
Roles of Type 1A Topoisomerases in Genome Maintenance in
DNA topoisomerases are ubiquitous enzymes that solve the topological problems associated with replication, transcription and recombination. Eukaryotic enzymes of the type 1A family work with RecQ-like helicases such as BLM and Sgs1 and are involved in genome maintenance. Interestingly, E. coli topo I, a type 1A enzyme and the first topoisomerase to be discovered, appears to have distinct cellular functions that are related to supercoiling regulation and to the inhibition of R-loop formation. Here we present data strongly suggesting that these cellular functions are required to inhibit inappropriate replication originating from either oriC, the normal origin of replication, or R-loops that can otherwise lead to severe chromosome segregation defects. Avoiding such inappropriate replication appears to be a key cellular function for genome maintenance, since the other E. coli type 1A topo, topo III, is also involved. Furthermore, our data suggest that bacterial type 1A topos, like their eukaryotic counterparts, can act with RecQ in genome maintenance. Altogether, our data provide new insight into the role of type 1A topos in genome maintenance and reveal an interplay between these enzymes and R-loops, structures that can also significantly affect the stability of the genome as recently shown in numerous studies.
Published in the journal:
. PLoS Genet 10(8): e32767. doi:10.1371/journal.pgen.1004543
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004543
Summary
DNA topoisomerases are ubiquitous enzymes that solve the topological problems associated with replication, transcription and recombination. Eukaryotic enzymes of the type 1A family work with RecQ-like helicases such as BLM and Sgs1 and are involved in genome maintenance. Interestingly, E. coli topo I, a type 1A enzyme and the first topoisomerase to be discovered, appears to have distinct cellular functions that are related to supercoiling regulation and to the inhibition of R-loop formation. Here we present data strongly suggesting that these cellular functions are required to inhibit inappropriate replication originating from either oriC, the normal origin of replication, or R-loops that can otherwise lead to severe chromosome segregation defects. Avoiding such inappropriate replication appears to be a key cellular function for genome maintenance, since the other E. coli type 1A topo, topo III, is also involved. Furthermore, our data suggest that bacterial type 1A topos, like their eukaryotic counterparts, can act with RecQ in genome maintenance. Altogether, our data provide new insight into the role of type 1A topos in genome maintenance and reveal an interplay between these enzymes and R-loops, structures that can also significantly affect the stability of the genome as recently shown in numerous studies.
Introduction
Type 1A topoisomerases (topos) are essential and ubiquitous enzymes found in bacteria, archaea and eukarya [1], [2]. They all require single-stranded DNA (ssDNA) regions for activity. Such substrates can already be present, for example, in negatively supercoiled DNA, at the replication fork and in R-loops, or can be generated by the action of proteins, such as helicases or RNA polymerases. E. coli topo I, the first topo to be discovered [3], is the prototype enzyme of this family. This enzyme binds to ssDNA close to double-stranded DNA (dsDNA) regions [4] and is therefore well suited to relax the excess negative supercoiling generated behind RNA polymerase molecules during transcription [5], or introduced by DNA gyrase, the enzyme that negatively supercoils DNA in bacteria [6].
The best evidence for a major role of topo I in the regulation of supercoiling came from the observation that topA null mutants accumulate compensatory mutations in gyrA or gyrB genes allowing them to grow [7]. These mutations decrease the supercoiling activity of gyrase which leads to a reduction in the global chromosome supercoiling level below that of wild-type cells [8]. The role of topo I in transcription is supported by the finding that it physically interacts with RNA polymerase [9]. One major consequence of excess negative supercoiling is R-loop formation [10]. This is supported by the observation that the growth defect of topA null mutant can be partially compensated by RNase HI overproduction [11]. Evidence for extensive R-loop formation in the absence of topo I has been provided both in vitro and in vivo [12]–[15]. It is believed that topo I prevents R-loop formation mainly by relaxing transcription-induced negative supercoiling [15].
After a temperature downshift to reactivate gyrase in a topA null mutant carrying a gyrB(Ts) allele, RNase HI overproduction was shown to prevent a transient growth arrest that correlated with the accumulation of excess negative supercoiling (hypernegative supercoiling) and extensive RNA degradation [16]. RNase HI overproduction was found both to reduce the accumulation of excess negative supercoils and to promote their rapid removal by topo IV [16], [17], the other enzyme that can relax negative supercoiling in E. coli [18]. Moreover, evidence for R-loops impeding transcription of ribosomal RNA genes (rrn operons) in topA null mutants has been reported [19]. Interestingly, R-loop-dependent gene expression inhibition related to RNA polymerase arrest and RNA degradation has also been reported for yeast cells lacking topo I, a type 1B topo [20]. Thus, R-loop-mediated impairment of gene expression appears to be a major mechanism by which excess negative supercoiling inhibits growth.
E. coli topo I is a relatively abundant protein being in the top 25% of the most abundant proteins in E. coli (134 ppm) [21]. The topA gene is under the control of promoters recognized by different sigma factors, σ32, σS and σ70 and its expression is important for E. coli's response to various stresses including heat and oxidative shock [22]. RNase HI overproduction was shown to partially restore the expression of σ32-regulated genes required for the heat shock response [23].
Although studies of topo I mostly focused on its role in supercoiling regulation and its effect on gene expression, evidence for the involvement of this enzyme in other DNA transactions such as chromosome segregation and replication initiation has been provided [17], [24]–[27]. Interestingly, one of the first functions to be proposed for topo I was a role as a specificity factor to inhibit replication initiation outside oriC, such as initiation from R-loops, that could occur in an in vitro reconstituted system for oriC-dependent replication [28]. However, there is no experimental evidence for such a role of topo I in vivo.
E. coli topo III, the second type 1A topo to be discovered, has a much higher preference for ssDNA than E. coli topo I [29]. As a consequence, topo III is very inefficient in relaxing DNA with a physiological supercoiling density and, in fact, this enzyme plays no role in supercoiling regulation [18], [30]. However, topo III was shown to be a very potent decatenase during replication in vitro provided that a ssDNA region was present on the DNA substrate for the binding of the enzyme [29]. The presence of a unique amino-acid sequence in the topo III protein named the “decatenation loop” (absent in eukaryotic type 1A enzymes), was found to be essential for the decatenation of replication intermediates [31].
Unlike topo I, topo III is a protein of low abundance (9.4 ppm) [21]. Moreover, as opposed to topA null mutants, cells lacking topo III activity display no growth defects [32]. Recently, it has been shown that topo III plays a role in chromosome segregation in vivo that is likely related to replication, as this function was shown to be mostly required when the activity of topo IV [33], the main cellular decatenase, or gyrase [34] was severely impaired. Topo III physically interacts with SSB protein and this interaction presumably allows topo III to act at the replication fork in the cell [35].
Similar to eukaryotic type 1A topos (see below), topo III activity was shown to be stimulated by RecQ helicase in vitro [35]–[37], but these two proteins do not physically interact. Evidence for RecQ acting with topo III in E. coli cells has been reported [30]. However, because some important properties of the strains used in this work could not be observed in an independent study, the conclusion that RecQ acts with topo III has been questioned [33].
Saccharomyces cerevisiae type 1A topo was the first enzyme of this family to be discovered in eukaryotic cells [38]. Being the third topo identified in this organism, it was named Top3. The existence of this topo was revealed following the isolation of a mutation, in top3, that stimulated recombination between repeated sequences [38]. Interestingly, phenotypes of top3 mutants including slow growth and sporulation deficiency were suppressed to different extents by inactivating SGS1, encoding the RecQ homolog of S. cerevisiae, or by overproducing E. coli topo I [38]–[40]. Moreover, deleting RAD51, encoding the RecA homolog of S. cerevisiae, was shown to rescue the slow growth phenotype of top3 mutants [41]. Altogether, these data suggested that Sgs1 processed recombination intermediates to generate structures that could only be resolved by a type 1A topo, such as Top3 or E. coli topo I.
Physical interactions between type 1A topos (named topo III in higher eukaryotes) and their RecQ-like partner from eukaryotic organisms (e.g BLM in humans and in Drosophila) have been demonstrated [1], [39], [42], [43]. It is now well established that these complexes can efficiently resolve homologous recombination intermediates (Double Holliday Junctions; DHJs) without genetic exchange [1], [44]–[46]. Reactions of BLM with topo III are often stimulated by the presence of RPA, the SSB homolog of eukaryotes that presumably stabilizes the BLM-generated ssDNA region, the substrate for topo III [44], [46]. Eukaryotic topo III enzymes have a higher requirement for ssDNA than E. coli topo I and, in fact, they are generally considered to be more closely related to E. coli topo III than topo I [1].
In E. coli, an interplay between topo I and III has been reported in two instances. In the first one, the topB gene was isolated as a multicopy suppressor of a topA null mutant [47]. Despite the significant correction of the growth defect of the topA null mutant by overproducing topo III, relaxation of the excess negative supercoiling introduced by gyrase was barely detected. This is consistent with our observation that topo III overproduction, unlike RNase HI overproduction, is unable to prevent the supercoiling-dependent transient growth arrest of a topA gyrB(Ts) strain, following a temperature downshift ([16]; Baaklini and Drolet, unpublished). These results might have suggested that topo III overproduction complemented a yet unknown function of topo I that was not directly related to excess supercoiling. Indeed, here we present genetic evidence for an important role of topo I acting with RecQ to resolve RecA-dependent recombination intermediates that otherwise inhibit chromosome segregation. Moreover, our data suggest that the requirement for this activity is related to over-replication mostly from oriC that takes place in the absence of topA, presumably due to excess negative supercoiling.
In the second instance, deleting topB from a topA null mutant carrying a gyrA or gyrB compensatory mutation, led to the formation of very long filaments with unsegregated nucleoids having abnormal structures and, eventually, to growth arrest [48]. Here, our data suggest that these phenotypes are exacerbated by excess negative supercoiling and are mostly related to over-replication from R-loops.
Overall, our data demonstrate that bacterial type 1A topos maintain the stability of the genome by preventing unregulated replication and at least one of its consequences, namely the inhibition of chromosome segregation.
Results
Supercoiling-dependent growth and chromosome segregation defects in cells lacking topo I activity
To look for chromosome segregation defects in a topA null mutant, cells of a ΔtopA gyrB(Ts) strain were stained with DAPI and prepared for microscopy such that both cell morphology and DNA content could be examined. By growing the cells at 30°C, the permissive temperature for gyrase, we could test the true effect of losing topA on nucleoid shape. As can be seen in Figure 1A, whereas nucleoids of gyrB(Ts) cells were well separated and compact, those of isogenic gyrB(Ts) ΔtopA cells were less compact and clearly not separated, thus showing chromosome segregation defects.

To verify if these problems were related to excess negative supercoiling, topA null cells were grown at 37°C so that gyrase activity was reduced. At this temperature the chromosome supercoiling level decreases below that of wild-type cells and, as a result, topA null cells can grow robustly [11], [49]. At 37°C, chromosome segregation in the gyrB(Ts) ΔtopA strain was significantly improved, as many cells had well separated and more compact nucleoids as compared to cells grown at 30°C (Figure 1A, gyrB(Ts) ΔtopA, 37°C vs 30°C). We tested the effect of RNase HI overproduction on chromosome segregation in the gyrB(Ts) ΔtopA strain grown at 30°C. It did not correct the chromosome segregation defect (gyrB(Ts) ΔtopA/pSK760). Thus, we conclude that topA null cells suffer from supercoiling-dependent chromosome segregation defects that are unrelated to R-loops.
RNase HI overproduction did not correct the chromosome segregation problem whereas it clearly stimulated the growth of gyrB(Ts) ΔtopA cells at 30°C (Figure 1B, gyrB(Ts) ΔtopA vs gyrB(Ts) ΔtopA/pSK760). Therefore, at this temperature the defect was not strong enough to offset the positive effect of overproducing RNase HI. We have previously shown that RNase HI overproduction could not complement the growth defect of topA null mutants at lower temperatures. In fact, it had a negative effect [47], [50]. The cold sensitivity of topA null mutants was found to be, at least in part, related to the inability of topo IV to efficiently relax negative supercoiling at low temperatures [16], [17]. As a result, hypernegative supercoiling accumulated.
We found that the chromosome segregation defect of our gyrB(Ts) ΔtopA strain was exacerbated at 24°C since the cells were generally longer and the DNA more diffuse as compared to cells grown at 30°C (Figure 1A, gyrB(Ts) ΔtopA, 30°C vs 24°C). Overproducing RNase HI further stimulated cell filamentation and produced cells with large DNA-free regions (Figure 1A, gyrB(Ts) ΔtopA/pSK760, 24°C). Growth of gyrB(Ts) ΔtopA cells on solid LB medium at 24°C was very poor whether RNase HI was overproduced or not (Figure 1B, 24°C). Thus, the cold sensitivity of topA null cells triggered by excessive hypernegative supercoiling correlates with a strong chromosome segregation defect that seems to be exacerbated by RNase HI overproduction.
Topo III overproduction and recA or recQ deletions correct both the growth and chromosome segregation defects in cells lacking topo I activity
Topo III overproduction was previously shown to correct the growth defect of topA null mutants at low temperatures [47]. In fact, unlike RNase HI, topo III overproduction was able to correct the growth defect of gyrB(Ts) ΔtopA cells at 21°C [47]. Since topo III is a potent decatenase and because the growth defect of gyrB(Ts) ΔtopA cells at 24°C correlates with a strong chromosome segregation problem (Figure 1A), topo III overproduction may have complemented by correcting this segregation defect.
This was confirmed by the observation that overproducing topo III almost fully, at 30°C, or partially, at 24°C, corrected the chromosome segregation defect of gyrB(Ts) ΔtopA cells (gyrB(Ts) ΔtopA/pPH1243; Figure 2A, at 30°C the nucleoids are well separated and compact; at 24°C some nucleoids separated, shorter cells and the DNA is more compact as compared to cells not overproducing topo III). As expected, topo III overproduction also promoted growth on solid media at these temperatures (Figure 2B). Thus, topo III overproduction corrects the growth defect of topA null mutants, at least in part, by facilitating chromosome segregation.
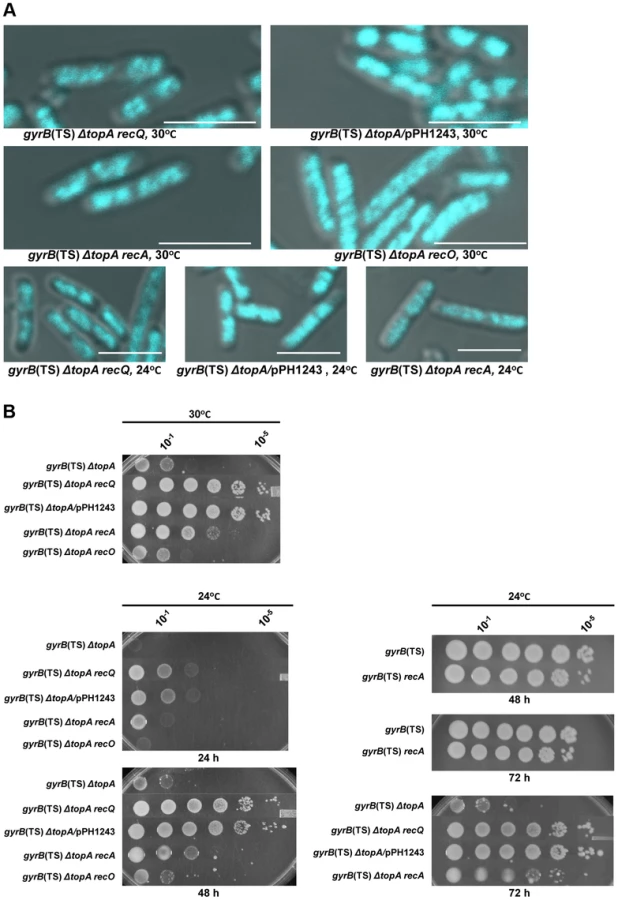
The next series of experiments were performed to test the hypothesis that, as is the case in eukaryotic cells, E. coli type 1A topos can act with RecQ to resolve RecA-generated recombination intermediates. We first tested the effect of deleting recQ on growth and chromosome segregation in topA null cells. We found that deleting recQ was as good as overproducing topo III in correcting the growth defect of our gyrB(Ts) ΔtopA strain at both 30 and 24°C (Figure 2B; gyrB(Ts) ΔtopA ΔrecQ; western blot experiments showed that topo IV was not overproduced in the topA null strain lacking recQ; Figure S3). Deleting recQ also partially corrected the chromosome segregation defect at these temperatures (Figure 2A). Thus, our results suggest that recQ and topB act in a pathway that is related to chromosome segregation in the absence of topA.
We next tested the effect of deleting recA on growth and chromosome segregation in topA null cells. The deletion of recA partially corrected the growth defect of our gyrB(Ts) ΔtopA strain at both 30 and 24°C (Figure 2B; gyrB(Ts) ΔtopA ΔrecA), though the effect was not as good as the one conferred by deleting recQ or overproducing topo III (Figure 2B). In fact, the positive effect of deleting recA on the growth of topA null cells at 24°C was more readily observed after three days of incubation (Figure 2B, 72 h). Deleting recA also partially alleviated the chromosome segregation defects of topA null cells at both temperatures (Figure 2A). These results demonstrate that the chromosome segregation defects of topA null mutants are largely RecA-dependent and therefore support the involvement of homologous recombination.
In E. coli, positive effects of deleting recQ on growth and chromosome segregation are often attributed to unnecessary RecA-mediated recombination via the RecFOR pathway at arrested replication forks [51], [52]. In this pathway, RecQ helicase acts with RecJ, a 5′-3′ exonuclease, to provide ssDNA regions on which RecF, O and R facilitate RecA nucleoprotein filament assembly. We found that deleting recJ, recO or recR had no effect on growth and chromosome segregation in our gyrB(Ts) ΔtopA strain (Figure 2, gyrB(Ts) ΔtopA ΔrecO; data not shown for recJ and recR). This indicated that the RecFOR pathway was not involved and therefore may suggest that RecQ and type 1A topos act together in a RecA-dependent recombination pathway.
If indeed RecQ acts on RecA-generated recombination intermediates to generate substrates for type 1A topos, neither topo III overproduction nor recQ deletion should improve the growth of gyrB(Ts) ΔtopA cells lacking recA. Moreover, overproducing topo III should also have no effects on the growth of gyrB(Ts) ΔtopA ΔrecQ cells. To test these predictions, the appropriate strains were constructed and spot assays were performed. As predicted, combinations of recQ and recA deletions or of topo III overproduction and recA mutation resulted in the same growth phenotype as the recA mutation alone (Figure 3A, compare gyrB(Ts) ΔtopA ΔrecQ, gyrB(Ts) ΔtopA ΔrecA and gyrB(Ts) ΔtopA ΔrecQ ΔrecA; Figure 3B, compare gyrB(Ts) ΔtopA/pPH1243, gyrB(Ts) ΔtopA ΔrecA and gyrB(Ts) ΔtopA ΔrecA/pPH1243). Furthermore, topo III overproduction did not improve the growth of gyrB(Ts) ΔtopA ΔrecQ cells (Figure 3C). These results are consistent with RecQ processing RecA-generated recombination intermediates in such a way that they can only be resolved by a type 1A topo. Since topo III needs to be overproduced, we believe that the much more abundant topo I enzyme is normally involved in the resolution of these intermediates (see Discussion).

Our microarray results indicated that the SOS response was constitutively expressed in our gyrB(Ts) ΔtopA strain and therefore that RecA was overproduced (not shown). The lexA3 allele was used to test the effect of the SOS response on the growth of the gyrB(Ts) ΔtopA strain. This allele makes the SOS response non-inducible and therefore considerably reduces the amount of RecA proteins produced. The lexA3 allele was found to be slightly better than deleting recA to improve the growth of the gyrB(Ts) ΔtopA strain (Figure 3D, compare gyrB(Ts) ΔtopA, gyrB(Ts) ΔtopA lexA3 and gyrB(Ts) ΔtopA ΔrecA; several lexA3 transductants were tested and were found to behave the same way). This result may suggest that the major effect of the recA mutation on the growth of the gyrB(Ts) ΔtopA strain was not related to the silencing of the SOS response but rather to the inactivation of the recombination function of RecA. If this is true, the lexA3 allele should behave differently from the recA mutation when combined with the recQ mutation in the gyrB(Ts) ΔtopA strain. Figure 3E shows that it was indeed the case. Whereas the recA growth phenotype was dominant over the recQ one (Figure 3A), the reverse was observed for the lexA3 allele i.e., the gyrB(Ts) ΔtopA ΔrecQ lexA3 strain grew like the gyrB(Ts) ΔtopA ΔrecQ one (Figure 3E). Thus, these results indicate that the lexA3 allele improved the growth of the gyrB(Ts) ΔtopA strain mostly by reducing the amount of RecA proteins, but at the same time that a minimal level of RecA-dependent recombination was required for the optimal growth of the gyrB(Ts) ΔtopA strain.
Our results showed that the RecF pathway for the loading of RecA on ssDNA was not involved in the RecA effects in the gyrB(Ts) ΔtopA strain (Figure 2). The RecBCD pathway is the other one involved in the loading of RecA on ssDNA in E. coli. The introduction of a recB::Tn10 mutation in our gyrB(Ts) ΔtopA strain resulted in a strain that grew very poorly. Growth inhibition was clearly observed at 37°C, a temperature normally fully permissive for the growth of the gyrB(Ts) ΔtopA strain (Figure 3F, compare gyrB(Ts) ΔtopA and gyrB(Ts) ΔtopA recB). At 30°C, growth was barely detected (Figure 3F) and the strain did not show grow at 24°C even after 6 days of incubation. These results indicate that the RecA effects in the gyrB(Ts) ΔtopA strain are most likely mediated through the RecBCD pathway and, more importantly, that a RecA-independent RecB function is required for the survival of the gyrB(Ts) ΔtopA strain. Such a RecB function has been linked to replication fork regression that can occur when forks are stalled [53], [54]. Thus, these results may suggest that replication is problematic in the gyrB(Ts) ΔtopA strain. This is supported by the results presented below.
An oriC::aph mutation still oriC competent in topA null but not in isogenic topA+ cells complements both the growth and chromosome segregation defects
Our data suggested that hypernegative supercoiling in topA null mutants triggered RecA-dependent recombination that led to the accumulation of RecQ-processed intermediates. Without a sufficient amount of type 1A topo activity to resolve these intermediates, chromosome segregation could not occur. However, how excess negative supercoiling stimulated RecA-dependent recombination to a level that caused chromosome segregation defects is unclear.
We have recently used a Tn5 mutagenesis system to isolate genetic suppressors of the growth defect of a gyrB(Ts) ΔtopA rnhA strain (Materials and Methods; Usongo and Drolet, manuscript in preparation). The growth defect of this strain was previously shown to be related to chromosome segregation problems that could be corrected by overproducing topo III [17]. Improving gyrase activity also suppressed the chromosome segregation defects [17]. Moreover, our study of replication in this mutant led us to speculate that unregulated replication either from oriC or R-loops, or from both, could contribute to the segregation defects [34]. In agreement with this hypothesis, insertion mutants were found in loci involved in replication.
In one mutant, the kanr cassette was found to be inserted within the oriC region, close to the middle (Figure 4A, aph). It was possible that the suppressed strain could survive without an active oriC region, as replication could occur from R-loops due to the absence of the rnhA gene (constitutive stable DNA replication, cSDR) [55]. Therefore, to verify if this oriC15::aph mutation was still competent for replication initiation, we tried to introduce it in wild-type (RFM443), gyrB(Ts) (RFM445) and gyrB(Ts) ΔtopA (RFM475) isogenic strains. Kanamycin resistant transductants were readily obtained for the gyrB(Ts) ΔtopA strain. Southern blot analysis confirmed that the gyrB(Ts) ΔtopA transductants carried the mutated but not the wild-type oriC region (Figure S4, RFM475 oriC15::aph). The few kanamycin resistant transductants of the wild-type and gyrB(Ts) strains were found to be false-positives as they kept the wild-type oriC region (Figure S4, a false positive RFM443 kanr is shown). Repeated transduction experiments yielded similar results. Therefore, we concluded that the oriC15::aph mutation was viable only when the topA gene was absent.

Our finding that overproducing RNase HI had no effect on the growth of the gyrB(Ts) ΔtopA strain carrying the oriC15::aph mutation (at 37 and 41°C, not shown), indicated that this strain does not replicate its chromosome via cSDR. This is in agreement with a previous report showing that, as opposed to an rnhA null mutant, a topA null mutant could not survive without a functional oriC/DnaA system [27]. Therefore, our topA null mutant most likely uses the oriC15::aph allele to initiate the replication of its chromosome. However, we can predict that this allele would be less active than a wild-type one and therefore should be able to complement the growth defect of a strain in which excess replication from oriC is growth inhibitory. The dnaAcos mutant, isolated as an intragenic suppressor of a dnaA46 mutant, fails to grow at 30°C and below, because of excessive replication initiation from oriC [56]. A dnaAcos strain carrying the oriC15::aph mutation showed good growth at both 36 and 30°C, whereas an isogenic strain with a wild-type oriC region did not (Figure S5, dnaAcos oriC15::aph vs dnaAcos). Thus, this result confirmed that (i) the oriC15::aph mutation is functional in replication initiation and (ii) it is less active than a wild-type oriC region.
Our results with the oriC15::aph mutation suggested that topo I may play an important role in regulating replication initiation from oriC. In a previous study, the left-half of the oriC region was shown to be essential for oriC function in vivo [57]. This section carries the DUE (DNA unwinding element, AT-rich) region from which oriC duplex melting is initiated (Figure 4A) [58]. The smallest oriC fragment found to be functional in vivo was a fragment encompassing nucleotide 1 to 163 of the oriC region (Figure 4A, oriC231). However, a wild-type strain carrying this fragment was sensitive to rich media (LB). It was concluded that the right-half of oriC was essential for multi-forked replication that is required to support high growth rates in rich media [57]. Therefore, the fact that the kanr cassette was inserted at position 142 in the oriC sequence (Figure 4A), likely explains why our oriC15::aph mutation was not functional in a wild-type strain. However, not only was the mutation oriC-competent in our topA null mutant, it apparently allowed multi-forked replication, since our gyrB(Ts) ΔtopA strain was able to grow robustly in rich media. Therefore, these results suggest that topo I plays an important regulatory role at oriC.
Flow cytometry was used in rifampicin run-out experiments with cells grown in M9 medium at 37°C to investigate the regulation of replication initiation in our strains. As recently shown [34], both wild-type and gyrB(Ts) cells contained 2n chromosome, thus indicating that replication initiation was well regulated in these strains (Figure 4B). Cells of the gyrB(Ts) ΔtopA strain had a near perfect 2n chromosomal pattern with one small additional peak, showing some asynchrony (Figure 4B). However, flow cytometry analysis revealed that replication initiation was not well regulated in the topA null mutant carrying the oriC15::aph mutation, as peaks reflecting 1, 2, 3, or 4 chromosomes were clearly observed (Figure 4B, gyrB(Ts) ΔtopA oriC). Highly asynchronous replication was also previously detected in a wild-type strain carrying the oriC231 mutation [57]. Flow cytometry analysis also revealed that the DNA/mass ratio was higher by roughly 40% in the gyrB(Ts) ΔtopA strain as compared to wild-type and gyrB(Ts) strains (Figure 4C). Introducing the oriC15::aph mutation into the topA null strain restored the DNA/mass ratio to the level seen in wild-type and gyrB(Ts) strains (Figure 4C, gyrB(Ts) ΔtopA oriC). Thus, the oriC15::aph mutation caused replication initiation to be less efficient in the gyrB(Ts) ΔtopA strain as shown by the loss of regulation and the lower DNA/mass ratio.
The oriC15::aph mutation was very effective in correcting the growth defect of our gyrB(Ts) ΔtopA strain at both 30 and 24°C (Figure 5B, compare gyrB(Ts) ΔtopA and gyrB(Ts) ΔtopA oriC). This mutation also significantly corrected the chromosome segregation defects of the topA null strain at both temperatures (Figure 5A, gyrB(Ts) ΔtopA oriC). Therefore, the recA-dependent chromosome segregation defects in the topA null mutant are likely related to excess replication from oriC. We conclude that one major role of E. coli topo I in genome maintenance is to prevent over-replication originating from oriC.

Supercoiling - and R-loop-dependent growth and chromosome segregation defects in a gyrB(Ts) ΔtopA ΔtopB strain
We have recently shown that deleting topA could complement the growth defect of our gyrB(Ts) strain at non-permissive temperatures (40 to 42°C) by partially correcting its replication initiation and chromosome segregation defects [34]. However, we found that the topB gene was required for chromosome segregation and overproducing topo IV, the main cellular decatenase, could not substitute for topB. These results, and others, allowed us to conclude that topo III plays a role in replication that becomes essential when gyrase activity is defective. Here, we have confirmed that recombination was not involved by showing that deleting recA or recQ did not correct the growth and chromosome segregation defects of the gyrB(Ts) ΔtopA ΔtopB strain at a non-permissive temperature (40°C, Figure S6). Moreover, RNase HI overproduction had no effect. Thus, at non-permissive temperatures for the gyrB(Ts) allele, the growth and chromosome segregation defects of the gyrB(Ts) ΔtopA ΔtopB strain [34] are unrelated to recombination and R-loops.
It was observed that the optimal temperature for the growth of the gyrB(Ts) ΔtopA ΔtopB strain was 37°C. Indeed, at 30°C the growth defect was found to be exacerbated (Figure S7a, compare 37 and 30°C for gyrB(Ts) ΔtopA ΔtopB/pSK762c). This strain also generated a higher proportion of longer cells at 30 than 37°C (Figure S7b, gyrB(Ts) ΔtopA ΔtopB, 37 vs 30°C). Since gyrase was re-activated at 30°C, we considered the possibility that deleting topB exacerbated topA phenotypes at this temperature. If this was true, overproducing RNase HI should have a positive effect on growth and chromosome segregation in our triple mutant. Indeed, this turned out to be true as the spot assay revealed that growth was better, by at least two logs, when RNase HI was overproduced (Figure 6b, compare gyrB(Ts) ΔtopA ΔtopB/pSK760, RNase HI overproduced and gyrB(Ts) ΔtopA ΔtopB/pSK762c, RNase HI not overproduced). Moreover, the strong chromosome segregation defects illustrated by the formation of very long filaments fully packed with diffuse DNA, were significantly corrected by overproducing RNase H. In this case, cells were shorter and the DNA was more compact (Figure 6A). Thus, R-loops-related problems of a topA null mutant were exacerbated by deleting topB and were mostly expressed as chromosome segregation defects.

RecA-dependent but RecQ-independent growth and chromosome segregation defects in a gyrB(Ts) ΔtopA ΔtopB strain at 30°C
The deletion of recA significantly improved the growth of the gyrB(Ts) ΔtopA ΔtopB strain at 30°C, though this was not as effective as overproducing RNase HI (Figure 6B, compare gyrB(Ts) ΔtopA ΔtopB ΔrecA/pSK762c and gyrB(Ts) ΔtopA ΔtopB/pSK760). However, deleting recA was at least as effective as overproducing RNase HI in correcting the chromosome segregation defects of the gyrB(Ts) ΔtopA ΔtopB strain (Figure 6A, compare gyrB(Ts) ΔtopA ΔtopB ΔrecA/pSK762c and gyrB(Ts) ΔtopA ΔtopB/pSK760). Furthermore, overproducing RNase HI had no effects on growth and chromosome segregation when recA was deleted (Figure 6A, compare gyrB(Ts) ΔtopA ΔtopB ΔrecA/pSK760 and gyrB(Ts) ΔtopA ΔtopB ΔrecA/pSK762c). These results demonstrate that the R-loop-dependent chromosome segregation defects in cells lacking type 1A topos, are also dependent on RecA.
Unlike inactivating recA, the deletion of recQ did not correct the phenotypes of the gyrB(Ts) ΔtopA ΔtopB strain (Figure 6, compare gyrB(Ts) ΔtopA ΔtopB ΔrecQ/pSK762c and gyrB(Ts) ΔtopA ΔtopB/pSK762c). However, RNase HI overproduction was still able to correct these phenotypes when recQ was absent (compare gyrB(Ts) ΔtopA ΔtopB ΔrecQ/pSK760 and gyrB(Ts) ΔtopA ΔtopB ΔrecQ/pSK762c). Thus, the RecA - and R-loop-dependent growth and chromosome segregation defects of the gyrB(Ts) ΔtopA ΔtopB strain are not caused by the accumulation of RecQ-processed recombination intermediates that are substrates for type 1A topos. As RecA was previously shown to be required for cSDR that initiates from R-loops [55], over-replication could possibly be the triggering event for the growth and chromosome segregation defects of cells lacking type 1A topos. This is supported by the genetic evidence presented below.
Suppressor mutations affecting R-loop - and/or oriC-dependent replication significantly correct the growth and chromosome segregation defects in gyrB(Ts) ΔtopB topA20::Tn10 strains at 30°C
One of the best suppressors of the growth defect of the gyrB(Ts) ΔtopA rnhA strain that displays cell filamentation and chromosome segregation phenotypes similar to our gyrB(Ts) ΔtopA ΔtopB strain, had the kanr cassette inserted within the promoter region of the dnaT gene (Figure S8A). DnaT is one of the various proteins that constitute the primosome (including PriA [59]). This protein complex allows the assembly of a replisome outside of oriC. Interestingly, the first mutation found to inhibit SDR mapped within dnaT [60]. The SOS-dependent form of stable DNA replication (iSDR) was shown to be inhibited in this case [55]. However, the involvement of dnaT in the R-loop-dependent form of SDR (cSDR) is still unknown [61]. To test this, the dnaT18::aph mutation was introduced in a dnaA46(Ts) strain also carrying an rnhA null mutation. The absence of RNase HI allows the dnaA46(Ts) strain to grow at 42°C as it can replicate its chromosome from R-loops (Figure 7A and B). Therefore, the fact that the dnaT18::aph allele inhibited the growth of the dnaA46(Ts) rnhA strain at 42°C, indicated that the dnaT gene was required for cSDR (Figure 7C, 42°C, compare rnhA dnaA46 and rnhA dnaA46 dnaT).

The dnaT18::aph mutation was also found to partially correct the chromosome segregation defects of the gyrB(Ts) ΔtopA rnhA strain (Figure S9). This suggested that replication from R-loops could, at least in part, be responsible for these problems. We therefore tested the ability of the dnaT18::aph mutation to correct similar defects in cells lacking type 1A topos. For this purpose, a different null allele of topA, the topA20::Tn10 allele that was previously shown to behave similarly to the ΔtopA allele used in the present study, was chosen [11]. A ΔtopB gyrB(Ts) strain was used in which the topA20::Tn10 allele was either immediately introduced to obtain the ΔtopB gyrB(Ts) topA20::Tn10 control strain, or introduced after the dnaT18::aph allele to obtain the ΔtopB gyrB(Ts) dnaT18::aph topA20::Tn10 strain. The chromosome segregation defects were found to be more severe in our new ΔtopB gyrB(Ts) topA20::Tn10 strain as compared to the other one carrying the ΔtopA allele (compare Figure 6A, gyrB(Ts) ΔtopA ΔtopB/pSK762c and Figure 8A, ΔtopB gyrB(Ts) topA20::Tn10 and data not shown). Indeed, the ΔtopB gyrB(Ts) topA20::Tn10 strain at 30°C produced almost exclusively extremely long filaments that were fully packed with diffuse DNA. This could be related to our previous observation that R-loop-related problems in the absence of topo I were more severe in strains carrying the topA20::Tn10 allele instead of the ΔtopA one [62]. RNase HI overproduction also significantly corrected both the growth and chromosome segregation defects of our ΔtopB gyrB(Ts) topA20::Tn10 strain (Figure 8A and B, compare ΔtopB gyrB(Ts) topA20::Tn10 and ΔtopB gyrB(Ts) topA20::Tn10/pSK760). However, at 24°C, RNase HI overproduction had no effect (Figure 8C, compare ΔtopB gyrB(Ts) topA20::Tn10/pSK760 and ΔtopB gyrB(Ts) topA20::Tn10/pSK762c). This was expected, as the cold-sensitivity of cells lacking topo I is not corrected by RNase HI overproduction (see above).

It was found that the dnaT18::aph mutation was at least as effective as RNase HI overproduction in correcting the chromosome segregation defects of the ΔtopB gyrB(Ts) topA20::Tn10 strain (Figure 8A, compare ΔtopB gyrB(Ts) dnaT topA20::Tn10, ΔtopB gyrB(Ts) topA20::Tn10/pSK762c and ΔtopB gyrB(Ts) topA20::Tn10/pSK760). However, RNase HI overproduction was slightly better than the dnaT18::aph mutation in correcting the growth defect (Figure 8B, compare ΔtopB gyrB(Ts) topA20::Tn10/pSK760 and ΔtopB gyrB(Ts) dnaT topA20::Tn10). The dnaT18::aph also had a negative effect on the growth of the ΔtopB gyrB(Ts) topA20::Tn10 strain at 37°C (Figure 8D, compare ΔtopB gyrB(Ts) dnaT topA20::Tn10 and ΔtopB gyrB(Ts) topA20::Tn10). This could be due to the presence of the gyrB(Ts) allele that was previously shown, at this semi-permissive temperature, to be incompatible with a mutation (priA null) inactivating the primosome [63]. Thus, our results support the hypothesis that the R-loop and RecA-dependent chromosome segregation defects in cells lacking type 1A topos are, at least in part, related to over-replication initiated from R-loops. The fact that the dnaT18::aph mutation slightly promoted the growth of our topA null mutant (Figure 5B, 30 and 24°C, compare gyrB(Ts) ΔtopA dnaT vs gyrB(Ts) ΔtopA), suggests that cSDR is primarily a problem for topA null cells that is exacerbated by deleting topB. This would be consistent with the assumption that topo I is the primary type 1A topo involved in the inhibition of R-loop formation [47].
Seven different kanr insertion mutations in the C-terminal region of RNase E, the main endoribonuclease in E. coli (Usongo and Drolet, manuscript in preparation), were found to suppress the growth defect of our topA rnhA gyrB(Ts) strain. Interestingly, experimental evidence for an interplay between RNase HI and RNase E in RNA degradation has been reported [64], [65]. One of these rne mutations (rne59::aph, Figure S8C) was introduced in a dnaA46(Ts) rnhA strain to test its effect on cSDR. The presence of the rne59::aph mutation significantly reduced the ability of the dnaA46(Ts) rnhA strain to grow at 42°C (by 2 to 3 logs; Figure 7D, 42°C, rnhA dnaA46 vs rnhA dnaA46 rne). This result shows that the mutated RNase E inhibited cSDR.
A topA topB gyrB(Ts) strain was constructed, with the topA20::Tn10 allele as described above, that carried the rne59::aph mutation. The rne59::aph mutation was found to be slightly better than RNAse HI overproduction to correct the growth defect of cells lacking type 1A topos (Figure 8B, compare ΔtopB gyrB(Ts) rne topA20::Tn10 and ΔtopB gyrB(Ts) topA20::Tn10/pSK760). Furthermore, it was at least as effective as RNase HI overproduction and the dnaT18::aph mutation to correct the chromosome segregation defects in these cells (Figure 8A). Thus, our results with the rne59::aph mutation lend further support to the hypothesis that cells lacking type 1A topos suffer from excess replication originating from R-loops.
The origins of replication for cSDR (oriKs) in rnhA null mutants are mostly found within or close to the ter region where bi-directional replication initiated at oriC normally terminates [55]. Thus, the origin to terminus (oriC/ter) ratio, is expected to be lowered by the occurrence of cSDR. This is indeed what was found for the rnhA null mutant (Figure S10, RFM443 vs RFM430 rnhA::cam). The ori/ter ratio was also similarly reduced in the topA null mutant, thus supporting the occurrence of cSDR in the absence of topo I (Figure S10, RFM475).
Several of our kanr insertion mutants were found to reduce the expression of the holC gene (Usongo and Drolet, manuscript in preparation). In a previous study, kanr insertion mutants that reduced the expression of the holC gene were also found to suppress the growth defect of a dnaAcos strain [66]. The holC gene encodes the χ subunit of DNA pol III, the replicative polymerase in E. coli [67]. The χ subunit interacts with SSB and this interaction was recently shown to play an important role in replisome establishment and maintenance [68]. The holC2::aph mutation was tested for its ability to suppress phenotypes of cells lacking type 1A topos. For this purpose, a topA topB gyrB(Ts) holC2::aph strain, carrying the topA20::Tn10 allele, was constructed. The holC2::aph mutation was shown to slightly correct the growth defect of cells lacking type 1A topos activity (Figure 8B and C, 30 and 24°C respectively, ΔtopB gyrB(Ts) holC topA20::Tn10 vs ΔtopB gyrB(Ts) topA20::Tn10). Both cell length and the amount of DNA were also slightly reduced (Figure 8A). The fact that holC mutations by themselves can cause filamentation and chromosome segregation defects [68], may explain why the holC2::aph mutation only partially corrected the phenotypes of the ΔtopB gyrB(Ts) topA20::Tn10 strain.
The holC2::aph mutation also partially corrected the growth defect of our topA null mutant (Figure 5B, 24°C, 48 h; compare gyrB(Ts) ΔtopA and gyrB(Ts) ΔtopA holC). Moreover, in rifampicin run-out experiments, replication did not appear to be well regulated in the topA null mutant carrying the holC2::aph mutation, as peaks reflecting 1, 2, 3, or 4 chromosomes were clearly observed (Figure S11, compare gyrB(Ts) ΔtopA and gyrB(Ts) ΔtopA holC). This result supports the hypothesis that the χ subunit of pol III plays a role in replication initiation [68] and therefore suggests that initiation from oriC could also be problematic in cells lacking both type 1A topos.
Discussion
The work described in this manuscript, which focused on the role of type 1A topos in genome maintenance in E. coli, revealed new important functions in replication and chromosome segregation for both topo I and III. Before this study, not much was known about the role of these enzymes in genome maintenance as opposed to the situation for eukaryotic type 1A topos. It was generally believed that topo I and III have distinct cellular functions, with topo I regulating supercoiling and R-loop formation and topo III being involved in chromosome segregation during replication. Here, our results show that E. coli type 1A topos play major roles in genome maintenance by inhibiting inappropriate replication and, as is the case for eukaryotic type 1A enzymes, by acting with RecQ to resolve RecA-generated recombination intermediates. Furthermore, while inhibiting replication from oriC is specific to topo I that regulates supercoiling, both the prevention of over-replication from R-loops and the activity with RecQ appear to be shared by both type 1A topos. This may suggest that these two functions are the major ones for type 1A topos and that they may have been conserved throughout evolution. Below, we discuss about the newly identified functions of E. coli type 1A topos in this work.
E. coli type 1A topos and RecQ
As stated in the introduction, the strand passage activity of E. coli topo III, but not topo I, was shown to be strongly stimulated by RecQ in vitro [35]–[37]. This would suggest that E. coli topo III and RecQ can act together to maintain the stability of the genome, as shown in eukaryotic cells [69]. However, no clear evidence for such a role of topo III has been reported in E. coli. Recent experimental evidence points to a role for topo III in chromosome segregation related to replication and independent of RecQ ([33], [34]; this work). In fact, the data presented here suggest that topo I, not topo III, is the primary type 1A topo acting with RecQ in E. coli. Indeed, the strong chromosome segregation and growth defects of topA null cells at low temperatures were shown to be partially corrected by deleting recQ or recA, independent of the RecFOR pathway and by overproducing topo III, a protein that is normally of very low abundance. Moreover, both deleting recQ and overproducing topo III were found to be epistatic to recA in correcting the growth problems. This is consistent with RecQ processing RecA-dependent recombination intermediates in such a way that they can only be resolved by a type 1A topo, as is the case in eukaryotic cells. In this context, topo III overproduction would substitute for topo I and perform the resolution, thus meaning that topo III can also perform this reaction in vivo.
Alternatively, in the absence of topA, DNA substrates for topo I may accumulate and some of them could be processed by topo III, thus leading to the depletion of this very low abundant protein. This situation would lead to the accumulation of RecQ-processed recombination intermediates, if topo III normally resolves them. However, we think that this is unlikely because while a topA recQ strain grow very well, deleting topB make this strain very sick with phenotypes identical to those of topA topB null cells. If recQ was acting with topB, then deleting topB should have had no effect on the growth of the topA recQ strain. Altogether, our results are more consistent with topo I being the primary type 1A topo working with RecQ in E. coli.
Despite the previously observed lack of stimulation of topo I activity by RecQ in vitro, we still believe that these two proteins can functionally interact. Indeed, it may be that the optimal experimental conditions and/or the appropriate substrate for their functional interaction have not yet been well defined. Alternatively or additionally, the much higher abundance of topo I in vivo as compared to topo III may compensate for its lower level of activity with RecQ. In fact, the finding that either E. coli topo I expression or a SGS1 mutation could compensate for the absence of Top3 in S. cerevisiae [38]–[40], supports the assumption that E. coli topo I can act with RecQ in vivo. Moreover, in an in vitro system for DHJs resolution by BLM helicase with a type 1A topo, E. coli topo I was shown to efficiently substitute for human topo IIIα [70]. Hsieh and co-workers have recently obtained experimental evidence for their “unravel and unlink” model whereby BLM first melts a DNA region to which RPA protein binds and topo IIIα acts to resolve a DHJ [1], [43]. Indeed, a topo IIIα mutant unable to physically interact with BLM was shown to partially resolve a DHJ in the presence of RPA, thus suggesting that the functions of the two proteins may be separated [43]. A similar model might also be proposed for RecQ acting with topo I, the activity of which can be stimulated by SSB [71], as the two proteins do not physically interact.
Interestingly, whereas topo I is present in all bacteria, topo III is present only in a few bacteria [72]. Therefore, if a collaboration between a type 1A topo and a RecQ-like helicase is also required in bacteria, it is not surprising that topo I performs this function. However, why such a function would be required in bacteria is currently unknown. In diploid organisms RecQ-like helicases act in concert with topo III to prevent the exchange of genetic material between DNA molecules involved in recombination (DHJ dissolution; [73]).
In the present study, it was found that inactivating recB almost completely inhibited the growth of our gyrB(Ts) ΔtopA mutant, while deleting recA improved its fitness. This indicated that a RecA-independent RecB function was required for the survival of the gyrB(Ts) ΔtopA strain. Such a RecB function has been linked to replication forks regression that can occur when forks are stalled [53], [54]. In the cell, RecB is present in the RecBCD complex that has both a recombination (RecA loading at χ sites) and a dsDNA degradation (exonuclease V) function. It has been hypothesized that upon fork reversal, a HJ forms and degradation of the dsDNA end is initiated by RecBCD. Following the encounter of a χ site and in the presence of RecA, homologous recombination can take place, leading to the formation of a second HJ. The involvement of homologous recombination is supported by the observation that, as opposed to recB mutants, recD mutants, that lack the dsDNA degradation activity of RecBCD, can survive under conditions that promote extensive replication forks reversal if the recA gene is present [74], [75]. Next, the two HJs can be resolved by RuvABC. Alternatively, as is the case during the process of genetic exchange in diploid organisms, we propose that RecQ can act on the two HJs to promote convergent branch migration. This would lead to the formation of a hemicatenane that must be unlinked by a type 1A topo to allow chromosome segregation. In the absence of RecA, the second HJ does not form and the dsDNA is instead degraded by the RecBCD complex up to the first HJ to produce a fork structure that is used to restart replication. In this context, RecQ does not promote the formation of hemicatenanes and, as a result, chromosomes segregation is not impeded by the lack of type 1A topos activity.
Experimental evidence for replication forks reversal has been reported in E. coli cells carrying defective DNA helicases involved in replication (DnaB and Rep; [53], [74]) and, more recently, following replication-transcription collisions [75]. We speculate that the high level of negative supercoiling in the gyrB(Ts) ΔtopA strain at low temperatures promotes the formation of alternative non-B DNA structures that may cause the stalling of replication forks and their reversal. Alternatively, such non-B DNA structures may first block transcription and the arrested RNA polymerases may, in turn, stop the progression of replication forks to cause their reversal. Furthermore, over-replication that occurs in this strain likely exacerbates the problem and makes the cell unable to adequately deal with the reversed forks. Clearly, more work will be required to fully characterize the role(s) of type 1A topos acting with RecQ in bacteria and to find out under which circumstances this activity would be required in various DNA transactions.
E. coli type 1A topos in replication
In E. coli, replication initiated at oriC is tightly regulated so that it occurs once and only once per cell cycle [58]. This process is synchronized with the “initiation mass”. DNA supercoiling is among the many elements, including DnaA that are required for replication initiation at oriC. Indeed, in vitro replication initiation necessitates that the oriC plasmid be negatively supercoiled [76]. In vivo, deleting topA was found to correct the thermo-sensitive growth of a dnaA(Ts) mutant [27] and altering gyrase supercoiling activity inhibited replication initiation from oriC [77]. Moreover, we have recently shown that a topA deletion could correct the replication initiation defect of a strain defective for gyrase supercoiling activity [34]. Interestingly, in a screen to isolate DnaA inhibitors a compound was recently found to rescue a dnaAcos mutant from lethal hyperinitiation by targeting gyrase [78]. Thus, in vitro and in vivo data demonstrate that negative DNA supercoiling is required for replication initiation from oriC.
The recent determination of the crystal structure of a truncated DnaA ortholog in complex with ssDNA supports a model whereby DnaA opens the oriC region by a direct ATP-dependent stretching mechanism [79]. This work provides the strongest evidence to date for a direct participation of DnaA in DNA melting at oriC, and is fully compatible with other elements, such as DNA supercoiling, also playing a role in this process. In a recent biochemical study, DNA fragments containing at least the left portion of oriC up to I1 or I2 (Figure 4a) were shown to be required for DnaA-ATP binding to ssDUE in the absence of torsional stress [80]. This result is totally consistent with our finding that an oriC region lacking these I1 and I2 sequences (oriC15::aph) is functional in a topA null mutant, where the negative supercoiling level is elevated, but not functional in an isogenic topA+ strain. Thus, our results, together with those reported in the two studies mentioned above, suggest that DNA supercoiling plays an important regulatory role at oriC.
When topB was deleted from a topA null mutant, a new growth inhibitory phenotype, again related to replication, appeared at temperatures where the oriC-related phenotype was attenuated. Our data suggest that this major phenotype in the absence of type 1A topos is related to replication from R-loops (cSDR). This is consistent with a major role of topo I in the inhibition of R-loop formation and with the identification of topo I, like RNase HI [81], as a specificity factor to inhibit replication initiation at sites other than oriC (e.g. R-loops), in an in vitro system [28]. Thus, although the strong phenotype expressed such as extensive cell filamentation, unsegregated nucleoids and growth inhibition, is triggered by deleting topB, cSDR is probably also activated in our single topA mutant. This is supported by the fact that the dnaT18::aph mutation improved the growth of our topA mutant and by the finding that, as was the case in an rnhA null mutant, the ori/ter ratio was lower in this topA mutant as compared to a wild-type strain. However, even if cSDR is activated in topA null mutants, the oriC/DnaA system is still required in these cells to replicate the chromosome. A similar situation has been described for recG mutants, in which cSDR is also activated but cannot support replication of the whole chromosome [82], [83].
As the strong phenotype is due to the simultaneous absence of both type 1A topos, it is likely related to similar functions performed by the two enzymes. We have previously shown that an R-loop was a hot-spot for topo III activity in vitro [47]. By acting on an R-looped plasmid, topo III was shown to destabilize the R-loop. As topo III can travel with the replication fork [35], it could possibly act by destabilizing R-loops blocking the progression of the replication forks. Interestingly, topo III was recently shown to prevent R-loop accumulation during transcription in mammalian cells [84]. Thus, inhibition of R-loop formation might be another important function of type 1A topos that has been conserved throughout evolution. The absence of a type 1A topo activity for decatenation (e.g. RecQ with topo I) also likely contributes to the strong chromosome segregation defects seen in cells lacking both topo I and III.
Materials and Methods
Bacterial strains and plasmids
Bacterial strains used in this study are all derivatives of E. coli K12 and are listed in Table S1. Details on their constructions as well as the list of plasmids used in this study are also given in Table S1. Transductions with P1vir were performed as described previously [34]. PCR was used to confirm that the expected gene transfer occurred in the selected transductants.
Insertional mutagenesis
Insertional mutagenesis with pRL27 was performed in a topA rnhA gyrB(Ts) strain and will be described in details elsewhere (Usongo and Drolet, manuscript in preparation). Briefly, pRL27 carries a hyperactive Tn5 transposase gene under the control of the tetA promoter, and an insertional cassette with a kanamycin resistance gene (aph) and a pir-dependent origin (oriR6K) bracketed by Tn5 inverted repeats [85]. Following electroporation of pRL27 in a pir- background, the kanr cassette inserts randomly into the chromosome. A topA rnhA gyrB(Ts)/pBAD18rnhA strain was electroporated with pRL27 and plated on LB containing 25 µg/ml kanamycin at 40°C, to select for suppressors that grew in the absence of arabinose (no RNase HI produced). At this temperature, the strain does not grow because of extensive inhibition of the supercoiling activity of gyrase [17], combined with over-replication ([34] and see below). P1vir was grown on the kanr clones that re-grew at 40°C and each phage lysate was used to infect a topA rnhA gyrB(Ts)/pPH1243 strain, that normally grows only in the presence of IPTG, to overproduce topo III from pPH1243 [17]. Transductants were selected on LB plates containing IPTG and kanamycin (50 µg/ml) at 37°C. Transductants that re-grew in the absence of IPTG were selected for further characterization. Four of the insertion mutants, described in Figure 4 and S8, were used in the present study.
Spot tests
Cells from glycerol stocks were resuspended in LB to obtain an OD600 of 0.6. Five µl of 10-fold serial dilutions were then spotted on LB plates that were incubated at the indicated temperatures. The experiments were performed with cells from glycerol stocks to minimize the chance of selecting cells with compensatory mutations. However, we eventually found that similar results were obtained whether the cells were from glycerol stocks or from overnight liquid cultures (not shown).
Microscopy
Cells were grown overnight at 37°C in liquid LB medium supplemented with the appropriate antibiotics. Overnight cultures were diluted in LB medium to obtain an OD600 of 0.01 and grown at the indicated temperature to an OD600 of 0.8. The cells were recovered and prepared for microscopy as previously described [17]. Pictures (fluorescence (DAPI) and DIC) were randomly taken with a LSM 510 Meta confocal microscope from Zeiss. The images were processed using Adobe Photoshop. Representative images are shown both in the Results and Supporting Information sections.
Flow cytometry
The procedure for flow cytometry in rifampicin run-out experiments with cells grown in M9 medium has been described [34]. The DNA/mass ratio was calculated has previously reported [34].
Supporting Information
Zdroje
1. ChenSH, ChanNL, HsiehTS (2013) New mechanistic and functional insights into DNA topoisomerases. Annu Rev Biochem 82 : 139–170.
2. ChampouxJJ (2001) DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem 70 : 369–413.
3. WangJC (1971) Interaction between DNA and an Escherichia coli protein omega. J Mol Biol 55 : 523–533.
4. KirkegaardK, WangJC (1985) Bacterial DNA topoisomerase I can relax positively supercoiled DNA containing a single-stranded loop. J Mol Biol 185 : 625–637.
5. LiuLF, WangJC (1987) Supercoiling of the DNA template during transcription. Proc Natl Acad Sci U S A 84 : 7024–7027.
6. GellertM, MizuuchiK, O'DeaMH, NashHA (1976) DNA gyrase: an enzyme that introduces superhelical turns into DNA. Proc Natl Acad Sci U S A 73 : 3872–3876.
7. DiNardoS, VoelkelKA, SternglanzR, ReynoldsAE, WrightA (1982) Escherichia coli DNA topoisomerase I mutants have compensatory mutations in DNA gyrase genes. Cell 31 : 43–51.
8. PrussGJ, ManesSH, DrlicaK (1982) Escherichia coli DNA topoisomerase I mutants: increased supercoiling is corrected by mutations near gyrase genes. Cell 31 : 35–42.
9. ChengB, ZhuCX, JiC, AhumadaA, Tse-DinhYC (2003) Direct interaction between Escherichia coli RNA polymerase and the zinc ribbon domains of DNA topoisomerase I. J Biol Chem 278 : 30705–30710.
10. DroletM (2006) Growth inhibition mediated by excess negative supercoiling: the interplay between transcription elongation, R-loop formation and DNA topology. Mol Microbiol 59 : 723–730.
11. DroletM, PhoenixP, MenzelR, MasseE, LiuLF, et al. (1995) Overexpression of RNase H partially complements the growth defect of an Escherichia coli delta topA mutant: R-loop formation is a major problem in the absence of DNA topoisomerase I. Proc Natl Acad Sci U S A 92 : 3526–3530.
12. DroletM, BiX, LiuLF (1994) Hypernegative supercoiling of the DNA template during transcription elongation in vitro. J Biol Chem 269 : 2068–2074.
13. PhoenixP, RaymondMA, MasseE, DroletM (1997) Roles of DNA topoisomerases in the regulation of R-loop formation in vitro. J Biol Chem 272 : 1473–1479.
14. MasseE, PhoenixP, DroletM (1997) DNA topoisomerases regulate R-loop formation during transcription of the rrnB operon in Escherichia coli. J Biol Chem 272 : 12816–12823.
15. MasseE, DroletM (1999) Escherichia coli DNA topoisomerase I inhibits R-loop formation by relaxing transcription-induced negative supercoiling. J Biol Chem 274 : 16659–16664.
16. BaakliniI, UsongoV, NolentF, SanscartierP, HraikyC, et al. (2008) Hypernegative supercoiling inhibits growth by causing RNA degradation. J Bacteriol 190 : 7346–7356.
17. UsongoV, NolentF, SanscartierP, TanguayC, BroccoliS, et al. (2008) Depletion of RNase HI activity in Escherichia coli lacking DNA topoisomerase I leads to defects in DNA supercoiling and segregation. Mol Microbiol 69 : 968–981.
18. ZechiedrichEL, KhodurskyAB, BachellierS, SchneiderR, ChenD, et al. (2000) Roles of topoisomerases in maintaining steady-state DNA supercoiling in Escherichia coli. J Biol Chem 275 : 8103–8113.
19. HraikyC, RaymondMA, DroletM (2000) RNase H overproduction corrects a defect at the level of transcription elongation during rRNA synthesis in the absence of DNA topoisomerase I in Escherichia coli. J Biol Chem 275 : 11257–11263.
20. El HageA, FrenchSL, BeyerAL, TollerveyD (2010) Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev 24 : 1546–1558.
21. WangM, WeissM, SimonovicM, HaertingerG, SchrimpfSP, et al. (2012) PaxDb, a database of protein abundance averages across all three domains of life. Mol Cell Proteomics 11 : 492–500.
22. RuiS, Tse-DinhYC (2003) Topoisomerase function during bacterial responses to environmental challenge. Front Biosci 8: d256–263.
23. ChengB, RuiS, JiC, GongVW, Van DykTK, et al. (2003) RNase H overproduction allows the expression of stress-induced genes in the absence of topoisomerase I. FEMS Microbiol Lett 221 : 237–242.
24. WeinreichMD, YigitH, ReznikoffWS (1994) Overexpression of the Tn5 transposase in Escherichia coli results in filamentation, aberrant nucleoid segregation, and cell death: analysis of E. coli and transposase suppressor mutations. J Bacteriol 176 : 5494–5504.
25. YigitH, ReznikoffWS (1999) Escherichia coli DNA topoisomerase I copurifies with Tn5 transposase, and Tn5 transposase inhibits topoisomerase I. J Bacteriol 181 : 3185–3192.
26. DanilovaO, Reyes-LamotheR, PinskayaM, SherrattD, PossozC (2007) MukB colocalizes with the oriC region and is required for organization of the two Escherichia coli chromosome arms into separate cell halves. Mol Microbiol 65 : 1485–1492.
27. LouarnJ, BoucheJP, PatteJ, LouarnJM (1984) Genetic inactivation of topoisomerase I suppresses a defect in initiation of chromosome replication in Escherichia coli. Mol Gen Genet 195 : 170–174.
28. KaguniJM, KornbergA (1984) Topoisomerase I confers specificity in enzymatic replication of the Escherichia coli chromosomal origin. J Biol Chem 259 : 8578–8583.
29. DiGateRJ, MariansKJ (1988) Identification of a potent decatenating enzyme from Escherichia coli. J Biol Chem 263 : 13366–13373.
30. LopezCR, YangS, DeiblerRW, RaySA, PenningtonJM, et al. (2005) A role for topoisomerase III in a recombination pathway alternative to RuvABC. Mol Microbiol 58 : 80–101.
31. LiZ, MondragonA, HiasaH, MariansKJ, DiGateRJ (2000) Identification of a unique domain essential for Escherichia coli DNA topoisomerase III-catalysed decatenation of replication intermediates. Mol Microbiol 35 : 888–895.
32. DiGateRJ, MariansKJ (1989) Molecular cloning and DNA sequence analysis of Escherichia coli topB, the gene encoding topoisomerase III. J Biol Chem 264 : 17924–17930.
33. Perez-CheeksBA, LeeC, HayamaR, MariansKJ (2012) A role for topoisomerase III in Escherichia coli chromosome segregation. Mol Microbiol 86 : 1007–1022.
34. UsongoV, TanguayC, NolentF, BessongJE, DroletM (2013) Interplay between type 1A topoisomerases and gyrase in chromosome segregation in Escherichia coli. J Bacteriol 195 : 1758–1768.
35. SuskiC, MariansKJ (2008) Resolution of converging replication forks by RecQ and topoisomerase III. Mol Cell 30 : 779–789.
36. HarmonFG, DiGateRJ, KowalczykowskiSC (1999) RecQ helicase and topoisomerase III comprise a novel DNA strand passage function: a conserved mechanism for control of DNA recombination. Mol Cell 3 : 611–620.
37. HarmonFG, BrockmanJP, KowalczykowskiSC (2003) RecQ helicase stimulates both DNA catenation and changes in DNA topology by topoisomerase III. J Biol Chem 278 : 42668–42678.
38. WallisJW, ChrebetG, BrodskyG, RolfeM, RothsteinR (1989) A hyper-recombination mutation in S. cerevisiae identifies a novel eukaryotic topoisomerase. Cell 58 : 409–419.
39. GangloffS, McDonaldJP, BendixenC, ArthurL, RothsteinR (1994) The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol Cell Biol 14 : 8391–8398.
40. GangloffS, de MassyB, ArthurL, RothsteinR, FabreF (1999) The essential role of yeast topoisomerase III in meiosis depends on recombination. EMBO J 18 : 1701–1711.
41. ShorE, GangloffS, WagnerM, WeinsteinJ, PriceG, et al. (2002) Mutations in homologous recombination genes rescue top3 slow growth in Saccharomyces cerevisiae. Genetics 162 : 647–662.
42. WuL, DaviesSL, NorthPS, GoulaouicH, RiouJF, et al. (2000) The Bloom's syndrome gene product interacts with topoisomerase III. J Biol Chem 275 : 9636–9644.
43. ChenSH, WuCH, PlankJL, HsiehTS (2012) Essential functions of C terminus of Drosophila Topoisomerase IIIalpha in double holliday junction dissolution. J Biol Chem 287 : 19346–19353.
44. PlankJL, WuJ, HsiehTS (2006) Topoisomerase IIIalpha and Bloom's helicase can resolve a mobile double Holliday junction substrate through convergent branch migration. Proc Natl Acad Sci U S A 103 : 11118–11123.
45. WuL, HicksonID (2003) The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426 : 870–874.
46. CejkaP, PlankJL, BachratiCZ, HicksonID, KowalczykowskiSC (2010) Rmi1 stimulates decatenation of double Holliday junctions during dissolution by Sgs1-Top3. Nat Struct Mol Biol 17 : 1377–1382.
47. BroccoliS, PhoenixP, DroletM (2000) Isolation of the topB gene encoding DNA topoisomerase III as a multicopy suppressor of topA null mutations in Escherichia coli. Mol Microbiol 35 : 58–68.
48. ZhuQ, PongpechP, DiGateRJ (2001) Type I topoisomerase activity is required for proper chromosomal segregation in Escherichia coli. Proc Natl Acad Sci U S A 98 : 9766–9771.
49. MasseE, DroletM (1999) Relaxation of transcription-induced negative supercoiling is an essential function of Escherichia coli DNA topoisomerase I. J Biol Chem 274 : 16654–16658.
50. MasseE, DroletM (1999) R-loop-dependent hypernegative supercoiling in Escherichia coli topA mutants preferentially occurs at low temperatures and correlates with growth inhibition. J Mol Biol 294 : 321–332.
51. MagnerDB, BlankschienMD, LeeJA, PenningtonJM, LupskiJR, et al. (2007) RecQ promotes toxic recombination in cells lacking recombination intermediate-removal proteins. Mol Cell 26 : 273–286.
52. LestiniR, MichelB (2008) UvrD and UvrD252 counteract RecQ, RecJ, and RecFOR in a rep mutant of Escherichia coli. J Bacteriol 190 : 5995–6001.
53. SeigneurM, BidnenkoV, EhrlichSD, MichelB (1998) RuvAB acts at arrested replication forks. Cell 95 : 419–430.
54. MirandaA, KuzminovA (2003) Chromosomal lesion suppression and removal in Escherichia coli via linear DNA degradation. Genetics 163 : 1255–1271.
55. KogomaT (1997) Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol Mol Biol Rev 61 : 212–238.
56. Kellenberger-GujerG, PodhajskaAJ, CaroL (1978) A cold sensitive dnaA mutant of E. coli which overinitiates chromosome replication at low temperature. Mol Gen Genet 162 : 9–16.
57. StepankiwN, KaidowA, BoyeE, BatesD (2009) The right half of the Escherichia coli replication origin is not essential for viability, but facilitates multi-forked replication. Mol Microbiol 74 : 467–479.
58. LeonardAC, GrimwadeJE (2011) Regulation of DnaA assembly and activity: taking directions from the genome. Annu Rev Microbiol 65 : 19–35.
59. GabbaiCB, MariansKJ (2010) Recruitment to stalled replication forks of the PriA DNA helicase and replisome-loading activities is essential for survival. DNA Repair (Amst) 9 : 202–209.
60. LarkCA, RiaziJ, LarkKG (1978) dnaT, dominant conditional-lethal mutation affecting DNA replication in Escherichia coli. J Bacteriol 136 : 1008–1017.
61. SandlerSJ (2005) Requirements for replication restart proteins during constitutive stable DNA replication in Escherichia coli K-12. Genetics 169 : 1799–1806.
62. BaakliniI, HraikyC, RalluF, Tse-DinhYC, DroletM (2004) RNase HI overproduction is required for efficient full-length RNA synthesis in the absence of topoisomerase I in Escherichia coli. Mol Microbiol 54 : 198–211.
63. GromponeG, EhrlichSD, MichelB (2003) Replication restart in gyrB Escherichia coli mutants. Mol Microbiol 48 : 845–854.
64. AnupamaK, LeelaJK, GowrishankarJ (2011) Two pathways for RNase E action in Escherichia coli in vivo and bypass of its essentiality in mutants defective for Rho-dependent transcription termination. Mol Microbiol 82 : 1330–1348.
65. BouvierM, CarpousisAJ (2011) A tale of two mRNA degradation pathways mediated by RNase E. Mol Microbiol 82 : 1305–1310.
66. NordmanJ, SkovgaardO, WrightA (2007) A novel class of mutations that affect DNA replication in E. coli. Mol Microbiol 64 : 125–138.
67. YaoNY, O'DonnellM (2008) Replisome dynamics and use of DNA trombone loops to bypass replication blocks. Mol Biosyst 4 : 1075–1084.
68. MarceauAH, BahngS, MassoniSC, GeorgeNP, SandlerSJ, et al. (2011) Structure of the SSB-DNA polymerase III interface and its role in DNA replication. EMBO J 30 : 4236–4247.
69. ChuWK, HicksonID (2009) RecQ helicases: multifunctional genome caretakers. Nat Rev Cancer 9 : 644–654.
70. WuL, BachratiCZ, OuJ, XuC, YinJ, et al. (2006) BLAP75/RMI1 promotes the BLM-dependent dissolution of homologous recombination intermediates. Proc Natl Acad Sci U S A 103 : 4068–4073.
71. SikderD, UnniramanS, BhaduriT, NagarajaV (2001) Functional cooperation between topoisomerase I and single strand DNA-binding protein. J Mol Biol 306 : 669–679.
72. ForterreP, GribaldoS, GadelleD, SerreMC (2007) Origin and evolution of DNA topoisomerases. Biochimie 89 : 427–446.
73. WuL, ChanKL, RalfC, BernsteinDA, GarciaPL, et al. (2005) The HRDC domain of BLM is required for the dissolution of double Holliday junctions. EMBO J 24 : 2679–2687.
74. MichelB, EhrlichSD, UzestM (1997) DNA double-strand breaks caused by replication arrest. EMBO J 16 : 430–438.
75. De SeptenvilleAL, DuigouS, BoubakriH, MichelB (2012) Replication fork reversal after replication-transcription collision. PLoS Genet 8: e1002622.
76. FunnellBE, BakerTA, KornbergA (1986) Complete enzymatic replication of plasmids containing the origin of the Escherichia coli chromosome. J Biol Chem 261 : 5616–5624.
77. FilutowiczM (1980) Requirement of DNA gyrase for the initiation of chromosome replication in Escherichia coli K-12. Mol Gen Genet 177 : 301–309.
78. JohnsenL, WeigelC, von KriesJ, MollerM, SkarstadK (2010) A novel DNA gyrase inhibitor rescues Escherichia coli dnaAcos mutant cells from lethal hyperinitiation. J Antimicrob Chemother 65 : 924–930.
79. DuderstadtKE, ChuangK, BergerJM (2011) DNA stretching by bacterial initiators promotes replication origin opening. Nature 478 : 209–213.
80. OzakiS, KatayamaT (2012) Highly organized DnaA-oriC complexes recruit the single-stranded DNA for replication initiation. Nucleic Acids Res 40 : 1648–1665.
81. OgawaT, PickettGG, KogomaT, KornbergA (1984) RNase H confers specificity in the dnaA-dependent initiation of replication at the unique origin of the Escherichia coli chromosome in vivo and in vitro. Proc Natl Acad Sci U S A 81 : 1040–1044.
82. HongX, CadwellGW, KogomaT (1995) Escherichia coli RecG and RecA proteins in R-loop formation. EMBO J 14 : 2385–2392.
83. RudolphCJ, UptonAL, StockumA, NieduszynskiCA, LloydRG (2013) Avoiding chromosome pathology when replication forks collide. Nature 500 : 608–611.
84. YangY, McBrideKM, HensleyS, LuY, ChedinF, et al. (2014) Arginine methylation facilitates the recruitment of TOP3B to chromatin to prevent R loop accumulation. Mol Cell 53 : 484–497.
85. LarsenRA, WilsonMM, GussAM, MetcalfWW (2002) Genetic analysis of pigment biosynthesis in Xanthobacter autotrophicus Py2 using a new, highly efficient transposon mutagenesis system that is functional in a wide variety of bacteria. Arch Microbiol 178 : 193–201.
86. KasaharaM, ClikemanJA, BatesDB, KogomaT (2000) RecA protein-dependent R-loop formation in vitro. Genes Dev 14 : 360–365.
87. ZaitsevEN, KowalczykowskiSC (2000) A novel pairing process promoted by Escherichia coli RecA protein: inverse DNA and RNA strand exchange. Genes Dev 14 : 740–749.
88. DugginIG, WakeRG, BellSD, HillTM (2008) The replication fork trap and termination of chromosome replication. Mol Microbiol 70 : 1323–1333.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 8
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Meta-Analysis of Genome-Wide Association Studies in African Americans Provides Insights into the Genetic Architecture of Type 2 Diabetes
- KDM6 Demethylase Independent Loss of Histone H3 Lysine 27 Trimethylation during Early Embryonic Development
- The RNA Helicases AtMTR4 and HEN2 Target Specific Subsets of Nuclear Transcripts for Degradation by the Nuclear Exosome in
- EF-P Dependent Pauses Integrate Proximal and Distal Signals during Translation
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy