
Variants Affecting Exon Skipping Contribute to Complex Traits
DNA variants that affect alternative splicing and the relative quantities of different gene transcripts have been shown to be risk alleles for some Mendelian diseases. However, for complex traits characterized by a low odds ratio for any single contributing variant, very few studies have investigated the contribution of splicing variants. The overarching goal of this study is to discover and characterize the role that variants affecting alternative splicing may play in the genetic etiology of complex traits, which include a significant number of the common human diseases. Specifically, we hypothesize that single nucleotide polymorphisms (SNPs) in splicing regulatory elements can be characterized in silico to identify variants affecting splicing, and that these variants may contribute to the etiology of complex diseases as well as the inter-individual variability in the ratios of alternative transcripts. We leverage high-throughput expression profiling to 1) experimentally validate our in silico predictions of skipped exons and 2) characterize the molecular role of intronic genetic variations in alternative splicing events in the context of complex human traits and diseases. We propose that intronic SNPs play a role as genetic regulators within splicing regulatory elements and show that their associated exon skipping events can affect protein domains and structure. We find that SNPs we would predict to affect exon skipping are enriched among the set of SNPs reported to be associated with complex human traits.
Published in the journal:
. PLoS Genet 8(10): e32767. doi:10.1371/journal.pgen.1002998
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002998
Summary
DNA variants that affect alternative splicing and the relative quantities of different gene transcripts have been shown to be risk alleles for some Mendelian diseases. However, for complex traits characterized by a low odds ratio for any single contributing variant, very few studies have investigated the contribution of splicing variants. The overarching goal of this study is to discover and characterize the role that variants affecting alternative splicing may play in the genetic etiology of complex traits, which include a significant number of the common human diseases. Specifically, we hypothesize that single nucleotide polymorphisms (SNPs) in splicing regulatory elements can be characterized in silico to identify variants affecting splicing, and that these variants may contribute to the etiology of complex diseases as well as the inter-individual variability in the ratios of alternative transcripts. We leverage high-throughput expression profiling to 1) experimentally validate our in silico predictions of skipped exons and 2) characterize the molecular role of intronic genetic variations in alternative splicing events in the context of complex human traits and diseases. We propose that intronic SNPs play a role as genetic regulators within splicing regulatory elements and show that their associated exon skipping events can affect protein domains and structure. We find that SNPs we would predict to affect exon skipping are enriched among the set of SNPs reported to be associated with complex human traits.
Introduction
Alternative Splicing (AS) is a eukaryotic-specific cellular mechanism that increases the diversity of mRNA and allows for the production of multiple proteins from one gene. As many as 40 to 90% of all human genes are estimated to be alternatively spliced [1]–[3], implicating this process in key aspects of biological complexity. AS has been found to play a role in developmental regulation [4], differentiation [5] and even in neuronal vulnerability [6]. Furthermore, AS is a biological system that leads to the production of tissue-specific and disease-specific expressed mRNA isoforms and may be a contributing factor to differences in normal and pathological physiology [2], [7], [8].
Alternative splicing is guided by the proper recognition of splice sites that allow for the removal of introns from mature mRNA. Intronic splicing enhancers (ISE), exonic splicing enhancers (ESE), intronic splicing silencers (ISS), and exonic splicing silencers (ESS) regulate the intron excision process by assisting in the recognition of the correct splice site to generate in the correct combination of exons in the final mRNA. Therefore, genetic variation in cis-acting splicing regulatory elements (SREs: ISE, ESE, ISS, and ESS) may lead to aberrant intron excision [9], resulting in exon skipping and varying protein products. Ultimately, the altered proteins may affect phenotypic traits and have been implicated in tumor progression and susceptibility to cancer [7], [10]–[13]. Recently, two studies have demonstrated that altered splicing patterns might be even more important than expression changes in determining risk for complex human trait [14], [15]. Furthermore, exon skipping interventions have been developed as potential molecular therapies for Duchenne Muscular Dystrophy [16]–[18] and effects of statins on alternative splicing have been suggested to contribute to efficacy variation in cardiovascular disease treatment [19].
Recently, intronic SNPs have been shown to be associated with AS events [19]–[21], which has initiated a new era of research that focuses on investigating how intronic SNPs affect alternative splicing. Importantly, previous studies have suggested that 15–50% of all human heritable disease may be affected by mutated canonical splice sites (GT…AG) or splicing regulatory elements [22]–[26]. Additionally, Genome Wide Association Studies (GWAS) have identified large numbers of intronic SNPs with significant and reproducible associations to diseases or traits. Furthermore, various high throughput data analyses, including exon tiling microarray [27], RNA-Seq [28] and whole exome sequencing [15], [29], have been designed to identify polymorphisms that have their effect primarily through alternative splicing. Most studies to date, however, have considered only the identification of genetic determinants of AS and have not focused on how these variations affect the alternative splicing machinery.
Here we hypothesize that the molecular roles of intronic SNPs in AS could be investigated by exploring their occurrences in ISEs, providing possible mechanistic links to their biological functionality. To investigate our hypothesis, we extend our previous work [30] to analyze exon-level expression microarrays of 176 HapMap lymphoblastoid cell lines (LCLs) which we combine with sequence-derived AS modeling data. We demonstrate significant enrichment of AS-associated intronic SNPs among the trait-associated SNPs identified through GWAS cataloged in the NHGRI web site (http://www.genome.gov/gwastudies [31]) and examine the biological function of the skipped exons by accounting for the location of the skipped exons in coding sequences and protein domains.
Results
Study Design
Figure 1 illustrates the study design and describes the five main components of the study, two for resource preparation processes and one each for data integration, analysis of genetic variations and evaluation of biological consequences. The main resource for identifying alternatively spliced exons was compiled using two data sources produced by high-throughput genome-wide techniques: 1) sequence-derived computed data and 2) expression-derived experimental data. Putative alternatively spliced exons were predicted using a sequence data-driven alternative splicing model (Figure 1A). We determined the genomic locations of ISEs by scanning the intronic regions nearby intronic SNPs for putative ISE motifs (6-mer in length) [32]. We then bioinformatically verified that the nearest exon on either side of an intronic SNP located within an ISE is skipped in at least one transcript isoform. Exon-level expression and cis-expression quantitative trait loci data (cis-eQTL, see Materials and Methods) were obtained from our previous work with HapMap LCLs (87 Utah residents with Northern and Western European ancestry (CEU) and 89 Yoruba from Ibadan, Nigeria (YRI) [30], [33] (Figure 1B). Combining sequence-based predictions of exon skipping and ISEs and exon array-based experimental expression data not only defines the regulatory relationship between ISE SNPs and exon skipping events but also identifies experimentally validated exon skipping events (Figure 1C). AS model-derived exons were mapped to exons present on the Affymetrix GeneChip Human Exon 1.0 ST microarray arrays and annotated as an identical exon when, based on physical genomic coordinates for exon start and exon end, more than 95% of the length of an exon in the exon array overlapped with more than 90% of the length of an exon identified in the AS model. This analysis resulted in 74 alternatively spliced exons and 151 ISE SNPs. A splicing index (SI, see Materials and Methods) was developed to determine which exons were alternatively skipped in each individual. By analyzing the resultant set, we sought to provide possible mechanistic insight into the functional effects of genetic variations in ISE sites that may be associated with variability of individual exon skipping (Figure 1D) and eventually to improve our ability to annotate SNPs involved in alternative splicing (Figure 1E).

Exon-Skipping ISE SNPs Are Significantly Enriched among Human Trait-Associated SNPs
We examined ISE SNPs, i.e. SNPs that we predicted to be within ISE motifs (6-mer in length and adjacent to predicted skipped exons) that are associated with exon skipping (denoted as ES ISE SNPs) in order to gain insight into the functionality of AS associated intronic SNPs. In total, 3,353 SNPs reproducibly associated with complex human traits from the NHGRI catalog (http://www.genome.gov/gwastudies) [31] were tested for enrichment for the predicted ISE SNPs (see Materials and Methods). Among the 3,353 trait-associated SNPs, 111 overlapped with our predicted ISE SNPs (n = 448,515), an enrichment that is significantly greater than expected by chance given the minor allele frequency (MAF) distribution of the trait-associated SNPs (p = 0.006, Figure S1, Table S1). Furthermore, to determine whether this finding is confounded by the distance to the nearest exon, we repeated the enrichment analyses while conditioning on both MAF and the distance to the nearest exon. The enrichment of ISE SNPs for trait-associated SNPs held robustly (p = 0.036, Figure 2A).

Subsequently, we examined 49 tissue-specific trait-associated SNPs derived from splicing quantitative trait locus (sQTL) analyses [15]. As illustrated in Figure 2B, among the 49 tissue-specific trait-associated sQTL SNPs, we found that 6 were recapitulated in our ISE SNPs, which is also more than expected by chance (p<0.001) given the MAF distribution of the trait-associated sQTLs. These results confirm that variants affecting alternative splicing may indeed play a role in the genetic component of complex diseases and related quantitative traits. Our data suggest that a subset of the trait-associated intronic variants affect phenotype by altering splicing enhancers.
Exon-Skipping ISE SNPs Are Likely to Be Cis-eQTLs
We obtained the set of 151 ISE SNPs associated with 74 putative skipped exons that resulted from integrating the experimental exon-array data with our ab initio data. An enrichment analysis validated the hypothesis that these 151 exon-skipping ISE SNPs were enriched for cis-eQTL SNPs (p<0.001, Figure 3). Indeed, 68 ISE SNPs were found to overlap with cis-eQTL SNPs, which is a significantly higher number than expected, from intronic SNPs, by chance given the MAF distribution of the exon-skipping ISE SNPs. When we conditioned on both MAF and distance to nearest exon, the level of enrichment was more pronounced (p<0.001). Thus, there is robust support for the conclusion that exon-skipping associated ISE SNPs are more likely than expected to be cis-eQTLs.

ISE SNPs Are Correlated with Higher Levels of Exon Skipping
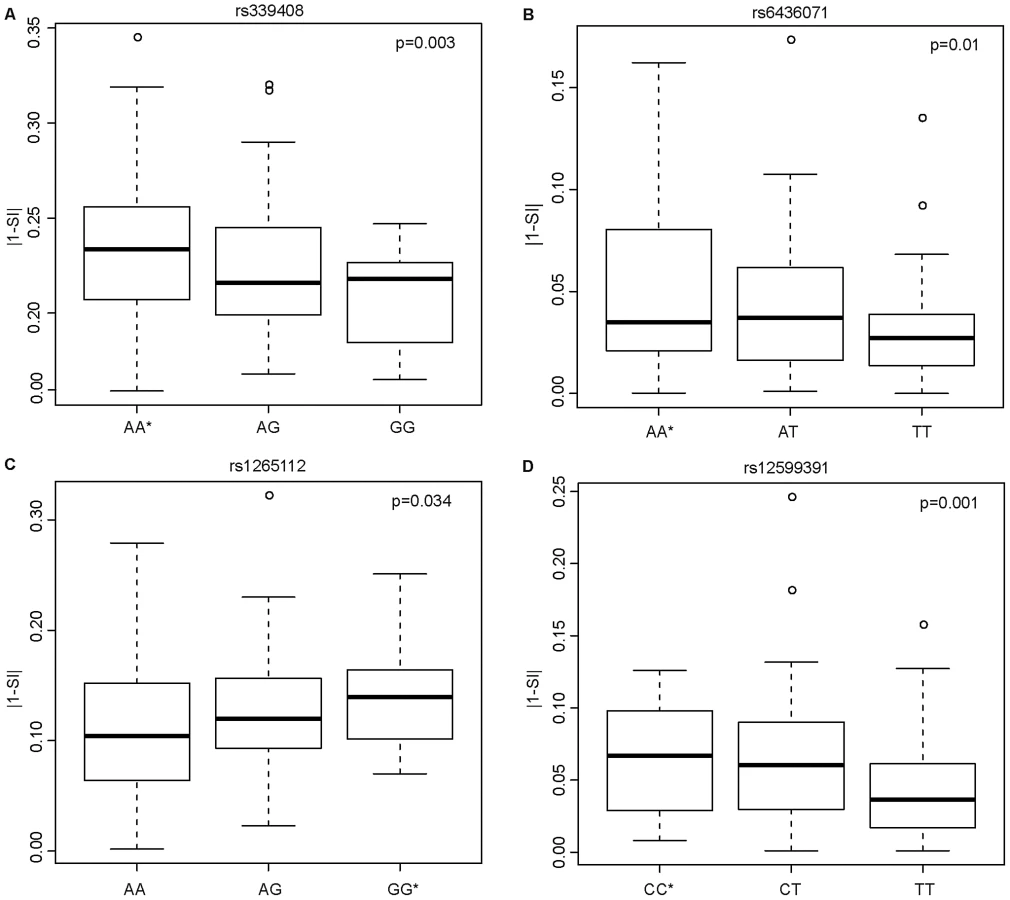
Having shown that SNPs within ISE motifs are enriched for cis-eQTLs, we systematically investigated their correlation with exon skipping events. We hypothesized that inter-individual variability in exon skipping could be explained by the genotypes at these SNPs. We therefore tested whether these ISE SNPs might “disrupt” the ISE motif sequence resulting in the loss of ISE function and, consequently, contributing to greater variability in the rate of exon skipping events. The SI is a statistic developed to identify exons with inclusion rates that differ from the expected value for the given gene (see Materials and Methods). Figure 4 shows the most significant SNP associations with SI using an additive model and the allele associated with an increase in exon skipping (p<0.05 (FDR<10%), R2>0.03). The latter is referred to as the “exon skipping associated ISE allele”, which in Figure 4 is identified with an asterisk.
Biological Consequences of Skipped Exons
We sought to further investigate the biological consequences of exon skipping on protein function. We selected 30 experimentally verified skipped exons that are skipped in more than 18 individuals (top 10% of 176 individuals ordered by SI) corresponding to 73 ISE SNPs and we examined their locations such as in coding regions, untranslated regions (UTRs), or protein function-related regions (Table 1). Of the 30 exons, 73% (22 exons) were located in coding regions, and 10% (3 exons) in 5′ UTRs. Among exons located in coding regions, only one exon was not located in a known domain. As described in Table 1, 67% of the skipped exons (20 out of 30) were located in protein domains, transmembrane helix or coiled-coil regions, suggesting that these skipped exons are likely to have biological consequences [34], [35]. More specifically, it has been reported that alternatively skipped exons are involved in or affect secondary structure and thus affect protein interactions with other protein partners or ligands [36]. Consistent with computational approaches that estimated that more than 50% of alternatively spliced regions in the human transcriptome affect protein interaction sites and more than 65% showed significant alteration in protein 3D structure [34], our study shows that 70% of skipped exons map to biologically functional regions.

To characterize the structural differences that may arise from the skipped exons, we selected 4 genes (RNF8, SLC25A15, PPP5C, NARF) from Table 1, each with a pair of known transcript isoforms that differ in the inclusion of a given exon according to Ensembl database [37]. For each gene, we conducted a three-dimensional protein structure comparison of the pair of transcript isoforms. Table S2 shows the C-score (see Materials and Methods) for each transcript isoform included in the analysis and the RMSD and TM-scores (see Materials and Methods) that quantify the structural similarity of the pair of transcript isoforms for any given gene. In PPP5C and NARF, the skipped exon is not likely to induce structural difference, as the corresponding pair of isoforms shows substantial structural similarity (RMSD = 0.66 Å, TM-score = 0.739 for PPP5C and RMSD = 2.37 Å, TM-score = 0.809 for NARF). However, in the pair of RNF8 and SLC25A15 transcript isoforms, the skipped exon is likely to induce structural difference, as the structure models of the RNF8 and SLC25A15 isoforms differ, as quantified by the high RMSD (7.34 Å for RNF8, 3.57 Å for SLC25A15) and low TM-score (0.199 for RNF8, 0.354 for SLC25A15). In summary, of four genes in which exons appear to be skipped, two of the genes (i.e., RNF8 and SLC25A15) yielded evidence for differences in the 3D structures for the protein with and without the skipped exon. Figure 5 illustrates the structural alignment of the pairs of transcript isoforms for SLC25A15 (panel A) and for RNF8 (panel B).

Other Dominant Genetic Variations and SREs May Act Together to Regulate Exon Skipping
While investigating the regulatory relationship between the ISE variants and exon-skipping events, we observed two exceptional phenomena in our exons. Some individuals were found who carried the exon skipping associated ISE allele but did not undergo exon skipping. Interestingly, we found that each of these alleles was present in multiple ISE motifs. Four such SNPs among our findings are shown in Figure S2. In each case, the SNP differs with regard to position in the motif. This finding suggests that these SNPs may be related to retention or gain of ISE motifs. Furthermore, we reasoned that this phenomenon may be also due to one of the following: 1) ESEs may exist in these exons, 2) splice sites of these exons may be canonical (strong) sequences (GT…AG) or 3) other genetic variations may exist that more dominantly affect the ISS or ESS motifs related to the exon. A second exceptional observation was that some individuals did not carry an exon skipping associated ISE allele but still underwent exon skipping according to their SI. While anomalous, this exon might be skipped for one of the following possible reasons: 1) ESSs may exist in this exon, 2) the splice site of the exon may be a non-canonical (weak) sequence, or 3) other genetic variations may exist that more dominantly affect the ISE or ESE motifs related to the exon.
ISE SNPs Are Less Likely to Be Closer to the Affected Exon Than Intronic SNPs Are, in General, to Nearest Exon
As the distance between ISE SNPs and the adjacent exons that they regulate may be very broad in range, we examined sets of ISE SNPs found within the following physical distances from their closest associated skipped exon: <60 bp, 200 bp, 1 Kbp, and 5 Kbp. We observed that the enrichment of complex trait associated SNPs among ISEs is uncorrelated with physical distance from the skipped exon's splice site junction and is consistent in all distance groups (Figure 6).

This raises the hypothesis that ISE SNPs (as function-implicated SNPs) are more proximal to the affected skipped exon than intronic SNPs in general are to the nearest exon. We found that the ISE SNPs are closer to the nearest exon than intronic SNPs in general, and indeed ISE SNPs and intronic SNPs have significantly different distributions of distance to nearest exon (p<2.2e-16, a two-sample Kolmogorov-Smirnov (KS) Test, Table 2, Figure S3). Unexpectedly, the skipped exons are farther from ISE SNPs than are the intronic SNPs from their nearest exon (with significant difference between the two distributions, p<2.2e-16, a two-sample KS Test); furthermore, the affected skipped exon for ISE SNPs is, in general, not the same as the nearest exon (and indeed, there is a significant difference between the distribution of distance to nearest exon and the distribution of distance to the skipped exon, p<2.2e-16, a two-sample KS Test). We observed similar findings for trait-associated SNPs (as curated in the NHGRI GWAS catalog). The skipped exons are farther from trait-associated ISE SNPs than are the trait-associated intronic SNPs from the nearest exon (p = 0.018, a two-sample KS Test, Figure 7).


Case Study: SNP rs12924138 Affects an Alternatively Spliced Exon in CDK10 and Is Associated with Human Disease Phenotype
The SNP rs12924138 (G/T) is located in the 1st intron of CDK10 gene and was found to be associated with the skipping of the 2nd exon in CDK10 (Figure S4A). As illustrated in Figure S4B, the T allele of rs12924138 was predicted to be in an ISE motif (AGGCCTG). However, the 6-mer sequence having the G was no longer an ISE motif. The GG genotype showed a higher level of exon skipping, as quantified by the SI, than the GT (p = 0.011, non-paired and two-tailed t-test) and TT (p = 0.009, non-paired and two-tailed t-test) genotypes (Figure S4C). We observed another SNP (rs258322) that is also near the skipped exon. As shown in Figure S4A, rs258322 is downstream of this exon and is in LD with rs12924138 (r2 = 0.331, D′ = 1, LOD = 6.78, HapMap CEU). The SNP rs258322 is known to be associated with melanoma [36], but was not found to be an ISE SNP. The affected exon (i.e., the skipped 2nd exon) might exert its functional consequence by being the first exon included in the “Tyrosine kinase, catalytic domain” of CDK10. Furthermore, CDK10 has been identified to play a role in cell cycle regulation and is located in a locus (16q24) harboring SNPs associated with melanoma risk [36].
Discussion
Rapid advances in high-throughput techniques have led to a deluge of biological datasets of a variety of types, including sequence, expression, structure, and ontology. One of our primary goals in this study was to develop an approach for integrating heterogeneous datasets and combining genome-wide associations studies with functional genomics. We developed a systematic approach to provide mechanistic or functional insight into how SNPs might affect alternative splicing and conversely to increase our understanding of the genetic regulation of alternative splicing. We investigated the functional implications of intronic SNPs on splicing regulatory elements that enhance splicing junction recognition, cause protein domain changes or are thought to contribute to human traits or diseases. Taken together, our results provide a primary resource for characterizing the functional role of genetic variations in the etiology of complex human traits or diseases. Additionally, we contribute to the compendium of primary functional knowledge of intronic SNPs. This latter point is particularly important as we move toward analyzing complex human disease sequence data, in which the knowledge of functional variants, including those involved in alternative splicing, will be key to making progress given the sea of genetic variations to be examined.
ISEs assist in splice-site recognition of adjacent exons and promote exon inclusion. ISE splicing control is dependent on many variables including variation in intron length, the density of ISEs in a given intron [32], and the strength of 5′/3′ splice sites sequences. Non-traditional splicing regulation may also occur without known splicing regulators [14], [38]. Furthermore, the relative position of the ISE to the downstream 5′ splice site has been shown to vary from 6 to 500 nucleotides [39], [40], [41], [42], [43], [44], [45], [46], [47], [48] and some ISEs with UGCAUG motifs may be more than a kilo-base downstream [49], [50].
Multiple distances (<60 bp, 200 bp, 1 Kbp, and 5 Kbp) between ISE and adjacent skipped exons were tested to determine if a particular distance exists in which the contained ISE SNPs are more likely to be human complex trait associated-SNPs, as defined by their inclusion in the NHGRI GWAS catalog (Figure 6 and Table 2). The observation that all distance boundaries for ISE-associated SNPs were found to exhibit enrichment of trait associated SNPs supports the claim that ISEs do not require proximity to their associated splice site in order to be efficacious. Thus their mechanism of action may extend beyond attracting splicing machinery to include other mechanisms not yet considered in this study. Additionally, this evidence may suggest the need for extended ISE motif predictions and analyses that include genomic regions that are farther from their exon of interest.
At the cellular level, the splicing regulatory mechanism is a multifaceted biological process that causes splicing to occur, initiated by splice site recognition and guided by SREs. The components of the spliceosome are then recruited to the correct splice junction, branch point or other splicing loci. However, while many elements that affect the splicing code have been identified, their combinatorial effects have yet to be comprehensively characterized. Since our study focuses only on ISE associated SNPs, it is important to mention this is a simplification and that ISEs are believed to work in conjunction with other SREs (ESS, ESE and ISS). This mechanism depends on the relative strength of the regulatory elements themselves, the strength or weakness of the actual splice site and the lengths of their associated introns/exons. Their synergistic antagonism or protagonism is believed to be contextual in that it relates not only to their sequence location but also to the relative location or existence of other SREs. The enhancer and silencer characteristics of these additional elements could be explored in a future study utilizing the method put forth in this paper.
Alternative splicing events have been shown to disrupt entire protein domains [35] and most often affect certain protein kinase domains and coiled-coil sequences embedded in transmembrane area [51]. Here, we evaluated the functional impact of alternative splicing on each relevant protein as aberrantly skipped exons can disrupt protein domains fully or partially or affect their final structure and function if the disruption is present in a critical area for biological activity, affinity or folding [36]. To this end, experimentally verified skipped exons that are regulated by intronic SNPs were investigated using protein domain analyses which provide a systematic molecular basis for determining how intronic SNPs affect their resultant AS variants. This analysis was also used to infer the functionality of intronic SNPs whose role in affecting final protein products has otherwise remained elusive.
Alternatively, exon-skipping events can further affect protein functionality if the excised piece of DNA is of a length that is not an exact multiple of 3. In this case, it is possible that all the exons downstream from the skipped splice junction would be greatly affected as their reading frame is shifted. This change can produce an entirely different amino acid sequence and may introduce premature termination codons, which (depending on where in the gene this occurred) could lead to nonsense-mediated decay (NMD) [52]. Thus, even though ISE SNPs are, by definition, not contained within a protein-coding region, such polymorphisms have the potential to cause physical changes in the resultant protein apart from the loss of an exon through reading frame.
Exon skipping is the first alternative splicing (AS) mode accounting for 40% of AS events in higher eukaryotes [53], [54]. Alternative 3′/5′ splice sites and intron retention are the second (18.4%) and third (7.9%) most common types, respectively. It has been shown that the exon array (Affymetrix GeneChip Human Exon 1.0 ST Array) is more useful for investigating exon skipping and retained intron events than other types of AS events such as alternative 5′/3′ intron splice site and alternative polyadenylation sites [55]. For this reason, the current study focused on analyzing exon skipping events.
In our study, this small set of experimentally validated skipped exons was tested to explain the possible functional mechanisms of intronic SNPs impacting exon skipping events. A larger data set of experimentally validated skipped exons and ISE SNPs should be studied in the future. Additionally, to further characterize inter-individual variability in exon skipping, individual sequence data should be integrated with our approach to investigate other SNPs located in the 5′ and 3′ splice sites of skipped exons. Although we have shown that the ISE SNPs are enriched for cis-eQTLs and have identified an association of genotype with exon skipping in the HapMap samples (using the SI statistic), it is possible that our findings are limited by eQTL tissue specificity [56].
In this paper we propose a systematic approach to integrate sequence, expression and genetics data (genotype/phenotype) in order to elucidate the impact of genetic variations on exon skipping and their importance in complex traits and diseases. Discerning genetic regulation of splicing is undeniably critical for understanding abnormal or physiological changes in AS since the number of currently characterized human splicing regulators cannot alone account for the tremendous number of splicing events known to occur in humans. We have shown not only that intronic SNPs are associated with exon skipping events but also that these SNPs are associated with complex traits, and that they are predicted to result in protein domain changes. While additional studies are needed to fully understand the role that genetic variation in SREs may play in alternative splicing, as well as how much AS-associated genetic variation contributes to common disease, this study provides support for continuing such investigations.
Materials and Methods
Identification of Exon Skipping-Associated ISE SNPs by Ab Initio Sequence-Based Data Analysis
Intronic SNP data
A list of SNPs and their genomic coordinates were obtained from the UCSC genome annotation database [57] (snp129.txt.gz downloaded on 8/11/2010 from http://hgdownload.cse.ucsc.edu/goldenPath/hg18/database/). This file is compatible with SNP information from dbSNP build 129, which is available at ftp://ftp.ncbi.nih.gov/snp. To create a list of intronic SNPS, we selected SNPs annotated as “intron”.
Resource of intronic splicing enhancer
Burge C. and colleagues previously developed methods to analyze the similarities and differences in the sequences and organization of splicing regulatory elements in humans (http://genes.mit.edu/acescan2/index.html) [32]. Their method predicted hexamer ISE motifs and they provided 127 and 266 predicted ISEs respectively located near the 5′ splice site (ss) and the 3′ ss.
Genome sequence
For our human reference genome map, we used the March 2006 (NCBI36/hg18) genome sequence downloaded from the UCSC genome Center (ftp://hgdownload.cse.ucsc.edu/goldenPath/hg18).
Identification of alternatively spliced exons
A comprehensive AS gene dataset was compiled with four typical AS resources downloaded from UCSC genome browser (http://hgdownload.cse.ucsc.edu/goldenPath/hg18/database/, 8/11/2010) [58]: 1) mRNAs from GenBank [59], 2) Ensembl Gene Predictions [60], 3) AceView Gene Models [61], and 4) UCSC known genes [62]. We utilized human genome alignment information from exons and introns to identify skipped exons by comparing all possible pairs of spliced transcripts for a given gene predicted by each AS model.
Identification of SNPs associated with ISEs (ISE SNP)
The sequences surrounding our intronic SNPs were extracted from the human reference genome assembly (hg18) by using, nibFrag, one of the Blat Suite programs [63] (downloaded from http://users.soe.ucsc.edu/~kent/src/). We obtained 5 bases upstream and downstream from the SNP's genomic location, producing an 11 base sequence for each given SNP's intron. To identify all possible 6-mer motifs that include the SNP of interest, each 11-mer sequence was fragmented into 6 individual hexamers by taking a 6 base long window that contained the SNP in the last position and successively shifting the frame upstream by one base at a time until the SNP was in the first position. All of the 6-mers with the embedded SNP were investigated using the predicted ISE motif sequences to identify whether each of the 6-mer scans of the 11-mer sequence is associated with an ISE motif. When the sequence of the intronic hexamers exactly matched one of the ISE motifs, the SNP was counted as an ISE SNP.
Identification of exon skipping–associated ISE SNPs
Using the genomic coordinates of the exons and introns of AS genes, we implemented the following steps: 1) AS variants of the gene embedding a given ISE SNP were extracted only if they occurred on the same strand as the SNP of interest. 2) Exons located adjacent to the intron in which the ISE SNP is embedded were examined. 3) The corresponding 3′ or 5′ exon (and splice site) was selected based on the location, upstream or downstream, respectively, of the ISE SNP in relation to the exon. 4) If the exon was found not to exist in at least one AS transcript isoform, this exon is defined as alternatively spliced. The first and last exons were ignored for exon skipping event identification since they by definition are not considered “skipped” or “included”. Throughout the methods, of the 14,574,533 SNPs in dbSNP129, 448,515 SNPs were identified to be within ISE sites adjacent to predicted skipped exons and comprise 3% of all SNPs, 7.3% of intragenic SNPs, and 8.1% of intronic SNPs, respectively. These SNPs were evaluated for enrichment among human complex trait-associated SNPs characterized in the NHGRI GWAS catalog and human tissue-specific-associated SNPs derived from splicing quantitative trait loci (sQTL) [15].
Detection of Exon-Skipping Events from Exon Array
We obtained exon-level expression and cis-eQTL (p<0.01) information from the SCAN database (SNP and Copy number Annotation database (http://www.scandb.org) [33]. The SCAN database serves the results of our eQTL studies; the initial dataset consisted of eQTLs for gene expression phenotypes assayed in HapMap LCLs (87 CEU and 89 YRI) using the Affymetrix GeneChip Human Exon 1.0 ST Array [30], [64]. The Splicing Index (SI) statistic aims to identify exons whose inclusion rates differ relative to the expected value at the gene-level [65]. The SI statistic is calculated as the log-ratio of the normalized exon-level probeset intensity to the gene-level transcript cluster intensity in each sample. In our study, an exon is considered spliced if its SI is >1.2 or <0.8.
Integration of Expression-Derived Data with Sequence-Derived Data
Mapping array-derived exons to splicing model-derived exons was done according to their genomic coordinates in order to identify exon skipping events found by the two distinct methods. By design, the exons tested in the array may not completely overlap the alternative splicing model-derived exons. To identify exons with evidence for exon skipping in both the expression array and the splicing models, we required that more than 95% of the length of an exon in the exon array overlap with more than 90% of the length of an exon identified in the AS model.
Cis-eQTL SNPs
As described previously, the eQTL data were downloaded from the SCAN database. The eQTLs SNPs [33], [66] that are within 4 Mb of target genes were defined as “cis-regulators” (cis-eQTL p<0.01). The cis-eQTL data were used to derive the regulatory relationships between the ISE SNPs (cis-eQTL SNPs p<0.01) and their associated mRNAs.
Calculation of Distance to the Nearest Exon
The distance to the nearest exon of intronic SNPs was calculated using Ensembl Gene data (ensGene.txt.gz) and the SNP annotation file (snp129.txt.gz) downloaded from the UCSC genome browser, http://hgdownload.cse.ucsc.edu/goldenPath/hg18/database/. To calculate the distance from the ISE SNP to the skipped exon, we used our predictions of exon skipping from the Ensembl Gene data.
Enrichment Analysis Method Conditional on Minor Allele Frequency and Distance to Nearest Exon
To conduct enrichment analyses [66], we generated 1000 random sets of SNPs matching either (a) the minor allele frequency spectrum or (b) both the minor allele frequency spectrum and distance to nearest exon of the SNP list of interest. The MAF information derived from the HapMap CEU population was used in our analyses, as most published GWAS to data have been conducted in populations of European ancestry. To test the exon-skipping ISE SNPs for enrichment of cis-eQTLs or trait-associated SNPs, the random sets were generated from intronic SNPs (based on dbSNP129 annotation).
Test for Association between SNP Genotype and Splicing Index
We conducted statistical analysis for correlation of genotype with exon skipping using linear regression. To adjust for multiple testing, we calculated FDR using the q-value R package [67]. FDR<0.10 was used as the statistical threshold for reporting results.
Location of Skipped Exons in Coding Regions and UTRs
To identify the locations of skipped exons in CDSs and UTRs as well as the protein domain encoded by these skipped exons, we used the Ensembl Human Genome Browser (http://useast.ensembl.org/index.html) [37]. For those transcripts of our genes that include the skipped exons of interest, we choose the most reliable Ensembl transcripts that include our predicted skipped exons that have been annotated with known protein sequences or full-length cDNA. The protein summary page visualizing the mapping of coding regions of alternative exons onto protein domains was utilized for determining if our putative skipped exons encode protein domains.
Protein Structure Prediction and Comparison of Transcript Isoforms
We used I-TASSER [68] to predict the three-dimensional protein structures of transcript isoforms. The resulting I-TASSER models for a pair of isoforms were compared using TM-align [69] to assess their structural similarity.
Analysis of Human Trait Associations
To identify the correlation of ISE SNPs with known human trait-SNPs, two datasets were utilized: human trait-associated SNPs and LD r2 scores (in CEU and YRI), downloaded from the NHGRI catalog (http://www.genome.gov/gwastudies/) and HapMap (http://hapmap.ncbi.nlm.nih.gov/downloads/ld_data/latest/) on Feb. 24 2011, respectively. We surveyed the LD r2 scores between the trait associated-SNPs and our exon-skipping ISE SNPs.
Supporting Information
Zdroje
1. JohnsonJM, CastleJ, Garrett-EngeleP, KanZ, LoerchPM, et al. (2003) Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 302 : 2141–2144.
2. WangET, SandbergR, LuoS, KhrebtukovaI, ZhangL, et al. (2008) Alternative isoform regulation in human tissue transcriptomes. Nature 456 : 470–476.
3. PanQ, ShaiO, LeeLJ, FreyBJ, BlencoweBJ (2008) Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nature genetics 40 : 1413–1415.
4. GraveleyBR, BrooksAN, CarlsonJW, DuffMO, LandolinJM, et al. (2011) The developmental transcriptome of Drosophila melanogaster. Nature 471 : 473–479.
5. GabutM, Samavarchi-TehraniP, WangX, SlobodeniucV, O'HanlonD, et al. (2011) An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell 147 : 132–146.
6. PolymenidouM, Lagier-TourenneC, HuttKR, HuelgaSC, MoranJ, et al. (2011) Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nature neuroscience 14 : 459–468.
7. VenablesJP, KlinckR, KohC, Gervais-BirdJ, BramardA, et al. (2009) Cancer-associated regulation of alternative splicing. Nature structural & molecular biology 16 : 670–676.
8. BarashY, CalarcoJA, GaoW, PanQ, WangX, et al. (2010) Deciphering the splicing code. Nature 465 : 53–59.
9. GaildratP, KriegerS, TheryJC, KillianA, RousselinA, et al. (2010) The BRCA1 c.5434C→G (p.Pro1812Ala) variant induces a deleterious exon 23 skipping by affecting exonic splicing regulatory elements. Journal of medical genetics 47 : 398–403.
10. VenablesJP (2004) Aberrant and alternative splicing in cancer. Cancer research 64 : 7647–7654.
11. LefaveCV, SquatritoM, VorlovaS, RoccoGL, BrennanCW, et al. (2011) Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. The EMBO journal 30 : 4084–4097.
12. ShapiroIM, ChengAW, FlytzanisNC, BalsamoM, CondeelisJS, et al. (2011) An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet 7: e1002218 doi:10.1371/journal.pgen.1002218
13. Allende-VegaN, DayalS, AgarwalaU, SparksA, BourdonJC, et al. (2012) p53 is activated in response to disruption of the pre-mRNA splicing machinery. Oncogene
14. YuY, MaroneyPA, DenkerJA, ZhangXH, DybkovO, et al. (2008) Dynamic regulation of alternative splicing by silencers that modulate 5′ splice site competition. Cell 135 : 1224–1236.
15. HeinzenEL, GeD, CroninKD, MaiaJM, ShiannaKV, et al. (2008) Tissue-specific genetic control of splicing: implications for the study of complex traits. PLoS Biol 6: e1000001 doi:10.1371/journal.pbio.1000001
16. YinH, LuQ, WoodM (2008) Effective exon skipping and restoration of dystrophin expression by peptide nucleic acid antisense oligonucleotides in mdx mice. Mol Ther 16 : 38–45.
17. MitrpantC, AdamsAM, MeloniPL, MuntoniF, FletcherS, et al. (2009) Rational design of antisense oligomers to induce dystrophin exon skipping. Mol Ther 17 : 1418–1426.
18. van DeutekomJC, JansonAA, GinjaarIB, FrankhuizenWS, Aartsma-RusA, et al. (2007) Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med 357 : 2677–2686.
19. MedinaMW, KraussRM (2009) The role of HMGCR alternative splicing in statin efficacy. Trends Cardiovasc Med 19 : 173–177.
20. DiFeoA, FeldL, RodriguezE, WangC, BeerDG, et al. (2008) A functional role for KLF6-SV1 in lung adenocarcinoma prognosis and chemotherapy response. Cancer Res 68 : 965–970.
21. NarlaG, DifeoA, ReevesHL, SchaidDJ, HirshfeldJ, et al. (2005) A germline DNA polymorphism enhances alternative splicing of the KLF6 tumor suppressor gene and is associated with increased prostate cancer risk. Cancer Res 65 : 1213–1222.
22. CaceresJF, KornblihttAR (2002) Alternative splicing: multiple control mechanisms and involvement in human disease. Trends in genetics : TIG 18 : 186–193.
23. CartegniL, ChewSL, KrainerAR (2002) Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 3 : 285–298.
24. FaustinoNA, CooperTA (2003) Pre-mRNA splicing and human disease. Genes & development 17 : 419–437.
25. PaganiF, BaralleFE (2004) Genomic variants in exons and introns: identifying the splicing spoilers. Nature reviews Genetics 5 : 389–396.
26. Sterne-WeilerT, HowardJ, MortM, CooperDN, SanfordJR (2011) Loss of exon identity is a common mechanism of human inherited disease. Genome research 21 : 1563–1571.
27. KwanT, BenovoyD, DiasC, GurdS, ProvencherC, et al. (2008) Genome-wide analysis of transcript isoform variation in humans. Nat Genet 40 : 225–231.
28. PickrellJK, MarioniJC, PaiAA, DegnerJF, EngelhardtBE, et al. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 464 : 768–772.
29. CirulliET, SinghA, ShiannaKV, GeD, SmithJP, et al. (2010) Screening the human exome: a comparison of whole genome and whole transcriptome sequencing. Genome Biol 11: R57.
30. DuanS, HuangRS, ZhangW, BleibelWK, RoeCA, et al. (2008) Genetic architecture of transcript-level variation in humans. American journal of human genetics 82 : 1101–1113.
31. HindorffLA, SethupathyP, JunkinsHA, RamosEM, MehtaJP, et al. (2009) Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A 106 : 9362–9367.
32. YeoG, HoonS, VenkateshB, BurgeCB (2004) Variation in sequence and organization of splicing regulatory elements in vertebrate genes. Proc Natl Acad Sci U S A 101 : 15700–15705.
33. GamazonER, ZhangW, KonkashbaevA, DuanS, KistnerEO, et al. (2010) SCAN: SNP and copy number annotation. Bioinformatics 26 : 259–262.
34. BowcockAM, KruegerJG (2005) Getting under the skin: the immunogenetics of psoriasis. Nature reviews Immunology 5 : 699–711.
35. Lango AllenH, EstradaK, LettreG, BerndtSI, WeedonMN, et al. (2010) Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467 : 832–838.
36. BishopDT, DemenaisF, IlesMM, HarlandM, TaylorJC, et al. (2009) Genome-wide association study identifies three loci associated with melanoma risk. Nature genetics 41 : 920–925.
37. FlicekP, AmodeMR, BarrellD, BealK, BrentS, et al. (2011) Ensembl 2011. Nucleic acids research 39: D800–806.
38. GraveleyBR (2009) Alternative splicing: regulation without regulators. Nat Struct Mol Biol 16 : 13–15.
39. BaraniakAP, ChenJR, Garcia-BlancoMA (2006) Fox-2 mediates epithelial cell-specific fibroblast growth factor receptor 2 exon choice. Molecular and cellular biology 26 : 1209–1222.
40. FagnaniM, BarashY, IpJY, MisquittaC, PanQ, et al. (2007) Functional coordination of alternative splicing in the mammalian central nervous system. Genome biology 8: R108.
41. HuhGS, HynesRO (1994) Regulation of alternative pre-mRNA splicing by a novel repeated hexanucleotide element. Genes & development 8 : 1561–1574.
42. JinY, SuzukiH, MaegawaS, EndoH, SuganoS, et al. (2003) A vertebrate RNA-binding protein Fox-1 regulates tissue-specific splicing via the pentanucleotide GCAUG. The EMBO journal 22 : 905–912.
43. LimLP, SharpPA (1998) Alternative splicing of the fibronectin EIIIB exon depends on specific TGCATG repeats. Molecular and cellular biology 18 : 3900–3906.
44. MinovitskyS, GeeSL, SchokrpurS, DubchakI, ConboyJG (2005) The splicing regulatory element, UGCAUG, is phylogenetically and spatially conserved in introns that flank tissue-specific alternative exons. Nucleic acids research 33 : 714–724.
45. PonthierJL, SchluepenC, ChenW, LerschRA, GeeSL, et al. (2006) Fox-2 splicing factor binds to a conserved intron motif to promote inclusion of protein 4.1R alternative exon 16. The Journal of biological chemistry 281 : 12468–12474.
46. UnderwoodJG, BoutzPL, DoughertyJD, StoilovP, BlackDL (2005) Homologues of the Caenorhabditis elegans Fox-1 protein are neuronal splicing regulators in mammals. Molecular and cellular biology 25 : 10005–10016.
47. ZhangW, LiuH, HanK, GrabowskiPJ (2002) Region-specific alternative splicing in the nervous system: implications for regulation by the RNA-binding protein NAPOR. RNA 8 : 671–685.
48. ZhuH, HasmanRA, YoungKM, KedershaNL, LouH (2003) U1 snRNP-dependent function of TIAR in the regulation of alternative RNA processing of the human calcitonin/CGRP pre-mRNA. Molecular and cellular biology 23 : 5959–5971.
49. GenettaT, MorisakiH, MorisakiT, HolmesEW (2001) A novel bipartite intronic splicing enhancer promotes the inclusion of a mini-exon in the AMP deaminase 1 gene. The Journal of biological chemistry 276 : 25589–25597.
50. GuoN, KawamotoS (2000) An intronic downstream enhancer promotes 3′ splice site usage of a neural cell-specific exon. The Journal of biological chemistry 2000/08/10 ed 33641–33649.
51. LiuS, AltmanRB (2003) Large scale study of protein domain distribution in the context of alternative splicing. Nucleic acids research 31 : 4828–4835.
52. LewisBP, GreenRE, BrennerSE (2003) Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proceedings of the National Academy of Sciences of the United States of America 100 : 189–192.
53. AlekseyenkoAV, KimN, LeeCJ (2007) Global analysis of exon creation versus loss and the role of alternative splicing in 17 vertebrate genomes. RNA 13 : 661–670.
54. SugnetCW, KentWJ, AresMJr, HausslerD (2004) Transcriptome and genome conservation of alternative splicing events in humans and mice. Pacific Symposium on Biocomputing Pacific Symposium on Biocomputing 66–77.
55. HaK, Coulombe-HuntingtonJ, MajewskiJ (2009) Comparison of Affymetrix Gene Array with the Exon Array shows potential application for detection of transcript isoform variation. BMC genomics 10 : 519.
56. NicaAC, PartsL, GlassD, NisbetJ, BarrettA, et al. (2011) The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS Genet 7: e1002003 doi:10.1371/journal.pgen.1002003
57. FujitaPA, RheadB, ZweigAS, HinrichsAS, KarolchikD, et al. The UCSC Genome Browser database: update 2011. Nucleic acids research 39: D876–882.
58. KuhnRM, KarolchikD, ZweigAS, WangT, SmithKE, et al. (2009) The UCSC Genome Browser Database: update 2009. Nucleic acids research 37: D755–761.
59. BensonDA, Karsch-MizrachiI, LipmanDJ, OstellJ, WheelerDL (2004) GenBank: update. Nucleic acids research 32: D23–26.
60. HubbardT, BarkerD, BirneyE, CameronG, ChenY, et al. (2002) The Ensembl genome database project. Nucleic acids research 30 : 38–41.
61. Thierry-MiegD, Thierry-MiegJ (2006) AceView: a comprehensive cDNA-supported gene and transcripts annotation. Genome Biol 7 Suppl 1 S12 11–14.
62. HsuF, KentWJ, ClawsonH, KuhnRM, DiekhansM, et al. (2006) The UCSC Known Genes. Bioinformatics 22 : 1036–1046.
63. KentWJ (2002) BLAT–the BLAST-like alignment tool. Genome Res 12 : 656–664.
64. ZhangW, DuanS, BleibelWK, WiselSA, HuangRS, et al. (2009) Identification of common genetic variants that account for transcript isoform variation between human populations. Human genetics 125 : 81–93.
65. IncA (2006) Identifying and validating alternative splicing events. Affymetrix Technical Note
66. NicolaeDL, GamazonE, ZhangW, DuanS, DolanME, et al. (2010) Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet 6: e1000888 doi:10.1371/journal.pgen.1000888
67. StoreyJD, TibshiraniR (2003) Statistical significance for genomewide studies. Proceedings of the National Academy of Sciences of the United States of America 100 : 9440–9445.
68. RoyA, KucukuralA, ZhangY (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nature protocols 5 : 725–738.
69. ZhangY, SkolnickJ (2005) TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic acids research 33 : 2302–2309.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- A Mutation in the Gene Causes Alternative Splicing Defects and Deafness in the Bronx Waltzer Mouse
- Mutations in (Hhat) Perturb Hedgehog Signaling, Resulting in Severe Acrania-Holoprosencephaly-Agnathia Craniofacial Defects
- Classical Genetics Meets Next-Generation Sequencing: Uncovering a Genome-Wide Recombination Map in
- Regulation of ATG4B Stability by RNF5 Limits Basal Levels of Autophagy and Influences Susceptibility to Bacterial Infection
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy