
Mutations in a P-Type ATPase Gene Cause Axonal Degeneration
Neuronal loss and axonal degeneration are important pathological features of many neurodegenerative diseases. The molecular mechanisms underlying the majority of axonal degeneration conditions remain unknown. To better understand axonal degeneration, we studied a mouse mutant wabbler-lethal (wl). Wabbler-lethal (wl) mutant mice develop progressive ataxia with pronounced neurodegeneration in the central and peripheral nervous system. Previous studies have led to a debate as to whether myelinopathy or axonopathy is the primary cause of neurodegeneration observed in wl mice. Here we provide clear evidence that wabbler-lethal mutants develop an axonopathy, and that this axonopathy is modulated by Wlds and Bax mutations. In addition, we have identified the gene harboring the disease-causing mutations as Atp8a2. We studied three wl alleles and found that all result from mutations in the Atp8a2 gene. Our analysis shows that ATP8A2 possesses phosphatidylserine translocase activity and is involved in localization of phosphatidylserine to the inner leaflet of the plasma membrane. Atp8a2 is widely expressed in the brain, spinal cord, and retina. We assessed two of the mutant alleles of Atp8a2 and found they are both nonfunctional for the phosphatidylserine translocase activity. Thus, our data demonstrate for the first time that mutation of a mammalian phosphatidylserine translocase causes axon degeneration and neurodegenerative disease.
Published in the journal:
. PLoS Genet 8(8): e32767. doi:10.1371/journal.pgen.1002853
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002853
Summary
Neuronal loss and axonal degeneration are important pathological features of many neurodegenerative diseases. The molecular mechanisms underlying the majority of axonal degeneration conditions remain unknown. To better understand axonal degeneration, we studied a mouse mutant wabbler-lethal (wl). Wabbler-lethal (wl) mutant mice develop progressive ataxia with pronounced neurodegeneration in the central and peripheral nervous system. Previous studies have led to a debate as to whether myelinopathy or axonopathy is the primary cause of neurodegeneration observed in wl mice. Here we provide clear evidence that wabbler-lethal mutants develop an axonopathy, and that this axonopathy is modulated by Wlds and Bax mutations. In addition, we have identified the gene harboring the disease-causing mutations as Atp8a2. We studied three wl alleles and found that all result from mutations in the Atp8a2 gene. Our analysis shows that ATP8A2 possesses phosphatidylserine translocase activity and is involved in localization of phosphatidylserine to the inner leaflet of the plasma membrane. Atp8a2 is widely expressed in the brain, spinal cord, and retina. We assessed two of the mutant alleles of Atp8a2 and found they are both nonfunctional for the phosphatidylserine translocase activity. Thus, our data demonstrate for the first time that mutation of a mammalian phosphatidylserine translocase causes axon degeneration and neurodegenerative disease.
Introduction
Neuronal loss and axonal degeneration are important pathological features of neurodegenerative diseases, such as Alzheimer disease, Parkinson's disease, amyotrophic lateral sclerosis and glaucoma. Axonopathies, conditions in which axon injury occurs first during disease progression, have been extensively studied, but the mechanism(s) underlying axon degeneration remain to be elucidated in most of these conditions. In order to rationally develop effective therapeutics for these conditions, it is critical to determine the molecular mechanisms underlying axonopathies.
Spontaneous mouse mutants have long been used to gain insight into human disease. Spontaneous mutants provide valuable platforms for identifying disease-associated pathways. Identifying the gene(s) that cause a certain phenotype in mice can lead to a greater understanding of the pathophysiology of a disease. Identification of a disease causing mutation in mice often precedes the identification of the orthologous gene as a cause of a corresponding disease in humans [1]–[10]. As with all forward genetic tools, the power of analyzing mutants is that it allows for the identification of pathways involved in disease that may not have been identified through experiments based on current knowledge. To understand axon pathophysiology better, we studied wabbler-lethal (wl) mutant mice that display axon degeneration. The autosomal recessive wl mutation arose spontaneously in a mouse colony at The Jackson Laboratory in 1952 [11]. Homozygous wl mice are characterized by severe neurological abnormalities that include ataxia and body tremors. Abnormalities are first apparent around twelve days of age and mutant mice generally die around 4 weeks of age [11]–[13]. Histopathology of the wl nervous system is consistent with wl being an axonopathy [12], [14], though it has also been suggested to be primarily a myelinopathy [11], [15]. The genetic defect that causes wl has been unknown.
Here, we performed an extensive analysis of wabbler lethal mice and show that they develop a progressive axonal degeneration in several different areas of the nervous system. The presence of prominent axon degeneration, without initial myelin damage, and the absence of obvious cell death point to an axonopathy. To gain further understanding of the molecular pathways that can underlie the axonopathy in wl mice, the autosomal recessive wl mutation was positionally cloned. Using a combination of genetic and biochemical approaches, we demonstrated that the pathological lesion of this mutation is due to loss of function mutations in the gene encoding the murine phosphatidylserine translocase (flippase) Atp8a2. Loss of phosphatidylserine flippase activity leads to decreased axonal transport as indicated by the accumulation of phosphorylated neurofilament in motor neurons and retinal ganglion cell bodies. These data establish a novel role for a phosphatidylserine flippase in maintaining axonal health in both the central and peripheral nervous systems.
Results
The wabbler-lethal (wl) phenotype is an axonopathy
Mice homozygous for the wl mutation (wl/wl; wl mutants) grow much slower than their littermate controls and are first phenotypically recognizable at about 12 days of age, due to their smaller body size (Figure 1A and B). Supplementation of dry food with a soft moist diet that was placed on the cage floor to allow easy access, allowed homozygous mutants to survive past the previously reported 30 days (Figure 1C) [11]. Even on this diet, however, twenty percent of wl/wl mice died by 65 days of age, and all died by 130 days. Homozygous mutant mice develop a body tremor, an abnormal gait (Figure 1D), and display an abnormal hind limb-clasping reflex indicative of a neurological deficit that is very obvious at two months of age (Figure 1E).
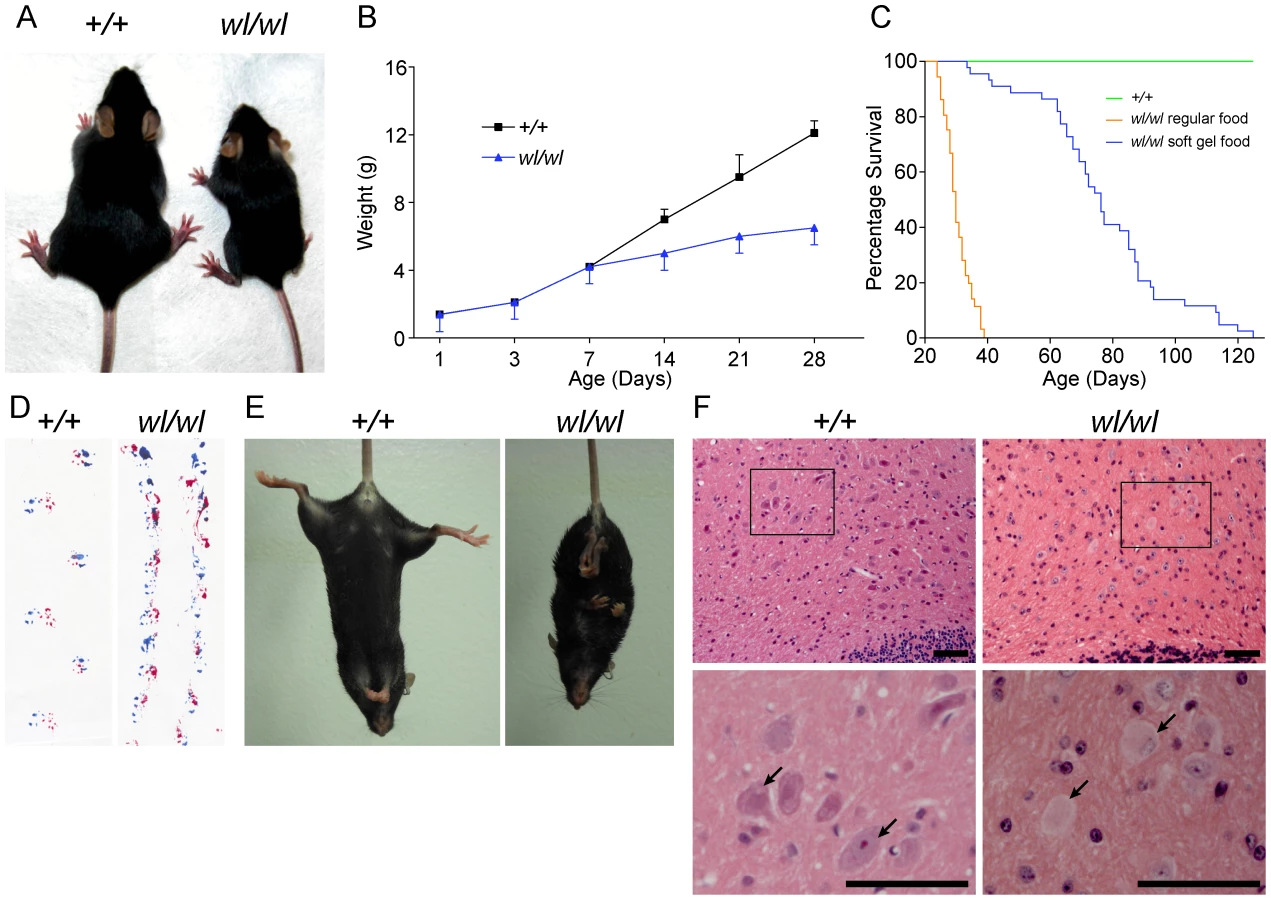
Central chromatolysis is regarded as a characteristic feature of axonopathies [16]. Here we documented chromatolysis in the lateral cerebellar nucleus (Figure 1F), medial cerebellar nucleus and lateral vestibular nucleus (Figure S1) in wl mutants but not controls. Affected neurons have pale staining and acentric nuclei in hematoxylin and eosin stained sections (Figure 1F, arrows). Importantly, despite cell bodies with obvious chromatolysis in the lateral cerebellar nucleus, intermediate nucleus, spinal cord and other regions of the cerebellum, no obvious cell loss was noted in any of these regions, and cleaved caspase 3 staining did not detect an increased number of apoptotic cells (Figure S2). Dystrophic axons were evident in the corticospinal tract, spinalcerebellar tract (Figure S3) and spinal white matter (Figure S4). These data are consistent with a primary axonopathy without cell loss.
Central chromatolysis was also observed in the spinal ventral horn at different spinal levels (Figure 2B and C), again without obvious cell loss. Spinal motor neurons are located in the ventral horns and their axons project into the ventral root and then the spinal nerves. Supporting an absence of cell loss, even at 3 months of age when the disease is very severe (see below), axon counts for the ventral root (close to the cell bodies) were indistinguishable between wl mutant (count 1090±14, n = 4) and control mice (count 1101±7, n = 4, P = 0.22).

The wabbler-lethal phenotype is a distal axonopathy
To determine if axon injury was first visible closer (proximal) to the cell body, or farther away (distal) from the cell body, we analyzed axons in the proximal ventral root and the distal femoral motor branch, which primarily consists of long motor axons. At two months of age, no obvious axon loss or morphological changes were present in the L4 ventral root (Figure 2D, E and H). However, the same wl/wl mice had lost 20% of axons in their distal femoral motor branch (Figure 2F, G, and I) with many axons having an irregular shape and having darkly stained axoplasm (Figure 2G). The femoral motor branch in 8-day old mutant and control mice were similar in both the total axon number and axon morphology, indicating the axon damage is not developmental and occurs with aging (Figure S5). The axonal degeneration in the femoral nerve is thus initially prominent in the distal part of the nerve with no apparent loss of axons in the ventral root. These data suggest that distal axonal degeneration is the main cause of the disease in wl mice.
Analysis of the distal sciatic nerve of wl mice at two months of age revealed that large diameter axons were preferentially lost (Figure 2J–L; Figure S6C). In general, large diameter motor neurons are known to have thicker myelin than smaller diameter motor neurons. Consistent with loss of large axons, myelin thickness is reduced in two-month old mutant mice from a mean value of 0.99±0.02 µm to a mean value of 0.57±0.01 µm (p<0.01). Despite severe neurological abnormalities at two months of age, no obvious demyelination defects were observed by assessing the ratio of inner axon diameter (inside myelin) to the total axon diameter (G-ratio, not shown). Additionally the condition of myelin in wl mutants and controls was indistinguishable by transmission electron microscopy at 2 month of age (Figure S6). These data suggest that demyelination is not the primary cause of the observed phenotype.
Wabbler-lethal mutants have disrupted axon transport of neurofilaments
The distal degeneration of motor axons in the wl mutants prompted us to examine phosphorylated neurofilament (pNF) as a marker of axonal transport in spinal motor neurons. The localization of intermediate to high molecular weight pNF has been widely used to assess axonal transport [17], [18]. Under normal conditions, pNF is rarely detected in the cell body as it is normally efficiently transported from the soma to the more distal axon. When axonal transport is disrupted, pNF accumulates in the cell body [17], [18]. Staining of lumbar motor neurons of one-month-old wild type mice with pNF antibody detected neurofilament in axons (Figure 3A and B), while accumulation in the somas was very rare (Figure 3E). In contrast, pNF was present in both the axons and somas of lumbar motor neurons in wl mutant mice (3C, D and E; 12±0.4 pNF positive soma in wl/wl mice; 0.2±0.2 pNF positive soma in +/+ control mice, p<0.01). Both motor neurons in the ventral horn and neurons in the dorsal gray column display accumulation of pNF in their cell bodies. In addition, pNF accumulation was observed in different regions of the brain, such as the medial cerebellar nucleus, intermediate reticular nucleus and raphe magnus nucleus (Figure S7), indicating a common defect in axonal transport of neurofilament.

The axons of retinal ganglion cells (RGCs) also degenerate in the optic nerves of wl mice [12]. By one month, RGCs from wl mutants showed disrupted axon transport as evidenced by pNF accumulation in their somas. This accumulation occurs mainly in the peripheral retina (Figure 3H, I and J; 364±7 pNF positive soma in wl/wl mice; 2±0.5 pNF positive soma in +/+ control mice, p<0.01). To determine if axon injury, evident as axon transport defects, occurs before or after morphological changes, the optic nerves of wl mutants were assessed for axon damage using a histochemical stain paraphenylenediamine (PPD). PPD stains all myelin sheaths but is very sensitive for detecting axon injury (Figure S8), as the axoplasm of injured axons stains darkly [19]–[23]. At 30 days of age, the optic nerves of wl mice are indistinguishable from those in wild type mice. Despite this normal appearance of their axons, the RGCs have axon transport defects as indicated by accumulation of pNF in their somas (Figure 3H–J). Together, the axon transport defects in neurons with structurally normal axons and myelin sheaths, the central chromatolysis and the distal axonal degeneration in the femoral and sciatic nerves are consistent with an axonopathy.
Wlds and Bax delay the axonopathy in wl mice
The Wallerian degeneration slow (Wlds) allele dominantly delays axonal degeneration after direct axonal trauma and in axonopathies [24]–[30]. To further examine the role of axonal injury in the wl mutant, the effect of the Wlds allele was tested by crossing the Wlds allele to wl mice. The Wlds mutation has a strong protective effect on the survival of femoral nerve axons (Figure 4A–C). At two months, wl mutant nerves displayed severe axon degeneration (Figure 4B and J). In comparison, the number of axons in wl Wlds nerves were nearly identical to the number of axons in controls (Figure 4C and J). To determine the effect of Wlds on early changes in motor neurons, we looked at pNF accumulation in lumbar spinal cord sections. Wlds prevented axon transport defects as assessed by pNF accumulation in wl mutants (for example, in the lumbar spinal cord; Figure 4D–I). This delay of axonopathy because of Wlds further argues that the wl phenotype is an axonopathy.

As a further functional evaluation of the protective effects of Wlds against spinal axonopathy, we assessed nerve conduction velocity (NCV) in the sciatic nerve. At two months of age NCV was decreased by 40% in the wl mice, falling from 30.7±3.1 m/s in control animals to 18.4±1.2 m/s (p<0.01) in wl mice. In contrast, wl Wlds mice retained normal conduction velocity (Figure 4K). These data indicate that Wlds delayed axon damage in wl mutants. However, at no point could wl Wlds mice be grossly distinguished from wl mice. Despite apparent healthy axon morphology, wl Wlds mice still displayed body tremor when walking. Their disease onset was still around 14 days after birth, they were smaller than wildtype controls, their locomotory performance did not improve, and they performed poorly in the wire hang test (Figure S9). In the latter, wl and wl Wlds mice were able to grip the cage top for an average of 12.2±3.4 and 13.2±2.9 seconds respectively (P>0.05), while wild type controls gripped for an average of 55.2±3 seconds (P = 0.01) compared to wl or wl Wlds mutants.
The Bax gene is best known for its role in somal apoptosis, but also has an independent and intra-axonal role in axon degeneration [31], [32]. Genetic ablation of Bax strikingly protects axons in the femoral motor branch in two-month old wl Bax−/− mutants. At this age, wl mutants had severe axon loss while wl Bax−/− mice had no axon loss (Figure 5C, D, E). As for Wlds, the Bax mutation had no effect on lifespan or the gross neurological phenotype.

The mutation causing the wl phenotype is in Atp8a2
Previous linkage analysis localized wl to chromosome 14 [33]. Analysis of 688 affected wl F2 mice narrowed the interval where the mutation resides to a 773 kb region on chromosome 14 between DLM14-10 (60.6 Mb) and DLM14-21 (61.4 Mb) (Figure 6A). This region is known to contain 10 protein-coding genes. The coding regions and splice sites of each of these genes were sequenced and a 21 bp deletion in exon 22 of Atp8a2 (Figure 6B and C) was identified. No mutations were found in the other 9 genes. This genomic deletion in Atp8a2 leads to the elimination of seven highly conserved amino acids (TAIEDRL) from the nucleotide-binding domain (N-domain) of ATP8A2 (Figure 6D, E). Two additional alleles of wl were available: wlvmd (vmd) and wl3J (3J). We found that the wlvmd mutation is a large 9167 bp genomic deletion that results in removal of the entire exon 32 of Atp8a2 (Figure 6C and Figure S10). This results in a 32 amino acid deletion in the ninth transmembrane domain of ATP8A2 (Figure 6D). wl3J mice were found to have a 641 bp deletion starting at the tenth base pair of exon 30 of Atp8a2, leading to the deletion of part of exon 30 and the whole exon 31. Furthermore, wl3J mice had a 10 bp duplication in exon 32 (Figure 6C and Figure S11). Genetic mapping and sequence analysis of these three alleles of wl clearly show that mutations in Atp8a2 cause the wl phenotype.

ATP8A2 is localized to the membrane
Atp8a2 is widely expressed in the central nervous system including the cerebrum, cerebellum, spinal cord, and retina (Figure 7A). Ectopic expression of ATP8A2 in HEK-293T or COS7 cells showed that ATP8A2 has the expected molecular weight of a 130 kDa (Figure 7B), as detected by a polyclonal antibody against mouse ATP8A2 developed in our laboratory (see methods). Western blot analysis of protein isolated from either cytosolic or membrane fractions showed that ATP8A2 is localized to the mouse brain membrane fraction (Figure 7C).

ATP8A2 is a phosphatidylserine translocase
Based on sequence comparison, we hypothesized that ATP8A2 is likely a phospholipid translocase. To test this hypothesis, full-length Atp8a2 cDNA was expressed in UPS-1 cells. These cells are defective in non-endocytic uptake of 7-nitrobenz-2-oxa-1,3-diazol-4-yl phosphatidylserine (NBD-PS) analogs and are thus optimal for testing phospholipid translocase (flippase) activity with NBD substrates [34], [35]. Compared to control cells transfected with empty vector, Atp8a2 transfected UPS-1 cells displayed significant phosphatidylserine translocase activity (1500% of control, Figure 8A, B). This phospolipid flippase activity is specific to phosphatidylserine (PS), as translocation of only NBD-phosphatidylserine and not of NBD-phosphatidyletholamine (PE), NBD-phosphatidylcholine (PC) or NBD-phosphatidylglycerol (PG) was observed (Figure 8B). To ascertain that PS translocation was due to ATP8A2 activity, we generated a mutant version of ATP8A2 in which Asp388 in the highly conserved core sequence DKTGTLT was replaced with an Ala. This Asp residue is phosphorylated and dephosphorylated during the catalytic cycle and is critical for the activity of all P-type ATPases. The mutant protein encoded by Atp8a2D388A failed to translocate NBD-PS into the inner leaflet of the plasma membrane (Figure 8C). Similarly, the chemical inhibitor sodium vanadate greatly reduced the flippase activity of ATP8A2 (Figure 8C).

To examine the activity of the mutant protein encoded by the wl mutant alleles, we introduced the 21 bp deletion identified in the wl mutant, and the 108 bp deletion of the vmd mutant into the Atp8a2 cDNA by site-direct mutagenesis. Although these mutant proteins were expressed (Figure 8D), neither protein displayed any flippase activity (Figure 8C). These data clearly demonstrate that the mutant proteins encoded by wl and vmd have no flippase activity.
Discussion
Axon degeneration and axonopathies are often observed in human neurodegenerative diseases, but their molecular causes are typically not known. To provide new insight into axon degeneration we studied the wabbler-lethal (wl) mouse. We show that wl mutant mice develop distal axon degeneration and neuronal chromatolysis in varying parts of the CNS and the PNS without cell death. Together with our finding that the Wlds mutation, which protects against axon injury, significantly delayed axonal degeneration [25]–[30], [36]–[39], this provides strong evidence that the wl mutation induces an axonopathy [11]–[15].
Although the Wlds mutation delays axonal degeneration in wl mice, it does not alter their gross phenotype or extend lifespan. Similar to the effect of Wlds in wl mice, Wlds inhibits axonal spheroid pathology in gracile axonal dystrophy (gad) mice with the Uchl1gad allele, but did not alleviate gad symptoms [38]. Compared to gad mice, both the gracile nucleus and cervical gracile fascicle contained fewer spheroids in Uchl1gad Wlds mice. However, similar to the previous observation that Wlds has a weaker effect on synapses than on axons, motor axon terminals at neuromuscular junctions continued to degenerate in Uchl1gad Wlds mice. This might contribute to the fact that Wlds did not alleviate gad symptoms. In contrast, in pmn mice, another motor neuron disease model, Wlds was able to delay axonal degeneration, extend life span, and improve motor performance [25]. The fact that Wlds did not appear to alter the gross behavior of wl mice suggests that there may be detrimental phenotypes in wl mice that are distinct from axonal degeneration.
Two recent reports demonstrated a requirement of Bax during axonal degeneration [31], [32], and Bax deficiency delayed axonal degeneration in a mouse model of glaucoma [21]. In wl mice, genetic ablation of Bax significantly delayed axonal degeneration, providing the first evidence for the role of Bax in an inherited mouse model of primary axonal degeneration. Our result indicates that Bax has a role in intrinsic axon degeneration, similar to a previous study using cultured sensory neurons [31]. As in cultured sensory neurons [31], it is possible that caspase 6 participates in this process. This will be the subject of future investigations.
While the wl phenotype has been investigated for over 60 years, the underlying mutation was not previously identified. We show that mutations in Atp8a2 underlie the phenotype for three independent wl alleles. Atp8a2 encodes a protein homologous to P4-type ATPases, putative phospholipid translocases that translocate aminophospholipids from the extracellular leaflet to the cytoplasmic leaflet of the plasma membrane bilayer [40]. They are important for the maintenance of phospholipid asymmetry in eukaryotic cell membranes, and play essential roles in many physiological conditions. Our cell-based PS translocation assay clearly showed that ATP8A2 is a phosphatidylserine translocase and both the wl and vmd mutant proteins do not retain PS flippase activity. These findings are in agreement with recent independent studies showing that ATP8A2 has PS flippase activity when reconstituted in liposomes [41], [42].
The importance of the P4-ATPase membrane protein family is increasingly evident through findings in which dysfunction of P4-ATPases is associated with developmental defect in animals and several human disorders [43]–[47]. In Caenorhabditis elegans loss of the P4-ATPase TAT-1 leads to the exposure of PS on the surface of germ cells and loss of certain neuronal cells [48]. In humans, mutations in the FIC1/ATP8B1 gene cause progressive familial cholestasis, a severe liver disease with defective bile secretion and hearing loss [43], [49]. Furthermore, Atp8b3 is exclusively expressed in the testis and has a role in sperm capacitation in mice [50]. Atp10a (also named Atp10c) is linked to diet-induced obesity and type II diabetes in mice [51]. Mutations in the murine Atp11c gene leads to cholestasis and a striking B cell differentiation defect [45]–[47]. Although Atp11c is ubiquitously expressed in different tissues, loss of ATP11C activity specifically affects adult B cell development, indicating cell and lineage-specific requirement of this transporter.
Based on our findings, we propose a number of possible mechanisms by which loss of ATP8A2 activity can lead to axonopathy. Like ATP8B1, ATP8A2 is thought to be important for maintaining phospholipid asymmetry of cell membranes by translocating phosphatidylserine from the outer leaflet to the inner leaflet of the membrane. PS asymmetry in the cell membrane has been shown to have an essential role in the mechanical stability of the red cell membrane [52]. In patients and mice with an ATP8B1/Atp8b1 deficiency, the canalicular membrane is not stable and extraction of lipid by the detergent action of bile salts leads to formation of granular bile and intrahepatic cholestasis. Similarly, loss of ATP8A2 activity may result in re-distribution of PS to the outer leaflet and loss of phospholipid asymmetry. Neuronal axon membranes may be unstable due to this abnormal PS distribution and thus become susceptible to degeneration. Alternately, loss of PS asymmetry might lead to a defect in intracellular sorting and transport of vesicular components similar to what was observed for the yeast homologue Drs2, which has a role in intracellular vesicular trafficking between the trans-Golgi network (TGN), the endosome and the plasma membrane [53].
Recent studies showed that disruption of phospholipid turnover and trafficking can lead to neurodegenerative diseases in both mice and people. For example, the spontaneous null mutation of mouse Fig4 in the pale tremor (plt) strain leads to neuronal loss, spongiform degeneration of the brain and loss of neurons from the dorsal root ganglia [54], [55]. Mutant animals had a severe peripheral neuropathy and a shorter life span. Fig4 encodes a phospholipid phosphatase with 5-phosphatase activity towards the 5-phosphate residue of PtdIns(3,5)P2 [56], [57]. Loss of the FIG4 phosphatase in humans leads to the autosomal recessive, demyelinating, Charcot-Marie-Tooth neuropathy (CMT), CMT type 4J (OMIM #611228) [54], [58]. The most common human mutation of FIG4 reduces the binding affinity of FIG4 for the PtdIns(3,5)P2 biosynthetic complex [59], while in mouse plt fibroblasts a significant decrease of PtdIns(3,5)P2 was observed [54]. Intracellular phosphoinositides (PIs) are essential regulators of membrane trafficking, including functions to promote recruitment and/or activation of spatially localized protein machinery on membranes. The production and turnover of PIs are tightly controlled by kinases and phosphatases [60], [61]. In the nervous system, neurons, especially neurons with long axons, depend on efficient membrane trafficking for maintenance of proper functions and health [62]. In humans and mice, autosomal recessive, demyelinating, Charcot-Marie-Tooth type B1 neuropathy [63]–[65] is also caused by an abnormality of phospholipid metabolism resulting from mutation of the myotubularin-related 2 gene (MTMR2). MTMR2 is a phospholipid phosphatase with 3-phosphatase activity towards the 3-phosphate residue of PtdIns(3,5)P2 and PtdIns3P [66], [67]. Therefore, Fig4, MTMR2 and Atp8a2 mutations may affect neurons by altering membrane protein trafficking. These mutant phenotypes highlight the importance phospholipid metabolism for neuronal health and the possible role of abnormal membrane trafficking in neurological diseases.
Loss of lipid asymmetry has an important impact on cell morphology, membrane protein activities, phagocytosis, apoptosis, endocytosis, and vesicle biogenesis [68]. In addition proper membrane lipid composition is also important for exocytosis and synaptic vesicle release in neuronal cells. PS content has been shown to affect PC12 cell exocytosis [69]. Altered PS distribution in wl neurons could affect release of synaptic vesicles and transduction of action potentials along nerve fibers.
Atp8a2 is specifically expressed in the nervous system and testis. In contrast, Atp8a1, a PS translocase and a close member of the P-type ATPase family, is expressed broadly in many tissues. Atp8a1 deficient mice have no grossly visible neurological phenotypes and have grossly normal brains ([70] and www.informatics.jax.org/external/ko/deltagen/1902.html). Recently, behavioral analysis of Atp8a1 deficient mice detected neurological abnormalities, including impaired hippocampus-dependent learning (Morris Water Maze test), hyperactivity, and poor maternal behavior [70]. Furthermore, deficiency of both Atp8a2 and Atp8a1 results in neonatal lethality (our unpublished data using the wl allele). Double mutant mice have labored breathing and die within a few hours after birth (unpublished observations). It is likely that ATP8A2 and ATP8A1 act redundantly in certain tissues to allow survival of single mutants by maintaining an adequate PS asymmetry. Consistent with this, no obvious neuronal death was detected in Atp8a2wl/wl mutants. Additionally, we could not detect any obvious disturbance of PS lipid asymmetry in cultured neurons and spermatogonia from these Atp8a2wl/wl mutants (not shown). Obvious loss of asymmetry has been well established to induce apoptosis [71]. Loss of both ATP8A2 and ATP8A1 leads to failure of tissue function and lethality. Conditional alleles of Atp8a2 and Atp8a1 will be helpful to pinpoint the temporal and tissue specific requirements of Atp8a2 and Atp8a1.
Interestingly, due to a de novo chromosome translocation, a patient with mental retardation and hypotonia was haploinsufficient for ATP8A2. No other genes were reported to be structurally altered by this genetic event. However, no gene expression studies for ATP8A2 or other genes flanking the translocation, whose expression may be affected by the translocation, were presented. Additionally, no mutations were found in thirty-eight other patients with similar phenotypes [72]. Thus, it is not yet clear if ATP8A2 plays a role in this disease. However, this patient together with the data presented in our current paper suggests that ATP8A2 should be further considered as a candidate gene for human neurological diseases.
In conclusion, normal ATP8A2 activity is indispensable for normal neuronal functions. To our knowledge, ATP8A2 is the first mammalian PS flippase identified to have a role in axon degeneration. Our experiments identify a new process that contributes to axonopathy and neurological disease.
Materials and Methods
Mouse strains and genotyping
The Association for Assessment and Accreditation of Laboratory Animal Care guidelines were followed for all animal procedures, and all procedures were approved by the Institutional Animal Care and Use Committee of The Jackson Laboratory. The original wabbler-lethal (wl) mutant arose in the pirouette strain of Mus musculus at The Jackson Laboratory [11]. The Vestibulo-Motor Degeneration (vmd) mutation arose in a C3H/H2SnJ colony at the Jackson Laboratory in 1987. Both wl and vmd mice were obtained from The Jackson Laboratory and backcrossed to C57BL/6J (B6) for at least ten generations. Before PCR based genotyping protocols were established, heterozygous mice of each strain (wl and vmd) were crossed and matings producing homozygous wl or vmd mutants were kept for strain production. As has become standard practice for these mutant mice, dry food was supplemented with a soft maintenance diet (DietGel 76A, ClearH2O). After identifying the mutations in Atp8a2 in the wl and vmd strains, PCR based protocols were established for genotyping. For wl, the primer pair wl-L 5′ - TGAACTGTCCCTTAACTGATGGTA - 3′ and wl-R, 5′ - TGGCTATGGTTTCTGGAACG - 3′ (Figure 6) were used. This primer pair spans the 21 base-pair deletion in exon 22 in wl mutant mice and produces a 108 bp amplicon in wild type controls, and an 87 bp amplicon in wl/wl mice. For the vmd genomic deletion, three primers flanking the region were used (Figure S10) to distinguish between the wild type and mutant alleles: vmdF, 5′ - CTAACTGTGGCTCACTTACCTCCT - 3′; vmdR1, 5′ - TCCTCCAGAACATTGAAGTGACTA - 3′; vmdR2, 5′ - TGCATCTTGATTTTTGCTTTGTAT - 3′. A 403 bp amplicon is produced in the presence of the wildtype allele using primer vmdF and vmdR1 and a 207 bp amplicon is produced in the presence of the mutant vmd allele using primers vmdF and vmdR2. wl3J arose in the CBA/J production colony at The Jackson Laboratory [73]. This strain is cryopreserved, and therefore genomic DNA of wl3J mutant and control animals was obtained from the DNA Resource at The Jackson Laboratory to determine the mutation.
To assess the effect of the Wlds allele on axonopathy in wl mice, the original Wlds allele (Wlds) [26]–[29], [74] was obtained from Harlan Sprague Dawley on a C57BL/10Hsd background. This allele is maintained by continuously backcrossing to C57BL/6J and was crossed to wl mice to generate the wl Wlds strain. All experiments assessing the effect of Wlds on axon degeneration were performed using mice hemizygous for Wlds. A Bax null allele (B6.129X2-Baxtm1Sjk/J, herein referred to as Bax−) was obtained from The Jackson Laboratory and crossed to wl mice to generate wl Bax−/− mice.
Histology
Anesthetized mice were fixed by transcardial perfusion with physiological saline (PBS), followed by freshly made 2% glutaraldehyde and 2% paraformaldehyde in 0.1 M cacodylate buffer (pH 7.4). Sciatic and femoral nerves were dissected under a dissecting microscope and post-fixed overnight in the same fixative. After post-fixing, nerves were rinsed twice with PBS and processed for plastic embedding, histological staining and transmission electron microscopy (TEM) by standard procedures [75]. Briefly, 0.5 µm semi-thin plastic sections were stained with toluidine blue and examined by light microscopy. The total number of myelinated axons in each nerve was counted using toluidine blue-stained plastic sections and a Leica DMRE microscope. For TEM, nerves were treated with uranyl acetate and standard embedding was done as previously described [75]. TEM images were collected on a Jeol 1230 microscope. Axon diameters of P30 and P60 mice were determined from six non-overlapping 5000× fields from each of three mutant and three littermate control samples. Axon diameters were measured using the associated software AMT Image Capture Engine. For axon counts, left and right nerves were taken, and counts obtained for both nerves were averaged. Thus each count sample represents the average count of the left and right nerve of one mouse.
For histological analysis of brain and spinal cord sections, anesthetized mice were fixed by transcardial perfusion with 2% glutaraldehyde and 2% paraformaldehyde in 0.1 M cacodylate buffer (pH 7.4). Tissues were dissected out, embedded in paraffin, serial sections obtained and stained using hematoxylin and eosin (H&E). To assess morphological structures of the retina, enucleated eyes were fixed overnight in 1.2% glutaraldehyde and 0.8% paraformaldehyde in 0.08 M phosphate buffer, embedded in Technovit resin, cut in 1.5 µm sections and stained with hematoxylin and eosin (H&E).
For analysis of the optic nerve sections with PPD staining, intracranial portions of optic nerves were processed and analyzed as previously described [19]–[23]. Briefly, optic nerves were fixed in situ in 1.2% glutaraldehyde and 0.8% paraformaldehyde in phosphate buffer for 48 hours, dissected free, processed, and embedded in plastic. One-micron-thick cross sections of optic nerve from behind the orbit were cut and stained with paraphenylenediamine (PPD). PPD darkly stains the myelin sheaths and axoplasm of sick or dying axons but not healthy axons.
Immunohistochemistry
For immunohistochemistry enucleated eyes or carefully dissected free spinal columns were fixed overnight in 4% paraformaldehyde in PBS, and then cryoprotected in sucrose. Tissues were embedded in optimal cutting temperature solution (OCT) and frozen on dry ice for sectioning. Neurofilament was stained with pNF antibody (Covance, 1∶500) and visualized with AlexaFluor488-conjugated anti-mouse IgG (Invitrogen). Nuclei were counterstained with DAPI (Sigma). Active caspase-3 (1∶200) was purchased from R&D Systems. Sections were analyzed and imaged on a Leica TCS SP5 II confocal microscope. Anti-Tubulin antibody was obtained from Sigma and used at a dilution of 1∶2000. Mouse anti-PSD95 was obtained from Neuromab (http://neuromab.ucdavis.edu/) and used at a dilution of 1∶1000.
Mapping the wl mutation
The wl mutation had originally been mapped to chromosome 14, close to the hairless (hr) locus [33]. To further narrow the interval, wl/+ male mice originally obtained from The Jackson Laboratory on a mixed background were crossed to C57BL/6J wild type females to generate F1 progeny. F1 progeny were intercrossed to generate F2 animals. Offspring from these intercrosses were examined at 3 weeks of age to identify those with abnormal gait and thus homozygous for the wl allele. Tail tissue was obtained from a total of 688 affected animals for DNA preparation and analysis. To narrow the region, MIT markers able to distinguish between C57BL/6J and wl on chromosome 14 were used [i.e. D14Mit154 (58.6Mb), D14Mit113 (60.4 Mb), DLM14-10 (60.6 Mb), DLM14-21 (61.4 Mb), D14Mit3 (61.5 Mb), and D14Mit37 (63.0 Mb)]. Primer sequences are listed in Table S1.
Identifying the mutation
Using the mapping strategy describe above, the region was narrowed to an interval of 773 Kb containing 10 genes. Primer pairs were designed to span the coding exons and splice sites for all 10 genes in the critical region. Those designed for Atp8a2 are given in Table S2. These primer pairs were utilized to obtain PCR products using Abgene 2X ReddyMix PCR Master Mix (1.5 mM MgCl2) with genomic DNA from wl/wl mutants as well as littermate controls as template. PCR products were purified using the QIAquick PCR Purification Kit (Qiagen) and subjected to sequencing using the BigDye Terminator Cycle Sequencing Kit (Applied Biosystems). Sequences were analyzed using the Sequencher 4.2 software, comparing publically available sequences for Atp8a2 with the sequences obtained for mutant and control mice. The same primer pairs and strategy were used to determine the mutation in wlvmd and wl3J mice.
Molecular cloning and site-directed mutagenesis of the Atp8a2 open reading frame (ORF)
Full-length cDNA of Atp8a2 (NM_015803.2) was synthesized using the de-novo DNA synthesis technique (GenScript Corporation, NJ, U.S.A.) and cloned into pUC57 with two flanking restriction enzyme sites EcoRV and SalI. The ORF itself is also flanked with AscI and MluI sites. The Atp8a2 ORF was sub-cloned into the pCMV6-AN-Flag (Origene) expression vector using the AscI and MluI restriction endonuclease sites to produce pCMV6-AN-Flag-ATP8A2. All sequences were confirmed using sequence analysis.
For the lipid translocation assays, the Atp8a2 ORF was amplified by PCR and sub-cloned into the pEntry1A dual selection vector (Invitrogen) at NotI and XhoI sites to generate pEntry1A-Atp8a2. PCR primers used were: Atp8a2-NotI: 5′-CACCAGTCCCGGGCCACGTCTGTTGGAGACC-3′; Atp8a2-XhoI: 5′-TTATTTCTTCCTTTCTCGAGTCTTTGGTGGTATCATAAGCGC-3′. The destination vector pcDNA6.2/EmGFP-Bsd/V5-DEST (Invitrogen Catalog no. V366-20) was used to generate the expression vector pcDNA6.2-Atp8a2 by performing an LR recombination reaction following the manufacture's instruction. pcDNA6.2/EmGFP-Bsd/V5-DEST contain the human cytomegalovirus immediate-early (CMV) promoter/enhancer for high-level expression of the gene of interest and the murine phosphoglycerate kinase-1 (PKG) promoter to drive expression of the Emerald GFP-Blasticidin (EmGFP-Bsd) fusion protein in mammalian cells. Transfected cells express both ATP8A2 and EmGFP-Bsd and can be detected by flow cytometry.
pcDNA6.2-Atp8a2wl was generated by introducing a 21 bp deletion into pcDNA6.2-Atp8a2 vector using the QuickChange II XL Site-Directed Mutagenesis Kit (Stratagene) according to manufacturer's instructions. Primers: wl-21bpdel-F, 5′-CTGTTACTTGGAG CTACAGCCGGCGTTCCAGAAACCATAGCCACTC-3′; wl-21bpdel-R, 5′-GAGTGGCTATGGTTTCTGGAACGGCTGTAGCTCCAAGTAACAG-3′. Similarly, pcDNA6.2-Atp8a2D388A was generated by the introduction of Asp→Ala (GAC→GCC) mutation into the pcDNA6.2-Atp8a2 vector. Primers: Atp8a2-changeD-F, 5′-GGGCAGGTAAAATACCTGTTTTCAGCCAAGACTGGAACTCTTACATGT -3′; Atp8a2-changeD-R, 5′-ACATGTAAGAGTTCCAGTCTTGGCTGAAAACAGGTATTTTACCTGCCC -3′. pcDNA6.2-Atp8a2vmd was generated by swapping the cDNA fragment (basepair 1489–3447th ) with the corresponding fragment amplified from vmd/vmd brain cDNA (HindIII and XhoI sites of pEntry1A-Atp8a2). Primers: vmdP1, 5′-AGCTAAGAAGCTTGGCTTTGTGTTTACCGGGAGG-3′; vmdP2, 5′-TTATTTCTTTGAATTCTCTTTGGTGGTATCATAAGCGC-3′. All mutant versions of Atp8a2 were confirmed by sequencing.
RT–PCR
Tissue from 1–2 months old animals were dissected free and placed into RNAlater (Ambion) at room temperature. Total RNA was prepared from these tissues using TRIzol Reagent (Life Technologies) according to the manufacturer's instructions. RNA samples were treated with RNase-free DNaseI (Ambion) and RNA concentration determined using a NanoDrop (ND-1000) spectrophotometer. 10 µg RNA was reverse transcribed using random primers and the MessageSensor RT Kit (Ambion). The primers used for PCR were: Atp8a2, Atp8a2F1 = 5′-GGAGCAGATCCTGGAGATTGACT-3′; Atp8a2R1 = 5′-CGGAAGCACTCTC-3′; beta-Actin, ActinbF1 = 5′-AGCCATGTACGTAGC CATCC-3′; ActinbR1 = 5′-CGGCCAGCCAGGTCCAGAC-3′.
Antibody production
The ATP8A2 antibody was produced by Proteintech Group, Inc (Chicago, IL) using an ATP8A2-GST fusion protein designed in our laboratory. Briefly, a cDNA fragment encoding amino acid 536–840 of the ATP8A2 P-domain was amplified using the following primers: Atp8a2-PDF1, 5′-TTTTGGATCCATGTCTGTCATTGTCCGACTG-3′, Atp8a2-PDR1, 5′ - TTTTCTCGAGTCAGTACAGGATACACTTGGTC-3′. The resulting PCR product was cloned in-frame with glutathione S-transferase (GST) in the pGEX-4T-1 vector (GE healthcare) using BamHI and XhoI restriction enzymes. The resulting plasmid was validated by sequencing and transformed into E. coli. BL21 was used for ATP8A2 fragment-GST fusion protein production. The fusion protein was purified with glutathione-Sepharose (GE Healthcare) using standard procedures. The purified fusion protein was used for immunization of rabbits. The ATP8A2 antibody was purified from serum by affinity purification using the GST-ATP8A2 fragment fusion protein.
Expression of Atp8a2 in HEK-293T cells
HEK-293T cells (American Type Culture Collection (ATCC), Manassas, VA) were cultured in DMEM medium with high glucose (HyClone) supplemented with 10% fetal bovine serum and 1% (vol/vol) penicillin/streptomycin at 37°C in a 5% CO2 atmosphere. Cells were seeded in six-well plates (Corning, NY) and transfected at 50% confluency with 1 µg of pCMV6-AN-Flag-Atp8a2 or empty vector using Lipofectimine 2000 (Invitrogen) according to the manufacturer's instructions. Cells were harvested after 48 hours.
UPS-1 cell culture
The CHO UPS-1 cell line is a mutant Chinese hamster ovary (CHO) cell line defective in the nonendocytic uptake of NBD-PS [34], and was kindly provided by Dr. Robert Pagano from Mayo Clinic (Rochester, MN). UPS-1 cells were grown in Ham's F12 medium supplemented with 10% fetal bovine serum and 1% glutamine (vol/vol), and 1% (vol/vol) penicillin/streptomycin at 37°C in a 5% CO2 atmosphere. Cells were seeded in 6-well plate and allowed to grow to 50% confluency. Transfection with empty vector or plasmid pcDNA62-Atp8a2 expressing ATP8A2 was carried out using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Lipid translocation assay
The following labeled phospholipid analogs were purchased from Avanti Polar Lipids: 16 : 0–06 : 0 NBD-PS [1-palmitoyl-2-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino] hexanoyl]-sn-glycero-3-phospho-L-serine (ammonium salt)]; 16 : 0–06 : 0 NBD-PE [1-palmitoyl-2-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino] hexanoyl]-sn-glycero-3-phospho-L-etholamine (ammonium salt)]; 16 : 0–06 : 0 NBD-PC [1-palmitoyl-2-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino] hexanoyl]-sn-glycero-3-phospho-choline (ammonium salt)]; and 16 : 0–06 : 0 NBD-PG [1-palmitoyl-2-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino] hexanoyl]-sn-glycero (ammonium salt)]. NBD-lipid powder stocks were dissolved in 95% ethanol and diluted to 20 µM with Hank's balanced salt solution with 15 mM MgCl2 and without phenol red (HBSS-15 mM MgCl2; Gibco). The translocation of NBD-lipid was determined as described with some modification [35]. In short, transfected UPS-1 cells grown to confluency in 6-well plates (Corning, NY) were washed, equilibrated in pre-warmed HBSS-15 mM MgCl2 for 15 minutes at 20°C, and incubated with 20 µM NBD-lipid for 20 minutes at 20°C. Subsequently, the cells were washed with HBSS-15 mM MgCl2 on ice. To quantify NBD-lipid translocated into the inner leaflet of the plasma membrane, lipid from the outer leaflet was removed by back-extraction. This was done by adding ice-cold HBSS supplemented with 2% bovine serum albumin (Sigma) to the cells for 10 min on ice, and repeating the process three times. Finally, the cells were washed with cold HBSS, treated with 0.25% Trypsin and suspended in HBSS. One microliter of 5 mg/ml DAPI in PBS was added to 3×106 cells in 0.5 ml HBSS just before FACS analysis. Flow cytometry of NBD-lipid labeled UPS-1 cells were performed on a Becton Dickinson LSR II cytometer equipped with an argon laser using FACSDiVa software. Ten thousand GFP positive cells were analyzed during the acquisition. Dead cells were excluded from the analysis by blue fluorescence (DAPI positive). The data were analyzed with FlowJo software (Tree Star Inc., Ashland, OR). The NBD-lipid fluorescence intensity of living UPS1 cells was plotted on a histogram to calculate the median fluorescence intensity. Lipid translocation activity was calculated as a ratio to vector control samples.
Western blotting
Brain and spinal cord were collected from 1 - to 2-month old mice and put directly into lysis buffer (10 mM Tris-HCL, pH 7.4, 100 mM NaCl, 1.5MgCl2, plus Complete Protease Inhibitor Cocktail (Roche)). Tissues were homogenized using a motor-driven Polytron homogenizer and the resulting lysates were centrifuged at 500 g to remove intact cells. To separate the cytosol and membrane fractions, the supernatant was subjected to centrifugation at 100,000 g for 45 min at 4°C using a Beckman SW40 rotor. The supernatant was collected as the cytosolic fraction and the pellet was resuspended in lysis buffer plus 0.1% Triton X-100 on ice for 20 min as membrane fraction. Total protein concentration was determined using the BCA Protein Assay Reagent (Pierce). Equal amounts of cytosol and membrane proteins were separated by electrophoresis using a 7.5% Mini-PROTEAN TGX Precast Gel (BioRad). Proteins were transferred to PVDF membranes (GE healthcare) and detected by immunoblotting using standard techniques.
Conduction Velocity (CV) recording
Conduction velocity of sciatic axons was determined by measuring the latency of compound motor action potentials recorded in the muscle of the left rear paw [76]. Mice were anesthetized with 1%–1.5% isoflurane and placed on a thermostatically regulated heating pad to maintain normal body temperature. Action potentials were produced by subcutaneous stimulation at two separate sites: proximal stimulation at the sciatic notch and by a second pair of needle electrodes placed distally at the ankle. For recording, a needle electrode was inserted in the center of the paw (active) and a second electrode was placed in the skin between the first and second digits. Conduction velocity was calculated as [(proximal latency – distal latency)/conduction distance]. Four animals of each genotype were tested.
Wire hang test
For the wire hang test each animal was placed on a wire cage top held approximately a foot above an empty cage. The wire cage top was then gradually inverted and the time each mouse was able to hang onto the cage top was recorded. If any mouse still gripped the wire top after 60 seconds, the animal was removed and the time was recorded as 60 seconds.
Statistical analysis
All data are presented as means ± SEM. Data were analyzed in JMP7.0 (SAS Campus Drive, Building T, Cary, NC). The differences between two groups was assessed by Student's t-test; p<0.05 was considered statistically significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BreedveldG, de CooIF, LequinMH, ArtsWF, HeutinkP, et al. (2006) Novel mutations in three families confirm a major role of COL4A1 in hereditary porencephaly. Journal of medical genetics 43 : 490–495.
2. FarberDB, LolleyRN (1974) Cyclic guanosine monophosphate: elevation in degenerating photoreceptor cells of the C3H mouse retina. Science 186 : 449–451.
3. GouldDB, PhalanFC, BreedveldGJ, van MilSE, SmithRS, et al. (2005) Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science 308 : 1167–1171.
4. GouldDB, PhalanFC, van MilSE, SundbergJP, VahediK, et al. (2006) Role of COL4A1 in small-vessel disease and hemorrhagic stroke. The New England journal of medicine 354 : 1489–1496.
5. KeelerCE (1924) The Inheritance of a Retinal Abnormality in White Mice. Proc Natl Acad Sci U S A 10 : 329–333.
6. LanfranconiS, MarkusHS (2010) COL4A1 mutations as a monogenic cause of cerebral small vessel disease: a systematic review. Stroke; a journal of cerebral circulation 41: e513–518.
7. McLaughlinME, SandbergMA, BersonEL, DryjaTP (1993) Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat Genet 4 : 130–134.
8. PittlerSJ, BaehrW (1991) Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase beta-subunit gene of the rd mouse. Proc Natl Acad Sci U S A 88 : 8322–8326.
9. PittlerSJ, KeelerCE, SidmanRL, BaehrW (1993) PCR analysis of DNA from 70-year-old sections of rodless retina demonstrates identity with the mouse rd defect. Proc Natl Acad Sci U S A 90 : 9616–9619.
10. SidmanRL, GreenMC (1965) Retinal degeneration in the mouse: location of the RD locus in linkage group XVII. J Hered 56 : 23–29.
11. DickieMM, SchneiderJ, HarmanPJ (1952) A juvenile wabbler lethal in the house mouse. Journal of Heredity 43 : 5.
12. CarrollEW, CurtisRL, SullivanDA, MelvinJL (1992) Wallerian degeneration in the optic nerve of the wabbler-lethal (wl/wl) mouse. Brain Res Bull 29 : 411–418.
13. LuseSA, ChenardC, FinkeEH (1967) The wabbler-lethal mouse. An electron microscopic study of the nervous system. Archives of Neurology 17 : 153–161.
14. BronsonRT, SweetHO, SpencerCA, DavissonMT (1992) Genetic and age related models of neurodegeneration in mice: dystrophic axons. J Neurogenet 8 : 71–83.
15. HarmanPJ (1954) Genetically Controlled Demyelination in the Mammalian Central Nervous System: Demyelination in Mammals. Annals of the New York Academy of Sciences 58 : 546–550.
16. Love S, Louis DN, Ellison DW (2008) Greenfield's Neuropathology. London Hodder Arnold. 2 p.
17. MizusawaH, MatsumotoS, YenSH, HiranoA, Rojas-CoronaRR, et al. (1989) Focal accumulation of phosphorylated neurofilaments within anterior horn cell in familial amyotrophic lateral sclerosis. Acta Neuropathologica 79 : 37–43.
18. SotoI, OglesbyE, BuckinghamBP, SonJL, RobersonEDO, et al. (2008) Retinal Ganglion Cells Downregulate Gene Expression and Lose Their Axons within the Optic Nerve Head in a Mouse Glaucoma Model. Journal of Neuroscience 28 : 548–561.
19. AndersonMG, LibbyRT, GouldDB, SmithRS, JohnSW (2005) High-dose radiation with bone marrow transfer prevents neurodegeneration in an inherited glaucoma. Proc Natl Acad Sci U S A 102 : 4566–4571.
20. LibbyRT, AndersonMG, PangIH, RobinsonZH, SavinovaOV, et al. (2005) Inherited glaucoma in DBA/2J mice: pertinent disease features for studying the neurodegeneration. Vis Neurosci 22 : 637–648.
21. LibbyRT, LiY, SavinovaOV, BarterJ, SmithRS, et al. (2005) Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet 1
22. SadunAA, SmithLE, KenyonKR (1983) Paraphenylenediamine: a new method for tracing human visual pathways. Journal of neuropathology and experimental neurology 42 : 200–206.
23. Smith RS, Zabaleta A, John SW, Bechtold LS, Ikeda S, et al. (2002) General and Specific Histopathology. In: Smith RS, editor. Systemic evaluation of the mouse eye. New York: CRC Press. pp. 265–297.
24. FengY, YanT, HeZ, ZhaiQ (2010) Wld(S), Nmnats and axon degeneration–progress in the past two decades. Protein & cell 1 : 237–245.
25. FerriA, SanesJR, ColemanMP, CunninghamJM, KatoAC (2003) Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Curr Biol 13 : 669–673.
26. LunnER, PerryVH, BrownMC, RosenH, GordonS (1989) Absence of Wallerian Degeneration does not Hinder Regeneration in Peripheral Nerve. Eur J Neurosci 1 : 27–33.
27. MackTG, ReinerM, BeirowskiB, MiW, EmanuelliM, et al. (2001) Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nature Neuroscience 4 : 1199–1206.
28. PerryVH, BrownMC, LunnER, TreeP, GordonS (1990) Evidence that Very Slow Wallerian Degeneration in C57BL/Ola Mice is an Intrinsic Property of the Peripheral Nerve. Eur J Neurosci 2 : 802–808.
29. PerryVH, LunnER, BrownMC, CahusacS, GordonS (1990) Evidence that the Rate of Wallerian Degeneration is Controlled by a Single Autosomal Dominant Gene. Eur J Neurosci 2 : 408–413.
30. RibchesterRR, TsaoJW, BarryJA, Asgari-JirhandehN, PerryVH, et al. (1995) Persistence of neuromuscular junctions after axotomy in mice with slow Wallerian degeneration (C57BL/WldS). Eur J Neurosci 7 : 1641–1650.
31. NikolaevA, McLaughlinT, O'LearyDD, Tessier-LavigneM (2009) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457 : 981–989.
32. SchoenmannZ, Assa-KunikE, TiomnyS, MinisA, Haklai-TopperL, et al. (2010) Axonal degeneration is regulated by the apoptotic machinery or a NAD+-sensitive pathway in insects and mammals. J Neurosci 30 : 6375–6386.
33. LanePW, DickieMM (1961) Linkage of wabbler-lethal and hairless in the mouse. Journal of Heredity 52 : 2.
34. HanadaK, PaganoRE (1995) A Chinese hamster ovary cell mutant defective in the non-endocytic uptake of fluorescent analogs of phosphatidylserine: isolation using a cytosol acidification protocol. J Cell Biol 128 : 793–804.
35. PaulusmaCC, FolmerDE, Ho-MokKS, de WaartDR, HilariusPM, et al. (2008) ATP8B1 requires an accessory protein for endoplasmic reticulum exit and plasma membrane lipid flippase activity. Hepatology 47 : 268–278.
36. HowellGR, LibbyRT, JakobsTC, SmithRS, PhalanFC, et al. (2007) Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol 179 : 1523–1537.
37. Meyer zu HorsteG, MiesbachTA, MullerJI, FledrichR, StassartRM, et al. (2011) The Wlds transgene reduces axon loss in a Charcot-Marie-Tooth disease 1A rat model and nicotinamide delays post-traumatic axonal degeneration. Neurobiol Dis 42 : 1–8.
38. MiW, BeirowskiB, GillingwaterTH, AdalbertR, WagnerD, et al. (2005) The slow Wallerian degeneration gene, WldS, inhibits axonal spheroid pathology in gracile axonal dystrophy mice. Brain 128 : 405–416.
39. PerryVH, BrownMC, LunnER (1991) Very Slow Retrograde and Wallerian Degeneration in the CNS of C57BL/Ola Mice. Eur J Neurosci 3 : 102–105.
40. TangX, HalleckMS, SchlegelRA, WilliamsonP (1996) A Subfamily of P-Type ATPases with Aminophospholipid Transporting Activity. Science 272 : 1495–1497.
41. ColemanJA, MoldayRS (2011) Critical role of the beta-subunit CDC50A in the stable expression, assembly, subcellular localization, and lipid transport activity of the P4-ATPase ATP8A2. The Journal of biological chemistry 286 : 17205–17216.
42. ColemanJA, KwokMC, MoldayRS (2009) Localization, purification, and functional reconstitution of the P4-ATPase Atp8a2, a phosphatidylserine flippase in photoreceptor disc membranes. The Journal of biological chemistry 284 : 32670–32679.
43. BullLN, EijkMJTv, PawlikowskaL, DeYoungJA, JuijnJA, et al. (1998) A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nature Genetics 18 : 219–224.
44. FolmerDE, ElferinkRP, PaulusmaCC (2009) P4 ATPases - lipid flippases and their role in disease. Biochim Biophys Acta 1791 : 628–635.
45. SiggsOM, ArnoldCN, HuberC, PirieE, XiaY, et al. (2011) The P4-type ATPase ATP11C is essential for B lymphopoiesis in adult bone marrow. Nat Immunol 12 : 434–440.
46. SiggsOM, SchnablB, WebbB, BeutlerB (2011) X-linked cholestasis in mouse due to mutations of the P4-ATPase ATP11C. Proc Natl Acad Sci U S A 108 : 7890–7895.
47. YabasM, TehCE, FrankenreiterS, LalD, RootsCM, et al. (2011) ATP11C is critical for the internalization of phosphatidylserine and differentiation of B lymphocytes. Nat Immunol 12 : 441–449.
48. Darland-RansomM, WangX, SunC-L, MapesJ, Gengyo-AndoK, et al. (2008) Role of C. elegans TAT-1 Protein in Maintaining Plasma Membrane Phosphatidylserine Asymmetry. Science 320 : 528–531.
49. StapelbroekJM, PetersTA, van BeurdenDHA, CurfsJHAJ, JoostenA, et al. (2009) ATP8B1 is essential for maintaining normal hearing. Proceedings of the National Academy of Sciences 106 : 9709–9714.
50. WangL, BeserraC, GarbersDL (2004) A novel aminophospholipid transporter exclusively expressed in spermatozoa is required for membrane lipid asymmetry and normal fertilization. Developmental Biology 267 : 203–215.
51. DharMS, SommardahlCarla S, KirklandTanisa, NelsonSarah, DonnellRobert, JohnsonDabney K, CastellaniLawrence W (2004) Mice Heterozygous for Atp10c, a Putative Amphipath, Represent a Novel Model of Obesity and Type 2 Diabetes. Journal of Nutrition 134 : 799–805.
52. MannoS, TakakuwaY, MohandasN (2002) Identification of a functional role for lipid asymmetry in biological membranes: Phosphatidylserine-skeletal protein interactions modulate membrane stability. Proceedings of the National Academy of Sciences of the United States of America 99 : 1943–1948.
53. ChenCY, IngramMF, RosalPH, GrahamTR (1999) Role for Drs2p, a P-type ATPase and potential aminophospholipid translocase, in yeast late Golgi function. The Journal of cell biology 147 : 1223–1236.
54. ChowCY, ZhangY, DowlingJJ, JinN, AdamskaM, et al. (2007) Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 448 : 68–72.
55. ZhangX, ChowCY, SahenkZ, ShyME, MeislerMH, et al. (2008) Mutation of FIG4 causes a rapidly progressive, asymmetric neuronal degeneration. Brain : a journal of neurology 131 : 1990–2001.
56. DuexJE, NauJJ, KauffmanEJ, WeismanLS (2006) Phosphoinositide 5-phosphatase Fig 4p is required for both acute rise and subsequent fall in stress-induced phosphatidylinositol 3,5-bisphosphate levels. Eukaryotic cell 5 : 723–731.
57. NicholsonG, LenkGM, ReddelSW, GrantAE, TowneCF, et al. (2011) Distinctive genetic and clinical features of CMT4J: a severe neuropathy caused by mutations in the PI(3,5)P(2) phosphatase FIG4. Brain : a journal of neurology 134 : 1959–1971.
58. DuexJE, TangF, WeismanLS (2006) The Vac14p-Fig4p complex acts independently of Vac7p and couples PI3,5P2 synthesis and turnover. The Journal of cell biology 172 : 693–704.
59. LenkGM, FergusonCJ, ChowCY, JinN, JonesJM, et al. (2011) Pathogenic mechanism of the FIG4 mutation responsible for Charcot-Marie-Tooth disease CMT4J. PLoS Genet 7: e1002104.
60. Di PaoloG, De CamilliP (2006) Phosphoinositides in cell regulation and membrane dynamics. Nature 443 : 651–657.
61. VicinanzaM, D'AngeloG, Di CampliA, De MatteisMA (2008) Function and dysfunction of the PI system in membrane trafficking. The EMBO journal 27 : 2457–2470.
62. VaccariI, DinaG, TronchereH, KaufmanE, ChicanneG, et al. (2011) Genetic interaction between MTMR2 and FIG4 phospholipid phosphatases involved in Charcot-Marie-Tooth neuropathies. PLoS Genet 7: e1002319
63. BolinoA, BolisA, PrevitaliSC, DinaG, BussiniS, et al. (2004) Disruption of Mtmr2 produces CMT4B1-like neuropathy with myelin outfolding and impaired spermatogenesis. The Journal of cell biology 167 : 711–721.
64. BolinoA, MugliaM, ConfortiFL, LeGuernE, SalihMA, et al. (2000) Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nature Genetics 25 : 17–19.
65. BolisA, CovielloS, BussiniS, DinaG, PardiniC, et al. (2005) Loss of Mtmr2 phosphatase in Schwann cells but not in motor neurons causes Charcot-Marie-Tooth type 4B1 neuropathy with myelin outfoldings. The Journal of neuroscience : the official journal of the Society for Neuroscience 25 : 8567–8577.
66. BegleyMJ, TaylorGS, KimSA, VeineDM, DixonJE, et al. (2003) Crystal structure of a phosphoinositide phosphatase, MTMR2: insights into myotubular myopathy and Charcot-Marie-Tooth syndrome. Molecular cell 12 : 1391–1402.
67. KimSA, TaylorGS, TorgersenKM, DixonJE (2002) Myotubularin and MTMR2, phosphatidylinositol 3-phosphatases mutated in myotubular myopathy and type 4B Charcot-Marie-Tooth disease. The Journal of biological chemistry 277 : 4526–4531.
68. UchidaY, HasegawaJ, ChinnapenD, InoueT, OkazakiS, et al. (2011) Intracellular phosphatidylserine is essential for retrograde membrane traffic through endosomes. Proc Natl Acad Sci U S A 108 : 15846–15851.
69. ZhangZ, HuiE, ChapmanER, JacksonMB (2009) Phosphatidylserine regulation of Ca2+-triggered exocytosis and fusion pores in PC12 cells. Mol Biol Cell 20 : 5086–5095.
70. LevanoK, PuniaV, RaghunathM, DebataPR, CurcioGM, et al. (2011) Atp8a1 Deficiency is Associated with Phosphatidylserine Externalization in Hippocampus and Delayed Hippocampus-Dependent Learning. J Neurochem
71. FadokVA, SavillJS, HaslettC, BrattonDL, DohertyDE, et al. (1992) Different populations of macrophages use either the vitronectin receptor or the phosphatidylserine receptor to recognize and remove apoptotic cells. Journal of Immunology 149 : 4029–4035.
72. CacciagliP, HaddadMR, Mignon-RavixC, El-WalyB, MonclaA, et al. (2010) Disruption of the ATP8A2 gene in a patient with a t(10;13) de novo balanced translocation and a severe neurological phenotype. Eur J Hum Genet 18 : 1360–1363.
73. CookS (1995) Spontaneous remutation (wl<3J>). Mouse Genome 93 : 862.
74. ColemanMP, ConfortiL, BuckmasterEA, TarltonA, EwingRM, et al. (1998) An 85-kb tandem triplication in the slow Wallerian degeneration (Wlds) mouse. Proc Natl Acad Sci U S A 95 : 9985–9990.
75. SeburnKL, NangleLA, CoxGA, SchimmelP, BurgessRW (2006) An Active Dominant Mutation of Glycyl-tRNA Synthetase Causes Neuropathy in a Charcot-Marie-Tooth 2D Mouse Model. Neuron 51 : 715–726.
76. OcchiS, ZambroniD, Del CarroU, AmadioS, SirkowskiEE, et al. (2005) Both Laminin and Schwann Cell Dystroglycan Are Necessary for Proper Clustering of Sodium Channels at Nodes of Ranvier. Journal of Neuroscience 25 : 9418–9427.
77. ThiessenD (1965) The wabbler-lethal mouse: A study in development. Animal Behavior 13 : 87–100.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 8
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Dissecting the Gene Network of Dietary Restriction to Identify Evolutionarily Conserved Pathways and New Functional Genes
- It's All in the Timing: Too Much E2F Is a Bad Thing
- A Quantitative Comparison of the Similarity between Genes and Geography in Worldwide Human Populations
- Variation of Contributes to Dog Breed Skull Diversity
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy