: the next generation. Cell 144 : 646–674.
6. GorgoulisVG, VassiliouLV, KarakaidosP, ZacharatosP, KotsinasA, et al. (2005) Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434 : 907–913.
7. JacksonJG, PostSM, LozanoG (2011) Regulation of tissue - and stimulus-specific cell fate decisions by p53 in vivo. J Pathol 223 : 127–136.
8. BeckermanR, PrivesC (2010) Transcriptional regulation by p53. Cold Spring Harb Perspect Biol 2: a000935.
9. GudkovAV, KomarovaEA (2003) The role of p53 in determining sensitivity to radiotherapy. Nat Rev Cancer 3 : 117–129.
10. LoweSW, SchmittEM, SmithSW, OsborneBA, JacksT (1993) P53 Is Required for Radiation-Induced Apoptosis in Mouse Thymocytes. Nature 362 : 847–849.
11. MacCallumDE, HuppTR, MidgleyCA, StuartD, CampbellSJ, et al. (1996) The p53 response to ionising radiation in adult and developing murine tissues. Oncogene 13 : 2575–2587.
12. SongS, LambertPF (1999) Different responses of epidermal and hair follicular cells to radiation correlate with distinct patterns of p53 and p21 induction. Am J Pathol 155 : 1121–1127.
13. KomarovaEA, ChernovMV, FranksR, WangK, ArminG, et al. (1997) Transgenic mice with p53-responsive lacZ: p53 activity varies dramatically during normal development and determines radiation and drug sensitivity in vivo. EMBO J 16 : 1391–1400.
14. RileyT, SontagE, ChenP, LevineA (2008) Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 9 : 402–412.
15. CoatesPJ, LorimoreSA, LindsayKJ, WrightEG (2003) Tissue-specific p53 responses to ionizing radiation and their genetic modification: the key to tissue-specific tumour susceptibility? Journal of Pathology 201 : 377–388.
16. BouvardV, ZaitchoukT, VacherM, DuthuA, CanivetM, et al. (2000) Tissue and cell-specific expression of the p53-target genes: bax, fas, mdm2 and waf1/p21, before and following ionising irradiation in mice. Oncogene 19 : 649–660.
17. FeiP, BernhardEJ, El-DeiryWS (2002) Tissue-specific induction of p53 targets in vivo. Cancer Res 62 : 7316–7327.
18. HamardPJ, BartheleryN, HogstadB, MungamuriSK, TonnessenCA, et al. (2013) The C terminus of p53 regulates gene expression by multiple mechanisms in a target - and tissue-specific manner in vivo. Genes Dev 27 : 1868–1885.
19. MehrotraS, MaqboolSB, KolpakasA, MurnenK, CalviBR (2008) Endocycling cells do not apoptose in response to DNA rereplication genotoxic stress. Genes Dev 22 : 3158–3171.
20. FoxDT, DuronioRJ (2013) Endoreplication and polyploidy: insights into development and disease. Development 140 : 3–12.
21. CalviBR (2013) Making big cells: one size does not fit all. Proc Natl Acad Sci U S A 110 : 9621–9622.
22. LeslieM (2014) Strength in numbers? Science 343 : 725–727.
23. DavoliT, de LangeT (2011) The causes and consequences of polyploidy in normal development and cancer. Annu Rev Cell Dev Biol 27 : 585–610.
24. StorchovaZ, PellmanD (2004) From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol 5 : 45–54.
25. HasselC, ZhangB, DixonM, CalviBR (2014) Induction of endocycles represses apoptosis independently of differentiation and predisposes cells to genome instability. Development 141 : 112–123.
26. PetersM, DeLucaC, HiraoA, StambolicV, PotterJ, et al. (2002) Chk2 regulates irradiation-induced, p53-mediated apoptosis in Drosophila. Proc Natl Acad Sci U S A 99 : 11305–11310.
27. SongYH, MireyG, BetsonM, HaberDA, SettlemanJ (2004) The Drosophila ATM ortholog, dATM, mediates the response to ionizing radiation and to spontaneous DNA damage during development. Curr Biol 14 : 1354–1359.
28. XuJ, XinS, DuW (2001) Drosophila Chk2 is required for DNA damage-mediated cell cycle arrest and apoptosis. Febs Letters 508 : 394–398.
29. BrodskyMH, NordstromW, TsangG, KwanE, RubinGM, et al. (2000) Drosophila p53 binds a damage response element at the reaper locus. Cell 101 : 103–113.
30. OllmannM, YoungLM, Di ComoCJ, KarimF, BelvinM, et al. (2000) Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell 101 : 91–101.
31. JinS, MartinekS, JooWS, WortmanJR, MirkovicN, et al. (2000) Identification and characterization of a p53 homologue in Drosophila melanogaster. Proc Natl Acad Sci U S A 97 : 7301–7306.
32. AbramsJM, WhiteK, FesslerLI, StellerH (1993) Programmed cell death during Drosophila embryogenesis. Development 117 : 29–43.
33. ChristichA, KauppilaS, ChenP, SogameN, HoSI, et al. (2002) The damage-responsive Drosophila gene sickle encodes a novel IAP binding protein similar to but distinct from reaper, grim, and hid. Curr Biol 12 : 137–140.
34. GretherME, AbramsJM, AgapiteJ, WhiteK, StellerH (1995) The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev 9 : 1694–1708.
35. SrinivasulaSM, DattaP, KobayashiM, WuJW, FujiokaM, et al. (2002) sickle, a novel Drosophila death gene in the reaper/hid/grim region, encodes an IAP-inhibitory protein. Curr Biol 12 : 125–130.
36. WingJP, KarresJS, OgdahlJL, ZhouL, SchwartzLM, et al. (2002) Drosophila sickle is a novel grim-reaper cell death activator. Curr Biol 12 : 131–135.
37. WhiteK, GretherME, AbramsJM, YoungL, FarrellK, et al. (1994) Genetic control of programmed cell death in Drosophila. Science 264 : 677–683.
38. AkdemirF, ChristichA, SogameN, ChapoJ, AbramsJM (2007) p53 directs focused genomic responses in Drosophila. Oncogene 26 : 5184–5193.
39. MaqboolSB, MehrotraS, KolpakasA, DurdenC, ZhangB, et al. (2010) Dampened activity of E2F1-DP and Myb-MuvB transcription factors in Drosophila endocycling cells. J Cell Sci 123 : 4095–4106.
40. McNameeLM, BrodskyMH (2009) p53-independent apoptosis limits DNA damage-induced aneuploidy. Genetics 182 : 423–435.
41. WichmannA, UyetakeL, SuTT (2010) E2F1 and E2F2 have opposite effects on radiation-induced p53-independent apoptosis in Drosophila. Dev Biol 346 : 80–89.
42. MoonNS, Di StefanoL, MorrisEJ, PatelR, WhiteK, et al. (2008) E2F and p53 induce apoptosis independently during Drosophila development but intersect in the context of DNA damage. PLoS Genet 4: e1000153.
43. van BergeijkP, HeimillerJ, UyetakeL, SuTT (2012) Genome-wide expression analysis identifies a modulator of ionizing radiation-induced p53-independent apoptosis in Drosophila melanogaster. PLoS One 7: e36539.
44. Tanaka-MatakatsuM, XuJ, ChengL, DuW (2009) Regulation of apoptosis of rbf mutant cells during Drosophila development. Dev Biol 326 : 347–356.
45. SherN, BellGW, LiS, NordmanJ, EngT, et al. (2012) Developmental control of gene copy number by repression of replication initiation and fork progression. Genome Res 22 : 64–75.
46. HendersonKD, AndrewDJ (2000) Regulation and function of Scr, exd, and hth in the Drosophila salivary gland. Dev Biol 217 : 362–374.
47. TschierschB, HofmannA, KraussV, DornR, KorgeG, et al. (1994) The protein encoded by the Drosophila position-effect variegation suppressor gene Su(var)3-9 combines domains of antagonistic regulators of homeotic gene complexes. EMBO J 13 : 3822–3831.
48. JenuweinT (2001) Re-SET-ting heterochromatin by histone methyltransferases. Trends Cell Biol 11 : 266–273.
49. StabellM, EskelandR, BjorkmoM, LarssonJ, AalenRB, et al. (2006) The Drosophila G9a gene encodes a multi-catalytic histone methyltransferase required for normal development. Nucleic Acids Res 34 : 4609–4621.
50. MisJ, NerSS, GrigliattiTA (2006) Identification of three histone methyltransferases in Drosophila: dG9a is a suppressor of PEV and is required for gene silencing. Mol Genet Genomics 275 : 513–526.
51. StankunasK, BergerJ, RuseC, SinclairDA, RandazzoF, et al. (1998) The enhancer of polycomb gene of Drosophila encodes a chromatin protein conserved in yeast and mammals. Development 125 : 4055–4066.
52. SotoMC, ChouTB, BenderW (1995) Comparison of germline mosaics of genes in the Polycomb group of Drosophila melanogaster. Genetics 140 : 231–243.
53. SinclairDAR, CleggNJ, AntonchukJ, MilneTA, StankunasK, et al. (1998) Enhancer of Polycomb is a suppressor of position-effect variegation in Drosophila melanogaster. Genetics 148 : 211–220.
54. PengJC, KarpenGH (2009) Heterochromatic genome stability requires regulators of histone H3 K9 methylation. PLoS Genet 5: e1000435.
55. MarygoldSJ, LeylandPC, SealRL, GoodmanJL, ThurmondJ, et al. (2013) FlyBase: improvements to the bibliography. Nucleic Acids Res 41: D751–757.
56. BourdonJC, FernandesK, Murray-ZmijewskiF, LiuG, DiotA, et al. (2005) p53 isoforms can regulate p53 transcriptional activity. Genes & Development 19 : 2122–2137.
57. Dichtel-DanjoyML, MaD, DourlenP, ChatelainG, NapoletanoF, et al. (2013) Drosophila p53 isoforms differentially regulate apoptosis and apoptosis-induced proliferation. Cell Death Differ 20 : 108–116.
58. FishMP, GrothAC, CalosMP, NusseR (2007) Creating transgenic Drosophila by microinjecting the site-specific phiC31 integrase mRNA and a transgene-containing donor plasmid. Nat Protoc 2 : 2325–2331.
59. BrodskyMH, WeinertBT, TsangG, RongYS, McGinnisNM, et al. (2004) Drosophila melanogaster MNK/Chk2 and p53 regulate multiple DNA repair and apoptotic pathways following DNA damage. Mol Cell Biol 24 : 1219–1231.
60. VenkenKJ, CarlsonJW, SchulzeKL, PanH, HeY, et al. (2009) Versatile P[acman] BAC libraries for transgenesis studies in Drosophila melanogaster. Nat Methods 6 : 431–434.
61. LiGY, FanB, SuGF (2009) Acute energy reduction induces caspase-dependent apoptosis and activates p53 in retinal ganglion cells (RGC-5). Exp Eye Res 89 : 581–589.
62. BeloteJM, FortierE (2002) Targeted expression of dominant negative proteasome mutants in Drosophila melanogaster. Genesis 34 : 80–82.
63. KimH, LeeJM, LeeG, BhinJ, OhSK, et al. (2011) DNA damage-induced RORalpha is crucial for p53 stabilization and increased apoptosis. Mol Cell 44 : 797–810.
64. NordstromW, AbramsJM (2000) Guardian ancestry: fly p53 and damage-inducible apoptosis. Cell Death Differ 7 : 1035–1038.
65. ChenS, WeiHM, LvWW, WangDL, SunFL (2011) E2 ligase dRad6 regulates DMP53 turnover in Drosophila. J Biol Chem 286 : 9020–9030.
66. BradyCA, JiangD, MelloSS, JohnsonTM, JarvisLA, et al. (2011) Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 145 : 571–583.
67. LaneDP, VermaC (2012) Mdm2 in evolution. Genes Cancer 3 : 320–324.
68. PerryME (2010) The regulation of the p53-mediated stress response by MDM2 and MDM4. Cold Spring Harb Perspect Biol 2: a000968.
69. FakharzadehSS, TruskoSP, GeorgeDL (1991) Tumorigenic potential associated with enhanced expression of a gene that is amplified in a mouse tumor cell line. EMBO J 10 : 1565–1569.
70. MomandJ, ZambettiGP, OlsonDC, GeorgeD, LevineAJ (1992) The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69 : 1237–1245.
71. AlltonK, JainAK, HerzHM, TsaiWW, JungSY, et al. (2009) Trim24 targets endogenous p53 for degradation. Proc Natl Acad Sci U S A 106 : 11612–11616.
72. RossAJ, LiM, YuB, GaoMX, DerryWB (2011) The EEL-1 ubiquitin ligase promotes DNA damage-induced germ cell apoptosis in C. elegans. Cell Death Differ 18 : 1140–1149.
73. WylieA, LuWJ, D'BrotA, BuszczakM, AbramsJM (2014) p53 activity is selectively licensed in the Drosophila stem cell compartment. Elife 3: e01530.
74. AndreyenkovaNG, KokozaEB, SemeshinVF, BelyaevaES, DemakovSA, et al. (2009) Localization and characteristics of DNA underreplication zone in the 75C region of intercalary heterochromatin in Drosophila melanogaster polytene chromosomes. Chromosoma 118 : 747–761.
75. PainterTS (1935) The Morphology of the Third Chromosome in the Salivary Gland of Drosophila Melanogaster and a New Cytological Map of This Element. Genetics 20 : 301–326.
76. ZhangY, LinN, CarrollPM, ChanG, GuanB, et al. (2008) Epigenetic blocking of an enhancer region controls irradiation-induced proapoptotic gene expression in Drosophila embryos. Dev Cell 14 : 481–493.
77. NordmanJ, LiS, EngT, MacalpineD, Orr-WeaverTL (2011) Developmental control of the DNA replication and transcription programs. Genome Res 21 : 175–181.
78. NordmanJ, Orr-WeaverTL (2012) Regulation of DNA replication during development. Development 139 : 455–464.
79. BelyaevaES, ZhimulevIF, VolkovaEI, AlekseyenkoAA, MoshkinYM, et al. (1998) Su(UR)ES: a gene suppressing DNA underreplication in intercalary and pericentric heterochromatin of Drosophila melanogaster polytene chromosomes. Proc Natl Acad Sci U S A 95 : 7532–7537.
80. BaehreckeEH (2005) Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol 6 : 505–510.
81. LeeCY, BaehreckeEH (2001) Steroid regulation of autophagic programmed cell death during development. Development 128 : 1443–1455.
82. McPheeCK, LoganMA, FreemanMR, BaehreckeEH (2010) Activation of autophagy during cell death requires the engulfment receptor Draper. Nature 465 : 1093–1096.
83. HongA, Narbonne-ReveauK, Riesgo-EscovarJ, FuH, AladjemMI, et al. (2007) The cyclin-dependent kinase inhibitor Dacapo promotes replication licensing during Drosophila endocycles. EMBO J 26 : 2071–2082.
84. LeachTJ, ChotkowskiHL, WotringMG, DilwithRL, GlaserRL (2000) Replication of heterochromatin and structure of polytene chromosomes. Mol Cell Biol 20 : 6308–6316.
85. LaghaM, BothmaJP, LevineM (2012) Mechanisms of transcriptional precision in animal development. Trends Genet 28 : 409–416.
86. EspinosaJM, VerdunRE, EmersonBM (2003) p53 functions through stress - and promoter-specific recruitment of transcription initiation components before and after DNA damage. Mol Cell 12 : 1015–1027.
87. ChenHZ, OusephMM, LiJ, PecotT, ChokshiV, et al. (2012) Canonical and atypical E2Fs regulate the mammalian endocycle. Nat Cell Biol 14 : 1192–1202.
88. PanditSK, WestendorpB, NantasantiS, van LiereE, TootenPC, et al. (2012) E2F8 is essential for polyploidization in mammalian cells. Nat Cell Biol 14 : 1181–1191.
89. SherN, Von StetinaJR, BellGW, MatsuuraS, RavidK, et al. (2013) Fundamental differences in endoreplication in mammals and Drosophila revealed by analysis of endocycling and endomitotic cells. Proc Natl Acad Sci U S A 110 : 9368–9373.
90. ZielkeN, KimKJ, TranV, ShibutaniST, BravoMJ, et al. (2011) Control of Drosophila endocycles by E2F and CRL4(CDT2). Nature 480 : 123–127.
91. MeserveJH, DuronioRJ (2012) Atypical E2Fs drive atypical cell cycles. Nat Cell Biol 14 : 1124–1125.
92. UllahZ, KohnMJ, YagiR, VassilevLT, DepamphilisML (2008) Differentiation of trophoblast stem cells into giant cells is triggered by p57/Kip2 inhibition of CDK1 activity. Genes Dev 22 : 3024–3036.
93. SolovevaV, LinzerDI (2004) Differentiation of placental trophoblast giant cells requires downregulation of p53 and Rb. Placenta 25 : 29–36.
94. ZhengL, DaiH, ZhouM, LiX, LiuC, et al. (2012) Polyploid cells rewire DNA damage response networks to overcome replication stress-induced barriers for tumour progression. Nat Commun 3 : 815.
95. VenkenKJ, HeY, HoskinsRA, BellenHJ (2006) P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science 314 : 1747–1751.
96. Harlow E, Lane D (1999) Using Antibodies: A Laboratory Manual. Cold Spring Harbor: Cold Spring Harbor Laboratory Press.
97. RothbauerU, ZolghadrK, MuyldermansS, SchepersA, CardosoMC, et al. (2008) A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol Cell Proteomics 7 : 282–289.
98. SchwedG, MayN, PecherskyY, CalviBR (2002) Drosophila minichromosome maintenance 6 is required for chorion gene amplification and genomic replication. Mol Biol Cell 13 : 607–620.
99. KhouryMP, BourdonJC (2010) The isoforms of the p53 protein. Cold Spring Harb Perspect Biol 2: a000927.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 9
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Admixture in Latin America: Geographic Structure, Phenotypic Diversity and Self-Perception of Ancestry Based on 7,342 Individuals
- Nipbl and Mediator Cooperatively Regulate Gene Expression to Control Limb Development
- Genome Wide Association Studies Using a New Nonparametric Model Reveal the Genetic Architecture of 17 Agronomic Traits in an Enlarged Maize Association Panel
- Histone Methyltransferase MMSET/NSD2 Alters EZH2 Binding and Reprograms the Myeloma Epigenome through Global and Focal Changes in H3K36 and H3K27 Methylation
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy