
Human Developmental Enhancers Conserved between Deuterostomes and Protostomes
The identification of homologies, whether morphological, molecular, or genetic, is fundamental to our understanding of common biological principles. Homologies bridging the great divide between deuterostomes and protostomes have served as the basis for current models of animal evolution and development. It is now appreciated that these two clades share a common developmental toolkit consisting of conserved transcription factors and signaling pathways. These patterning genes sometimes show common expression patterns and genetic interactions, suggesting the existence of similar or even conserved regulatory apparatus. However, previous studies have found no regulatory sequence conserved between deuterostomes and protostomes. Here we describe the first such enhancers, which we call bilaterian conserved regulatory elements (Bicores). Bicores show conservation of sequence and gene synteny. Sequence conservation of Bicores reflects conserved patterns of transcription factor binding sites. We predict that Bicores act as response elements to signaling pathways, and we show that Bicores are developmental enhancers that drive expression of transcriptional repressors in the vertebrate central nervous system. Although the small number of identified Bicores suggests extensive rewiring of cis-regulation between the protostome and deuterostome clades, additional Bicores may be revealed as our understanding of cis-regulatory logic and sample of bilaterian genomes continue to grow.
Published in the journal:
. PLoS Genet 8(8): e32767. doi:10.1371/journal.pgen.1002852
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002852
Summary
The identification of homologies, whether morphological, molecular, or genetic, is fundamental to our understanding of common biological principles. Homologies bridging the great divide between deuterostomes and protostomes have served as the basis for current models of animal evolution and development. It is now appreciated that these two clades share a common developmental toolkit consisting of conserved transcription factors and signaling pathways. These patterning genes sometimes show common expression patterns and genetic interactions, suggesting the existence of similar or even conserved regulatory apparatus. However, previous studies have found no regulatory sequence conserved between deuterostomes and protostomes. Here we describe the first such enhancers, which we call bilaterian conserved regulatory elements (Bicores). Bicores show conservation of sequence and gene synteny. Sequence conservation of Bicores reflects conserved patterns of transcription factor binding sites. We predict that Bicores act as response elements to signaling pathways, and we show that Bicores are developmental enhancers that drive expression of transcriptional repressors in the vertebrate central nervous system. Although the small number of identified Bicores suggests extensive rewiring of cis-regulation between the protostome and deuterostome clades, additional Bicores may be revealed as our understanding of cis-regulatory logic and sample of bilaterian genomes continue to grow.
Introduction
The bilaterian tree unites two major clades, deuterostomes (e.g. humans) and protostomes (e.g. flies) [1]. Protostome species such as insects, nematodes, annelids, and mollusks have served as invaluable model organisms. Much of the utility of these model systems stems from fundamental homologies between the two clades. Across bilaterians, early embryos undergo gastrulation to form three germ layers. These germ layers are patterned along dorsal-ventral and anterior-posterior axes. Underlying these processes are ancient conserved signaling pathways and transcription factors, often interacting as part of conserved genetic circuits. In both deuterostomes and protostomes, the precise expression of each circuit component depends on cis-regulatory elements [2], [3]. Cis-regulatory elements are genomic regions that transcription factors bind in order to modify the expression of a target gene [4].
Cis-regulatory elements are often identified as conserved non-coding elements (CNEs) [5]–[8]. Among closely related species, CNEs can show extreme conservation. For example, the human genome contains hundreds of non-coding ultraconserved elements that align to mouse and rat with 100 percent identity across 200 bases or more [5]. Many of these elements function as developmental enhancers [7]. Protostome genomes contain a distinct set of similarly ultraconserved elements [9]. Strikingly, in contrast to the genes they regulate, no cis-regulatory elements have previously been found to be conserved between deuterostomes and protostomes [5], [6], [10]–[12] (see Text S1). Even the oldest known enhancer, conserved between deuterostomes and the cnidarian sea anemone, has not been found to be conserved in protostomes [12]. These observations may suggest that the cis-regulatory component of genetic circuits has been completely rewired between deuterostomes and protostomes. Alternatively, it may be that some ancestral regulatory regions are conserved between these clades and have remained elusive due to limitations in our tools and our sample of bilaterian genomes. If conserved cis-regulatory elements do exist, such elements offer a new avenue for exploring how developmental logic is encoded in the genome and how this logic evolves.
Here we present the first examples of cis-regulatory elements conserved between deuterostomes and protostomes. These elements have conserved sequence and gene synteny. The conserved sequence reflects conservation of a series of transcription factor binding sites, and we show that these elements function as developmental enhancers.
Results/Discussion
Discovery of bilaterian conserved regulatory elements
Conservation of non-coding sequence is rare compared to coding sequence, even over much shorter evolutionary distances than that between deuterostomes and protostomes. For example, nearly a third of human coding bases (11 Mb out of 34 Mb) align to the amphioxus genome. In stark contrast, less than one percent of CNE bases (<0.3 Mb out of 34 Mb) align to amphioxus (Figure 1).

To screen for enhancers conserved between deuterostome and protostome species, we first defined a set of vertebrate conserved non-coding elements (vertCNEs) which are human non-coding regions well-conserved in at least a subset of vertebrates (see methods). In total, we constructed a conservative set of 8,069 vertCNEs. We were especially stringent in filtering out sequences with evidence for potential coding functions (exonic or other functional RNA). vertCNEs show exceptional enrichment in GREAT [13] for clustering near transcription factors and developmental genes (Table S1). We used lastz [14] to query these elements against three previously published non-vertebrate deuterostome genomes – ciona [15], amphioxus [16], and sea urchin [17] (Figure S1). We filtered hits for both quality of alignment and for conserved gene synteny. We thus found five candidate elements showing conservation between vertebrates and at least one invertebrate (Table S2). In order to comprehensively define the extent of conservation of these elements, we searched each against all publicly available sequence data for non-vertebrate metazoans. This search included genome assemblies for 47 species (Table S3) (∼16 Gb) and NCBI trace data for 134 species (∼219 Gb). We also searched all Genbank sequence data, which included 530 species with at least 100 kilobases of sequence (∼21 Gb). Of the five candidates, two elements stood out as having conservation of sequence and gene synteny in protostome species. We call these elements bilaterian conserved regulatory elements (Bicores).
Bicore1 is found in two protostome species - Aplysia californica (sea hare) and Lottia gigantea (owl limpet). Bicore2 is found in the protostome Ixodes scapularis (tick) (Figure 2). Together, Bicore1 and Bicore2 are the first examples of human CNEs conserved between deuterostome and protostome species.

Bicore1
Each instance of Bicore1 is conserved upstream of a conserved Id (Inhibitor of DNA binding) ortholog (Figure 3C; Table S4). Id genes encode helix-loop-helix proteins that bind bHLH transcription factors, acting as transcriptional repressors. Id factors are known to inhibit cells from terminally differentiating, promoting progenitor states [18]. Mammalian genomes contain four Id genes. Bicore1 occurs upstream of Id1. In addition, we found mammalian paralogs of Bicore1 upstream of Id2 and Id4 (Figure S2; Table S4).
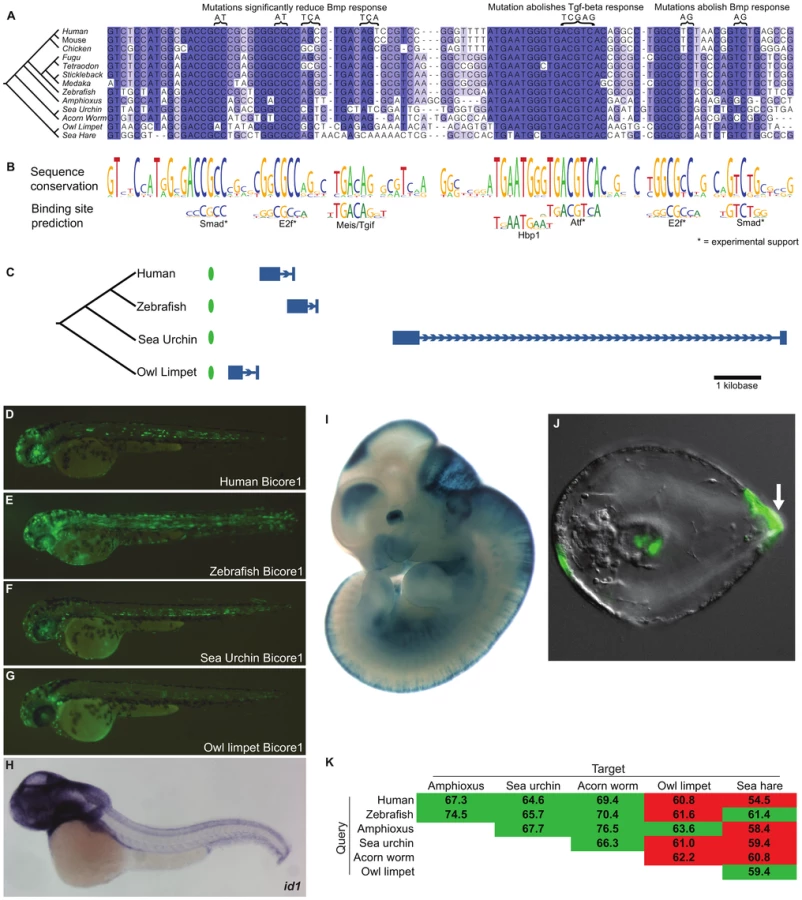
A multiple alignment of Bicore1 shows a striking pattern of sequence conservation (Figure 3A–3B). Short stretches of 5–10 base pairs are highly conserved, separated by stretches of non-conserved sequence. The highly conserved 5–10mers have allowed few substitutions and have completely resisted indels across species. These short conserved sequences match closely to the known binding preference of transcription factors (Figure 3B).
Two conserved sites match the primary and secondary binding preference of Smad transcription factors. Smad factors act downstream of Tgf-beta/Bmp signaling [19], suggesting that Bicore1 may be responsive to this pathway. In fact, previous studies have shown that, in a mammalian cell line, these conserved Smad sites are bound by smad transcription factors in response to Tgf-beta/Bmp signaling. These studies further showed that a region containing Bicore1 drives expression of a luciferase reporter in response to Bmp. Mutation of the Smad sites reduces or abolishes this Bmp response [20], [21]. We also see conserved matches to the E2f binding motif. E2f factors are known to act as Smad cofactors in response to Tgf-beta [22]. Mutations of these conserved E2f sites reduce Bmp responsiveness in a luciferase assay [20], [21]. ChIP-seq data also supports binding of E2f factors to this region [23]. The most highly conserved region of Bicore1 corresponds to an 8 base pair palindromic sequence that is perfectly conserved across clades. This sequence matches the binding preference of Atf factors. In mammalian cells, Atf forms a complex with Smad and directly binds Bicore1 [24]. This binding event occurs in response to Tgf-beta signaling and leads to the repression of a luciferase reporter. Mutation of the Atf site abolishes the ability of Bicore1 to repress luciferase expression in response to Tgf-beta [24]. Altogether, cell line experiments show that Bicore1 is a Tgf-beta/Bmp responsive cis-regulatory element; Smad, E2f, and Atf factors bind the element, and the conserved Smad, E2f, and Atf sites are necessary for its function. Thus, Bicore1 has conserved a series of transcription factor binding sites that have maintained order, orientation, and spacing for over more than 600 million years [25] of evolution.
To test if instances of Bicore1 have a conserved capacity to function as an enhancer in vivo, we used a zebrafish transient transgenic enhancer assay. We tested the human, zebrafish, sea urchin, and owl limpet Bicore1 sequences. These short ∼100 base pair sequences are fairly diverged from one another, with all but the human-zebrafish pairwise similarity at 60–65 percent identity. At 21 hours post-fertilization (hpf), the constructs drove strong expression throughout the embryo. Zebrafish id1 expression at this time point, measured by whole-mount in situ hybridization (WISH), is similar (Figure S3). At 48 hours, we found that all four constructs drove scattered expression throughout the central nervous system (CNS) (Figure 3D–3G and Table S6), congruent with id1 expression at this time [26] (Figure 3H). In addition to CNS, we saw expression in the notochord, a structure in which previous WISH experiments have not detected id1 expression. It is possible that Bicore1 enhances the weak notochord background of our expression vector (see methods; Figure S4).
Further support for Bicore1 functioning as a CNS enhancer in vertebrates is provided by mouse experiments. A human construct containing Bicore1 was previously tested in a mouse transgenic enhancer assay [8]. At embryonic day 11.5, this construct drives expression in the forebrain, midbrain, hindbrain, neural tube, and eye (Figure 3I), closely matching Id1 expression in mouse [27].
Among the protostome species that have conserved Bicore1 (owl limpet and sea hare), transgenic enhancer assays are not yet well developed. However, such assays have been described in sea urchin [28]. We tested a construct containing the sea urchin Bicore1 in a transgenic sea urchin assay. The construct drove expression in the aboral ectoderm of developing sea urchin embryos (75% of embryos) (Figure 3J). Whole-mount in situ hybridization experiments in a closely related sea urchin species have shown that id is expressed specifically in the aboral ectoderm during sea urchin development. Moreover, overexpressing Bmp expands the id expression pattern, and blocking Bmp signaling greatly diminishes id expression [29]. These data provide evidence that Bicore1 functions as a developmental enhancer of id in sea urchins.
Bicore2
Bicore2 is conserved upstream of Znf503 (Figure 4C; Table S5), a gene which encodes a zinc-finger transcription factor predicted to act as a transcriptional repressor in deuterostomes and protostomes [30], [31]. Znf503 functions as a regulator of vertebrate hindbrain development [32], [33]. Transcription factor binding site analysis shows that Bicore2 has conserved several sequences matching the Meis/Tgif family binding preference as well as sequences matching Hox and Pbx binding preferences (Figure 4B). Among vertebrates, these factors are known to form a complex that functions during hindbrain development [34], [35]. These factors are also known to interact in protostome species [36], [37]. Bicore2 also contains highly conserved Tcf/Lef family binding sites that are supported by ChIP-seq data in mammalian cell lines [23]. Tcf/Lef transcription factors act downstream of Wnt signaling. In both deuterostomes and protostomes, Wnt signaling defines the anterior-posterior axis during development [38]. In vertebrates, Wnt signaling has also been shown to be necessary in defining the midbrain-hindbrain boundary [39].

We examined human, zebrafish, sea urchin, and tick versions of Bicore2 in a zebrafish transient transgenic enhancer assay. All four constructs drove consistent expression in the hindbrain (Figure 4D–4G), recapitulating the zebrafish znf503 expression pattern [26] (Figure 4H). Interestingly, the zebrafish Bicore2 stood out as having the weakest expression. Zebrafish also stands out in the multiple alignment as having mutated three highly conserved bases, interrupting a predicted Meis-Pbx-Hox binding site (Figure 4A–4B). Two other sequenced fish, tetraodon and fugu, have deleted a highly conserved site predicted to bind Tcf/lef and Pbx. It is possible that in some fish, Bicore2 function has been modulated.
We also tested a human construct containing Bicore2 in a mouse assay. The construct drives expression in the hindbrain (5/6 embryos) and apical ectoderm of the limb (4/6 embryos) at embryonic day 11.5 (Figure 4I). This pattern of hindbrain and limb expression matches previously reported Znf503 expression in mouse [40].
Perspectives
In this study, we have identified the first examples of cis-regulatory sequence conserved between deuterostomes and protostomes. These bilaterian conserved regulatory elements (Bicores) are developmental enhancers that encode complex patterns of transcription factor binding sites. Bicore1 is an enhancer of Id. Binding site analysis predicts that it functions as a Bmp responsive element, and several lines of experimental evidence support this prediction. In vertebrates, Bicore1 drives expression in the developing central nervous system. In sea urchin, Bicore1 drives expression in the aboral ectoderm, a structure that goes on to form the squamous epithelium of the late larval wall [41]. Although the vertebrate nervous system and the urchin aboral ectoderm are unlikely to be homologous structures, it is reasonable to hypothesize that they might utilize similar genetic circuits. Both are ectodermal structures that define analogous axes (dorsal-ventral and aboral-oral) through Bmp signaling.
Further work will be needed to determine how Bicore1 functions in those protostome species that have conserved the sequence. We suspect that protostomes like owl limpet and sea hare also use Bicore1 as a Bmp responsive enhancer to drive id expression in ectodermal structures. As enhancer assays are developed in these species, we can begin testing this hypothesis. It is interesting to note that in Drosophila melanogaster, Emc (ortholog to Id) is not expressed in the ectoderm during development [42]. Further, constitutively active Dpp (ortholog to Bmp) signaling does not alter Emc expression [43]. Loss of Bicore1 in drosophila is consistent with these observations.
Bicore2 is an enhancer of Znf503. We predict from binding site analysis that it acts as a response element to Wnt signaling. In vertebrate embryos, Bicore2 drives expression in the hindbrain, an ectoderm derived structure. It may seem surprising that CNS enhancers are conserved in species that lack central nervous systems. In fact, we can infer that Bicores existed in the urbilaterian ancestor, long before the process of neurulation and the vertebrate central nervous system ever emerged. It is well established that as the vertebrate nervous system evolved, it took advantage of preexisting transcription factors and signaling pathways [44], [45]. We can now appreciate that in addition to these ancient genes, ancient cis-regulatory integrators, in the form of rigid enhancers, were also coopted into vertebrate nervous system development. In fact, it is expected that when key regulatory genes and pathways are activated in a new context, they initially affect downstream targets via pre-existing cis-regulatory regions.
Several past studies have searched for cis-regulatory elements conserved between deuterostomes and protostomes (see Text S1). However, these studies focused on searching the genomes of the most commonly used protostome model organisms, drosophila and caenorhabditis. These model organisms correspond to two of the three lineages (tunicates, insects, and nematode) in which we could detect no Bicore homologies. Our search of these lineages was extensive and included genome assemblies for 3 species of tunicate (∼0.4 Gb) and trace data for 4 species (∼7.4 Gb), genome assemblies for 21 species of insect (∼6.0 Gb) and trace data for 45 species (∼81.2 Gb), and genome assemblies for 11 species of nematode (∼1.4 Gb) and trace data for 21 species (∼15.7 Gb). Thus, it is possible that Bicores have been lost in the tunicate, insect, and nematode lineages. Corroborating this possibility, genomics studies have shown that these three lineages are perhaps the most diverged among bilaterians [46]–[48].
Even within the protostome genomes that have clear conservation of Bicores, these homologies lie at the cusp of what current computational tools can detect. For example, using human Bicore1 as a query, we can detect Bicore1 deuterostome orthologs in amphioxus, sea urchin, and acorn worm. However, we miss the critical protostome owl limpet and sea hare elements. Using zebrafish Bicore1 as the query, we detect the owl limpet element but miss the sea hare. Using amphioxus Bicore1, we detect the sea hare element but miss the owl limpet (Figure 3K). We see similar results for Bicore2 (Figure 4J). When one considers the alignments of Figure 3A and Figure 4A, it is easy to imagine how variations in spacing between co-linearly conserved binding sites may drop the overall sequence conservation below 60% identity and below the detection ability of our current tools.
While the full-length Bicores are not identical (or ultraconserved) even between human and rodents, the binding sites they encode have resisted substitutions, insertions, deletions, and rearrangements for over 600 million years in highly diverged deuterostome and protostome species. At least two fundamental questions are raised by these observations: First, have Bicores conserved their ancestral sequence while being independently co-opted in different lineages to serve unrelated contexts, or do these conserved sequences also conserve a common ancestral function (e.g. in early lineage differentiation) yet to be revealed? The second closely related question is what makes Bicores unique? The small number of identified Bicores implies extensive cis-regulatory rewiring. If the Bicores are indeed the only examples of cis-regulatory elements conserved between deuterostomes and protostomes, we are left asking what makes these enhancers different from others.
It is, however, currently hard to know what the true number of Bicores is. The initial discovery of the conservation of hox genes across bilateria ignited the field of “evo-devo” [49]. Now, scores of other bilaterian conserved developmental genes have been characterized. Over a decade later, the discovery of let-7 revealed the first example of a bilaterian conserved micro-RNA [50]. It is now appreciated that ∼30 other bilaterian conserved miRNAs exist [51]. Here we have presented the first examples of developmental enhancers conserved between deuterostomes and protostomes. We predict that as more genomes are sequenced, our understanding of cis-regulatory logic improves, and our screening is refined, more bilaterian conserved regulatory elements will be discovered. As our catalog of such genomic regions and our ability to experimentally probe these distant relatives grow, so will our understanding of the true extent of cis-regulatory sequence and function conservation underlying animal development.
Materials and Methods
Coding versus non-coding aligning bases
The base set of “coding exons” for Figure 1 was defined as the union of all coding bases in the Human (hg18) UCSC Genes track [52].
The base set of “placental mammal conserved non-coding elements (CNEs)” for Figure 1 was defined using the UCSC PhastCons placental mammal most conserved track [53]. From this set, we strictly removed regions that show any evidence for coding potential. To do so, we excluded any region annotated as an exon by UCSC knownGene [52], refSeq [54], or Ensembl [55]. We also excluded any region predicted to be exonic by Exoniphy [56]. Further, we excluded any region that aligns to a vertebrate or invertebrate mRNA from Genbank [57], an mRNA from the Mammalian Gene Collection [58], or a human spliced EST from Genbank [57]. Potential functional non-coding RNAs were removed by eliminating all non-coding genes annotated by Ensembl and UCSC [52], [55]. We excluded all miRNAs and snoRNAs found in miRBase and snoRNABase [59], [60]. Next, we removed all pseudogenes based on annotations from the Yale Pseudogene Database [61] and the Vertebrate Genome Annotation (VEGA) database [62]. For each potentially coding region we removed with any of the above filters, we also removed the 150 bases upstream and the 150 bases downstream. We added this stringency because the regions immediately flanking exons are often conserved, and we wish to focus on conservation that cannot be accounted for by coding or splicing related functions. Lastly, we required each CNE to be at least 50 base pairs in length and to align syntenically in mouse (mm9).
For each species comparison of either coding or non-coding bases, we counted aligning bases using the UCSC whole-genome alignments between hg18 and the following assemblies: mm9, monDom4, galGal3, xenTro1, danRer5, petMar1, braFlo1. This resulted in 34,116,513 base pairs of (1.18% of the human genome) “coding exons” conserved to mouse, and a conservative (non-overlapping) 34,124,419 base pairs (1.18% as well) of “placental mammal CNEs” conserved to mouse in Figure 1.
Vertebrate CNEs
We used the UCSC phastCons placental mammal most conserved and vertebrate most conserved tracks to find regions of the human genome (hg18) that are the most conserved in comparisons of 32 placental mammals as well as across 44 vertebrates [53]. PhastCons elements combine the level of base pair conservation with depth of species conservation, and are not necessarily found in all genomes in either alignment. We then used the UCSC nets (a subset of the full alignments between any pair of species most likely to be orthologous) to require each element to align to at least two non-amniote vertebrates (xenTro2, tetNig1, fr2, oryLat2, gasAcu1, danRer5, petMar1), as well as to mouse (mm9).
We applied the same stringent non-coding filters used to generate our placental mammal CNE set (see above). As an added stringency, we also compared each region to the full RefSeq database using blastx and removed any element that hit a validated protein with any e-value. Lastly, we looked for overlap between our elements and regions of the genome that have predicted conserved RNA secondary structure [63]. Elements with ≥60% of bases overlapping such regions were removed. We required each element to be at least 50 base pairs, and in total we generated a conservative set of 8,069 vertebrate CNEs, covering 1.7 Mb (0.06%) of the human genome.
Screen for bilaterian conserved regulatory elements
To identify elements most likely to be conserved across bilaterians, we searched for elements with the strongest signatures of conservation within deuterostomes. We compared all 8,069 of our vertebrate CNEs to the published Ciona (ci2) [15], Amphioxus (braFlo1) [16], and Sea urchin (strPur2) [17] genomes. First, we soft masked low complexity sequences. We then used lastz [14] to search each element against each genome. We ran lastz using very low thresholds (hspthresh = 1500, gappedthresh = 2500) [64]. We find these to increase sensitivity but also result in many dubious alignments. We thus further filtered the lastz hits, keeping only alignments with at least 65% identity and an entropy score of 1.7 or greater. The entropy of the alignment is calculated as -∑fblog(fb) for all bases b such that fb>0, where fb is the fraction of aligning bases of base b.
For each element with at least one hit passing these filters, we manually inspected the alignment and the surrounding genomic landscape. As a final filter, we only kept elements that have maintained synteny with the same target gene in vertebrates and the aligning invertebrates. We associated each vertebrate CNE with the two nearest genes in the human genome (hg18). For any hit to an invertebrate genome, we found the nearest annotated mRNAs and compared these to the database of validated Refseq proteins using blastx [65]. If the top hit for this search was an ortholog of the appropriate human target gene, then we called the hit syntenic (Table S4 and S5). Five vertebrate CNEs had at least one such syntenic hit (Table S2).
To more fully characterize the evolution of these five elements across the metazoan tree of life, we then performed a second more comprehensive and more sensitive search. For each of the five CNEs, we extracted all vertebrate instances of the element using the UCSC 44-way multiple alignment [66] on the hg18 genome browser. As a query to our search, we used all vertebrate instances of each element as well as the previously discovered invertebrate instances. Each query was searched against all available non-vertebrate metazoan sequence data (Figure S1).
Each new hit found with this comprehensive search was then used as a query to repeat the search process until no new hits were found. We manually inspected all hits, checking for the quality of the alignment and for gene synteny. For hits to genomes without an annotated set of mRNAs, we checked for synteny using nearby spliced ESTs (Table S4 and S5). Of the five elements, one was conserved in chordates, two were conserved in deuterostomes, and two were conserved in both deuterostomes and protostomes (Table S2).
Other computational analysis
Multiple alignments were generated using ClustalW [67] and manually edited using JalView [68]. Conservation profiles of the multiple alignments were generated using WebLogo [69]. Conservation profiles were compared to a library of position weight matrices (PWMs) from Uniprobe [70], TRANSFAC [71], and GENOMATIX. PWMs that best match the substitution pattern in the multiple alignment were manually chosen and aligned to the conservation profile.
Zebrafish transgenics
Human, zebrafish, sea urchin, owl limpet and tick sequences were PCR amplified from genomic DNA samples or synthesized (Genescript, Piscataway, NJ) (Dataset S1 and Table S7). All sequences were cloned into the E1B-GFP Tol2 vector [72] using the D-TOPO and Gateway cloning systems (Invitrogen, Life Technologies Corporation). Wildtype AB strain zebrafish were bred according to standard methods and 1-cell stage embryos were injected with the enhancer assay vector and Tol2 transposase mRNA according to previously described methods [73]. Embryos were examined for GFP expression at 6 hours post fertilization (hpf), 21hpf and 48hpf. A minimum of two independent transgenic experiments was done for each construct, and over 60 morphologically healthy embryos were scored for GFP expression at each time point.
To test for background patterns that may be generated by our expression vector, we injected the empty vector, lacking any added enhancer sequence. We could appreciate weak expression in about 50 percent of fish. However, expression was too minimal to determine cell-type using the dissecting microscope. Using confocal microscopy, we could identify expression in single cells. The most common cell types that showed such weak expression were notochord, skeletal muscle, and heart (Figure S4).
Mouse transgenics
The human genomic region encompassing Bicore2 was synthesized with flanking NotI restriction sites (Genescript, Piscataway, NJ) and cloned into the Not5'hsp68lacZ minimal promoter expression vector [74]. The construct was linearized with SalI prior to injection. Transgenic mice were generated by pronuclear injections of FVB embryos (Xenogen Biosciences, Cranberry, NJ). Embryos were harvested at embryonic day 11.5, fixed, and whole mount stained for lacZ as described [74].
Sea urchin transgenics
The sea urchin Bicore1 sequence was PCR amplified from Strongylocentrotus purpuratus genomic DNA. The product was digested and cloned into the EcoRI-BglII sites of the EpGFPII vector [28]. Carrier DNA was prepared using HindIII digestion of S. purpura sperm, followed by phenol chloroform extraction and precipitation with sodium acetate. The carrier DNA was brought to 0.5–1 ug/uL, spun, and filtered with a 0.2 uM filter. The construct DNA was mixed with carrier DNA at a 1∶3 molar ratio. 50% glycerol was added to the mixture to make the construct DNA a final concentration of 2,000 molecules/2pl. Over 1,000 fertilized eggs were injected and over 700 embryos were scored.
Ethics
All animals were treated under protocols #18487 and #21758 approved by Stanford University Institutional Animal Use and Care Committee.
Supporting Information
Zdroje
1. DunnCWHejnolAMatusDQPangKBrowneWE 2008 Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 452 745 749 doi:10.1038/nature06614
2. CarrollSB 2008 Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell 134 25 36 doi:10.1016/j.cell.2008.06.030
3. PeterISDavidsonEH 2011 Evolution of gene regulatory networks controlling body plan development. Cell 144 970 985 doi:10.1016/j.cell.2011.02.017
4. MastonGAEvansSKGreenMR 2006 Transcriptional regulatory elements in the human genome. Annu Rev Genomics Hum Genet 7 29 59 doi:10.1146/annurev.genom.7.080505.115623
5. BejeranoGPheasantMMakuninIStephenSKentWJ 2004 Ultraconserved elements in the human genome. Science 304 1321 1325 doi:10.1126/science.1098119
6. WoolfeAGoodsonMGoodeDKSnellPMcEwenGK 2005 Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol 3 e7 doi:10.1371/journal.pbio.0030007
7. PennacchioLAAhituvNMosesAMPrabhakarSNobregaMA 2006 In vivo enhancer analysis of human conserved non-coding sequences. Nature 444 499 502 doi:10.1038/nature05295
8. ViselAMinovitskySDubchakIPennacchioLA 2007 VISTA Enhancer Browser–a database of tissue-specific human enhancers. Nucleic Acids Res 35 D88 92 doi:10.1093/nar/gkl822
9. GlazovEAPheasantMMcGrawEABejeranoGMattickJS 2005 Ultraconserved elements in insect genomes: a highly conserved intronic sequence implicated in the control of homothorax mRNA splicing. Genome Res 15 800 808 doi:10.1101/gr.3545105
10. International Chicken Genome Sequencing Consortium 2004 Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432 695 716 doi:10.1038/nature03154
11. VavouriTWalterKGilksWRLehnerBElgarG 2007 Parallel evolution of conserved non-coding elements that target a common set of developmental regulatory genes from worms to humans. Genome Biol 8 R15 doi:10.1186/gb-2007-8-2-r15
12. RoyoJLMaesoIIrimiaMGaoFPeterIS 2011 Transphyletic conservation of developmental regulatory state in animal evolution. Proc Natl Acad Sci USA 108 14186 14191 doi:10.1073/pnas.1109037108
13. McLeanCYBristorDHillerMClarkeSLSchaarBT 2010 GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28 495 501 doi:10.1038/nbt.1630
14. HarrisRS 2007 Improved pairwise alignment of genomic DNA. The Pennsylvania State University
15. DehalPSatouYCampbellRKChapmanJDegnanB 2002 The draft genome of Ciona intestinalis: insights into chordate and vertebrate origins. Science 298 2157 2167 doi:10.1126/science.1080049
16. PutnamNHButtsTFerrierDEKFurlongRFHellstenU 2008 The amphioxus genome and the evolution of the chordate karyotype. Nature 453 1064 1071 doi:10.1038/nature06967
17. SodergrenEWeinstockGMDavidsonEHCameronRAGibbsRA 2006 The genome of the sea urchin Strongylocentrotus purpuratus. Science 314 941 952 doi:10.1126/science.1133609
18. YokotaY 2001 Id and development. Oncogene 20 8290 8298 doi:10.1038/sj.onc.1205090
19. MizutaniCMBierE 2008 EvoD/Vo: the origins of BMP signalling in the neuroectoderm. Nat Rev Genet 9 663 677 doi:10.1038/nrg2417
20. López-RoviraTChalauxEMassaguéJRosaJLVenturaF 2002 Direct binding of Smad1 and Smad4 to two distinct motifs mediates bone morphogenetic protein-specific transcriptional activation of Id1 gene. J Biol Chem 277 3176 3185 doi:10.1074/jbc.M106826200
21. KorchynskyiOten DijkeP 2002 Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J Biol Chem 277 4883 4891 doi:10.1074/jbc.M111023200
22. ChenC-RKangYSiegelPMMassaguéJ 2002 E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell 110 19 32
23. RaneyBJClineMSRosenbloomKRDreszerTRLearnedK 2011 ENCODE whole-genome data in the UCSC genome browser (2011 update). Nucleic Acids Res 39 D871 875 doi:10.1093/nar/gkq1017
24. KangYChenC-RMassaguéJ 2003 A self-enabling TGFbeta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell 11 915 926
25. PetersonKJCottonJAGehlingJGPisaniD 2008 The Ediacaran emergence of bilaterians: congruence between the genetic and the geological fossil records. Philos Trans R Soc Lond, B, Biol Sci 363 1435 1443 doi:10.1098/rstb.2007.2233
26. RauchGJLyonsDAMiddendorfIFriedlanderBAranaN 2003 Submission and Curation of Gene Expression Data. ZFIN Direct Data Submission
27. GrayPAFuHLuoPZhaoQYuJ 2004 Mouse brain organization revealed through direct genome-scale TF expression analysis. Science 306 2255 2257 doi:10.1126/science.1104935
28. Revilla-i-DomingoRMinokawaTDavidsonEH 2004 R11: a cis-regulatory node of the sea urchin embryo gene network that controls early expression of SpDelta in micromeres. Dev Biol 274 438 451 doi:10.1016/j.ydbio.2004.07.008
29. SaudemontAHaillotEMekpohFBessodesNQuirinM 2010 Ancestral regulatory circuits governing ectoderm patterning downstream of Nodal and BMP2/4 revealed by gene regulatory network analysis in an echinoderm. PLoS Genet 6 e1001259 doi:10.1371/journal.pgen.1001259
30. CheahPYMengYBYangXKimbrellDAshburnerM 1994 The Drosophila l(2)35Ba/nocA gene encodes a putative Zn finger protein involved in the development of the embryonic brain and the adult ocellar structures. Mol Cell Biol 14 1487 1499
31. RunkoAPSagerströmCG 2004 Isolation of nlz2 and characterization of essential domains in Nlz family proteins. J Biol Chem 279 11917 11925 doi:10.1074/jbc.M310076200
32. RunkoAPSagerströmCG 2003 Nlz belongs to a family of zinc-finger-containing repressors and controls segmental gene expression in the zebrafish hindbrain. Dev Biol 262 254 267
33. HoyleJTangYPWielletteELWardleFCSiveH 2004 nlz gene family is required for hindbrain patterning in the zebrafish. Dev Dyn 229 835 846 doi:10.1002/dvdy.20001
34. VlachakisNChoeSKSagerströmCG 2001 Meis3 synergizes with Pbx4 and Hoxb1b in promoting hindbrain fates in the zebrafish. Development 128 1299 1312
35. ChoeS-KVlachakisNSagerströmCG 2002 Meis family proteins are required for hindbrain development in the zebrafish. Development 129 585 595
36. RyooHDMartyTCasaresFAffolterMMannRS 1999 Regulation of Hox target genes by a DNA bound Homothorax/Hox/Extradenticle complex. Development 126 5137 5148
37. JiangYShiHLiuJ 2009 Two Hox cofactors, the Meis/Hth homolog UNC-62 and the Pbx/Exd homolog CEH-20, function together during C. elegans postembryonic mesodermal development. Dev Biol 334 535 546 doi:10.1016/j.ydbio.2009.07.034
38. PetersenCPReddienPW 2009 Wnt signaling and the polarity of the primary body axis. Cell 139 1056 1068 doi:10.1016/j.cell.2009.11.035
39. RhinnMLunKLuzMWernerMBrandM 2005 Positioning of the midbrain-hindbrain boundary organizer through global posteriorization of the neuroectoderm mediated by Wnt8 signaling. Development 132 1261 1272 doi:10.1242/dev.01685
40. McGlinnERichmanJMMetzisVTownLButterfieldNC 2008 Expression of the NET family member Zfp503 is regulated by hedgehog and BMP signaling in the limb. Dev Dyn 237 1172 1182 doi:10.1002/dvdy.21508
41. DavidsonEHCameronRARansickA 1998 Specification of cell fate in the sea urchin embryo: summary and some proposed mechanisms. Development 125 3269 3290
42. CubasPModolellJRuiz-GómezM 1994 The helix-loop-helix extramacrochaetae protein is required for proper specification of many cell types in the Drosophila embryo. Development 120 2555 2566
43. TomoyasuYNakamuraMUenoN 1998 Role of dpp signalling in prepattern formation of the dorsocentral mechanosensory organ in Drosophila melanogaster. Development 125 4215 4224
44. HollandLZ 2009 Chordate roots of the vertebrate nervous system: expanding the molecular toolkit. Nat Rev Neurosci 10 736 746 doi:10.1038/nrn2703
45. ArendtDDenesASJékelyGTessmar-RaibleK 2008 The evolution of nervous system centralization. Philos Trans R Soc Lond, B, Biol Sci 363 1523 1528 doi:10.1098/rstb.2007.2242
46. RaibleFTessmar-RaibleKOsoegawaKWinckerPJubinC 2005 Vertebrate-type intron-rich genes in the marine annelid Platynereis dumerilii. Science 310 1325 1326 doi:10.1126/science.1119089
47. PutnamNHSrivastavaMHellstenUDirksBChapmanJ 2007 Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 317 86 94 doi:10.1126/science.1139158
48. DenoeudFHenrietSMungpakdeeSAuryJ-MDa SilvaC 2010 Plasticity of animal genome architecture unmasked by rapid evolution of a pelagic tunicate. Science 330 1381 1385 doi:10.1126/science.1194167
49. McGinnisWKrumlaufR 1992 Homeobox genes and axial patterning. Cell 68 283 302
50. PasquinelliAEReinhartBJSlackFMartindaleMQKurodaMI 2000 Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 408 86 89 doi:10.1038/35040556
51. ProchnikSERokhsarDSAboobakerAA 2007 Evidence for a microRNA expansion in the bilaterian ancestor. Dev Genes Evol 217 73 77 doi:10.1007/s00427-006-0116-1
52. HsuFKentWJClawsonHKuhnRMDiekhansM 2006 The UCSC Known Genes. Bioinformatics 22 1036 1046 doi:10.1093/bioinformatics/btl048
53. SiepelABejeranoGPedersenJSHinrichsASHouM 2005 Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15 1034 1050 doi:10.1101/gr.3715005
54. PruittKDTatusovaTMaglottDR 2007 NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35 D61 65 doi:10.1093/nar/gkl842
55. HubbardTBarkerDBirneyECameronGChenY 2002 The Ensembl genome database project. Nucleic Acids Res 30 38 41
56. SiepelAHausslerD 2004 Computational identification of evolutionarily conserved exons. Proceedings of the eighth annual international conference on Resaerch in computational molecular biology. RECOMB '04 New York, NY, USA ACM 177 186 Available:http://doi.acm.org/10.1145/974614.974638. Accessed 22 December 2011
57. BensonDAKarsch-MizrachiILipmanDJOstellJWheelerDL 2004 GenBank: update. Nucleic Acids Res 32 D23 26 doi:10.1093/nar/gkh045
58. GerhardDSWagnerLFeingoldEAShenmenCMGrouseLH 2004 The status, quality, and expansion of the NIH full-length cDNA project: the Mammalian Gene Collection (MGC). Genome Res 14 2121 2127 doi:10.1101/gr.2596504
59. Griffiths-JonesSSainiHKvan DongenSEnrightAJ 2008 miRBase: tools for microRNA genomics. Nucleic Acids Res 36 D154 158 doi:10.1093/nar/gkm952
60. LestradeLWeberMJ 2006 snoRNA-LBME-db, a comprehensive database of human H/ACA and C/D box snoRNAs. Nucleic Acids Res 34 D158 162 doi:10.1093/nar/gkj002
61. KarroJEYanYZhengDZhangZCarrieroN 2007 Pseudogene.org: a comprehensive database and comparison platform for pseudogene annotation. Nucleic Acids Res 35 D55 60 doi:10.1093/nar/gkl851
62. AshurstJLChenC-KGilbertJGRJekoschKKeenanS 2005 The Vertebrate Genome Annotation (Vega) database. Nucleic Acids Res 33 D459 465 doi:10.1093/nar/gki135
63. PedersenJSBejeranoGSiepelARosenbloomKLindblad-TohK 2006 Identification and classification of conserved RNA secondary structures in the human genome. PLoS Comput Biol 2 e33 doi:10.1371/journal.pcbi.0020033
64. ChiaromonteFYapVBMillerW 2002 Scoring pairwise genomic sequence alignments. Pac Symp Biocomput 115 126
65. AltschulSFGishWMillerWMyersEWLipmanDJ 1990 Basic local alignment search tool. J Mol Biol 215 403 410 doi:10.1006/jmbi.1990.9999
66. BlanchetteMKentWJRiemerCElnitskiLSmitAFA 2004 Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res 14 708 715 doi:10.1101/gr.1933104
67. LarkinMABlackshieldsGBrownNPChennaRMcGettiganPA 2007 Clustal W and Clustal X version 2.0. Bioinformatics 23 2947 2948 doi:10.1093/bioinformatics/btm404
68. WaterhouseAMProcterJBMartinDMAClampMBartonGJ 2009 Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25 1189 1191 doi:10.1093/bioinformatics/btp033
69. CrooksGEHonGChandoniaJ-MBrennerSE 2004 WebLogo: a sequence logo generator. Genome Res 14 1188 1190 doi:10.1101/gr.849004
70. NewburgerDEBulykML 2009 UniPROBE: an online database of protein binding microarray data on protein-DNA interactions. Nucleic Acids Res 37 D77 82 doi:10.1093/nar/gkn660
71. MatysVKel-MargoulisOVFrickeELiebichILandS 2006 TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res 34 D108 110 doi:10.1093/nar/gkj143
72. LiQRitterDYangNDongZLiH 2010 A systematic approach to identify functional motifs within vertebrate developmental enhancers. Dev Biol 337 484 495 doi:10.1016/j.ydbio.2009.10.019
73. FisherSGriceEAVintonRMBesslingSLUrasakiA 2006 Evaluating the biological relevance of putative enhancers using Tol2 transposon-mediated transgenesis in zebrafish. Nat Protoc 1 1297 1305 doi:10.1038/nprot.2006.230
74. DiLeoneRJRussellLBKingsleyDM 1998 An extensive 3′ regulatory region controls expression of Bmp5 in specific anatomical structures of the mouse embryo. Genetics 148 401 408
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 8
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Dissecting the Gene Network of Dietary Restriction to Identify Evolutionarily Conserved Pathways and New Functional Genes
- It's All in the Timing: Too Much E2F Is a Bad Thing
- A Quantitative Comparison of the Similarity between Genes and Geography in Worldwide Human Populations
- Variation of Contributes to Dog Breed Skull Diversity
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy