
The Translational Regulators GCN-1 and ABCF-3 Act Together to Promote Apoptosis in
Apoptosis, also referred to as programmed cell death, is a crucial cellular process that eliminates unwanted cells during animal development and tissue homeostasis. Abnormal regulation of apoptosis can cause developmental defects and a variety of other human disorders, including cancer, neurodegenerative diseases and autoimmune diseases. Therefore, it is important to identify regulatory mechanisms that control apoptosis. Previous studies have demonstrated that the transcriptional induction of apoptotic genes can be crucial to initiating an apoptotic program. Less is known about translational controls of apoptosis. Here we report that the evolutionarily conserved C. elegans translational regulators GCN-1 and ABCF-3 promote apoptosis generally and act independently of the anti-apoptotic BCL-2 homolog CED-9. GCN-1 and ABCF-3 physically interact and maintain the phosphorylation level of eukaryotic initiation factor 2α, suggesting that GCN-1 and ABCF-3 act together to regulate the initiation of translation. We propose that the translational regulators GCN-1 and ABCF-3 maternally contribute to the proper execution of the apoptotic program.
Published in the journal:
. PLoS Genet 10(8): e32767. doi:10.1371/journal.pgen.1004512
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004512
Summary
Apoptosis, also referred to as programmed cell death, is a crucial cellular process that eliminates unwanted cells during animal development and tissue homeostasis. Abnormal regulation of apoptosis can cause developmental defects and a variety of other human disorders, including cancer, neurodegenerative diseases and autoimmune diseases. Therefore, it is important to identify regulatory mechanisms that control apoptosis. Previous studies have demonstrated that the transcriptional induction of apoptotic genes can be crucial to initiating an apoptotic program. Less is known about translational controls of apoptosis. Here we report that the evolutionarily conserved C. elegans translational regulators GCN-1 and ABCF-3 promote apoptosis generally and act independently of the anti-apoptotic BCL-2 homolog CED-9. GCN-1 and ABCF-3 physically interact and maintain the phosphorylation level of eukaryotic initiation factor 2α, suggesting that GCN-1 and ABCF-3 act together to regulate the initiation of translation. We propose that the translational regulators GCN-1 and ABCF-3 maternally contribute to the proper execution of the apoptotic program.
Introduction
Apoptosis is a naturally occurring process that eliminates unwanted cells during development and maintains tissue homeostasis [1], [2]. For example, apoptosis removes most larval tissues of insects during metamorphosis, sculpts the future inner ear in chicks, eliminates the interdigital web in mammals and shapes the endocardial cushion into valves and septa to generate the four-chamber architecture of the mammalian heart [1], [2]. Apoptosis also culls nearly 80% of oocytes prior to birth in humans and eliminates cells that receive insufficient cell-survival signals to maintain homeostasis [1]. The improper regulation of an apoptotic program can result in either too much or too little cell death, leading to developmental abnormalities and a wide variety of human disorders, such as cancer, neurodegenerative diseases, autoimmune diseases and developmental disorders [3], [4]. It is important to identify mechanisms that regulate apoptosis to understand both animal development and human disorders caused by the dysregulation of apoptosis.
The precise spatial and temporal expression of regulators of apoptosis is known to be crucial for initiating the apoptotic cell-killing program during development and in response to environmental stresses, including ionizing radiation, temperature change, nutrient limitation, oxidative stress and viral infection [1], [2]. Many examples of the transcriptional control of apoptosis have been described. For example, in mammals the genes that encode the pro-apoptotic BCL-2 family member BAX, the BH3-only proteins NOXA, PUMA and BID, the apoptotic protease-activating factor-1 APAF-1 and the death receptor 5 DR5 protein are transcriptionally upregulated by the tumor suppressor p53 transcription factor in response to DNA damage or to the induced expression of p53 [5]–[11], resulting in an induction of apoptosis. The Drosophila apoptotic activator gene reaper is upregulated by multiple transcriptional regulators, including Hox transcription factors, nuclear hormone receptors, AP-1, Polycomb, p53, and histone-modifying enzymes, to promote the morphogenesis of segment boundaries, metamorphosis, and DNA damage responses [1]. In C. elegans, the transcription of the pro-apoptotic BH3-only gene egl-1 is directly regulated in a cell-specific manner by transcription factors that include the Hox family proteins MAB-5, CEH-20, LIN-39 and CEH-34, the E2F protein EFL-3, the Snail family zinc finger protein CES-1, the Gli family transcription factor TRA-1, and the basic helix-loop-helix proteins HLH-2 and HLH-3 [12]–[15]. The caspase gene ced-3 is also upregulated by the Hox transcription factor PAL-1 in the tail spike cell before its death [16]. Recently, we showed that the Sp1 transcription factor SPTF-3 directly drives the transcription of both the pro-apoptotic BH3-only gene egl-1, which mediates a caspase-dependent apoptotic pathway, and the AMPK-related gene pig-1, which mediates a caspase-independent apoptotic pathway [17]. The transcriptional regulation of apoptotic genes clearly plays a crucial role in determining whether specific cells live or die during development.
Translational control is also important for the apoptotic process. In mammals, expression of the pro-apoptotic protein APAF-1 and the anti-apoptotic protein X-chromosome-linked inhibitor of apoptosis XIAP are regulated at the translational level by internal ribosome-entry sites (IRES) [18]. Exposure of cultured mammalian cells to etoposide or UV light induces APAF-1 expression via IRES-mediated translation, resulting in the activation of the caspase-dependent apoptotic program [19]. The protein level of XIAP is increased via IRES-mediated translation under stress conditions, such as serum starvation [20]. However, the specific translational regulators involved in IRES-mediated translation of APAF-1 and XIAP are unknown. In C. elegans, the RNA-binding protein GLD-1, which is highly expressed in the transition zone and early pachytene regions of the hermaphrodite gonad, inhibits translation of the mRNA of the p53 homolog cep-1 by directly binding to the cep-1 3′ UTR, thereby preventing cep-1-dependent apoptosis in response to DNA damage [21]. Translational initiation factors have also been reported to be involved in the control of apoptosis in C. elegans. For example, RNAi knockdown of the C. elegans eukaryotic initiation factor-4G IFG-1 induces CED-4 expression in the gonad and increases the frequency of germ-cell death [22], [23]. The eukaryotic initiation factor-3 subunit-k eIF-3.K is partially required for the deaths of somatic cells and acts through the caspase CED-3 to promote those cell deaths [24]. Although many studies have shown that both transcriptional and translational regulation of apoptotic genes is crucial for controlling apoptotic programs, how transcriptional and translational mechanisms are coordinated to promote apoptosis remains elusive.
Here we show that the maternally-contributed translational regulators GCN-1 and ABCF-3 act together to promote the cell deaths of possibly all somatic cells and of germ cells in response to ionizing radiation in a pathway distinct from the BCL-2 homolog CED-9-regulated canonical cell-death execution pathway of C. elegans. GCN-1 and ABCF-3 are required to maintain the basal level of phosphorylation of eukaryotic initiation factor 2 (eIF2α). The functions of GCN-1 and ABCF-3 in the promotion of programmed cell death are evolutionarily conserved between C. elegans and Saccharomyces cerevisiae. We show that GCN-1 and ABCF-3 cooperate with the transcriptional regulators CEH-34, EYA-1 and SPTF-3 and the protein kinase PIG-1 to promote the death of a specific somatic cell, the sister cell of the pharyngeal M4 motor neuron. We propose that the evolutionarily-conserved translational regulators GCN-1 and ABCF-3 contribute to apoptosis in general.
Results
The translational regulators GCN-1 and ABCF-3 are required for M4 sister cell death
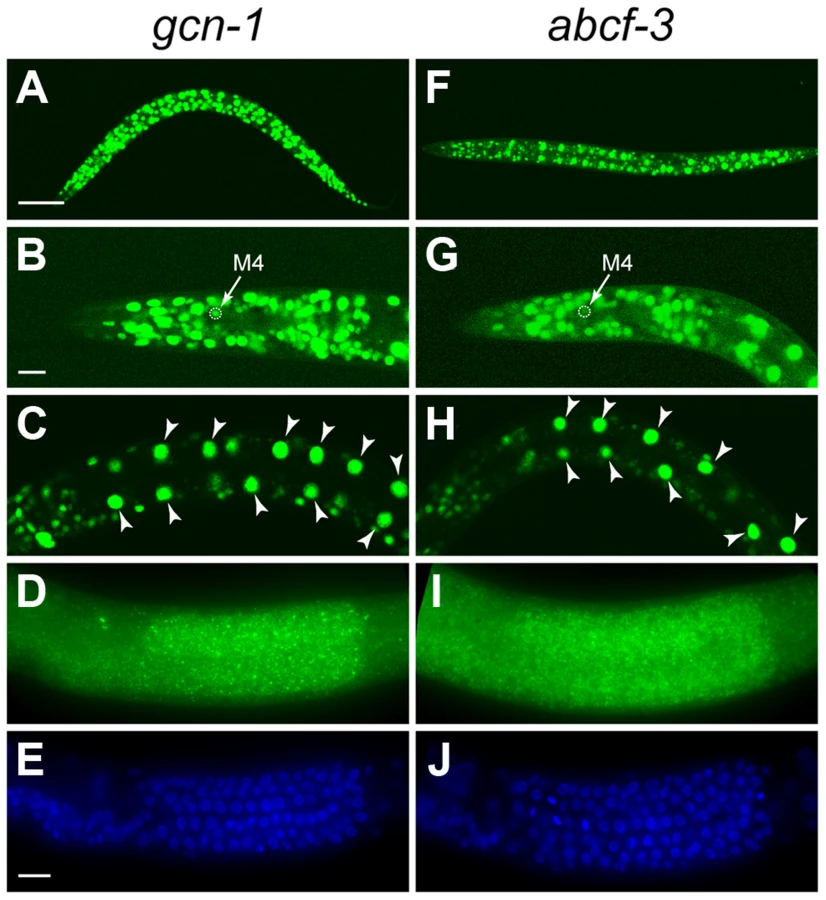
The C. elegans pharyngeal M4 motor neuron is generated during embryonic development and survives to regulate pharyngeal muscle contraction in feeding behavior, whereas the M4 sister cell dies by programmed cell death soon after its generation (Figure 1A) [25], [26]. We created a Pceh-28::gfp reporter transgene that expresses GFP specifically in the M4 neuron of wild-type animals and in both the M4 neuron and the surviving M4 sister of ced-3 caspase mutants defective in programmed cell death (Figure 1B–1C). This reporter allowed us to easily identify mutants with a defect in M4 sister cell death. [15]. Using this reporter, we performed a genetic screen for mutations that cause a defect in M4 sister cell death. Among our isolates were two non-allelic mutations, n4827 and n4927, that caused M4 sister survival in 12% of n4827 mutants and 13% of n4927 mutants (Figure 1D–1F).

We mapped n4827 to a 175 kb interval of chromosome III containing 18 predicted genes (Figure S1A). We used whole-genome sequencing to identify four strain-specific unique homozygous mutations within this interval in n4827 animals (Figure S1A) [27]. Of the four mutations, only one was exonic. This mutation was located in the third exon of gcn-1, which encodes a homolog of the S. cerevisiae Gcn1p protein. The n4827 mutation is predicted to change the tryptophan 164 codon to an opal stop codon, generating a small truncated protein (Figure 1G). A deletion mutation of gcn-1, nc40Δ, phenocopied the n4827 mutation [28]: 11% of gcn-1(nc40Δ) mutants and 12% of n4827 mutants had a surviving M4 sister, respectively (Figure 1F). The cell-death defect of n4827 mutants was partially rescued by a transgene that express gcn-1 cDNA under the control of the gcn-1 promoter (Figure 1F). These results indicate that n4827 is likely a null allele of gcn-1 and that loss of gcn-1 function causes a defect in M4 sister cell death.
We mapped n4927 to a 5.3 Mb interval of chromosome III (Figure S1B). This interval contains the gene abcf-3, which encodes a homolog of the S. cerevisiae Gcn20p protein [29]. Gcn20p physically interacts with Gcn1p, the S. cerevisiae homolog of GCN-1 [30]. We determined the sequence of abcf-3 in n4927 animals and identified a mutation that changes the arginine 206 codon to an opal stop codon (Figure 1G). A deletion mutation of abcf-3, ok2237Δ, that removes most of the abcf-3 coding region phenocopied the n4927 mutation: 13% of abcf-3(ok2237Δ) mutants and 13% of n4927 mutants had a surviving M4 sister (Figure 1F). Furthermore, the cell-death defect of n4927 mutants was completely rescued by a transgene carrying only the abcf-3 genomic locus. We concluded that n4927 is likely a null allele of abcf-3 and that loss of abcf-3 function causes a defect in M4 sister cell death.
abcf-3 encodes an AAA ATPase protein with two AAA domains (Figure 1G). In many proteins AAA domains have ATPase activity. To determine whether ATPase activity is important for ABCF-3 to promote M4 sister cell death, we generated abcf-3 transgenes carrying mutations that presumably inactivate the ATPase activity of each AAA domain by altering the lysine residues known to be catalytically essential for other AAA ATPases [31]. A wild-type abcf-3 transgene as well as mutant abcf-3 transgenes that changed lysine 217 of the first AAA domain to methionine [abcf-3 (K217M)], lysine 536 of the second AAA domain to methionine [abcf-3 (K536M)] or both lysine residues [abcf-3 (K217M, K536M)] completely rescued the defect in M4 sister cell death of abcf-3(n4927) mutants (Figure 1F). These results support the idea that the ATPase activity of ABCF-3 is dispensable for M4 sister cell death. This result is consistent with studies of S. cerevisiae Gcn20p, the homolog of C. elegans ABCF-3. Gcn20p that lacks the ATPase activities of both AAA domains because of mutations in conserved glycine residues (Gly371 and Gly654) or because of the deletion of two AAA domains still retains Gcn20p function comparable to that of wild-type Gcn20p [32].
GCN-1 and ABCF-3 act together to promote M4 sister cell death
GCN-1 and ABCF-3 are evolutionarily conserved among S. cerevisiae, C. elegans and humans (Figure 1H, Figure S2 and S3). Expression of S. cerevisiae GCN1, the homolog of C. elegans gcn-1, and GCN20, the homolog of C. elegans abcf-3, under the control of the abcf-3 promoter rescued the defect in M4 sister cell death of C. elegans gcn-1 and abcf-3 mutants, respectively, indicating that S. cerevisiae GCN1 and GCN20 are functional homologs of C. elegans gcn-1 and abcf-3, respectively (Figure 1F).
S. cerevisiae Gcn1p has a domain (amino acids 1350–2152) similar to that of translation elongation factor 3 (EF3). The EF3-like domain is highly conserved among species (Figure 1H and Figure S2) and is necessary and sufficient for binding to Gcn20p [32]. We therefore tested whether C. elegans GCN-1 can physically interact with ABCF-3 using the yeast two-hybrid assay (Figure 2A). Full-length GCN-1 (1–2634) interacted with full-length ABCF-3 (1–712). To identify the protein domains important for GCN-1 to bind to ABCF-3, we generated a series of deletion constructs of GCN-1 and assayed each for ABCF-3-binding activity using the yeast two-hybrid assay. GCN-1 fragments not containing entire the EF3 domain (1–1760, 1–880, 880–1760 and 1760–2634) or containing only the EF3 domain (1350–2150) failed to bind ABCF-3, whereas GCN-1 fragments containing the EF-3 domain and surrounding regions (880–2634) bound ABCF-3. These results suggest that GCN-1 physically interacts with ABCF-3 but that unlike in yeast the EF3-like domain is not sufficient for GCN-1 to bind to ABCF-3.

We also defined the domains of ABCF-3 important for ABCF-3 to bind to GCN-1 (Figure 2A). ABCF-3 fragments lacking the N-terminal region (202–712, 512–712 and 202–512) failed to bind GCN-1, whereas ABCF-3 fragments containing the N-terminal region (1–712, 1–512 and 1–202) bound GCN-1, suggesting that the N-terminal portion of ABCF-3 (which does not include the first AAA domain) is necessary and sufficient for binding to GCN-1, just as the N-terminal region of S. cerevisiae Gcn20p is necessary and sufficient for binding to Gcn1p, the S. cerevisiae homolog of GCN-1.
To determine whether GCN-1 and ABCF-3 interact in vivo, we generated antibodies against GCN-1 and ABCF-3 and performed co-immunoprecipitation experiments. We first tested whether these antibodies specifically recognize GCN-1 or ABCF-3 protein using western blot analysis. The antibodies against GCN-1 or ABCF-3 recognized proteins of the sizes predicted for the GCN-1 or ABCF-3 proteins in wild-type animals but not in gcn-1(n4827) or abcf-3(n4927) animals, respectively, confirming the specificity of these antibodies (Figure 2C). Then we tested whether GCN-1 could be co-immunoprecipitated with ABCF-3. Whole-protein extracts from wild-type animals were subjected to immunoprecipitation using an anti-ABCF-3 antibody (or normal IgG as a control), and then immunocomplexes were analyzed by western blotting using antibodies against ABCF-3 or GCN-1. Both ABCF-3 and GCN-1 were recovered in an immunocomplex purified with the anti-ABCF-3 antibody, whereas neither ABCF-3 nor GCN-1 was recovered in an immunocomplex purified with normal IgG (Figure 2B). We conclude that GCN-1 and ABCF-3 are present in the same protein complex in vivo.
Since GCN-1 and ABCF-3 form a complex in vivo, we suspected that deletion of either protein might affect the stability of the other protein [29], [33]. To test this hypothesis, we examined the levels of GCN-1 and ABCF-3 proteins by western blot analyses of whole-protein extracts prepared from wild-type, gcn-1(n4827) and abcf-3(n4927) animals using antibodies against ABCF-3 or GCN-1. The steady-state level of ABCF-3 protein was decreased in gcn-1(n4827) animals by 3.6 fold compared to that of wild-type animals. Similarly, the steady-state level of GCN-1 protein was decreased in abcf-3(n4927) animals by 4.4 fold (Figure 2C). These results suggest that a lack of ABCF-3 or GCN-1 protein affects the stability of the other protein and support our conclusion that GCN-1 and ABCF-3 are in a protein complex together in vivo.
If GCN-1 and ABCF-3 physically interact in vivo to promote M4 sister cell death, GCN-1 and ABCF-3 should act together in the same pathway. Since gcn-1(n4827) and abcf-3(n4927) are likely null mutations, the gcn-1(n4827) mutation would not enhance the M4 sister cell-death defect of abcf-3(n4927) mutants if gcn-1 and abcf-3 function in the same process or pathway. Indeed, we observed no enhancement of the M4 sister cell-death defect of gcn-1(n4827) abcf-3(n4927) double mutants compared to that of either single mutant: there was 13% M4 sister survival in gcn-1(n4827) abcf-3(n4927) double mutants, 12% M4 sister survival in gcn-1(n4827) mutants and 13% M4 sister survival in abcf-3(n4927) mutants (Figure 1F). We conclude that gcn-1 and abcf-3 function together in the same process of pathway to promote M4 sister cell death, consistent with our finding that GCN-1 and ABCF-3 physically interact in vivo.
In S. cerevisiae, Gcn1p and Gcn20p are required for the efficient phosphorylation of eukaryotic initiation factor 2 (eIF2α) under both normal conditions and conditions of amino-acid starvation [29], [30]. Gcn1p and Gcn20p form a protein complex that activates the serine-threonine protein kinase Gcn2p, which then phosphorylates an evolutionarily conserved serine residue of eIF2α. The amino acid sequences surrounding the eIF2α phosphorylation site are identical in S. cerevisiae, C. elegans and humans, suggesting a conserved regulatory mechanism of eIF2α [28]. We tested whether GCN-1 and ABCF-3 promote the phosphorylation of eIF2α in C. elegans using an antibody that specifically recognizes eIF2α that is phosphorylated at serine 49 (P-eIF2α). From wild-type animals cultivated under normal physiological conditions, a single band of eIF2α was detected in western blotting analyses using either the anti-P-eIF2α antibody or an antibody that recognized total eIF2α (Figure 2D). In gcn-1(n4827) and abcf-3(n4927) mutants, the phosphorylation levels of eIF2α in physiological conditions were 52% and 54% of the levels in wild-type animals, respectively (Figure 2D and 2E). We conclude that gcn-1 and abcf-3 are required to maintain the steady-state level of the phosphorylation of eIF2α.
The regulation of phosphorylation of eIF2α plays an essential role in the initiation of translation. We therefore directly tested whether gcn-1 and abcf-3 affect gene expression at the translational level. Since gcn-1 and abcf-3 are highly expressed in the gonads at the fourth larval stage, maternally contribute to the death of the M4 sister and affect most programmed cell deaths (see below, Figure 3D and 3I, Table 1 and Table S5), we isolated both wild-type animals and gcn-1 and abcf-3 mutants at the fourth larval stage and performed mRNA-seq and ribosome profiling (Ribo-seq) to generate quantitative genome-wide information concerning mRNA abundance and the locations of mRNAs occupied by ribosomes [34]. Parallel analyses of data from Ribo-seq and mRNA-seq studies allowed us to distinguish differences in mRNA abundance from differences in translational control and to generate a quantitative and comprehensive list of genes the expression of which is likely regulated by gcn-1 and abcf-3 at the translational level. Loss of gcn-1 or abcf-3 function affected the expression of a large number of genes at either the transcriptional or translational level or at both (Figure S4A and S4B and Table S1). Since GCN-1 and ABCF-3 very likely function in translational control, their effects on transcript levels are likely indirect. Changes in gene expression compared to wild-type animals were similar between gcn-1 and abcf-3 mutants, supporting our conclusion that gcn-1 and abcf-3 act together (Figure S4C and S4D). The expression of 464 genes or 217 genes changed in both gcn-1 and abcf-3 mutants compared to wild-type animals at least two-fold (p<0.1) in mRNA-seq or Ribo-seq analyses, respectively (Figure S4E and S4F). Of the 217 genes altered in translational expression, 98 genes showed no alterations in mRNA levels using our standards of a two-fold change and p<0.1 (Table S2 and Table S3). These genes are candidates for being directly regulated by both gcn-1 and abcf-3 translationally. These results suggest that gcn-1 and abcf-3 function together in the translational control of many genes.

In mammals, eIF2α phosphorylation is mediated by at least four different protein kinases: PKR-like endoplasmic reticulum kinase (PERK), general control non-derepresessible-2 (GCN2), double-stranded RNA-activated protein kinase (PKR) and heme-regulated inhibitor kinase (HRI); each of these kinases is activated by a distinct stress signal [18]. These kinases share homology in their kinase catalytic domains, but their effector domains are distinct and are subject to different regulatory mechanisms. Homologs of genes encoding two of these protein kinases exist in the C. elegans genome: the PERK homolog PEK-1 and the GCN2 homolog GCN-2. Y38E10A.8 has a kinase domain similar to that of mammalian eIF2α kinases but does not have an obvious homolog. We tested whether these three protein kinases are required for the programmed cell death of the M4 sister. Neither single mutants of each kinase gene nor the triple mutant was defective in M4 sister cell death (Table S4), suggesting that one or more unidentified protein kinase(s) regulated by GCN-1 and ABCF-3 are responsible for phosphorylating eIF2α in the regulation of M4 sister cell death. Alternatively, it is possible GCN-1 and ABCF-3 promote M4 sister cell death through one or more targets other than eIF2α.
gcn-1 and abcf-3 are expressed ubiquitously
To determine the expression patterns of gcn-1 and abcf-3, we generated transgenes expressing a reporter GFP under the control of the endogenous gcn-1 or abcf-3 promoter. Both gcn-1 and abcf-3 were expressed in most cells during all stages of development. We observed gcn-1 and abcf-3 expression in head neurons, hypodermal cells, intestinal cells, body wall muscles, and pharyngeal neurons, including the M4 neuron (Figure 3A–3C and 3F–3H). We also used the technique of fluorescence in situ hybridization (FISH) with a level of sensitivity sufficient to detect single mRNA molecules [35] to observe endogenous gcn-1 and abcf-3 transcripts. Consistent with the expression of the GFP reporter transgenes, gcn-1 and abcf-3 mRNAs were observed in most somatic cells. In addition, gcn-1 and abcf-3 mRNAs were abundant in the germ cells in the hermaphrodite gonad (Figure 3D, 3E, 3I and 3J). The similar expression patterns of gcn-1 and abcf-3 are consistent with our observations that GCN-1 and ABCF-3 physically interact and act together to promote the death of the M4 sister.
Since gcn-1 and abcf-3 are ubiquitously expressed and required to broadly maintain the basal level of phosphorylation of eIF2α, we tested whether gcn-1 and abcf-3 might be involved in other biological processes. We did not observe abnormalities in the morphologies of the hermaphrodite vulva, the male tail or the neurite processes of the M4, I2 and PVQ neurons. However, the growth rate of gcn-1 and abcf-3 mutants from embryogenesis to the fourth larval stage was around 24 hours longer than that of wild-type animals, and the mitotic pachytene region of the hermaphrodite gonad was expanded over the loop regions of the gonads. (data not shown). These observations suggest that gcn-1 and abcf-3 affect biological processes in addition to programmed cell death.
GCN-1 and ABCF-3 promote the deaths of most somatic cells during development and of germ cells in response to ionizing radiation
Given the ubiquitous expression patterns of gcn-1 and abcf-3, we tested whether gcn-1 and abcf-3 promote programmed cell deaths in addition to that of the M4 sister. We examined gcn-1(n4827) and abcf-3(n4927) mutants for defects in the deaths of the NSM sisters, the PVQ sisters, the g1A sisters, the RIM and RIC sisters and multiple cells in the anterior pharynx. gcn-1(n4827) and abcf-3(n4927) single mutants did not exhibit defects in the deaths of these cells (Table 1). However, when either the gcn-1(n4827) or the abcf-3(n4927) mutation was combined with the partial loss-of-function ced-3(n2427) mutation, which sensitizes strains to weak defects in cell death [36], we observed significant cell-death defects for all cell types tested (Table 1). For example, the gcn-1(n4827) and abcf-3(n4927) mutations enhanced the ced-3(n2427) defect from 16% to 38% and 34%, respectively, for the NSM sister and from 13% to 36% and 45%, respectively, for the g1A sister. We conclude that gcn-1 and abcf-3 promote programmed cell death generally rather than specifically affecting the M4 sister cell death.
We next tested whether gcn-1 and abcf-3 are involved in the deaths of germ cells in the gonad of the adult hermaphrodite. More than half of germ cells stochastically undergo programmed cell death under normal conditions during oocyte differentiation [37]. We scored the number of apoptotic germ cells using the vital dye acridine orange (AO), which stains nucleic acids within apoptotic cells in living animals [38]. gcn-1(n4827) and abcf-3(n4927) mutants had 9.1 and 9.5 apoptotic germ cells per gonadal arm on average, respectively, similar to wild-type animals, which had 8.6 apoptotic germ cells per gonadal arm (Figure 4I). We also scored the number of apoptotic germ cells by direct observation of the gonads of engulfment-defective ced-1(e1735) mutants, in which cell corpses accumulate because of a defect in cell-corpse engulfment, facilitating a sensitive assay for the deaths of germ cells [37]. ced-1(e1735) mutants had an average of 14.4 cell corpses per gonadal arm (Figure S5). ced-1(e1735) double mutants with gcn-1(n4827) or abcf-3(n4927) had nearly identical numbers of cell corpses per gonadal arm, 13.9 and 14.0, respectively (Figure S5). These results indicate that gcn-1 and abcf-3 are dispensable for germ-cell death under physiological conditions.

Since many germ cells undergo apoptosis in response to genotoxic stresses such as ionizing radiation [39], we tested whether gcn-1 and abcf-3 mediate ionizing radiation damage-induced germ cell death. As assayed with AO, wild-type animals normally contained an average of 8.6 apoptotic germ cells per gonadal arm, while wild-type animals exposed to ionizing radiation contained on average 27.1 apoptotic germ cells (Figure 4A, 4E and 4I). This germ-cell death was completely blocked by a mutation in the caspase gene ced-3 in wild-type, gcn-1(n4837) and abcf-3(n4927) animals (Figure 4B, 4F and 4I). Strikingly, ionizing radiation failed to increase the number of apoptotic germ cells in gcn-1(n4827) and abcf-3(n4927) mutants (10.9 and 10.4 apoptotic germ cells per gonadal arm in gcn-1 and abcf-3 mutants, respectively, 24 hours after gamma ray irradiation) (Figure 4C, 4D, 4G, 4H and 4I). These results indicate that gcn-1 and abcf-3 are required for ionizing radiation-induced germ cell death but not for the stochastic germ cell death that occurs in physiological conditions.
gcn-1 and abcf-3 gene dosage affects programmed cell death
The gcn-1(n4827) and abcf-3(n4927) mutations partially blocked both the programmed cell deaths of somatic cells (Table 1) and the deaths of germ cells in response to ionizing radiation (Figure 4). Both somatic and ionizing radiation-induced germ cell deaths involve the canonical cell-death execution pathway consisting of the BH3-only gene egl-1, the BCL-2 homolog ced-9, the pro-apoptotic APAF-1 homolog ced-4, and the caspase gene ced-3 [40]. Interestingly, animals doubly heterozygous for gcn-1 and ced-3, ced-4 or egl-1 had a defect in M4 sister cell death (gcn-1/+; ced-3/+ 18%, gcn-1/+; ced-4/+ 12% or gcn-1/+; egl-1/+ 17%, respectively) significantly higher than that of singly heterozygous animals (gcn-1/+ 4%, ced-3/+ 0%, ced-4/+ 0% or egl-1/+ 1%, respectively) (Table S5). These results indicate that the simultaneous reduction by half of the dosage of gcn-1 and of genes in the canonical cell-death execution pathway causes a significant defect in M4 sister cell death. We observed a similar genetic interaction in animals heterozygous for abcf-3 and ced-3, ced-4 or egl-1 (Table S5).
gcn-1 and abcf-3 maternally contribute to programmed cell death
We observed that maternal gcn-1 and abcf-3 contribute to zygotic programmed cell death. While gcn-1(−) animals generated by gcn-1(−) hermaphrodites and gcn-1(−) males exhibited a defect in M4 sister cell death (12% of M4 sister survival), gcn-1(−) animals produced from gcn-1/+ hermaphrodites and gcn-1(−) males did not (0% of M4 sister survival) (Table S5), indicating that maternal gcn-1 is sufficient to promote programmed cell death. gcn-1(−) animals generated by gcn-1(−) hermaphrodites and gcn-1/+ males exhibited a defect in M4 sister cell death (13% of M4 sister survival). gcn-1/+ animals generated by gcn-1(−) hermaphrodites and gcn-1(+) males exhibited a very weak defect in M4 sister cell death (4% of M4 sister survival) compared to 12% of M4 sister survival in gcn-1(−) self-progeny of gcn-1(−) hermaphrodites. By contrast, gcn-1/+ animals produced from gcn-1(+) hermaphrodites and gcn-1(−) males exhibited no defect in M4 sister cell death (0% of M4 sister survival) (Table S5). These results indicate that maternal gcn-1 is partially required for the M4 sister to undergo programmed cell death. We observed a similar maternal requirement and sufficiency for abcf-3 (Table S5). We conclude that maternal gcn-1 and abcf-3 are sufficient and partially required for the M4 sister to undergo programmed cell death.
GCN-1 and ABCF-3 act independently of the BCL-2 homolog CED-9 to promote programmed cell death and can act in the cell fated to die
To examine interactions between gcn-1 and abcf-3 and the canonical cell-death execution pathway, we performed epistasis analyses between gcn-1 or abcf-3 and ced-9, which functions downstream of egl-1 and upstream of ced-4 and ced-3 in the cell-death execution pathway [40]. Because the ced-9(n2812) null mutation causes ectopic cell deaths and organismic inviability, we used the ced-3 partial loss-of-function mutation n2446 to suppress ced-9(n2812) lethality [41]. We observed that 50% of ced-9(n2812) animals had a surviving M4 sister in the ced-3(n2446) mutant background. This increase over the 5% frequency of M4 sister survival in ced-3(n2446) mutants is consistent with the proposal that ced-9 has a cell-killing activity [42]. We observed that gcn-1 ced-9 and abcf-3 ced-9 double mutants were more highly penetrant for M4 sister survival (90% and 89%, respectively) than either single mutant in the ced-3(n2446) mutant background: gcn-1 (45%), abcf-3 (40%) and ced-9 (50%), respectively (Table 2). These results indicate that ced-9 is not required for gcn-1 and abcf-3 to promote programmed cell death. Thus, gcn-1 and abcf-3 function downstream of or in parallel to ced-9 in the regulation of programmed cell death.

We next tested whether the activity of gcn-1 and abcf-3 can act cell-autonomously to promote programmed cell death. Previous studies showed that expression of a ced-3, ced-4 or egl-1 cDNA under the control of the mec-7 promoter can act cell-autonomously to cause the deaths of a set of touch neurons, including the PLML and PLMR cells. We expressed gcn-1 and abcf-3 cDNAs in the PLM neurons under the control of the mec-7 promoter. We observed that 100% of the PLM neurons survived in wild-type animals, whereas only 50% or 89% of the PLM neurons survived in animals expressing gcn-1 or abcf-3, respectively, under the control of the mec-7 promoter (Figure 5A). Expression of both gcn-1 and abcf-3 also reduced a survival of the PLM neurons: 61% of the PLM neurons survived. These results indicate that expression of gcn-1 and abcf-3 are sufficient to induce cell death and suggest that gcn-1 and abcf-3 acts cell-autonomously to promote programmed cell death.

Our genetic screen for mutants defective in M4 sister cell death identified other genes in addition to gcn-1 and abcf-3: ceh-34, eya-1, sptf-3 and pig-1 [15], [17]. We previously showed that the Six family homeodomain protein CEH-34 and the Eyes absent homolog EYA-1 directly drive the transcription of the BH3-only gene egl-1 in the M4 sister to promote M4 sister cell-type specific death [15] and that the SP1 family transcription factor SPTF-3 directly drives the transcription of both egl-1 and the AMPK-related protein kinase gene pig-1, which also promotes M4 sister cell death [17]. We determined how gcn-1 and abcf-3 interact with these genes by examining double mutants. The partial loss-of-function alleles ceh-34(n4796) and sptf-3(n4850) and the null allele pig-1(gm344Δ) enhanced the M4 sister-cell death defect of gcn-1(n4827) and abcf-3(n4927) null mutants (Table 3). These results indicate that ceh-34, sptf-3 and pig-1 function in pathways distinct from that of gcn-1 and abcf-3 to promote M4 sister cell death.

Discussion
The maternally-contributed translational regulators GCN-1 and ABCF-3 act together to promote the death of the M4 sister in a pathway distinct from the CED-9-mediated cell-death execution pathway
We demonstrated that the translational regulators GCN-1 and ABCF-3 are pro-apoptotic factors that maternally contribute to the programmed cell death of the M4 sister in C. elegans. GCN-1 and ABCF-3 promote the deaths of all somatic cells tested. Essentially all somatic cell deaths are mediated by an evolutionarily conserved cell-death execution pathway consisting of the BH3-only gene egl-1, the BCL-2 homolog ced-9, the APAF-1 homolog ced-4 and the caspase gene ced-3 [40]. How do gcn-1 and abcf-3 interact with this pathway to regulate apoptosis? We propose that gcn-1 and abcf-3 likely act in a novel pathway distinct from the canonical cell-death execution pathway. First, gcn-1 and abcf-3 promote apoptosis in the absence of ced-9 activity, indicating that gcn-1 and abcf-3 function independently of ced-9 in the regulation of apoptosis and hence do not regulate either ced-9 or egl-1. Second since ced-3 and ced-4 function downstream of ced-9 in the cell-death execution pathway, gcn-1 and abcf-3 could act through these genes to promote cell death. However, our mRNA-seq and Ribo-seq results indicate that gcn-1 and abcf-3 do not have major effects on mRNA abundance or the ribosome footprint density of ced-3 and ced-4 (Figure S6). Our preferred model is that GCN-1 and ABCF-3 function in a pathway that acts in parallel to the canonical cell-death execution pathway, although we cannot preclude the possibility that GCN-1 and ABCF-3 translationally regulate unidentified factors that act through ced-3 or ced-4 without changing the transcriptional and translational levels of the products of these genes. Also, since we used whole animals for our mRNA-seq and Ribo-seq analyses, we would not have detected alterations in CED-3 or CED-4 levels that were specific to a small subset of cells, including the M4 sister.
Our genetic analyses revealed that gcn-1 and abcf-3 maternally contribute to the death of the M4 sister, which undergoes programmed cell death during embryogenesis. Maternally-contributed factors might act to ensure the rapid deaths of cells during embryogenesis; perhaps zygotic expression of apoptotic genes would be too slow. Also, the maternal effects of gcn-1 and abcf-3 might explain why we discovered new general cell-death genes, despite the fact that many genetic screens have been performed in search of C. elegans mutants defective in somatic cell deaths. Most such genetic screens have examined F2 animals after mutagenesis, and would have missed maternally-contributed genes that affect general cell death. Perhaps, additional maternal-effect genes with functions in apoptosis exist in C. elegans. Such genes might be efficiently identified by screening in the third generation after mutagenesis.
gcn-1(n4827) and abcf-3(n4927) single mutations appeared to cause a defect in only M4 sister cell death, and these mutations both affected other cell deaths to differing extents in strains sensitized to weak defects in cell death. For example, the death of the M4 sister was most sensitive and the deaths of the PVQ sisters were least sensitive to the gcn-1(n4827) and abcf-3(n4927) mutations among the cells we tested (Table 1). We speculate that sensitivity to perturbation of cell-death genes is different among different cell types. This hypothesis is supported by the observations that penetrance of cell-death defects varies among different cell types in partial loss-of-function ced-3(n2427) mutants and that the extent of the cell-death defect of ced-3(n2427) mutants is well correlated with that of gcn-1(n4827) and abcf-3(n4927) mutants.
Our genetic and biochemical data strongly suggest that GCN-1 and ABCF-3 physically interact in a complex in vivo to promote apoptosis. First, GCN-1 and ABCF-3 interacted in the yeast two-hybrid system. Second, GCN-1 co-immunoprecipitated with ABCF-3 from a total protein extract from C. elegans. Third, the absence of either the GCN-1 or ABCF-3 protein decreased the steady-state level of the other protein (ABCF-3 or GCN-1, respectively), indicating that an interaction between GCN-1 and ABCF-3 is likely important for the stability of both proteins. Fourth, gcn-1(n4827) abcf-3(n4927) double mutants were not enhanced in the defect in apoptosis compared to each single mutant.
GCN-1 and ABCF-3 play an essential role in germ-cell death in response to ionizing radiation
Although GCN-1 and ABCF-3 promote the deaths of most somatic cells, in gcn-1 or abcf-3 mutants only 12% or 13% of animals are defective in M4 sister cell death, respectively, and the cell-death defect of most other cells was observed only in a partial loss-of-function ced-3 mutant background, which is sensitized to weak defects in cell death. Furthermore, loss-of-function of gcn-1 and abcf-3 did not affect the deaths of germ cells under physiological conditions. By striking contrast, we found that GCN-1 and ABCF-3 play an essential role in germ-cell deaths induced by ionizing radiation. These results suggest that translational control by GCN-1 and ABCF-3 plays a more important role in germ-cell deaths induced by ionizing radiation than in somatic cell deaths. The hypothesis that translational control is particularly important for cell deaths induced by ionizing radiation is supported by a recent report that a mutation in RNA polymerase I (rpoa-2), which synthesizes ribosomal RNAs, causes a defect in germ-cell deaths induced by ionizing radiation [43].
Ionizing radiation causes DNA double-strand breaks, which lead to the progressive accumulation of mutations and chromosomal aberrations as damaged cells undergo division, resulting in apoptosis and the demise of genetically damaged cells. In C. elegans, ionizing radiation causes massive deaths of the germ cells during the late pachytene stage of oocyte development in adult gonads, resulting in the elimination of the damaged oocytes [39]. Germ-cell deaths induced by ionizing radiation specifically involve activation of the p53 homolog CEP-1 by the DNA damage response pathway and subsequent CEP-1 - dependent transcriptional induction of the BH3-only gene egl-1, which activates the cell-death execution pathway regulated by CED-9 [44], [45]. How might GCN-1 and ABCF-3 interact with the known DNA-damage response and cell-death execution pathways in the regulation of germline cell deaths induced by ionizing radiation? Our genetic results suggest that GCN-1 and ABCF-3 function independently of CED-9, at least in the regulation of the death of the M4 sister cell. We suggest that as is the case for somatic cell deaths, GCN-1 and ABCF-3 function in a novel pathway independently of CED-9 in regulating the germ-cell deaths induced by ionizing radiation. Alternatively, if GCN-1 and ABCF-3 regulate germ-cell deaths induced by ionizing radiation via a mechanism different from that of somatic cell deaths, it is possible that GCN-1 and ABCF-3 act through egl-1 and its target ced-9, since egl-1 is involved in somatic programmed cell deaths and germ-cell deaths induced by ionizing radiation but not in the stochastic germ-cell deaths that occur under physiological conditions.
The functions of GCN-1 and ABCF-3 in the control of translation are conserved between S. cerevisiae and C. elegans
GCN-1 and ABCF-3 are conserved proteins from yeast to humans. The C. elegans GCN-1 protein has 43% and 53% similarities (23% and 32% identities) to the homologs of S. cerevisiae and humans, respectively, and the C. elegans ABCF-3 proteins has 57% and 69% similarities (40% and 49% identities) to the homologs of S. cerevisiae and humans, respectively (Figure 1H). The yeast GCN-1 homolog Gcn1p and ABCF-3 homolog Gcn20p are required to maintain the basal level of the phosphorylation of eukaryotic initiation factor 2α (eIF2α) in the physiological condition and to increase the phosphorylation of eIF2α in response to amino-acid starvation. Gcn1p and Gcn20p activate the serine-threonine protein kinase Gcn2p, which phosphorylates an evolutionarily conserved serine residue of eIF2α. The phosphorylation of eIF2α results in both the inhibition of global translation and the translational activation of the GCN4 mRNA, which encodes a basic leucine zipper transcription factor. Translation of GCN4 mRNA is regulated by four short upstream open reading frames (uORFs) in the 5′ UTR with start codons that are out-of-frame with the main coding sequence and which generally reduce translation from the main reading frame [46].
We speculate that the mechanistic roles of C. elegans GCN-1 and ABCF-3 in translational control are conserved between yeast and C. elegans. First, the amino-acid sequences of GCN-1 and ABCF-3 proteins are conserved between yeast and C. elegans, particularly in functionally important domains (Figure 1H). Second, the functions of GCN-1 and ABCF-3 can be substituted with those of S. cerevisiae GCN1 and GCN20, respectively, for the promotion of M4 sister cell death. Third, like their yeast counterparts, C. elegans GCN-1 and ABCF-3 are required to maintain the basal level of phosphorylation of eIF2α and physically interact through an EF3-like domain-containing region of GCN-1 and an N-terminal ABCF-3 domain [43]. Fourth, like Gcn20p, the AAA domain ATPase activity of ABCF-3 is not required for its function [32]. Fifth, the atf-5 gene, the C. elegans homolog of S. cerevisiae GCN4, has two upstream ORFs that have been shown to inhibit the translation of the atf-5 mRNA [47].
A regulatory network involving transcription, translation and protein phosphorylation specifies the death of the M4 sister
We have shown that in addition to gcn-1 and abcf-3, ceh-34, eya-1, sptf-3 and pig-1 function in M4 sister cell death [15], [17]. We previously reported that the Six family homeodomain protein CEH-34 and the Eyes absent homolog EYA-1 physically interact to directly drive expression of the pro-apoptotic BH3-only gene egl-1 in the M4 sister, leading to the death of the M4 sister (Figure 5B) [15]. We found that the SP1 family transcription factor SPTF-3 directly drives the transcription of the gene egl-1, which encodes a BH3-only protein that promotes apoptosis via the CED-3 caspase-mediated canonical cell-death execution pathway [17]. SPTF-3 also directly drives the transcription of the AMPK-related gene pig-1, which encodes a protein kinase that functions in a pathway in parallel to the CED-3-mediated canonical cell-death execution pathway. These interactions are shown in Figure 5B.
Our analyses indicate that gcn-1 and abcf-3 likely function in a pathway that acts in parallel to those of pig-1, ceh-34 and sptf-3. These results are consistent with a model in which GCN-1 and ABCF-3 act independently of CED-9 to promote M4 sister cell death. In short, we propose that the regulatory network for the death of the M4 sister includes at least three different pathways involving translation, transcription and protein phosphorylation (Figure 5B). Each gene in this network (gcn-1, abcf-3, sptf-3, pig-1, egl-1, ceh-34 and eya-1) has a human counterpart, some of which are implicated in human diseases, including developmental disorders and cancer. We anticipate that further analyses of this regulatory network will both reveal an evolutionarily conserved mechanism of apoptosis shared between C. elegans and humans and provide insights concerning how abnormalities in this apoptotic network can lead to human disease.
Materials and Methods
C. elegans strains
C. elegans strains were cultured at 20°C as described [48]. The N2 strain was used as the wild type. The following mutations, integrations and extrachromosomal arrays were used.
LGI: sptf-3(n4850), eya-1(ok654Δ), nIs177[Pceh-28::gfp, lin-15AB(+)], nIs180[Ptdc-1::gfp, lin-15AB(+)], zdIs5[Pmec-4::gfp, lin-15AB(+)].
LGII: rol-1(e91), gcn-2(ok871Δ), Y38E10A.8(tm4094Δ).
LGIII: ced-4(n1162), ced-9(n2812), gcn-1(n4827, nc40Δ), abcf-3(n4927, ok2237Δ), unc-45(r450), dpy-18(e364), nIs176[Pceh-28::gfp, lin-15AB(+)].
LGIV: ced-3(n717, n2427, n2446), pig-1(gm344Δ), nIs175[Pceh-28::gfp, lin-15AB(+)].
LGV: egl-1(n1084 n3082), ceh-34(n4796), oyIs14[sra-6::gfp].
LGX: lin-15(n765), pek-1(ok275Δ), nIs106[Plin-11::gfp, lin-15AB(+)], nIs429[Pphat-5::gfp, lin-15AB(+)], bcIs24[Ptph-1::gfp, lin-15AB(+)].
Unmapped: nIs460[Pgcn-1::gfp], nIs488[Pabcf-3::gfp], nIs645 and nIs646[Pmec-7::gcn-1 cDNA, Pmec-7::abcf-3 cDNA, Pmec-3::mCherry, rol-6(su1006)], nIs648 and nIs649[Pmec-7::gcn-1 cDNA, Pmec-3::mCherry, rol-6(su1006)], nIs651 and nIs652[Pmec-7::abcf-3 cDNA, Pmec-3::mCherry, rol-6(su1006)]. Extrachromosomal arrays: nEx1817 and nEx1818[Pgcn-1::gcn-1 cDNA::gcn-1 3′ UTR, Plin-44::gfp], nEx1925 and nEx1926[abcf-3(+), Plin-44::gfp], nEx1928 and nEx1929[abcf-3 K217M, Plin-44::gfp], nEx1931 and nEx1932[abcf-3 K536M, Plin-44::gfp], nEx1934 and nEx1935[abcf-3 K217M K536M, Plin-44::gfp], nEx2223 and nEx2224[Pceh-34::eIF2α S49A, Plin-44::gfp]
Genetic screen and mapping of gcn-1(n4827) and abcf-3(n4927)
gcn-1(n4827) and abcf-3(n4927) were isolated from a genetic screen for mutations that cause an extra GFP-positive M4-like cell in animals carrying the Pceh-28::gfp transgene [15]. Mutagenesis was performed as described [48]. Mutagenized P0 animals were allowed to lay eggs, and 144,000 synchronized F2 animals were screened with a fluorescence-equipped dissecting microscope. Single nucleotide polymorphisms were used to map gcn-1(n4827) and abcf-3(n4927) to a 175 kb interval (III: 2,044,521–2,220,200) and a 5.3 Mb interval (III: 5,346,407–10,613,191), respectively [49]. Whole-genome sequencing of gcn-1(n4827) mutants was performed using an Illumina/Solexa GAII, according to the instructions of the manufacture. DNA sequencing of the abcf-3 locus of abcf-3(n4927) mutants was performed using an Applied Biosystems 3130×.
Analyses of defects in programmed cell deaths of specific cells
The programmed cell deaths of specific cells were scored at the indicated stages using the following strains, which express GFP in specific cells. A fluorescence-equipped compound microscope was used to score the programmed cell deaths. M4 sister cell death, nIs175, nIs176 or nIs177 at the L1 stage. g1A sister cell death, nIs429 at the L1 stage. PVQ sister cell death, oyIs14 at the L4 stage [50]. NSM sister cell death, bcIs24 at the L1 stage [51]. RIM and RIC sister cell death, nIs180 at the L1 stage. Extra cells in the anterior pharynx were scored using a compound microscope equipped with Nomarski differential interference contrast optics. For physiological germ-cell deaths, germ-cell corpses in gonads of animals 24 hours after the fourth-larval stage were counted by direct observation using Nomarski optics. For ionizing radiation-induced germ-cell deaths, fourth-larval stage animals were exposed to 120 Gy of ionizing radiation, and germ-cell deaths were scored using acridine orange at 24 hours post-irradiation as described [38].
Plasmid construction
The transgenes Pceh-28::gfp, Ptph-1::gfp and sra-6::gfp are described [15], [50], [51]. The phat-5 promoter sequence in pGD48 was cloned in pPD122.56 to generate the Pphat-5::gfp transgene [52]. The Pflp-15::gfp transgene contained 2.4 kbp of the 5′ promoter of flp-15 in pPD122.56. The Pgcy-37::gfp transgene contained 1.1 kbp of 5′ promoter of gcy-37 in pPD122.56. The Ptdc-1::gfp transgene contained 4.5 kbp of 5′ promoter of tdc-1 in pPD121.83. The Pgcn-1::gcn-1 cDNA::gcn-1 3′UTR transgene (pTH gcn-1 cDNA) contained 4.2 kbp of 5′ promoter of gcn-1, a full-length gcn-1 cDNA and 1.0 kbp 3′ of the stop codon of gcn-1. The 5′ promoter of gcn-1, a full-length gcn-1 cDNA and the 3′ promoter of gcn-1 were generated by PCR and fused in pBluescript II using the In-Fusion cloning system (Clontech). The abcf-3(+) transgene contained 1.6 kbp of 5′ promoter, the coding region and 0.8 kbp 3′ of the stop codon of abcf-3 in pBluescript II. The QuickChange II XL Site-Directed Mutagenesis Kit (Stratagene) was used to generate transgenes of abcf-3 K217M, abcf-3 K536M and abcf-3 K217M K536M. gcn-1 cDNA corresponding to amino acids 1–2634, 1–1760, 1–880, 880–2634, 880–1760 or 1760–2634 of GCN-1 was cloned in pGBKT7. abcf-3 cDNA corresponding to amino acids 1–712, 1–512, 1–202, 202–712, 512–712 or 202–512 was cloned in pGADT7. The Pgcn-1::gfp transgene contained 4.2 kbp of 5′ promoter of gcn-1 in pPD122.56. The Pabcf-3::gfp transgene contained 1.6 kbp of 5′ promoter of abcf-3 in pPD122.56. The Pceh-34::eIF2α S49A transgene contained 3.8 kbp of 5′ promoter of ceh-34 and eIF2α with a replacement of serine 49 with alanine in pPD49.26. For the Pmec-7::gcn-1 cDNA and Pmec-7::abcf-3 cDNA transgenes, full-length cDNA of gcn-1 and abcf-3 were cloned in pPD96.41. Primer sequences used are available from the authors.
Germline transformation
Germline transformation was performed as described [53]. The gfp reporter transgene was injected at 50 µg/ml into lin-15(n765ts) animals with 50 µg/ml of pL15EK as a coinjection marker [54]. To rescue the defect in M4 sister cell death, the transgenes pTH gcn-1 cDNA, abcf-3(+), abcf-3 K217M, abcf-3 K536M and abcf-3 K217M K536M described above were injected at 20 µg/ml into gcn-1(n4827) or abcf-3(n4927) animals with 50 µg/ml of Plin-44::gfp as a coinjection marker [55]. To establish transgenic lines carrying the Pceh-34::eIF2α S49A transgene, the Pceh-34::eIF2α S49A transgene was injected at 50 µg/ml into nIs175 animals with 50 µg/ml of Plin-44::gfp as a coinjection marker. The Pmec-7::gcn-1 cDNA and Pmec-7::abcf-3 cDNA transgenes were injected at 50 µg/ml, respectively, into zdIs5 animals with 50 µg/ml of pRF4[rol-6(su1006)] and 20 µg/ml of the Pmec-3::mCherry transgene as coinjection markers.
Yeast two-hybrid binding assay
GAL4 fusion constructs were introduced into yeast strain PJ649A as described [56]. Single colonies were streaked and cultured for two days at 30°C on SD plates containing minimal supplements without tryptophan and leucine. Then yeast strains were streaked and cultured for three days at 30°C on SD plates containing minimal supplements without tryptophan, leucine and histidine to test yeast growth.
Antibody production
Protein fragments corresponding to amino acids 753–857 of GCN-1 and 74–185 of ABCF-3 fused to glutathione S - transferase (GST) were expressed, purified using glutathione Sepharose 4B (Amersham Biosciences) and used to raise rabbit anti-GCN-1 or anti-ABCF-3 antibodies, respectively. Antisera were generated by Pocono Rabbit Farm and Laboratory. Specific antibodies were affinity-purified using identical GCN-1 or ABCF-3 protein fragments fused to maltose-binding protein (MBP) and coupled to Affigel 10 (Bio-Rad).
Western blots and immunoprecipitation analysis
Protein extracts were prepared from nIs175, gcn-1(n4827); nIs175 and abcf-3(n4927); nIs175 animals synchronized at the fourth larval stage as described [57]. 10 µg of total protein was loaded onto a 7.5% SDS PAGE gel and then transferred to nitrocellulose membranes. The membranes were probed with anti-GCN-1 or anti-ABCF-3 antibody. Immunocomplexes were detected using HRP-conjugated anti-rabbit IgG secondary antibodies (Invitrogen) followed by chemiluminescence (Western Lightning ECL, PerkinElmer). To determine the level of phosphorylated eIF2α, protein extracts were prepared from nIs175, gcn-1(n4827); nIs175 and abcf-3(n4927); nIs175 animals synchronized at the fourth larval stage as described [57]. 15 µg of total protein was loaded on a 10% SDS PAGE gel and then transferred to nitrocellulose membranes. The membranes were probed with anti-phospho-eIF2α (Cell Signaling Technology) and anti-eIF2α antibodies [28]. Immunocomplexes were detected as described above.
For immunoprecipitation experiments, protein extracts were prepared from mixed-staged wild-type animals in TNE buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, 1% NP-40, 5 mM β-mercaptoethanol, 10% glycerol. Protein extracts were mixed with either an affinity-purified anti-ABCF-3 antibody or a control IgG at 4°C for 2 hours. Immunocomplexes were recovered using Protein A Sepharose 4 Fast Flow (GE Healthcare Life Sciences) and washed with TNE buffer four times. The recovered immunocomplexes were subjected to western blot analysis using anti-GCN-1 or anti-ABCF-3 antibody.
Fluorescence in situ hybridization
Fluorescence in situ hybridization was performed as described [58]. The gcn-1 and abcf-3 probes (Biosearch Technologies, Inc) were conjugated to the fluorophore Cy5 using the Amersham Cy5 Mono-reactive Dye pack (GE Healthcare). DNA was visualized using 4′,6-diamidino-2-phenylindole (DAPI). The probe sequences used are shown in Tables S3 and S4. Figures 4D and H are maximum intensity projections of a Z-stack of images processed with the FFT Bandpass Filter operations in the image processing program Fiji. Oligonucleotides used for gcn-1 and abcf-3 FISH probe were described in Table S6 and S7).
mRNA-seq and ribosome profiling (Ribo-seq)
For mRNA-seq [59], total RNA was purified using an RNAeasy Mini kit (Qiagen) from synchronized L4 animals of wild-type animals and gcn-1 and abcf-3 mutants. The purified RNA was subjected to oligo (dT) selection, fragmentation and first - and double-strand synthesis with an Illumina Tru-Seq kit according to the manufacturer's instructions. DNA fragments longer than 30 bp were purified using SPRI-TE beads (Beckmann Coulter) according to the manufacturer's instructions. The purified DNA was end-repaired and single A bases were added for adaptor ligations. The adaptor-ligated DNA was then subjected to double SPRI-TE purification to select for 200 bp fragments. These fragments were enriched and barcoded by PCR for multiplexing. A final SPRI-TE purification was performed to purify the barcoded RNA-Seq libraries for Illumina DNA sequencing using HiSeq 2000. RNA-seq data were aligned against the C. elegans reference genome (ce10) using the Burrows-Wheeler Aligner (BWA) and Tophat.
Ribosome profiling was performed as described [60] with modifications. Synchronized L4 wild type animals and gcn-1 and abcf-3 mutants were collected and washed with M9 buffer three times. Animals were homogenized using a dounce homogenizer in lysis buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 5 mM MgSO4, 1 mM DTT, 100 µg/ml cycloheximide, 1% Triton X-100 and 25 U/ml Turbo DNase (Invitrogen) and centrifuged at 20,000 g for 20 min at 4°C. The absorbance of the extract was measured at 260 nm. 40 absorbance units of extract were incubated with 300 units of RNase I at 25°C for an hour, and then 200 units of SUPERase In RNase Inhibitor (Invitrogen) were added. Digested extracts were loaded on 10–50% linear sucrose gradients containing 20 mM Tris (pH 7.5), 150 mM NaCl, 5 mM MgSO4, 1 mM DTT and 100 µg/ml cycloheximide and centrifuged for three hours at 35, 000 rpm at 4°C using a SW-40 rotor to isolate a monosome fraction. RNA from the monosome fraction was purified by phenol-chloroform extraction followed by miRNeasy Mini Kit (Qiagen) and separated using a 15% TBE-Urea gel (BioRad) to isolate ribosome-protected fragments (RPFs). The RPFs were eluted from gels by incubating in RNA elution buffer containing 300 mM sodium acetate (pH 5.5), 1 mM EDTA and 0.25% SDS. RPFs were 3′ dephosphorylated with T4 polynucleotide kinase (New England Labs) and ligated to Universal miRNA Cloning Linker (New England Labs) using T4 RNA ligase 2, truncated (New England Labs) according to the manufacturer's instructions. RPFs ligated with the linker were separated from an unligated linker using a 15% TBE-Urea gel (BioRad) and eluted from gels using RNA extraction buffer followed by phenol-chloroform extraction. RFPs were reverse-transcribed by Superscript III (Invitrogen) with a reverse transcription primer according to the manufacturer's instructions. The products of reverse transcripts (RT) were purified using a 15% TBE-Urea gel (BioRad) and eluted from a gel by incubating in DNA elution buffer containing 300 mM NaCl, 10 mM Tris (pH 8.0) and 1 mM EDTA followed by phenol-chloroform extraction. The RT products were circularized by CircLigase (Epicentre) according to the manufacturer's instructions. About a quarter of the RT products were used in PCR reactions containing 1× Phusion HF buffer, 0.2 mM dNTP, 0.5 µm forward library primer, 0.5 µm reverse indexed primer and 0.02 units/µl Phusion polymerase (New England Labs), and PCR was performed with a 30 second initial denaturation at 98°C, followed by 6, 8, 10, 12 and 14 cycles of 98°C for 10 second, 65°C for 10 second and 72°C for 5 second. PCR products were separated using a 8% TBE gel (BioRad) and eluted from gels by incubating in DNA elution buffer followed by phenol-chloroform extraction. PCR products were suspended in 20 µl of 10 mM Tris (pH 8.0) and sequenced by HiSeq 2000. The adaptor sequences (CTGTAGGCACCATC) from 3′ end of the ribosome footprint reads were removed, then trimmed reads were mapped using BWA to distinguish the reads from ribosomal RNAs. About 60% of the reads were filtered out, and the remaining reads (non-ribosomal) were aligned to the C. elegans reference genome (ce10) using BWA and Tophat. Because translational initiation is thought to be blocked rapidly by the stress animals encounter during harvesting, many ribosomes are stalled at the beginning of each transcript in the presence of cycloheximide, which prevents translation elongation [34]. Hence, high frequencies of reads at the beginning of each transcript might not correspond to high rates of translation. For this reason, the reads that mapped to the first 25 nucleotides of each transcript were not counted in evaluating gene expression in the Ribo-seq analyses.
Supporting Information
Zdroje
1. FuchsY, StellerH (2011) Programmed cell death in animal development and disease. Cell 147 : 742–758 doi:10.1016/j.cell.2011.10.033
2. ConradtB (2009) Genetic control of programmed cell death during animal development. Annu Rev Genet 43 : 493–523 doi:10.1146/annurev.genet.42.110807.091533
3. HymanBT, YuanJ (2012) Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat Rev Neurosci 13 : 395–406 doi:10.1038/nrn3228
4. HipfnerDR, CohenSM (2004) Connecting proliferation and apoptosis in development and disease. Nat Rev Mol Cell Biol 5 : 805–815 doi:10.1038/nrm1491
5. MiyashitaT, ReedJC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80 : 293–299.
6. OdaE, OhkiR, MurasawaH, NemotoJ, ShibueT, et al. (2000) Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288 : 1053–1058.
7. SaxJK, FeiP, MurphyME, BernhardE, KorsmeyerSJ, et al. (2002) BID regulation by p53 contributes to chemosensitivity. Nat Cell Biol 4 : 842–849 doi:10.1038/ncb866
8. MoroniMC, HickmanES, Lazzerini DenchiE, CapraraG, ColliE, et al. (2001) Apaf-1 is a transcriptional target for E2F and p53. Nat Cell Biol 3 : 552–558 doi:10.1038/35078527
9. WuGS, BurnsTF, McDonaldER3rd, JiangW, MengR, et al. (1997) KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet 17 : 141–143 doi:10.1038/ng1097-141
10. NakanoK, VousdenKH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7 : 683–694.
11. SchulerM, GreenDR (2005) Transcription, apoptosis and p53: catch-22. Trends Genet 21 : 182–187 doi:10.1016/j.tig.2005.01.001
12. NehmeR, ConradtB (2008) egl-1: a key activator of apoptotic cell death in C. elegans. Oncogene 27 Suppl 1: S30–40 doi:10.1038/onc.2009.41
13. WinnJ, CarterM, AveryL, CameronS (2011) Hox and a newly identified E2F co-repress cell death in Caenorhabditis elegans. Genetics 188 : 897–905 doi:10.1534/genetics.111.128421
14. PottsMB, WangDP, CameronS (2009) Trithorax, Hox, and TALE-class homeodomain proteins ensure cell survival through repression of the BH3-only gene egl-1. Dev Biol 329 : 374–385 doi:10.1016/j.ydbio.2009.02.022
15. HiroseT, GalvinBD, HorvitzHR (2010) Six and Eya promote apoptosis through direct transcriptional activation of the proapoptotic BH3-only gene egl-1 in Caenorhabditis elegans. Proc Natl Acad Sci USA 107 : 15479–15484 doi:10.1073/pnas.1010023107
16. MaurerCW, ChiorazziM, ShahamS (2007) Timing of the onset of a developmental cell death is controlled by transcriptional induction of the C. elegans ced-3 caspase-encoding gene. Development 134 : 1357–1368 doi:10.1242/dev.02818
17. HiroseT, HorvitzHR (2013) An Sp1 transcription factor coordinates caspase-dependent and -independent apoptotic pathways. Nature 500 : 354–358 doi:10.1038/nature12329
18. HolcikM, SonenbergN (2005) Translational control in stress and apoptosis. Nat Rev Mol Cell Biol 6 : 318–327 doi:10.1038/nrm1618
19. NevinsTA, HarderZM, KornelukRG, HolcíkM (2003) Distinct regulation of internal ribosome entry site-mediated translation following cellular stress is mediated by apoptotic fragments of eIF4G translation initiation factor family members eIF4GI and p97/DAP5/NAT1. J Biol Chem 278 : 3572–3579 doi:10.1074/jbc.M206781200
20. HolcikM, LefebvreC, YehC, ChowT, KornelukRG (1999) A new internal-ribosome-entry-site motif potentiates XIAP-mediated cytoprotection. Nat Cell Biol 1 : 190–192 doi:10.1038/11109
21. SchumacherB, HanazawaM, LeeM-H, NayakS, VolkmannK, et al. (2005) Translational repression of C. elegans p53 by GLD-1 regulates DNA damage-induced apoptosis. Cell 120 : 357–368 doi:10.1016/j.cell.2004.12.009
22. ContrerasV, FridayAJ, MorrisonJK, HaoE, KeiperBD (2011) Cap-independent translation promotes C. elegans germ cell apoptosis through Apaf-1/CED-4 in a caspase-dependent mechanism. PLoS ONE 6: e24444 doi:10.1371/journal.pone.0024444
23. ContrerasV, RichardsonMA, HaoE, KeiperBD (2008) Depletion of the cap-associated isoform of translation factor eIF4G induces germline apoptosis in C. elegans. Cell Death Differ 15 : 1232–1242 doi:10.1038/cdd.2008.46
24. HuangC-Y, ChenJ-Y, WuS-C, TanC-H, TzengR-Y, et al. (2012) C. elegans EIF-3.K promotes programmed cell death through CED-3 caspase. PLoS ONE 7: e36584 doi:10.1371/journal.pone.0036584
25. AveryL, HorvitzHR (1987) A cell that dies during wild-type C. elegans development can function as a neuron in a ced-3 mutant. Cell 51 : 1071–1078.
26. SulstonJE, SchierenbergE, WhiteJG, ThomsonJN (1983) The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol 100 : 64–119.
27. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282 : 2012–2018.
28. NukazukaA, FujisawaH, InadaT, OdaY, TakagiS (2008) Semaphorin controls epidermal morphogenesis by stimulating mRNA translation via eIF2alpha in Caenorhabditis elegans. Genes Dev 22 : 1025–1036 doi:10.1101/gad.1644008
29. Vazquez de AldanaCR, MartonMJ, HinnebuschAG (1995) GCN20, a novel ATP binding cassette protein, and GCN1 reside in a complex that mediates activation of the eIF-2 alpha kinase GCN2 in amino acid-starved cells. EMBO J 14 : 3184–3199.
30. MartonMJ, CrouchD, HinnebuschAG (1993) GCN1, a translational activator of GCN4 in Saccharomyces cerevisiae, is required for phosphorylation of eukaryotic translation initiation factor 2 by protein kinase GCN2. Mol Cell Biol 13 : 3541–3556.
31. HansonPI, WhiteheartSW (2005) AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol 6 : 519–529 doi:10.1038/nrm1684
32. MartonMJ, Vazquez de AldanaCR, QiuH, ChakraburttyK, HinnebuschAG (1997) Evidence that GCN1 and GCN20, translational regulators of GCN4, function on elongating ribosomes in activation of eIF2alpha kinase GCN2. Mol Cell Biol 17 : 4474–4489.
33. DavisonEM, SafferAM, HuangLS, DeModenaJ, SternbergPW, et al. (2011) The LIN-15A and LIN-56 transcriptional regulators interact to negatively regulate EGF/Ras signaling in Caenorhabditis elegans vulval cell-fate determination. Genetics 187 : 803–815 doi:10.1534/genetics.110.124487
34. IngoliaNT, GhaemmaghamiS, NewmanJRS, WeissmanJS (2009) Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324 : 218–223 doi:10.1126/science.1168978
35. RajA, RifkinSA, AndersenE, Van OudenaardenA (2010) Variability in gene expression underlies incomplete penetrance. Nature 463 : 913–918 doi:10.1038/nature08781
36. ReddienPW, CameronS, HorvitzHR (2001) Phagocytosis promotes programmed cell death in C. elegans. Nature 412 : 198–202 doi:10.1038/35084096
37. GumiennyTL, LambieE, HartwiegE, HorvitzHR, HengartnerMO (1999) Genetic control of programmed cell death in the Caenorhabditis elegans hermaphrodite germline. Development 126 : 1011–1022.
38. LettreG, KritikouEA, JaeggiM, CalixtoA, FraserAG, et al. (2004) Genome-wide RNAi identifies p53-dependent and -independent regulators of germ cell apoptosis in C. elegans. Cell Death Differ 11 : 1198–1203 doi:10.1038/sj.cdd.4401488
39. GartnerA, MilsteinS, AhmedS, HodgkinJ, HengartnerMO (2000) A conserved checkpoint pathway mediates DNA damage–induced apoptosis and cell cycle arrest in C. elegans. Mol Cell 5 : 435–443.
40. MetzsteinMM, StanfieldGM, HorvitzHR (1998) Genetics of programmed cell death in C. elegans: past, present and future. Trends Genet 14 : 410–416.
41. HengartnerMO, EllisRE, HorvitzHR (1992) Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature 356 : 494–499 doi:10.1038/356494a0
42. HengartnerMO, HorvitzHR (1994) Activation of C. elegans cell death protein CED-9 by an amino-acid substitution in a domain conserved in Bcl-2. Nature 369 : 318–320 doi:10.1038/369318a0
43. EberhardR, StergiouL, HofmannER, HofmannJ, HaenniS, et al. (2013) Ribosome synthesis and MAPK activity modulate ionizing radiation-induced germ cell apoptosis in Caenorhabditis elegans. PLoS Genet 9: e1003943 doi:10.1371/journal.pgen.1003943
44. SchumacherB, HofmannK, BoultonS, GartnerA (2001) The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr Biol 11 : 1722–1727.
45. HofmannER, MilsteinS, BoultonSJ, YeM, HofmannJJ, et al. (2002) Caenorhabditis elegans HUS-1 is a DNA damage checkpoint protein required for genome stability and EGL-1-mediated apoptosis. Curr Biol 12 : 1908–1918.
46. MillerPF, HinnebuschAG (1990) cis-acting sequences involved in the translational control of GCN4 expression. Biochim Biophys Acta 1050 : 151–154.
47. RousakisA, VlassisA, VlantiA, PateraS, ThireosG, et al. (2013) The general control nonderepressible-2 kinase mediates stress response and longevity induced by target of rapamycin inactivation in Caenorhabditis elegans. Aging Cell 12 : 742–51 doi:10.1111/acel.12101
48. BrennerS (1974) The genetics of Caenorhabditis elegans. Genetics 77 : 71–94.
49. WicksSR, YehRT, GishWR, WaterstonRH, PlasterkRH (2001) Rapid gene mapping in Caenorhabditis elegans using a high density polymorphism map. Nat Genet 28 : 160–164 doi:10.1038/88878
50. TroemelER, ChouJH, DwyerND, ColbertHA, BargmannCI (1995) Divergent seven transmembrane receptors are candidate chemosensory receptors in C. elegans. Cell 83 : 207–218.
51. ThellmannM, HatzoldJ, ConradtB (2003) The Snail-like CES-1 protein of C. elegans can block the expression of the BH3-only cell-death activator gene egl-1 by antagonizing the function of bHLH proteins. Development 130 : 4057–4071.
52. SmitRB, SchnabelR, GaudetJ (2008) The HLH-6 transcription factor regulates C. elegans pharyngeal gland development and function. PLoS Genet 4: e1000222 doi:10.1371/journal.pgen.1000222
53. MelloCC, KramerJM, StinchcombD, AmbrosV (1991) Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J 10 : 3959–3970.
54. ClarkSG, LuX, HorvitzHR (1994) The Caenorhabditis elegans locus lin-15, a negative regulator of a tyrosine kinase signaling pathway, encodes two different proteins. Genetics 137 : 987–997.
55. HermanMA, VassilievaLL, HorvitzHR, ShawJE, HermanRK (1995) The C. elegans gene lin-44, which controls the polarity of certain asymmetric cell divisions, encodes a Wnt protein and acts cell nonautonomously. Cell 83 : 101–110.
56. KnopM, SiegersK, PereiraG, ZachariaeW, WinsorB, et al. (1999) Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast 15 : 963–972 doi:;10.1002/(SICI)1097-0061(199907)15 : 10B<963::AID-YEA399>3.0.CO;2-W
57. YoungmanMJ, RogersZN, KimDH (2011) A decline in p38 MAPK signaling underlies immunosenescence in Caenorhabditis elegans. PLoS Genet 7: e1002082 doi:10.1371/journal.pgen.1002082
58. RajA, Van den BogaardP, RifkinSA, Van OudenaardenA, TyagiS (2008) Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5 : 877–879 doi:10.1038/nmeth.1253
59. SubramanianV, MazumderA, SurfaceLE, ButtyVL, FieldsPA, et al. (2013) H2A.Z acidic patch couples chromatin dynamics to regulation of gene expression programs during ESC differentiation. PLoS Genet 9: e1003725 doi:10.1371/journal.pgen.1003725
60. IngoliaNT, BrarGA, RouskinS, McGeachyAM, WeissmanJS (2012) The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc 7 : 1534–1550 doi:10.1038/nprot.2012.086
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 8
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Meta-Analysis of Genome-Wide Association Studies in African Americans Provides Insights into the Genetic Architecture of Type 2 Diabetes
- KDM6 Demethylase Independent Loss of Histone H3 Lysine 27 Trimethylation during Early Embryonic Development
- The RNA Helicases AtMTR4 and HEN2 Target Specific Subsets of Nuclear Transcripts for Degradation by the Nuclear Exosome in
- EF-P Dependent Pauses Integrate Proximal and Distal Signals during Translation
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy