
KDM6 Demethylase Independent Loss of Histone H3 Lysine 27 Trimethylation during Early Embryonic Development
H3K27me3 represses developmental genes at initial embryonic stages. The KDM6 family, comprised of UTX and JMJD3, are the only known proteins that demethylate H3K27me3 and they are hypothesized to catalyze the rapid removal of repressive chromatin in early mammalian development. However, we report that male embryos carrying mutations in both Utx and Jmjd3 survive to term and appear phenotypically normal at mid-gestation. We utilize several cell culture models to demonstrate that H3K27me3 is lost from repressed promoters in the absence of active KDM6 demethylation. Our data indicate that KDM6 H3K27me3 demethylation is not essential in the early embryo and that H3K27me3 loss from developmental genes occurs via novel mechanisms.
Published in the journal:
. PLoS Genet 10(8): e32767. doi:10.1371/journal.pgen.1004507
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004507
Summary
H3K27me3 represses developmental genes at initial embryonic stages. The KDM6 family, comprised of UTX and JMJD3, are the only known proteins that demethylate H3K27me3 and they are hypothesized to catalyze the rapid removal of repressive chromatin in early mammalian development. However, we report that male embryos carrying mutations in both Utx and Jmjd3 survive to term and appear phenotypically normal at mid-gestation. We utilize several cell culture models to demonstrate that H3K27me3 is lost from repressed promoters in the absence of active KDM6 demethylation. Our data indicate that KDM6 H3K27me3 demethylation is not essential in the early embryo and that H3K27me3 loss from developmental genes occurs via novel mechanisms.
Introduction
The mammalian embryo undergoes drastic changes in cellular specification and gene expression programs throughout development. These changes are facilitated by post-translational modifications to histones, which provide an epigenetic mechanism to coordinate initiation and maintenance of lineage specific transcriptional profiles that can be inherited through multiple cellular divisions. In embryonic stem cells and other pluripotent progenitors, crucial developmental genes are maintained in a quiescent state. A bivalent epigenetic signature defines this large class of genes. These promoters are maintained in a repressive chromatin state through histone H3 lysine 27 trimethylation (H3K27me3), however the presence of an active chromatin modification (H3K4me3) suggests that these genes are poised for rapid induction as development dictates [1]–[4]. Bivalent promoters have been identified in ES cells, the early embryo, lineage progenitors, and the germline [4]–[15]. With specification or differentiation, these bivalent promoters can be resolved to either a univalent H3K4me3 active state or a H3K27me3 repressed state. In numerous cell culture model systems, histone demethylases are required to remove H3K27me3 to promote gene activation, suggesting that H3K27me3 demethylation is essential in embryonic development [16]–[30].
H3K27me3 demethylases are members of the KDM6, Jumonji-C (JmjC) domain family of histone demethylases. The three KDM6 proteins, JMJD3 (KDM6B, encoded by an autosomal gene), UTX (KDM6A, X-chromosome), and UTY (Y-chromosome), all share a well-conserved JmjC histone demethylation domain [31]. Within this protein family, JMJD3 and UTX demethylate H3K27 tri-methyl and di-methyl residues, whereas human UTY demonstrates greatly reduced catalytic activity [27], [31]–[34]. Mouse UTY, despite maintaining 82% similarity to the X-chromosome homologue UTX, does not demethylate H3K27me3 due to mutations in the catalytic active site of its JmjC domain [31].
UTX and JMJD3 are individually involved in early embryonic specification events in cell culture [17]–[19], [22], [32], leading to the hypothesis that H3K27me3 demethylases function in early embryonic differentiation events. However, mouse mutagenesis suggests otherwise, as embryos deficient for individual demethylases survive to term. Jmjd3−/− homozygotes exhibit post-natal lethality due to neonatal respiratory deficits [35]. Utx−/y hemizygous males survive to adulthood and exhibit a normal lifespan [31]. In contrast, mutation of the Polycomb Repressive Complex 2 (PRC2) that methylates H3K27 yields precocious expression of early embryonic developmental genes and arrest in gastrulation [36]–[39]. Utx−/− homozygous females and Utx−;Uty− hemizygous males are both mid-gestational lethal with developmental delay and defects in embryonic heart development [31]. Therefore, the mid-gestational cardiovascular lethality that is driven by loss of UTX/UTY is due to demethylase independent function of these proteins. It is not clear if an early embryonic demethylase dependent function exists for the KDM6 family as some redundancy may exist between JMJD3 and UTX.
To study the role of the KDM6 family in early embryonic development we generated mutations designed to eliminate all KDM6 H3K27me3 demethylase activity in the developing mouse embryo. Male Utx−/y;Jmjd3−/− embryos devoid of KDM6 H3K27 demethylation survived to term. Mid-gestational Utx−/y;Jmjd3−/− embryos appeared phenotypically normal with characteristic features of embryonic day 10.5 (E10.5) embryos. We utilized several model systems (embryoid body, retinoic acid, mouse embryonic fibroblasts) to demonstrate that H3K27me3 can be removed from the promoters of repressed genes in the absence of active KDM6 demethylation. We conclude that KDM6 demethylases are not essential for early embryonic development and that H3K27me3 repression can be alleviated in the absence of active KDM6 demethylation.
Results
Mouse embryos devoid of KDM6 demethylation survive to term and display normal early embryonic phenotypes
To remove H3K27 demethylase activity in the mouse embryo we generated mutant alleles in both Utx and Jmjd3. We previously characterized the generation of the Utxfl allele that flanks exon 3 with loxP sites [31]. Cre mediated deletion of exon 3 (UtxΔ) created a frameshift in the coding sequence and is null for UTX protein. We now characterize a targeted allele, Jmjd3tm1Mag (Jmjd3fl) that integrates loxP sites 5′ to exon 14 and 3′ to exon 20 (Figure S1A). As verified by Southern blotting, PCR genotyping, and RT-PCR (Figure S1B, S1C, and S1D), Cre mediated deletion of this portion of the coding sequence (Jmjd3Δ) removed the JmjC catalytic H3K27 demethylase domain (Figure S1A). Similar to published reports, Jmjd3Δ/Δ homozygous pups died at birth with respiratory defects (Figure S1E). Jmjd3Δ/Δ homozygous embryos appeared phenotypically normal at mid-gestation (Figure S1F); however, several phenotypes manifested late in embryonic development which will be described elsewhere. While Jmjd3Δ/Δ homozygous pups were not observed at weaning, they were readily recovered at E18.5 (Figure 1A).
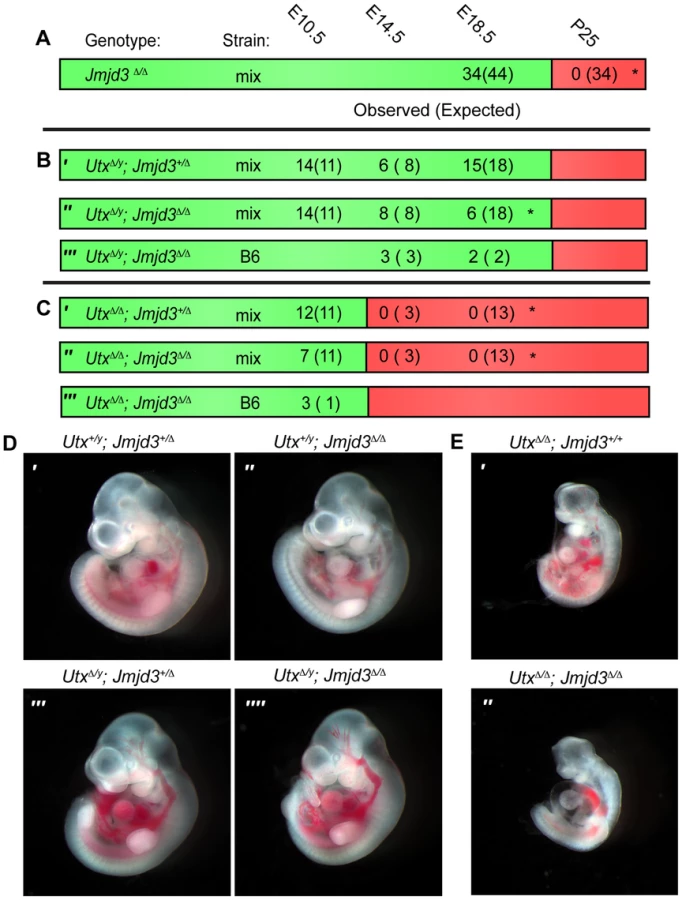
We next attempted to derive UtxΔ/y;Jmjd3Δ/Δ embryos whereby all KDM6 H3K27 demethylation is lost, while retaining the demethylase independent function of wild-type UTY. Similar to Utx−/y mutation alone [31], both UtxΔ/y;Jmjd3+/Δ and UtxΔ/y;Jmjd3Δ/Δ embryos survived to E18.5 (Figure 1B′ and 1B″). However, there was some reduction in observed UtxΔ/y;Jmjd3Δ/Δ embryos relative to expected Mendelian frequencies. Expected genotype frequencies of UtxΔ/y;Jmjd3Δ/Δ embryos were obtained at E14.5, so some redundancy may exist between Utx and Jmjd3 in late embryonic viability. At mid-gestation, all combinations of male Utx and Jmjd3 mutation were largely indistinguishable from controls (Figure 1D). UtxΔ/y;Jmjd3Δ/Δ embryos demonstrated normal features of E10.5 embryos, such as normal size and somite numbers (35-40), prominent fore and hind-limb buds, and developed branchial arches (including separation of arch 1 into maxilar and mandibular components, Figure 1D″″). As Utx−/y post-natal lethality is more pronounced on the C57BL6/J (B6) background [31], we backcrossed UtxΔ and Jmjd3Δ alleles. On a B6 background, UtxΔ/y;Jmjd3Δ/Δ embryos remained viable at both E14.5 and E18.5 timepoints (Figure 1B′″). Given that deposition of maternal UTX into the Drosophila embryo contributes to demethylation activity in early development [40], we tested if deletion of the UTX and JMJD3 maternal pool enhances mouse phenotypes. Utxfl/Δ;Jmjd3fl/Δ;VasaCre female mice (with oocytes carrying deletion of Utx and Jmjd3) were crossed with Utxfl/y;Jmjd3fl/Δ;VasaCre male mice (with sperm carrying deletion of Utx and Jmjd3) and resulting E10 UtxΔ/y;Jmjd3Δ/Δ embryos (Figure S2A′) had completely recombined Utx and Jmjd3 floxed alleles (Figure S2B) and phenocopied those derived from Utx+/Δ;Jmjd3+/Δ heterozygous mothers (Figure 1D″″).
Utx and Jmjd3 knockout was confirmed by quantitative RT-PCR (Figure S3A), and at E10.5, UtxΔ/y;Jmjd3Δ/Δ embryos did not exhibit altered Hox expression levels or elevated global levels of H3K27me3 (Figure S3B,C,D). Comparative H3K27me3 immunofluorescence of E10.5 Utx+/y;Jmjd3+/Δ and UtxΔ/y;Jmjd3Δ/Δ embryos sectioned onto the same slide revealed similar H3K27me3 levels within heart myocardium (Figure S3E) and ISL1 positive motor neurons within the proximal spinal chord (Figure S3F). However, mouse embryonic fibroblasts (MEFs) derived from these embryos had minor, yet statistically significant elevations in H3K27me3 levels (Figure S3G,H). Overall, in the absence of KDM6 H3K27 demethylation, embryos can clearly survive through gastrulation and exhibit normal patterning at E10.5. Notably, the phenotypes of UtxΔ/Δ;Jmjd3+/Δ and UtxΔ/Δ;Jmjd3Δ/Δ female embryos were similar to UtxΔ/Δ homozygous mutation alone, as these embryos are all lethal after E10.5 (Figure 1C) and exhibit similar features of developmental delay (Figure 1E). Additionally, maternal loss of UTX and JMJD3 demethylation had no contribution to phenotypic severity (Figure S2A″). Taken together, our data indicate that Utx/Uty are epistatic to Jmjd3, which primarily functions in later developmental stages.
ES cells with no KDM6 H3K27me3 demethylation have female specific differentiation defects
We established ES cell differentiation models to study the time-course of H3K27me3 demethylation in the absence of UTX and JMJD3. We utilized a CAGGCre-ER transgenic system that will induce allelic recombination with the addition of tamoxifen [41]. Following 2 days of tamoxifen treatment (+TX), Utxfl/y;Jmjd3fl/fl;CreER male or Utxfl/fl;Jmjd3fl/fl;CreER female ES lines demonstrated complete deletion of floxed exons and loss of endogenous protein (Figure S4A and S4B). A 140-KD background band is present in Figure S4B and is not lost in Utxfl/y;Jmjd3fl/fl;CreER ES +TX. To ensure that this is not an alternative Utx product, we analyzed its presence in UtxGT1/y ES cells [31], where any alternative products should be gene trapped. Even though UtxGT1/y ES cells did trap Utx transcripts preventing expression across the JmjC domain (Figure S4C) it did not affect the level of background bands (Figure S4B), indicating that these are indeed non-specific bands. Furthermore, western blot with a second, independent UTX antibody produced a clean blot with no UTX band in Utxfl/y;Jmjd3fl/fl;CreER ES +TX samples (Figure S4D).
We induced embryoid body (EB) differentiation as outlined in Figure 2A. By 4 days in culture, Utxfl/y;Jmjd3fl/fl;CreER +TX EBs looked identical to untreated controls (Figure 2B). Utxfl/fl;Jmjd3fl/fl;CreER female EBs +TX were small and displayed a disorganized outer endodermal layer with cells protruding or sloughing off of the EB (Figure 2B). In contrast to aggregate EB differentiation, hanging drop EB differentiation utilized smaller starting ES cell numbers in a defined drop volume, but still produced similar EB phenotypes (Figure S5A). Embryoid bodies exhibit a characteristic decrease in global H3K27me3 levels as differentiation progresses [32], [42]. Histones were extracted from EBs to determine if this process occurs in the absence of UTX and JMJD3 demethylation. Relative to global levels of H3K27me3 in ES cells, all EBs, even Utxfl/fl;Jmjd3fl/fl;CreER female EBs +TX demonstrated a reduction in H3K27me3 (Figure 3C). Fluorescent quantitative western blotting verified that both male and female +TX EBs exhibit loss of H3K27me3 levels (Figure S5B,C). Therefore, early EB differentiation events coincide with downregulation of H3K27me3 levels in the absence of all KDM6.


RT-PCR across deleted exons verified that even after 8 days in culture, wild type Utx and Jmjd3 expression was absent, and Uty expression was not diminished in TX treated cells (Figure 2D). Male Utxfl/y;Jmjd3fl/fl;CreER EBs +TX demonstrated normal activation of primitive ectoderm (Fgf5 expression), mesoderm (Flk1), and endoderm (Gata6, Figure 2E). In contrast, Utxfl/fl;Jmjd3fl/fl;CreER female EBs +TX initiated differentiation and induced primitive ectoderm (Fgf5), but failed to specify meso-endoderm (Brachyury T, illustrated as T), mesoderm (Flk1), and endoderm (Gata6, Figure 2E). Utxfl/y;Jmjd3fl/fl;CreER ES +TX were plated to derive single cell colonies of mutant clones, and constitutive propagation of this mutant ES line over several weeks did not affect the ability of the cells to differentiate into EBs (Figure S5D,E). Overall, the severe deficits of EB differentiation in Utxfl/fl;Jmjd3fl/fl;CreER +TX cells does not recapitulate the mild phenotypes of UtxΔ/Δ;Jmjd3Δ/Δ female embryos (Figure 1E″).
ES cell H3K27me3 removal and gene activation does not require KDM6 demethylases
To study the role of KDM6 in H3K27me3 demethylation, we utilized Retinoic acid (+RA) differentiation of ES cells. As Utxfl/fl;Jmjd3fl/fl;CreER EB +TX appeared capable of initiating ectoderm specification, RA differentiation towards a neuro-ectodermal lineage can be studied in this cellular model whereby all active demethylation by KMD6 members has been removed. We utilized a 2 day RA differentiation timecourse outlined in Figure 3A. H3K27me3 ChIP was performed on 4 individual replicates of WT ES (Utxfl/fl;Jmjd3fl/fl;CreER ES −TX), KO ES (Utxfl/fl;Jmjd3fl/fl;CreER ES +TX), WT RA (Utxfl/fl;Jmjd3fl/fl;CreER RA −TX), and KO RA (Utxfl/fl;Jmjd3fl/fl;CreER RA +TX). Two of the 4 replicates of each group were pooled together and the resulting 2 replicates of each group were subject to high throughput sequencing. H3K4me3 ChIP-seq was also performed on 2 replicates of WT RA and KO RA. The model-based analysis for ChIP-seq (MACS) algorithm identified enrichment peaks of H3K27me3 and H3K4me3 in each group, and edgeR statistical analysis software identified genes undergoing H3K27me3 demethylation in WT (WT ES vs. WT RA) and KO (KO ES vs. KO RA) RA differentiation. Overall, 1044 WT ES promoters (Transcription Start Site: TSS +/−1 KB) and 1141 KO ES promoters demonstrated H3K27me3 peaks. Of these promoters, 945 and 1055 (WT and KO respectively) also had a RA H3K4me3 peak, signifying that the majority of these promoters are bivalent.
EdgeR identified 54 WT promoters (from 50 unique genes, some having alternative promoters) and 109 KO promoters (103 genes) that demonstrated significant loss of H3K27me3 with RA differentiation (Figure 3B and Table S1) with many genes overlapping in both datasets. Many Hox genes lost H3K27me3 in both WT and KO RA differentiation. Tracks of H3K27me3 and H3K4me3 ChIP-seq were uploaded into the UCSC genome browser. With RA differentiation, the proximal Hoxa cluster (Figure 3C) from Hoxa1 through Hoxa6 demonstrated widespread loss of H3K27me3 in ES to RA differentiation of both WT and KO cells. This shift in histone profile correlated with large peaks of H3K4me3 at proximal Hoxa promoters, demonstrating gene activation events. Similar large-scale loss of H3K27me3 occurs from the proximal Hoxb (Hoxb1-Hoxb6), Hoxc (Hoxc4-Hoxc6), and Hoxd (Hoxd1-Hoxd4) clusters (Figure S6A,B,C). In addition to Hox genes, many other transcription factors such as Foxa1, Gata3, Meis2, and Nr2f2 demonstrated loss of promoter H3K27me3 in KO RA treatment (Figure 4A and Table S1). H3K27me3 ChIP-qPCR confirmed the loss of H3K27me3 in the absence of KDM6 demethylation (Figure 4B). Notably, all genes tested exhibited similar WT and KO loss of H3K27me3 with RA differentiation. Only Hoxb1 demonstrated a slight, significant increase in H3K27me3 for KO RA compared to WT RA, but overall Hoxb1 did exhibit a tremendous decrease in H3K27me3 in KO RA compared to KO ES. Furthermore, RT-PCR expression analysis confirmed that the genes demonstrating loss of H3K27me3 efficiently induced transcriptional activation in RA KO cells (Figure 4C). Utxfl/fl;Jmjd3fl/fl;CreER ES +TX were plated to derive single cell colonies of mutant clones, and constitutive propagation of this mutant ES line over several weeks did not affect the ability of the cells to activate transcription (Figure S5F). H3K27 demethylases physically associate with the MLL complex family of H3K4 methyl-transferases. Two members of this complex (ASH2L and RBBP5) were expressed at normal levels in KO cells (Figure S7A). One alternative explanation for loss of H3K27me3 in the absence of demethylation is that the PRC2 H3K27 methylation complex is down-regulated in differentiated cells or displaced from targeted promoters in a demethylase independent manner. While the EZH2 H3K27 methyl-transferase maintained high expression in RA differentiated cells (Figure S7B), the protein is displaced from promoters experiencing H3K27me3 loss in the absence of KDM6 demethylation (). In summary, repressed genes can demonstrate loss of H3K27me3 and initiate expression in the absence of KDM6.

Loss of KDM6 reduces H3K4 methylation and transcriptional activation in a small subset of H3K4me3 regulated genes
MACs analysis of H3K4me3 ChIP-seq of WT RA and KO RA cells identified enrichment peaks at 19,140 and 19,367 respective promoters (TSS +/−1 KB). EdgeR comparison of WT RA to KO RA identified 161 promoters (147 genes) that exhibited significant (FDR<0.05) reduction in KO H3K4me3 (Figure 5B, Table S2). The majority of these were small fold changes across a wide range in overall peak intensity. Of these 161 promoters, only 27 (26 genes) had an ES WT or KO H3K27me3 peak, so the majority of these genes are not regulated by H3K27 methylation. A few genes that exhibited KO H3K4me3 reductions and had a H3K27me3 peak are illustrated in the UCSC genome browser image of Figure 5A. RT-PCR confirmed that several genes with reduced KO H3K4me3 demonstrated reduced expression (Crabp2, Pax6, Wnt6, Ccnd2, Figure 5C). Several genes that had been denoted as experiencing normal H3K27me3 loss and gene activation in KO RA samples (Hoxb1, Foxa1) experienced normal KO RA H3K4me3 up-regulation relative to the ES cell state (Figure 5D). However, ChIP-qPCR did confirm genes identified in ChIP-seq to be deficient in RA KO H3K4me3 (Crabp2, Wnt6, Figure 5D). We next examined whether KO RA H3K4me3 affected genes can have associated increases in H3K27me3. ChIP-qPCR of Crabp2 and Wnt6 demonstrated increased KO RA H3K27me3 to ES comparable levels (Figure 5E).

Meta-analysis of categorized gene subsets reveals low-level changes in H3K27me3 in the absence of KDM6 demethylation
To better analyze the distribution of H3K27me3 and K3K4me3 we performed a meta-analysis examining the overall distribution of these histone modifications across all promoters with a corresponding MACS peak. These meta analyses (Figure 6A–G) were plotted with 95% confidence intervals centered on the mean normalized read counts. ES cells and RA differentiated cells both had a broad K27me3 distribution across promoters with a drop-off near the TSS (Figure 6A,B). H3K4me3 enrichment peaked downstream of the TSS (Figure 6C), and there was very close overlap between WT and KO profiles of both H3K27me3 and H3K4me3. Genes with significant reductions in H3K27me3 after RA differentiation, had visible differences in relative H3K27me3 sequence reads between ES and RA samples for both WT and KO (Figure 6D). In comparing WT RA to KO RA, there was very close overlap near the TSS (+/−1 KB); however, upstream of the TSS (−2 KB to −1 KB) KO RA exhibited a significant enrichment in the mean H3K27me3 levels per gene. Comparison of WT ES to KO ES across this same region also demonstrated significantly increased H3K27me3 levels (Figure 6D). Although H3K27me3 was elevated for these KO promoters, there was a complete overlap in H3K4me3 distribution (Figure 6E), further supporting data that this gene category was efficiently activating transcription in KO RA cells (Figure 4C). Thus, with RA differentiation, KO cells can experience dramatic reductions in H3K27me3 at specific promoters, and although there is minor H3K27me3 accumulation upstream of the TSS, transcription is not compromised.

Meta-analysis of promoters exhibiting H3K4me3 reductions in KO RA cells verified that this dataset was significantly deficient in H3K4me3 downstream of the TSS (Figure 6F). The H3K27me3 profile of this dataset revealed that these genes were not subject to dramatic K3K27me3 loss in WT ES to WT RA differentiation (Figure 6G). However, there was a small, but significant H3K27me3 elevation in RA KO relative to RA WT from TSS +0.5 KB to +1.5 KB. Because identifying a small subset of data from a graph for statistical analysis amounts to cherry-picking and is not without bias, we performed a genome-wide comparison of WT RA to KO RA H3K27me3 levels with edgeR. Genome-wide cross-comparison of WT RA vs. KO RA did not identify any promoters (TSS +/−1 KB) exhibiting a significant increase in KO H3K27me3. We also compared all KO RA H3K27me3 MACS peaks (peak center +/−0.5 KB), including those not found at TSSs, for normalized sequence read enrichment over WT RA. This analysis identified 74 KO RA H3K27me3 MACS peaks (out of 4504 total MACS peaks) that were enriched in sequence reads over WT (Table S3). These peaks resided in varying proximity to 64 unique genes; however, only 7 of these genes demonstrated compromised transcription based on a reduction in TSS H3K4me3 levels (Table S3). Overall, with KDM6 loss of demethylation, a small subset of genes have minor reductions in H3K4me3 and reduced transcription, and a fraction of these experience elevated H3K27me3.
MEFs exhibit KDM6 independent loss of H3K27me3 from promoters
To examine loss of H3K27me3 repression in a differentiated primary tissue, we utilized mouse embryonic fibroblasts (MEFs). Primary MEFs were cultured from E13.5 Utxfl/fl;Jmjd3fl/fl;CreER embryos and treated with tamoxifen as indicated in Figure 7A. Relative to ES cells, a panel of representative Hox genes (Hoxa3, Hoxc4, Hoxa13 and Hoxd13) demonstrated elevated expression levels in MEFs (Figure S8A). Following tamoxifen treatment, the growth of Utxfl/fl;Jmjd3fl/fl;CreER MEFs slowed dramatically, while MEFs without Cre continued to proliferate. Regardless, Utxfl/fl;Jmjd3fl/fl;CreER MEFs +TX largely did not experience reductions in Hox expression relative to untreated controls (Figure 7B). Only the most distal genes within the Hox A and D clusters (Hoxa13 and Hoxd13) demonstrated a slight but significant reduction in expression with loss of UTX and JMJD3. Hox H3K27 methylation in Utxfl/fl;Jmjd3fl/fl;CreER MEFs +TX matched their expression profile as the few distal genes that demonstrated mild expression deficiencies (Hoxa13 and Hoxd13) also exhibited a significant increase in H3K27me3 (Figure 7C).

Primary MEFs were also cultured from E13.5 UtxΔ/y;Jmjd3Δ/Δ embryos to assay function in both establishment and maintenance of a H3K27 demethylated state. Similar to transient TX induced KDM6 loss, UtxΔ/y;Jmjd3Δ/Δ MEFs had significantly reduced expression of more distal Hox genes (Hoxa13, Hoxd13, Figure 7D) relative to Utx+/y;Jmjd3+/Δ controls. H3K27me3 ChIP of UtxΔ/y;Jmjd3Δ/Δ MEFs only revealed some accumulation on Hoxa13 while other Hox genes were unaffected (Figure 7E). Notably, several Hox genes demonstrated significant reduction in H3K27me3 levels relative to ES cells (Hoxa3, Hoxc4, Hoxa6, Hoxd12) in both control and UtxΔ/y;Jmjd3Δ/Δ MEFs (Figure 7E). Some proximal Hox genes actually demonstrated an increase in KO MEF Hox expression (Hoxa3, Hoxc4 and Hoxa6), but this may be an artifact of the decreased growth rate of these cells as the H3K27me3 profile of these genes is unaffected. Furthermore, several other transcription factors (Wnt5a, Zeb2, Smarcd3) also exhibited near-complete loss of H3K27me3 even though all KDM6 demethylases had been removed throughout embryonic development. Notably these genes experience loss of localized H3K27me3 even though total H3K27me3 protein levels (Figure S3G,H) and EZH2 levels (Figure S8B,C) were elevated (H3K4me3 levels were not affected, Figure S8C). Therefore, H3K27me3 repressed genes can establish promoter states cleared of this repressive chromatin in the absence of KDM6 demethylases.
Discussion
A tremendous dichotomy exists in the field of H3K27 demethylases. H3K27me3 results in gene repression throughout the early embryo, yet enzymes that catalyze its removal are individually not essential for male embryonic viability. These findings are unexpected given that numerous genes crucial for early embryonic gastrulation events [43]–[52] experience H3K27me3 de-repression during development [4], [53], [54] including but not limited to GATA, TGF-β/BMP, WNT, FGF, and T-box transcription factor networks. Cell culture model systems have implicated that UTX and JMJD3 function in activation of several of these pathways [17]–[19], [22], [32]. We now demonstrate UtxΔ/y;Jmjd3Δ/Δ embryos devoid of all KDM6 demethylation remarkably survive to term and appear phenotypically normal at mid-gestation. UtxΔ/Δ;Jmjd3Δ/Δ embryos (lacking the demethylase independent function of UTY) survive through gastrulation and albeit smaller in size, can develop E10.5 features. Therefore, KDM6 is not crucial for alleviating the H3K27me3 repression of genes needed for early embryonic gastrulation events.
Even within individual cellular and organismal models, several studies have produced conflicting reports. UTX mediated H3K27 demethylation is reported to function in cellular reprogramming and germ cell development [55], however surviving Utx mutant male mice are fertile [31], [56]. UTX is reported to be essential for appropriate expression of germ layer markers in male ES cell differentiation [19], [56], yet differentiation deficits can largely be rescued by UTY or a catalytically inactive form of UTX [32], [57]. UTX is essential for efficient Hox H3K27me3 demethylation and gene activation [27], [29], [30], [57], [58], yet Utx null male ES cells can largely remove Hox H3K27me3 and demonstrate normal transcriptional activation [56]. The discrepancies in these reports may be accounted for by differences in cell type, genetic background, intrinsic growth differences in ES cell clones, differential JMJD3 redundancy, or differential UTY expression [as has been reported [31], [32], [56], [59]. We take advantage of mutant inducible alleles to enable comparison of control and knockout of the entire KDM6 family within the same ES cell clone with identical growth rate and genetic background. Similar to the normal appearance of mid-gestation UtxΔ/y;Jmjd3Δ/Δ embryos, KDM6 mutant male ES cells could differentiate normally into all EB germ layers. Mutant female ES cells null for any KDM6 demethylation demonstrated dramatic reduction in both global levels of H3K27me3 (with EB differentiation) and local levels of H3K27me3 from proximal Hox clusters and from promoters of other transcription factors (with RA differentiation). While there was a low level significant H3K27me3 accumulation upstream of these promoters in RA KO cells, these genes were largely cleared of H3K27me3 and experienced normal transcriptional activation, similar to findings in Utx male knockout studies alone [56]. Utx and Jmjd3 mutant MEFs demonstrated mild H3K27me3 accumulation and reduced expression only within the most distal regions of the Hox cluster. Similarly, Zebrafish UTX loss of function produces modest deficiencies in distal Hox expression [27]. EZH2 protein levels were mildly upregulated in Kdm6 mutant MEFs and may account for altered distal Hox gene regulation. Relative to ES cells, UtxΔ/y;Jmjd3Δ/Δ MEFs exhibited substantial reductions in promoter H3K27me3 of several Hox genes and developmental transcription factors. Thus in the absence of KDM6 H3K27 demethylation, H3K27me3 loss can be both initiated and maintained in developmental situations.
Our study raises intriguing questions regarding early embryonic removal of H3K27me3. Thus far, only UTX and JMJD3 have demonstrated the ability to demethylate H3K27me3. It is unlikely that another JmjC protein can demethylate H3K27me3. The KDM7 family including JHDM1D and PHF8 can demethylate both H3K9 and H3K27 dimethyl residues, but does not demethylate trimethyl residues [60], [61]. The PHF8 active site cannot sterically accommodate trimethyl residues [62]. In contrast, UTX positions H3K27me3 farther from the active site to properly position the larger residue modifications [63]. Furthermore, UTX amino acid Y1135 bonds with a methyl group of H3K27me3 and is essential for demethylation [31], [63]. This residue is not conserved in the KDM7 family. A tyrosine at this position is conserved for members of the KDM4 family of H3K9 and H3K36 trimethyl demethylases. However, this family of proteins has a catalytic core buried within a deep pocket, and residues downstream of H3K27 do not encode enough flexibility to fit this modification in the active site [64]. It is possible that a novel family of proteins may actively demethylate H3K27me3 utilizing distinct chemistry.
Alternatively, H3K27me3 in the early embryo may be replaced by passive mechanisms, as histones can be turned over multiple times within each cell cycle [65]. PRC complexes remain bound to chromatin during DNA replication and associate with the replication fork in dividing cells to direct methylation of H3K27me3 on newly incorporated daughter strand histones [66]–[68]. Thus, in a passive model for H3K27me3 replacement, displacement of the PRC2 complex during replication allows for incorporation of un-methylated H3K27. In a similar fashion, DNA methylation in the mouse pre-implantation embryo and germline may be removed via passive replication dependent mechanisms via displacement of DNA methyl-transferase from sites of replication [69], [70].
The role of the KDM6 family in development is not clear. Passive H3K27me3 removal may dominate in rapidly dividing cells such as in the early embryo. Active H3K27me3 demethylation may prove more essential for rapid response to specific environmental or developmental cues, particularly in more static cellular populations. Alternatively, rather than facilitating drastic gene induction, H3K27 demethylases may act to fine-tune transcriptional activity to promote accurate robust temporal and spatial patterns of gene expression. Accordingly, UTY associates with a wide array of chromatin and transcriptional machinery that may promote proper gene expression output [31]. In this sense, KDM6 members may encode a reader function to recruit transcriptional complexes to repressed genes and/or to displace PRC2 without the need for active demethylation. Point mutagenesis in the UTX and JMJD3 catalytic domains further emphasizes demethylase independent roles for the KDM6 family [32], [71], [72]. Going forward, genetic experiments analyzing crosstalk between chromatin modifying factors and structure/function analysis within the mammalian embryo are required to define specific function of the KDM6 family in embryonic development.
Materials and Methods
Mice
All mouse experimental procedures were approved by the University of North Carolina Institutional Animal Care and Use Committee. The Utxfl allele is described [31]. The Jmjd3tm1Mag (Jmjd3fl) allele was derived by targeting E14 ES cells. The Jmjd3 targeting construct was generated by BAC recombineering to insert a LoxP site in intron 13 and a FRT-Neomycin-FRT-LoxP cassette in intron 20 of Jmjd3. After successful targeting, the Neomycin cassette was removed by electroporation of a pCAGG-FLP construct. The resulting Jmjd3+/fl ES cells were injected into C57BL/6J blastocysts. Resulting chimeras were mated to CD1 females to assess germline transmission, and were maintained on either a mixed CD1 background or were backcrossed to C57BL/6J. UtxΔ and Jmjd3Δ alleles were generated by crossing floxed alleles to the VasaCre transgene that restricts Cre activity to the germline [73]. These progeny were then mated to propagate the UtxΔ and Jmjd3Δ alleles. To generate higher proportions of desired genotypes in the Utx Jmjd3 genetic interaction cross, Utx+/Δ;Jmjd3+/Δ female mice were mated with either Utxfl/y;Jmjd3fl/Δ;VasaCre or Utx+/y;Jmjd3fl/Δ;VasaCre males. Embryos were PCR genotyped from yolk sac samples for Utx or Jmjd3 and were sexed by a PCR genotyping scheme to distinguish Utx from Uty. All primer sequences are listed in Table S4.
Cell culture
Utxfl/fl;Jmjd3fl/fl;CreER and Utxfl/y;Jmjd3fl/fl;CreER ES cell lines were generated from E3.5 blastocysts in crosses utilizing CAGGCre-ER transgenic mouse line [41]. ES cells were cultured as described [36]. ES cells were split off of feeder MEFs and treated with 1 µg/mL 4-hydroxytamoxifen (4-OHT) for 3 days and were allowed to recover for 1 day. In EB differentiation experiments, 106 ES cells were cultured in 10 mL of ES culture media lacking LIF on agarose coated Petri dishes. Hanging drop EBs were set up with 25 µL drops of ES cells at a concentration of 20,000 cells per mL. In RA differentiation experiments, ES cells were plated following tamoxifen recovery, and the next day were cultured without LIF in 1 µM Retinoic Acid. E13.5 MEFs were generated as described [31]. MEFs were passaged 1x and treated with 0.5 µM 4-OHT for 2 days, then were passaged and cultured for an additional 3 days.
RT-PCR, western blotting, and ChIP
RNA was isolated with Trizol and cDNA was synthesized with Multiscribe reverse transcriptase. Gene expression was analyzed by qRT-PCR (Bio-Rad SsoFast EvaGreen, CFX96 real time system). All RT-PCR was normalized to Gapdh expression and graphed relative to control samples. All primer sequences are listed in Table S4. Nuclear lysates, histone extracts, and western blotting was performed as described [31] utilizing anti-RBBP5 (Bethyl Labs A300-109A, 1∶5000), anti-ASH2L (Bethyl Labs A300-107A, 1∶3000), anti-H3K27me3 (Millipore 07-449, 1∶2000), anti-H3 (Abcam ab1791, 1∶10,000), anti-GAPDH (Sigma G9545, 1∶10000), anti-UTX [29], or anti-UTX (Bethyl Labs A302-374A, 1∶4000) antibodies. ChIP was performed as described [74] and graphed relative to % of total ChIP input DNA for each immunoprecipitation. 5×106 cells were sonicated by a Branson Sonifier at 15% duty cycle (0.7 s on 0.3 s off). For some experiments, chromatin was sonicated in a chilled water bath by a Bioruptor sonifier on high setting for 30 s with 60 s rest. Rabbit IgG (Sigma, I5006), anti-H3K27me3 (Abcam ab6002, 2.5 µl), anti-H3K4me3 (Abcam ab8580, 2 µl), or EZH2 (Cell Signaling 5246, 3 µl) antibodies were used for ChIP. All ChIP primers are listed in Table S4. Immunofluorescence experiments were performed as described [31] with anti H3K27me3 (Cell Signaling 9733S, 1∶500) or ISL1 (DSHB 39.4D5-S, 1∶200)
Preparation of ChIP-seq libraries and data analysis
ChIP DNA and Input DNA were ligated to Truseq adapters as described [75]. Samples were multiplexed and sequenced with the HiSeq 2000 Analyzer (UNC High Throughput Sequencing Facility). The quality of the sequences reads was evaluated with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were then mapped to the B6 genome using Bowtie (http://bowtie-bio.sourceforge.net/index.shtml). Significant enrichment (peaks) were called using MACS (http://liulab.dfci.harvard.edu/MACS/index.html), with the input ChIP-seq datasets for the background model, on pooled replicates. Peak lists were filtered using an FDR cutoff of 0.05. We then identified peaks that were within 1 Kb of transcriptional start sites (TSSs), as annotated in the UCSC genome browser for mm9. Read counts for a particular locus were normalized to the total number of sequence reads generated for each sample. We used edgeR (http://www.bioconductor.org/packages/2.12/bioc/html/edgeR.html) to determine if there was a bias in the number of H3K27me3 or H3K4me3 reads at MACS positive promoters between various samples. Metaplots were drawn using custom Python scripts and R, and t-tests were performed using the average read count per gene within the ranges specified in the text. ChIP-seq datasets were submitted to GEO (accession GSE58391: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE58391).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. AzuaraV, PerryP, SauerS, SpivakovM, JorgensenHF, et al. (2006) Chromatin signatures of pluripotent cell lines. Nat Cell Biol 8 : 532–538.
2. BernsteinBE, MikkelsenTS, XieX, KamalM, HuebertDJ, et al. (2006) A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125 : 315–326.
3. KuM, KocheRP, RheinbayE, MendenhallEM, EndohM, et al. (2008) Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 4: e1000242.
4. MikkelsenTS, KuM, JaffeDB, IssacB, LiebermanE, et al. (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448 : 553–560.
5. Rugg-GunnPJ, CoxBJ, RalstonA, RossantJ (2010) Distinct histone modifications in stem cell lines and tissue lineages from the early mouse embryo. Proc Natl Acad Sci U S A 107 : 10783–10790.
6. SachsM, OnoderaC, BlaschkeK, EbataKT, SongJS, et al. (2013) Bivalent Chromatin Marks Developmental Regulatory Genes in the Mouse Embryonic Germline In Vivo. Cell Rep 3 : 1777–84 doi: 10.1016/j.celrep.2013.04.032
7. HammoudSS, NixDA, ZhangH, PurwarJ, CarrellDT, et al. (2009) Distinctive chromatin in human sperm packages genes for embryo development. Nature 460 : 473–478.
8. LindemanLC, AndersenIS, ReinerAH, LiN, AanesH, et al. (2011) Prepatterning of developmental gene expression by modified histones before zygotic genome activation. Dev Cell 21 : 993–1004.
9. DahlJA, ReinerAH, KlunglandA, WakayamaT, CollasP (2010) Histone H3 lysine 27 methylation asymmetry on developmentally-regulated promoters distinguish the first two lineages in mouse preimplantation embryos. PLoS One 5: e9150.
10. AlderO, LavialF, HelnessA, BrookesE, PinhoS, et al. (2010) Ring1B and Suv39h1 delineate distinct chromatin states at bivalent genes during early mouse lineage commitment. Development 137 : 2483–2492.
11. CuiK, ZangC, RohTY, SchonesDE, ChildsRW, et al. (2009) Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell 4 : 80–93.
12. van ArensbergenJ, Garcia-HurtadoJ, MoranI, MaestroMA, XuX, et al. (2010) Derepression of Polycomb targets during pancreatic organogenesis allows insulin-producing beta-cells to adopt a neural gene activity program. Genome Res 20 : 722–732.
13. RohTY, CuddapahS, CuiK, ZhaoK (2006) The genomic landscape of histone modifications in human T cells. Proc Natl Acad Sci U S A 103 : 15782–15787.
14. ZhaoXD, HanX, ChewJL, LiuJ, ChiuKP, et al. (2007) Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell 1 : 286–298.
15. PanG, TianS, NieJ, YangC, RuottiV, et al. (2007) Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell 1 : 299–312.
16. WangAH, ZareH, MousaviK, WangC, MoravecCE, et al. (2013) The histone chaperone Spt6 coordinates histone H3K27 demethylation and myogenesis. EMBO J 32 : 1075–1086.
17. KartikasariAE, ZhouJX, KanjiMS, ChanDN, SinhaA, et al. (2013) The histone demethylase Jmjd3 sequentially associates with the transcription factors Tbx3 and Eomes to drive endoderm differentiation. EMBO J 32 : 1393–1408.
18. RamadossS, ChenX, WangCY (2012) Histone demethylase KDM6B promotes epithelial-mesenchymal transition. J Biol Chem 287 : 44508–44517.
19. JiangW, WangJ, ZhangY (2012) Histone H3K27me3 demethylases KDM6A and KDM6B modulate definitive endoderm differentiation from human ESCs by regulating WNT signaling pathway. Cell Res 23 : 122–130.
20. WangJK, TsaiMC, PoulinG, AdlerAS, ChenS, et al. (2010) The histone demethylase UTX enables RB-dependent cell fate control. Genes Dev 24 : 327–332.
21. SeenundunS, RampalliS, LiuQC, AzizA, PaliiC, et al. (2010) UTX mediates demethylation of H3K27me3 at muscle-specific genes during myogenesis. EMBO J 29 : 1401–1411.
22. BurgoldT, SpreaficoF, De SantaF, TotaroMG, ProsperiniE, et al. (2008) The histone H3 lysine 27-specific demethylase Jmjd3 is required for neural commitment. PLoS One 3: e3034.
23. SatohT, TakeuchiO, VandenbonA, YasudaK, TanakaY, et al. (2010) The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol 11 : 936–944.
24. SenGL, WebsterDE, BarraganDI, ChangHY, KhavariPA (2008) Control of differentiation in a self-renewing mammalian tissue by the histone demethylase JMJD3. Genes Dev 22 : 1865–1870.
25. BarradasM, AndertonE, AcostaJC, LiS, BanitoA, et al. (2009) Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev 23 : 1177–1182.
26. AggerK, CloosPA, RudkjaerL, WilliamsK, AndersenG, et al. (2009) The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene - and stress-induced senescence. Genes Dev 23 : 1171–1176.
27. LanF, BaylissPE, RinnJL, WhetstineJR, WangJK, et al. (2007) A histone H3 lysine 27 demethylase regulates animal posterior development. Nature 449 : 689–694.
28. De SantaF, TotaroMG, ProsperiniE, NotarbartoloS, TestaG, et al. (2007) The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 130 : 1083–1094.
29. AggerK, CloosPA, ChristensenJ, PasiniD, RoseS, et al. (2007) UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449 : 731–734.
30. LeeMG, VillaR, TrojerP, NormanJ, YanKP, et al. (2007) Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 318 : 447–450.
31. ShpargelKB, SengokuT, YokoyamaS, MagnusonT (2012) UTX and UTY demonstrate histone demethylase-independent function in mouse embryonic development. PLoS Genet 8: e1002964.
32. WangC, LeeJE, ChoYW, XiaoY, JinQ, et al. (2012) UTX regulates mesoderm differentiation of embryonic stem cells independent of H3K27 demethylase activity. Proc Natl Acad Sci U S A 109 : 15324–15329.
33. HongS, ChoYW, YuLR, YuH, VeenstraTD, et al. (2007) Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc Natl Acad Sci U S A 104 : 18439–18444.
34. WalportLJ, HopkinsonRJ, VollmarM, MaddenSK, GileadiC, et al. (2014) Human UTY(KDM6C) is a Male-Specific N-Methyl Lysyl-Demethylase. J Biol Chem 289 : 18302–18313.
35. BurgoldT, VoituronN, CaganovaM, TripathiPP, MenuetC, et al. (2012) The H3K27 demethylase JMJD3 is required for maintenance of the embryonic respiratory neuronal network, neonatal breathing, and survival. Cell Rep 2 : 1244–1258.
36. ChamberlainSJ, YeeD, MagnusonT (2008) Polycomb repressive complex 2 is dispensable for maintenance of embryonic stem cell pluripotency. Stem Cells 26 : 1496–1505.
37. FaustC, SchumacherA, HoldenerB, MagnusonT (1995) The eed mutation disrupts anterior mesoderm production in mice. Development 121 : 273–285.
38. PasiniD, BrackenAP, JensenMR, Lazzerini DenchiE, HelinK (2004) Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J 23 : 4061–4071.
39. O'CarrollD, ErhardtS, PaganiM, BartonSC, SuraniMA, et al. (2001) The polycomb-group gene Ezh2 is required for early mouse development. Mol Cell Biol 21 : 4330–4336.
40. CopurO, MullerJ (2013) The histone H3-K27 demethylase Utx regulates HOX gene expression in Drosophila in a temporally restricted manner. Development 140 : 3478–3485.
41. HayashiS, McMahonAP (2002) Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol 244 : 305–318.
42. WalkerE, ChangWY, HunkapillerJ, CagneyG, GarchaK, et al. (2010) Polycomb-like 2 associates with PRC2 and regulates transcriptional networks during mouse embryonic stem cell self-renewal and differentiation. Cell Stem Cell 6 : 153–166.
43. ZhangH, BradleyA (1996) Mice deficient for BMP2 are nonviable and have defects in amnion/chorion and cardiac development. Development 122 : 2977–2986.
44. MeyersEN, LewandoskiM, MartinGR (1998) An Fgf8 mutant allelic series generated by Cre - and Flp-mediated recombination. Nat Genet 18 : 136–141.
45. MolkentinJD, LinQ, DuncanSA, OlsonEN (1997) Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev 11 : 1061–1072.
46. MorriseyEE, TangZ, SigristK, LuMM, JiangF, et al. (1998) GATA6 regulates HNF4 and is required for differentiation of visceral endoderm in the mouse embryo. Genes Dev 12 : 3579–3590.
47. ArnoldSJ, HofmannUK, BikoffEK, RobertsonEJ (2008) Pivotal roles for eomesodermin during axis formation, epithelium-to-mesenchyme transition and endoderm specification in the mouse. Development 135 : 501–511.
48. RussAP, WattlerS, ColledgeWH, AparicioSA, CarltonMB, et al. (2000) Eomesodermin is required for mouse trophoblast development and mesoderm formation. Nature 404 : 95–99.
49. HerrmannBG (1991) Expression pattern of the Brachyury gene in whole-mount TWis/TWis mutant embryos. Development 113 : 913–917.
50. WinnierG, BlessingM, LaboskyPA, HoganBL (1995) Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev 9 : 2105–2116.
51. LiuP, WakamiyaM, SheaMJ, AlbrechtU, BehringerRR, et al. (1999) Requirement for Wnt3 in vertebrate axis formation. Nat Genet 22 : 361–365.
52. CarverEA, JiangR, LanY, OramKF, GridleyT (2001) The mouse snail gene encodes a key regulator of the epithelial-mesenchymal transition. Mol Cell Biol 21 : 8184–8188.
53. XieW, SchultzMD, ListerR, HouZ, RajagopalN, et al. (2013) Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 153 : 1134–1148.
54. GiffordCA, ZillerMJ, GuH, TrapnellC, DonagheyJ, et al. (2013) Transcriptional and Epigenetic Dynamics during Specification of Human Embryonic Stem Cells. Cell 153 : 1149–1163.
55. MansourAA, GafniO, WeinbergerL, ZviranA, AyyashM, et al. (2012) The H3K27 demethylase Utx regulates somatic and germ cell epigenetic reprogramming. Nature 488 : 409–413.
56. WelsteadGG, CreyghtonMP, BilodeauS, ChengAW, MarkoulakiS, et al. (2012) X-linked H3K27me3 demethylase Utx is required for embryonic development in a sex-specific manner. Proc Natl Acad Sci U S A 109 : 13004–13009.
57. Morales TorresC, LaugesenA, HelinK (2013) Utx is required for proper induction of ectoderm and mesoderm during differentiation of embryonic stem cells. PLoS One 8: e60020.
58. ShahhoseiniM, TaghizadehZ, HatamiM, BaharvandH (2013) Retinoic acid dependent histone 3 demethylation of the clustered HOX genes during neural differentiation of human embryonic stem cells. Biochem Cell Biol 91 : 116–122.
59. LeeS, LeeJW, LeeSK (2012) UTX, a Histone H3-Lysine 27 Demethylase, Acts as a Critical Switch to Activate the Cardiac Developmental Program. Dev Cell
60. HuangC, XiangY, WangY, LiX, XuL, et al. (2010) Dual-specificity histone demethylase KIAA1718 (KDM7A) regulates neural differentiation through FGF4. Cell Res 20 : 154–165.
61. LoenarzC, GeW, ColemanML, RoseNR, CooperCD, et al. (2010) PHF8, a gene associated with cleft lip/palate and mental retardation, encodes for an Nepsilon-dimethyl lysine demethylase. Hum Mol Genet 19 : 217–222.
62. YuL, WangY, HuangS, WangJ, DengZ, et al. (2010) Structural insights into a novel histone demethylase PHF8. Cell Res 20 : 166–173.
63. SengokuT, YokoyamaS (2011) Structural basis for histone H3 Lys 27 demethylation by UTX/KDM6A. Genes Dev 25 : 2266–2277.
64. ChenZ, ZangJ, KapplerJ, HongX, CrawfordF, et al. (2007) Structural basis of the recognition of a methylated histone tail by JMJD2A. Proc Natl Acad Sci U S A 104 : 10818–10823.
65. DealRB, HenikoffJG, HenikoffS (2010) Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science 328 : 1161–1164.
66. HansenKH, BrackenAP, PasiniD, DietrichN, GehaniSS, et al. (2008) A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol 10 : 1291–1300.
67. PetrukS, SedkovY, JohnstonDM, HodgsonJW, BlackKL, et al. (2012) TrxG and PcG proteins but not methylated histones remain associated with DNA through replication. Cell 150 : 922–933.
68. FrancisNJ, FollmerNE, SimonMD, AghiaG, ButlerJD (2009) Polycomb proteins remain bound to chromatin and DNA during DNA replication in vitro. Cell 137 : 110–122.
69. InoueA, ZhangY (2011) Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science 334 : 194.
70. OhnoR, NakayamaM, NaruseC, OkashitaN, TakanoO, et al. (2013) A replication-dependent passive mechanism modulates DNA demethylation in mouse primordial germ cells. Development 140 : 2892–903 doi: 10.1242/dev.093229
71. MillerSA, MohnSE, WeinmannAS (2010) Jmjd3 and UTX play a demethylase-independent role in chromatin remodeling to regulate T-box family member-dependent gene expression. Mol Cell 40 : 594–605.
72. VandammeJ, LettierG, SidoliS, Di SchiaviE, Norregaard JensenO, et al. (2012) The C. elegans H3K27 demethylase UTX-1 is essential for normal development, independent of its enzymatic activity. PLoS Genet 8: e1002647.
73. GallardoT, ShirleyL, JohnGB, CastrillonDH (2007) Generation of a germ cell-specific mouse transgenic Cre line, Vasa-Cre. Genesis 45 : 413–417.
74. RahlPB, LinCY, SeilaAC, FlynnRA, McCuineS, et al. (2010) c-Myc regulates transcriptional pause release. Cell 141 : 432–445.
75. CalabreseJM, SunW, SongL, MugfordJW, WilliamsL, et al. (2012) Site-specific silencing of regulatory elements as a mechanism of X inactivation. Cell 151 : 951–963.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 8
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Meta-Analysis of Genome-Wide Association Studies in African Americans Provides Insights into the Genetic Architecture of Type 2 Diabetes
- KDM6 Demethylase Independent Loss of Histone H3 Lysine 27 Trimethylation during Early Embryonic Development
- The RNA Helicases AtMTR4 and HEN2 Target Specific Subsets of Nuclear Transcripts for Degradation by the Nuclear Exosome in
- EF-P Dependent Pauses Integrate Proximal and Distal Signals during Translation
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy