
Use of Allele-Specific FAIRE to Determine Functional Regulatory Polymorphism Using Large-Scale Genotyping Arrays
Following the widespread use of genome-wide association studies (GWAS), focus is turning towards identification of causal variants rather than simply genetic markers of diseases and traits. As a step towards a high-throughput method to identify genome-wide, non-coding, functional regulatory variants, we describe the technique of allele-specific FAIRE, utilising large-scale genotyping technology (FAIRE-gen) to determine allelic effects on chromatin accessibility and regulatory potential. FAIRE-gen was explored using lymphoblastoid cells and the 50,000 SNP Illumina CVD BeadChip. The technique identified an allele-specific regulatory polymorphism within NR1H3 (coding for LXR-α), rs7120118, coinciding with a previously GWAS-identified SNP for HDL-C levels. This finding was confirmed using FAIRE-gen with the 200,000 SNP Illumina Metabochip and verified with the established method of TaqMan allelic discrimination. Examination of this SNP in two prospective Caucasian cohorts comprising 15,000 individuals confirmed the association with HDL-C levels (combined beta = 0.016; p = 0.0006), and analysis of gene expression identified an allelic association with LXR-α expression in heart tissue. Using increasingly comprehensive genotyping chips and distinct tissues for examination, FAIRE-gen has the potential to aid the identification of many causal SNPs associated with disease from GWAS.
Published in the journal:
. PLoS Genet 8(8): e32767. doi:10.1371/journal.pgen.1002908
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002908
Summary
Following the widespread use of genome-wide association studies (GWAS), focus is turning towards identification of causal variants rather than simply genetic markers of diseases and traits. As a step towards a high-throughput method to identify genome-wide, non-coding, functional regulatory variants, we describe the technique of allele-specific FAIRE, utilising large-scale genotyping technology (FAIRE-gen) to determine allelic effects on chromatin accessibility and regulatory potential. FAIRE-gen was explored using lymphoblastoid cells and the 50,000 SNP Illumina CVD BeadChip. The technique identified an allele-specific regulatory polymorphism within NR1H3 (coding for LXR-α), rs7120118, coinciding with a previously GWAS-identified SNP for HDL-C levels. This finding was confirmed using FAIRE-gen with the 200,000 SNP Illumina Metabochip and verified with the established method of TaqMan allelic discrimination. Examination of this SNP in two prospective Caucasian cohorts comprising 15,000 individuals confirmed the association with HDL-C levels (combined beta = 0.016; p = 0.0006), and analysis of gene expression identified an allelic association with LXR-α expression in heart tissue. Using increasingly comprehensive genotyping chips and distinct tissues for examination, FAIRE-gen has the potential to aid the identification of many causal SNPs associated with disease from GWAS.
Introduction
The proliferation of genome-wide association studies (GWAS) has achieved considerable advances concerning the identification of novel genetic loci associated with phenotypic traits and diseases, and also confirmed many established genetic associations. Following GWAS, the next objective in genetics will be identification of the causal variants marked by current GWAS, and determination of the molecular mechanisms altered by these genetic variants. This step will be another major milestone towards realisation of the fundamental goal for GWAS, in developing novel drug targets based on this new genetic information.
Only a small percentage of GWAS hits are themselves non-synonymous coding SNPs, with their expected causality by changing protein structure and function. The majority of GWAS hits occur within intronic and intergenic regions of the genome and are likely to exert their effects at the level of gene regulation [1]. Due to the complex nature of gene regulation [2], with regulatory elements commonly occurring up to 100 kb from a transcription start site (TSS), identifying the causal SNP from potentially hundreds of other SNPs that are simply in near or complete linkage disequilibrium (LD) with one identified from GWAS, is a challenging undertaking.
The ENCODE project has significantly increased our understanding of the location of regulatory elements throughout the genome [3]. Using techniques such as chromatin immunoprecipitation followed by sequencing (ChIP-seq), we now know the genomic binding sites for some of the key transcription factors (TF) involved in gene regulation in a number of experimental tissues. This technique relies on the existence of a ChIP-grade antibody to recognise each DNA-bound transcription factor, and is the major limitation towards the complete characterisation of all human TF binding sites [4]. A more widespread use of ChIP-seq has been the annotation of the genome for histone methylation signatures, such as H3K4me1 and H3K4me3, strong markers of enhancers and promoters [5]. Other sequencing techniques have been used to map the genome for open chromatin, including DNase I hypersensitivity (DNase-seq) [6] and formaldehyde-assisted isolation of regulatory elements (FAIRE-seq) [7]. These regions of open chromatin correlate extremely highly with both histone methylation signatures and TF ChIP-seq, but in contrast to ChIP-seq, are able to identify regulatory regions without prior knowledge of a specific transcription factor involved.
If a non-coding SNP associated with gene regulation were to be functional, it would be expected to alter not only transcription factor binding, but also histone methylation signatures and chromatin accessibility. We have applied this hypothesis to identify the functionality of SNPs on a larger scale than has previously been possible, using gene chip technology. In this paper we describe a method for allele-specific FAIRE using gene chip technology, we term FAIRE-gen, to identify possible candidate functional SNPs in loci related to cardiovascular disease.
Results
Use of FAIRE-gen to Determine Allele-Specific Enrichment of Regulatory Regions
To examine the potential to use gene chips to assess allele-specific FAIRE, three lymphoblastoid cell lines were examined following IL-1β stimulation to induce cell proliferation [8]. Subsequent to cell fixing, chromatin extraction and sonication, the fragmented chromatin was divided into two groups for each cell line: a control DNA and a FAIRE-enriched DNA sample. For the control DNA, the crosslinks were reversed and the DNA purified; for the FAIRE-enriched DNA, the chromatin underwent three rounds of phenol:chloroform extraction to enrich the sample for open chromatin, followed by reversal of crosslinks and DNA purification. Both samples were standardised to 50 ng/µl and genotyping performed using the Illumina CVD BeadChip, a custom-designed chip containing 49,094 SNPs from gene loci selected to play a potential role in cardiovascular disease (Figure 1).

Genotyping call frequencies for sonicated control DNA were comparable to non-fragmented DNA (97.2% vs 98.1%); whereas those for FAIRE-enriched DNA were significantly lower (56.7%). Using an existing lymphoblastoid FAIRE-seq dataset, the level of enrichment at the location of the CVD BeadChip SNPs was compared with the FAIRE-gen samples. The logR ratio output from the Illumina GenomeStudio was used to indicate the level of allelic amplification and therefore FAIRE-gen enrichment, compared to the respective control samples. A strong association of mean FAIRE-gen-enriched allelic intensity with FAIRE-seq peak intensity was observed (p = 2.34×10−82, Figure 2). The reduced amplification of alleles outside of open chromatin results in decreased genotype clustering and a lower call-rate in the FAIRE-enriched samples.

Following FAIRE, an allelic effect on open chromatin would enrich one allele over the other in a heterozygous individual. To examine whether this small dataset was large enough to identify an allele-specific effect on open chromatin, each sonicated control sample and its respective FAIRE-enriched sample was examined using the B allele frequency (BAF), which measures the proportion of the genotype from an individual attributed to the B allele (often the minor allele). To ensure a consistent allelic effect was found, only SNPs that were heterozygous in all three cell lines were examined. This reduced the number of SNPs under analysis to 3,129.
These 3,129 heterozygous SNPs were examined for allelic enrichment, where the control BAF and FAIRE-enriched BAF were compared for each cell line. One SNP showed a statistical significant difference with all three cell lines after applying the Bonferroni correction: rs7120118 (Figure 3), where the C allele was enriched in open chromatin. The fact that only a single association was identified was not unexpected for such a genotyping chip, where the SNP coverage per gene is low and concentrated within coding regions, where the majority of genes covered do not overlap with eQTLs or GWAS studies, and considering the very small number of cell lines examined. Despite only one SNP reaching the Bonferroni cut-off, there was overall enrichment in the study for p-values<0.05 (Figure S1), highlighting the potential for a greater number of significant results with a larger sample.

The SNP that did show statistical significance is located within intron 6 of NR1H3, coding for LXR-α. Examining genomic annotations for this SNP on the UCSC Genome Browser, it can be seen that not only is this SNP located in a region of open chromatin by DNase I-seq [9], [10], FAIRE-seq [7], [11] and with enhancer-specific histone methylation signatures [5], [12] (Figure 4), it has also been identified as a GWAS SNP for HDL-C levels [13].

Replication of rs7120118 FAIRE-gen Using 200K SNP Illumina Metabochip
To confirm the effects seen using the Illumina CVD BeadChip on rs7120118 with allele-specific FAIRE, the study was replicated using the Illumina Metabochip, a consortia custom-designed genotyping chip, containing 196,726 SNPs to primarily examine associations identified by GWAS for cardiometabolic traits and diseases, those in strong LD, and also a number of rare variants. The Metabochip contains rs7120118, and seven out of the eight further SNPs identified as in complete LD with rs7120118 from the CEU panel in the 1000 Genome Project.
A total of 20 lymphoblastoid cells were examined, including new FAIRE preparations for the original three cell lines. 6 additional cell lines were heterozygous for rs7120118, excluding the three previously examined. Comparing BAF between sonicated controls and FAIRE DNA for these 6 cell lines, the C allele was again enriched in the FAIRE sample (control BAF = 0.44, FAIRE-enriched BAF = 0.67, p = 0.0036).
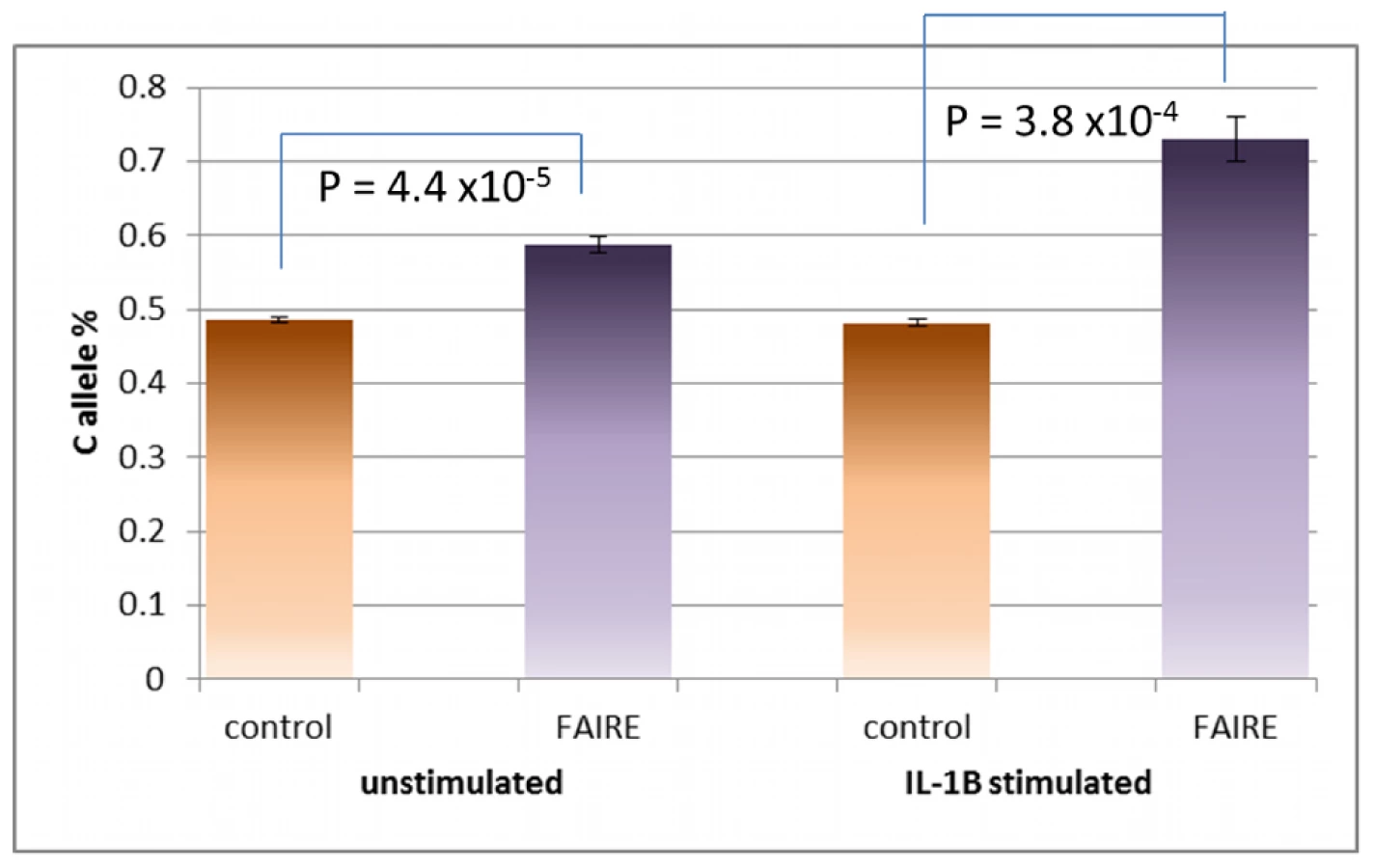
The seven SNPs in complete LD with rs7120118 were examined by the same analysis from the Metabochip using all 9 heterozygous cell lines. Unlike the original Illumina CVD BeadChip assay, Metabochip FAIRE-gen was performed on both unstimulated and IL-1β-stimulated lymphoblastoid cell lines, allowing a direct comparison of IL-1β stimulation on allele-specific open chromatin. The results for all analyses are shown in Table 1. The rs7120118 C allele was enriched with and without IL-1β stimulation by 15.5% (p = 0.008) and 4.4% (p = 0.022), respectively. No other SNPs from the seven in complete LD with rs7120118 in the IL-1β-stimulated cell lines showed allelic enrichment. From the stimulated cell lines there was a trend towards BAF enrichment from the adjacent SNP rs2279239 (11.3%, p = 0.01, Figure 5), contained within the same region of open chromatin, but this did not reach statistical significance when correcting for multiple comparisons. Examining the unstimulated cell lines, two further SNPs showed modest allelic imbalance following FAIRE: rs2167079 (exon 1 of ACP2), with a 10.3% reduction in BAF (p = 0.003) and rs326222 (intron 8 of DDB2) with a 4.9% reduction in BAF (p = 0.003).


Use of TaqMan Allelic Discrimination to Confirm Allele-Specific Genotyping
To confirm the allele-specific enrichment from the C allele of rs7120118, genotyping of the 20 sonicated control and FAIRE samples was carried out using the TaqMan platform for allelic discrimination. Allelic ratios were determined by extrapolation from a standard curve of the vic/fam ratio from samples of known genotype. The allelic ratios do not differ significantly from the Metabochip data, confirming the ability for gene chips to provide a suitable high-throughput method for FAIRE-gen (Figure 6).
Confirmation of rs7120118 as a Marker for HDL-C Plasma Levels
The SNP showing the greatest and most consistent allelic effect for open chromatin, and confirmed in two subsequent genotyping platforms, rs7120118, has been identified using GWAS as being associated with plasma HDL-C levels [13]. The SNP was associated with a beta coefficient of 0.04 (0.0073 SE, p = 6.7×10−8), but this finding has not been replicated in further GWAS, and not reported in a recent meta-analysis of lipid traits comprising >100,000 individuals [14]. To confirm the original association, we examined this SNP in a prospective UK cohort of 4724 individuals from the Whitehall II study. Baseline characteristics of the study are shown in Table 2. This data replicated the reported association with an HDL-C raising effect from the C allele (beta = 0.016, p = 0.0059). No other SNPs in strong LD with this SNP (r2>0.5) showed significantly greater effect sizes (Table 3). An additional cohort, the Copenhagen City Heart Study (CCHS; n = 10,322, baseline characteristics shown in Table 2) was genotyped for rs7120118, and this also showed a similar effect size (beta = 0.015, p = 0.041, Table 4). Combining the two datasets in a meta-analysis using a fixed-effects model did not alter the effect size (beta = 0.016) although increased the significance (p = 0.0006). As there is an association between gender and HDL-C levels in the general population, we also carried out stratification for gender. This showed a similar direction of effect in both studies, showing that the effect seen with rs7120118 functionality is unlikely to be gender-specific. This correlates with the functional in vivo findings, where the rs7120118 C allele is associated with open chromatin in cells from both male and female origin (data not shown).



Effects of rs7120118 on Gene Expression
To determine if the association of rs7120118 with both HDL-C levels and open chromatin was also associated with an intermediate phenotype of NR1H3 gene expression, this SNP was examined in five tissue samples from 316 patients undergoing aortic valve surgery. A significant allele-specific effect was observed in heart tissue (p = 0.0127) (Figure 7), with a trend towards significance in aortic adventitia (P = 0.154). In both cases the C allele of rs7120118 was associated with an upregulation of NR1H3 expression.

Discussion
We have examined the possibility of using high-throughput gene chips to examine the allele-specific nature of open chromatin using FAIRE (illustrated in Figure 1). The study identified a functional SNP, rs7120118, where the minor C allele is enriched in open chromatin and associated with increased HDL-C. Although the level of significance for HDL-C levels was adequate for a SNP with an a priori hypothesis, this would be much lower than required for genome-wide significance, highlighting the importance of combining functional studies with GWAS to identify candidate SNPs for disease or trait associations, particularly those with lower effect sizes, rare SNPs or small cohorts. Indeed, examining a recent meta-analysis of lipid traits in >100,000 individuals, rs7120118 did show a strong association with HDL-C levels (p = 1.297×10−14, Figure 8) although this was not reported as significant in the study [14], perhaps due to the strong LD in the region, with the association signals covering >29 genes. We have shown that the minor allele is associated with increased NR1H3 gene expression in heart tissue and aortic adventitia, adding to a previous genome-wide study revealing a significant association with rs7120118 and gene expression of NR1H3 and ACP2 in lymphoblast cells [15]. From this data it can be postulated that rs7120118 directly affects a long-range regulatory element (>15 kb from NR1H3 TSS) in a non-tissue-specific manner, altering gene expression and HDL-C levels.

The principle of allele-specific FAIRE was previously applied by Gaulton et al to examine the functionality of a single type II diabetes (T2D) GWAS SNP in TCF7L2 [16]. The authors used FAIRE-seq to determine global tissue-specific regions of open chromatin in pancreatic tissue, followed by TaqMan allelic discrimination to ascertain the effect of a single putative functional SNP on open chromatin. They found that the allele conferring increased risk of T2D and higher gene expression was also associated with enrichment for open chromatin. Although successfully demonstrating the use of FAIRE to identify a causal SNP from a GWAS, the use of TaqMan would not be applicable for examining a large number of potentially functional SNPs. FAIRE-gen, in contrast is only restricted by the number of SNPs that can fit on a genotyping chip.
The action of IL-1β on chromatin structure, a cytokine known to induce proliferation of EBV-transformed lymphoblasts [8], was examined in this study to reveal further potential allele-specific differences in open chromatin under different environmental conditions. For rs7120118, an allele-specific effect was observed in both unstimulated and IL-1β-stimulated cell lines, although the effects were stronger in the IL-1β stimulated samples. The action of IL-1β activates NF-κB, potentially altering expression of transcription factors that bind to the regulatory region surrounding rs7120118. Indeed, a nearby cluster of transcription factor binding sites determined by ChIP-seq includes a site for c-Jun binding (Figure 4); the JUN promoter contains several NF-κB binding sites (UCSC Genome Browser hg19/NCBI37) [3], which may explain this enhanced effect. It could be hypothesised that the C allele that favours open chromatin allows for preferential access for known, or as yet uncharacterised, transcription factors, which would act as an enhancer for NR1H3 gene expression, and increased HDL-C levels.
In contrast, a potential allelic effect was observed with the promoter SNP rs2167079 (in complete LD with rs7120118), only in unstimulated cells. IL-1β is known to reduce expression of NR1H3 in HK-2 cells [17], and it could be postulated that IL-1β may lead to chromatin remodelling and a decrease in open chromatin at the NR1H3 promoter in lymphoblasts, accounting for the lack of allelic effect in the IL-1β-stimulated cells. Alternatively, the modest allele-specific chromatin effects from the unstimulated cell lines could simply represent false-positive findings.
Haplotype structure may also affect local chromatin, particularly where more than one SNP occurs in the same region of open chromatin. We have examined the variation surrounding rs7120118 using HapMap-derived genotypes for the lymphoblasts used in the Metabochip study. No further SNPs at the locus provided additional haplotypic information for the effects on open chromatin, suggesting that rs7120118, rather than a haplotype, is responsible for this observation.
To assess the reproducibility of the FAIRE-gen methodology, the two Metabochip datasets were examined, considering the second IL-1β-treated study as a replicate. Examining the SNPs showing an allele-specific effect on open chromatin from the untreated samples, following Bonferroni correction (p<5.2×107; n = 127), 100% were replicated in the treated sample with significance set at p<0.05, (91% replicated with Pc<3.9×10−4; n = 116), indicating the sensitivity of the assay. The sensitivity and specificity of the assay to identify true functional variants can only be accurately determined by further functional analysis of each putative SNP. The smallest detectable difference in allele-specific open chromatin for the SNPs reaching genome-wide Bonferroni cut-off in the Metabochip was 10% (rs75106522).
One limitation with FAIRE-gen, as opposed to FAIRE-seq is the dependence of the gene chip to contain all relevant SNPs for the trait under examination. For the recent custom-designed chips which contain dense markers and aim to include all SNPs that tag GWAS-identified markers for diseases and related traits, such as the Illumina Metabochip and Immunochip, this is less of a problem. Future genotyping chips containing all common SNPs associated with diseases/traits could potentially resolve this drawback. For determining the location of potential causal SNPs from a number of SNPs acting as proxies, FAIRE-gen is only able to identify single allele-specific SNPs if other proxies are not located within the same region of open chromatin. This can be illustrated for rs7120118, where a nearby SNP, rs2279239, is located only 4.6 kb away, and close to the same region of open chromatin (Figure S2). This SNP shows a similar trend for allelic-specificity, although somewhat reduced due to the distance from the putative causal SNP.
Since the assay includes data from SNPs that are not present in open chromatin, there may also be a number of false-positive associations from the methodology, where amplification from background (non-open) chromatin may, in theory, preferentially occur for one allele. For this reason, replication using FAIRE-gen or FAIRE-seq in a separate study, and in vitro methodologies would be desirable to confirm functionality.
In conclusion, FAIRE-gen shows promise as an economical, high-throughput method to enable targeted unbiased detection of allele-specific regulatory elements, which may help to refine GWAS disease-association signals to identify disease-causing variants.
Materials and Methods
Ethics Statement
The Whitehall II study was approved by the UCL Research Ethics Committee, and participants gave informed consent to each aspect of the study. The CCHS was approved by institutional review boards and Danish ethical committees, and conducted according to the Declaration of Helsinki. Written informed consent was obtained from all participants.
Cell Lines and Culture
20 EBV-transformed lymphoblastoid cell lines, derived from the Centre d'Etude du Polymorphism Humain (CEPH) panel (Coriell Cell Repositories, identifiers listed in table S1), were cultured in RPMI 1640 (PAA) with 2 mM L-glutamine and 15% fetal bovine serum (PAA) at 37°C, 5% CO2. Cell viability was verified using the ADAM-MC cell counter (Digital Bio), and minimum cell viability for experiments was ≥99%. Stimulation of cells was carried out by an overnight incubation in serum-free media, and addition of 5 ng/ml IL-1β, two hours prior to cell fixing.
Chromatin Fixing, Isolation, and Sonication
1×108 cells were cultured for each experiment and incubated with 1/10 volume of fresh 11% formaldehyde for 20 min. 1/20 volume of 2.5 M glycine was added to quench formaldehyde. Cells were washed 3 times in PBS and resuspended in 10 ml lysis buffer 1 (50 mM HEPES-KOH, pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton-X-100, 1× protease inhibitors) for 10 min. After centrifugation, the supernatant was discarded and pellet resuspended in 10 ml lysis buffer 2 (10 mM Tris-HCL, pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1× protease inhibitors) for 10 mins. The nuclei were pelleted and resuspended in 3.5 ml lysis buffer 3 (10 mM Tris-HCL, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% NA Deoxycholate, 0.5% N-lauroylsarcosine, 1× protease inhibitors). Sonication was performed using the Bioruptor sonicator (Wolflabs, York, UK) and optimized to produce maximum enrichment of fragments 100–1000 bp, prior to downstream analysis. 1/10 volume of 10% Triton X was added to the sonicated sample, the sample centrifuged at 20,000 g and the lysate stored on ice.
Formaldehyde-Assisted Isolation of Regulatory Elements (FAIRE) and Genotyping
Following chromatin fixing, isolation and sonication, the sheared lysate was subject to three rounds of phenol:chloroform extraction, followed by a final chloroform extraction. The DNA was ethanol precipitated and the pellet resuspended in TE buffer. The DNA solution was treated with 0.2 mg/ml RNase A and incubated at 37°C, and 0.2 mg/ml proteinase K at 55°C for two hours. Samples were incubated at 65°C overnight to remove crosslinks. The samples were subjected to a further phenol:chloroform extraction and ethanol precipitation and standardised to 50 ng/ml for Illumina genotyping chips. For each respective control sample, 10% of the fixed and sonicated chromatin was reverse-crosslinked at 65°C overnight, treated with 0.2 mg/ml RNase A and incubated at 37°C for two hours and 0.2 mg/ml proteinase K at 55°C for 2 hours. The samples underwent 3 rounds of phenol:chlororom extraction followed by ethanol precipitation and standardisation to 50 ng/ml for Illumina genotyping. Genotyping was carried out using the Illumina CVD BeadChip and Illumina Metabochip. Genotype calls for control samples were generated using Illumina GenomeStudio software. Call rates for control and FAIRE samples are described in the Results.
Whitehall II Study (WHII)
DNA was extracted from whole blood. Genotyping for 6,156 samples and laboratory analysis of has been described previously [18]. 5529 samples were genotyped using the Illumina CVD BeadChip [19] and 3,413 samples were genotyped using the Illumina Metabochip. Genotype calls were generated using Illumina GenomeStudio software. After filtering for duplicates, cryptic relatedness, ambiguous gender, self-reported non-Caucasians, outliers based on the genome-wide identity-by-state analysis implemented in PLINK, sample call rate>80% and SNP call rate>98%, 5059 CVD BeadChip and 3126 Metabochip genotyped samples were available for analysis.
Copenhagen City Heart Study (CCHS)
The CCHS [20], [21] is a prospective study of the Danish general population initiated in 1976–78 with follow-up examinations in 1981–84, 1991–94, and 2001–03. Individuals were randomly selected to represent the Danish general population aged 20 to 80+ years. We included 10,322 participants who gave blood for DNA analysis at the 1991–94 and/or 2001–03 examinations. The study was approved by institutional review boards and Danish ethical committees, and conducted according to the Declaration of Helsinki. Written informed consent was obtained from all participants. Plasma levels of total cholesterol, LDL cholesterol, HDL cholesterol, and triglycerides were measured using standard hospital assays (Konelab, Helsinki, Finland, and Boehringer Mannheim, Mannheim, Germany). LDL cholesterol was calculated using the Friedewald equation if the triglyceride level was less than 4 mmol per liter (354 mg per deciliter) and was measured directly for higher triglyceride levels. Follow-up studies of rs7120118 in the samples from Copenhagen were performed using an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems Inc, Foster City, California, USA) and a TaqMan-based assay.
Expression Studies
Tissue biopsies (mammary artery, ascending thoracic aorta and liver) were taken from patients undergoing aortic valve surgery as part of the Advanced Study of Aortic Pathology (ASAP) study [22]. Aortic biopsies were divided into intimal-medial and adventitial halves. Peri-aortic fat was removed from the adventitial specimens where present. RNA from the tissue biopsies was hybridized to Affymetrix ST 1.0 Exon arrays and obtained scans were RMA normalized and log2 transformed. eQTL analysis was performed with an imputed genotype from circulating blood DNA (Illumina 610w-Quad BeadArrays). The full methods for this study have been described previously [22].
Statistical Analysis
Comparison of the GM12878 lymphoblast FAIRE-seq data track was obtained from the UCSC Genome Browser (http://hgdownload.cse.ucsc.edu/goldenPath/hg18/encodeDCC/wgEncodeChromatinMap/wgEncodeUncFAIREseqZinbaGm12878.narrowPeak.gz) and compared to (mean log R ratio of SNPs following FAIRE-enrichment) - (mean log R ratio for the respective control SNPs). The mean SNP log R ratios stratified by strength of FAIRE-seq signal were compared by ANOVA. A paired two-sided t-test was used to compare the control BAF with the respective FAIRE-enriched BAF. Visualisation of Manhattan plots and data management from the UCSC Genome Browser was carried out using Galaxy software [23]–[25]. In WHII, linear regression analysis of log-transformed HDL-C with SNPs using an additive model was performed using PLINK 1.0.7. Analysis was carried out in all individuals and stratified by gender. Regression analysis was performed unadjusted for covariates as well as gender (only in analysis of all individuals) and age added as covariates. Stata software, version 10 (Stata Corp, College Station, Texas) was used for all analyses in the CCHS. Trend tests were by Cuzick's nonparametric test for trend. Linear regression was used to determine per-allele β-coefficients. For trend tests and linear regression analysis, rs7120118 TT, TC and CC genotypes were coded as 0, 1, and 2, respectively. Statistical analysis of gene expression was carried out using R-2.13.0 and Bioconductor 2.8 [26]. Association between gene expression and genotype was calculated using an additive linear model as implemented in the lm-function in R.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. HindorffLA, SethupathyP, JunkinsHA, RamosEM, MehtaJP, et al. (2009) Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proceedings of the National Academy of Sciences of the United States of America 106 : 9362–9367.
2. DimasAS, DermitzakisET (2009) Genetic variation of regulatory systems. Current opinion in genetics & development 19 : 586–590.
3. BirneyE, StamatoyannopoulosJA, DuttaA, GuigoR, GingerasTR, et al. (2007) Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447 : 799–816.
4. ParkPJ (2009) ChIP-seq: advantages and challenges of a maturing technology. Nature reviews Genetics 10 : 669–680.
5. MikkelsenTS, KuM, JaffeDB, IssacB, LiebermanE, et al. (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448 : 553–560.
6. CrawfordGE, HoltIE, WhittleJ, WebbBD, TaiD, et al. (2006) Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome research 16 : 123–131.
7. GiresiPG, KimJ, McDaniellRM, IyerVR, LiebJD (2007) FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res 17 : 877–885.
8. GordonJ, GuyG, WalkerL, NathanP, ExleyR, et al. (1986) Autocrine growth of human B lymphocytes: maintained response to autostimulatory factors is the special feature of immortalization by Epstein-Barr virus–a hypothesis. Med Oncol Tumor Pharmacother 3 : 269–273.
9. SaboPJ, HawrylyczM, WallaceJC, HumbertR, YuM, et al. (2004) Discovery of functional noncoding elements by digital analysis of chromatin structure. Proceedings of the National Academy of Sciences of the United States of America 101 : 16837–16842.
10. SaboPJ, KuehnMS, ThurmanR, JohnsonBE, JohnsonEM, et al. (2006) Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nature methods 3 : 511–518.
11. GiresiPG, LiebJD (2009) Isolation of active regulatory elements from eukaryotic chromatin using FAIRE (Formaldehyde Assisted Isolation of Regulatory Elements). Methods 48 : 233–239.
12. BernsteinBE, KamalM, Lindblad-TohK, BekiranovS, BaileyDK, et al. (2005) Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120 : 169–181.
13. SabattiC, ServiceSK, HartikainenAL, PoutaA, RipattiS, et al. (2009) Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat Genet 41 : 35–46.
14. TeslovichTM, MusunuruK, SmithAV, EdmondsonAC, StylianouIM, et al. (2010) Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466 : 707–713.
15. VeyrierasJB, KudaravalliS, KimSY, DermitzakisET, GiladY, et al. (2008) High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet 4: e1000214 doi:10.1371/journal.pgen.1000214.
16. GaultonKJ, NammoT, PasqualiL, SimonJM, GiresiPG, et al. (2010) A map of open chromatin in human pancreatic islets. Nat Genet 42 : 255–259.
17. WangY, MoserAH, ShigenagaJK, GrunfeldC, FeingoldKR (2005) Downregulation of liver X receptor-alpha in mouse kidney and HK-2 proximal tubular cells by LPS and cytokines. J Lipid Res 46 : 2377–2387.
18. TalmudPJ, DrenosF, ShahS, ShahT, PalmenJ, et al. (2009) Gene-centric association signals for lipids and apolipoproteins identified via the HumanCVD BeadChip. American journal of human genetics 85 : 628–642.
19. KeatingBJ, TischfieldS, MurraySS, BhangaleT, PriceTS, et al. (2008) Concept, design and implementation of a cardiovascular gene-centric 50 k SNP array for large-scale genomic association studies. PLoS ONE 3: e3583 doi:10.1371/journal.pone.0003583.
20. Frikke-SchmidtR, NordestgaardBG, SteneMC, SethiAA, RemaleyAT, et al. (2008) Association of loss-of-function mutations in the ABCA1 gene with high-density lipoprotein cholesterol levels and risk of ischemic heart disease. JAMA : the journal of the American Medical Association 299 : 2524–2532.
21. StenderS, Frikke-SchmidtR, AnestisA, KardassisD, SethiAA, et al. (2011) Genetic variation in liver X receptor alpha and risk of ischemic vascular disease in the general population. Arteriosclerosis, thrombosis, and vascular biology 31 : 2990–2996.
22. FolkersenL, van't HooftF, ChernogubovaE, AgardhHE, HanssonGK, et al. (2010) Association of genetic risk variants with expression of proximal genes identifies novel susceptibility genes for cardiovascular disease. Circulation Cardiovascular genetics 3 : 365–373.
23. BlankenbergD, Von KusterG, CoraorN, AnandaG, LazarusR, et al. (2010) Galaxy: a web-based genome analysis tool for experimentalists. Current protocols in molecular biology/edited by Frederick M Ausubel [et al] Chapter 19: Unit 19 10 11–21.
24. GiardineB, RiemerC, HardisonRC, BurhansR, ElnitskiL, et al. (2005) Galaxy: a platform for interactive large-scale genome analysis. Genome research 15 : 1451–1455.
25. GoecksJ, NekrutenkoA, TaylorJ (2010) Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome biology 11: R86.
26. GentlemanRC, CareyVJ, BatesDM, BolstadB, DettlingM, et al. (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome biology 5: R80.
27. PruimRJ, WelchRP, SannaS, TeslovichTM, ChinesPS, et al. (2010) LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26 : 2336–2337.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 8
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Dissecting the Gene Network of Dietary Restriction to Identify Evolutionarily Conserved Pathways and New Functional Genes
- It's All in the Timing: Too Much E2F Is a Bad Thing
- A Quantitative Comparison of the Similarity between Genes and Geography in Worldwide Human Populations
- Variation of Contributes to Dog Breed Skull Diversity
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy