
Topoisomerase II– and Condensin-Dependent Breakage of -Sensitive Fragile Sites Occurs Independently of Spindle Tension, Anaphase, or Cytokinesis
Fragile sites are loci of recurrent chromosome breakage in the genome. They are found in organisms ranging from bacteria to humans and are implicated in genome instability, evolution, and cancer. In budding yeast, inactivation of Mec1, a homolog of mammalian ATR, leads to chromosome breakage at fragile sites referred to as replication slow zones (RSZs). RSZs are proposed to be homologous to mammalian common fragile sites (CFSs) whose stability is regulated by ATR. Perturbation during S phase, leading to elevated levels of stalled replication forks, is necessary but not sufficient for chromosome breakage at RSZs or CFSs. To address the nature of additional event(s) required for the break formation, we examined involvement of the currently known or implicated mechanisms of endogenous chromosome breakage, including errors in replication fork restart, premature mitotic chromosome condensation, spindle tension, anaphase, and cytokinesis. Results revealed that chromosome breakage at RSZs is independent of the RAD52 epistasis group genes and of TOP3, SGS1, SRS2, MMS4, or MUS81, indicating that homologous recombination and other recombination-related processes associated with replication fork restart are unlikely to be involved. We also found spindle force, anaphase, or cytokinesis to be dispensable. RSZ breakage, however, required genes encoding condensin subunits (YCG1, YSC4) and topoisomerase II (TOP2). We propose that chromosome break formation at RSZs following Mec1 inactivation, a model for mammalian fragile site breakage, is mediated by internal chromosomal stress generated during mitotic chromosome condensation.
Published in the journal:
. PLoS Genet 8(10): e32767. doi:10.1371/journal.pgen.1002978
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002978
Summary
Fragile sites are loci of recurrent chromosome breakage in the genome. They are found in organisms ranging from bacteria to humans and are implicated in genome instability, evolution, and cancer. In budding yeast, inactivation of Mec1, a homolog of mammalian ATR, leads to chromosome breakage at fragile sites referred to as replication slow zones (RSZs). RSZs are proposed to be homologous to mammalian common fragile sites (CFSs) whose stability is regulated by ATR. Perturbation during S phase, leading to elevated levels of stalled replication forks, is necessary but not sufficient for chromosome breakage at RSZs or CFSs. To address the nature of additional event(s) required for the break formation, we examined involvement of the currently known or implicated mechanisms of endogenous chromosome breakage, including errors in replication fork restart, premature mitotic chromosome condensation, spindle tension, anaphase, and cytokinesis. Results revealed that chromosome breakage at RSZs is independent of the RAD52 epistasis group genes and of TOP3, SGS1, SRS2, MMS4, or MUS81, indicating that homologous recombination and other recombination-related processes associated with replication fork restart are unlikely to be involved. We also found spindle force, anaphase, or cytokinesis to be dispensable. RSZ breakage, however, required genes encoding condensin subunits (YCG1, YSC4) and topoisomerase II (TOP2). We propose that chromosome break formation at RSZs following Mec1 inactivation, a model for mammalian fragile site breakage, is mediated by internal chromosomal stress generated during mitotic chromosome condensation.
Introduction
Unintended double strand breaks (DSBs) arise during the unchallenged life of the cell. These breaks do not arise randomly throughout the genome, but occur preferentially at loci referred to as fragile sites. Fragile sites exist in all organisms examined to date including bacteria, yeast, flies, plants, and mammals. Examples include the bacterial ter [1], budding yeast replication slow zones (RSZs) [2], and mammalian common - and rare - fragile sites [3], [4]. Some fragile sites are loci of specialized DNA/chromosomal processes. For example, the bacterial ter function as preferred loci of replication fork termination [5]. For the majority of fragile sites however, their precise function, assuming that it exists, remains elusive.
The term “fragile site” was first used to describe a heritable locus of recurrent chromosome breakage on metaphase spreads of human lymphocytes [3]. Currently, there are more than 120 fragile sites identified in the human genome [6]. Notably, not all fragile sites form breaks at the same frequency, and some are more prone to breakage than others. For example, FRA3B at 3p14.2 is the most fragile site in the human genome, exhibiting breaks in 50% of metaphases after a mild replication stress [6], [7]. Reason(s) for the differential tendencies for breakage among mammalian fragile sites is not known.
Mammalian fragile sites are classified as either rare or common, depending on their frequency within the population. Rare fragile sites are seen in <5% of the population. Most rare fragile sites are tri-nucleotide repeats, whose increased breakage is caused by expansion of the repeats [6]. Common fragile sites (CFSs), on the other hand, are present in every chromosome and in all individuals. Furthermore, common fragile sites are conserved throughout mammalian evolution [8], [9] suggesting that they might be a normal component of the chromosome [4].
Mammalian fragile sites are said to be “expressed” when they display signs of breaks or gaps on metaphase chromosome spreads. Studies have identified several conditions that may play a role in mammalian fragile site expression. These include: (i) the time at which a locus is replicated during normal S phase, based on the early observations that the vast majority of mammalian fragile sites replicate late [10]; (ii) mild inhibition of DNA replication, contributing to elevated levels of stalled replication forks and further delays in the replication of the normally late replicating fragile loci [11]; (iii) inactivation of checkpoint proteins such as ATR [12] or ATM [13]; (iv) inactivation of proteins involved in DSB repair and/or replication fork restart [14]; (v) premature onset of mitosis [15]; and (vi) anaphase and/or cytokinesis [16], [17]. These observations led to a number of models regarding the mechanism underlying fragile site expression. In all cases, replication fork stalling is proposed to be the initiating event, with stalled forks ultimately giving rise to a DSB by a process or processes whose exact nature remains unresolved. The uncertainly is, in part, due to the fact that equally plausible hypotheses have not yet been tested in a suitable model system.
The budding yeast genome, like that of other organisms, contains different types of fragile sites. These differ with respect to their structure, distribution in the genome, and genetic requirement for their stability or breakage [2], [18]–[23]. The Replication Slow Zone (RSZ) is a fragile site that was identified based on its sensitivity to the loss of Mec1 function [2]. Mec1, like its mammalian counterpart ATR, is an essential protein [24] involved in a number of fundamental processes, including genome duplication, DNA repair, recombination, meiosis, and checkpoint regulation [24]–[27]. It promotes dNTP synthesis during every G1-S transition to ensure that the cell has sufficient levels of dNTPs for genome duplication [28], [29]. Mec1 up-regulation of dNTP synthesis is also essential during replication stress - or DNA damage - checkpoint responses [30], [31]. Additional checkpoint functions of Mec1 include stabilization of stalled forks, coordination of repair, and preventing cell cycle progression until the damage situation is resolved [27], [32], [33].
The name RSZ was based on the observation that replication forks moved notably slower through these regions than through other loci during normal S phase [2]. In MEC1 cells, forks continue to progress through RSZs, eventually completing their duplication. In mec1-4 cells, replication forks progress more or less normally until they reach RSZs. At RSZs, the forks remain stalled for about 90 minutes, until the appearance of DSBs at these loci some time during G2/M [2]. Analysis of eleven RSZs identified on chromosomes III and VI suggests that they do not occur randomly along the chromosome, but occur between highly active replication origins along the entire length of the chromosome; a notable exception, however, is the centromeric region, which lacks a RSZ [2; N Hashash and R Cha, unpublished data].
RSZs and mammalian CFSs are both large genetic determinants, each comprising about 0.1% of the respective genome (i.e. ∼10 kb RSZ of 1.5×107 bp budding yeast genome and ∼1 Mb CFS of the 3×109 bp mammalian genome). Some studies reported a correlation between the occurrence of some CFSs or RSZs and certain features of the genome, including high flexibility, high AT content, hairpin structure, and/or hotspots for ectopic genome integration [2], [6], [34], [35]. Currently however, there are no structural or functional features that can be utilized for their a priori identification. RSZs and CFSs are both late replicating loci [2], [10] and exhibit sensitivity to mild replication stress or deficiencies in Mec1 or ATR [2], [12], [19]. Largely based on these similarities, RSZs were proposed to be homologous to mammalian CFSs [2], [19], [35].
Here, we investigate the mechanism of RSZ breakage following Mec1 inactivation. Specifically, we tested involvement of each of the five processes implicated in mammalian fragile site expression (above). Results showed that RSZ breakage following Mec1 inactivation required functions of topoisomerase II (Top2) and the condensin complex; in the absence of Top2 or condensins, RSZs did not break, even though replication forks still stalled. In contrast, replication fork restart, spindle tension, anaphase, or cytokinesis were all dispensable for RSZ breakage. Based on these observations, we propose that internal chromosomal stress, generated during mitotic chromosome condensation, promotes the conversion of stalled forks at RSZs to DSBs.
Results
Key genes required for processing of stalled replication forks are dispensable for RSZ breakage
Replication forks stall during unchallenged S phase either as a part of normal replication program [2], [21], [36], [37] or incidentally, upon encountering a damaged template [33] or due to insufficient levels of dNTPs [19], [30], [32]. In either case, the resumption of DNA synthesis from stalled forks or the rescue by the firing of cryptic origins, while maintaining the integrity of stalled forks, is essential for cell's survival. Homologous recombination plays a key role in replication fork restart [38]. During this process, DSBs can be generated as an intermediate and contribute to endogenous chromosome breakage. To test whether breakage at RSZs is generated via homologous recombination or recombination-related process, we utilized a previously characterized temperature sensitive mec1 strain, mec1-4 [2], and assessed the effects of eliminating relevant proteins; homologous recombination proteins (Rad50, Rad51, Rad52, Rad54, Rad55, and Mre11), the Sgs1BLM-Top3 complex, the Srs2 helicase, and the Mus81-Mms4 endonuclease, a putative resolvase [38]–[42]. Thermal inactivation of Mec1-4 results in prolonged replication fork stalling at RSZs, followed by chromosome breakage at these loci. The breaks appear typically around 90–120 minutes after alpha-factor arrest/release, corresponding to the G2/M phase of the first cell cycle after the release.
MEC1 and mec1-4 strains carrying a null allele of one of the genes mentioned above were arrested with a-factor at 23°C and released into fresh YPD media at 37°C, a restrictive temperature for mec1-4 [2]. Samples were collected 3 hours after the release, and the status of chromosome III (ChrIII) was assessed by pulse field gel electrophoresis (PFGE) followed by Southern hybridization using a telomere-proximal probe, CHA1 (Figure 1A, 1B). As shown previously [2], DSBs enriched for RSZs in ChrIII were observed in the mec1-4 culture (Figure 1C, 1D). Elimination of various proteins with a role in the replication fork restart did not prevent break formation at RSZs (Figure 1; data not shown) indicating that the involvement of this process was unlikely.

Inappropriate mitosis does not contribute to RSZ breakage
The spindle assembly checkpoint (SAC) is an evolutionarily conserved mechanism responsible for ensuring that every pair of sister-chromatids is under spindle tension prior to anaphase. The SAC monitors this process by assessing microtubule occupancy of the kinetochores and/or tension generated across chromosomes/kinetochores [43]. Like the other checkpoint systems, the SAC is a signal transduction cascade and is mediated by the Mad1, 2, 3 (mitotic arrest deficient) and Bub1, 2, 3 (budding uninhibitied by benzimidazole) proteins. In the absence of these proteins, cells proceed through mitosis irrespective of whether all sister kinetochores are under spindle tension, resulting in frequent chromosome mis-segregation and cell death.
Although the SAC was originally thought to operate independently of the DNA damage checkpoint, recent evidence suggests an interplay between the two. For example, Mec1/Tel1 have been shown to inhibit anaphase by utilizing Mad/Bub proteins independently of the kinetochores in response to DNA damage [44], which might account for the earlier observation that about 50% of mec1-4 cells undergo mitosis despite the presence of unresolved replication forks [2]. These considerations raise the possibility that RSZ breakage might occur as a result of inappropriate mitosis. If this was the case, we reasoned that inactivation of the SAC might increase RSZ breakage by allowing a greater proportion of mec1-4 cells to proceed through mitosis with unresolved replication forks at RSZs. We tested this possibility by assessing the impact of deleting MAD2 or BUB2 on RSZ breakage.
MEC1 and mec1-4 strains in mad2Δ BUB2, MAD2 bub2Δ, or MAD2 BUB2 backgrounds were arrested with alpha-factor at 23°C and released into fresh YPD media at a restrictive temperature for mec1-4. Samples were collected 3 hours after the release, and the status of ChrIII was assessed. As expected, RSZ breakage was observed in mec1-4 control culture (Figure 2A). RSZ breakage was also observed in the mec1-4 mad2Δ or mec1-4 bub2Δ cultures (Figure 2A). Furthermore, the extent of breakage was comparable in the presence or absence of MAD2/BUB2, suggesting that RSZ breakage was unlikely to be caused by compromised SAC function, leading to inappropriate mitosis.

Spindle tension is dispensable for RSZ breakage
Another implication of the lack of an impact of mad2Δ or bub2Δ is that RSZ breakage might be independent of spindle tension. Assuming that inactivation of the SAC would have allowed some cells to proceed through mitosis in the absence, or with a reduced level, of spindle tension, we reasoned that mad2Δ or bub2Δ may have resulted in a reduction in RSZ breakage if spindle tension played a role. To directly address the involvement of spindle tension, we investigated the effects of microtubule depolymerising drugs such as methyl1-2-benzimidazolecarbamate (MBC) or nocodazole. First, we confirmed that spindle poison was effective in preventing elongation of spindles in mec1-4 cells (Figure 2D). In the absence of spindle poison, the elongation in mec1-4 cells occurred reproducibly earlier than in MEC1 (Figure 2Di). The reason for this remains unknown but is likely to be related to the role(s) of Mec1/Tel1/Rad53 in regulating spindle status in response to DNA damage or replication stress [45], [46]. We also found that MBC blocked Clb2 degradation, a readout for mitotic exit [47], in mec1-4 (Figure 2E). The latter suggested that although mec1-4 cells were compromised in preventing the onset of mitosis in the presence of stalled forks [2], they were competent in mediating a spindle damage-dependent SAC response.
To ensure that we assessed the impact of spindle depolymerisation on RSZ breakage, rather than the impact of SAC response to the depolymerisation, we decided to examine the effects of spindle poison in the absence of the SAC. mec1-4 mad2Δ or mec1-4 bub2Δ strains were released from alpha-factor arrest as described above, except that they were released into YPD media containing either MBC or nocodazole (Materials and Methods). Samples were collected 3 hours after the release and assessed for RSZ breakage. Results showed chromosome breakage in the presence of either drug (Figure 2B; data not shown), demonstrating that mitotic spindles are dispensable for RSZ breakage.
RSZ breakage requires Top2 and condensin function
The dispensability of mitotic spindles prompted us to consider whether internal chromosomal stress might be involved in RSZ breakage. During mitotic prophase, the duplicated genome undergoes dramatic structural reorganization, leading to sister chromatid individualisation and chromosome compaction in preparation for segregation during anaphase [48], [49]. To test whether the intra-chromosomal stress generated during these processes might have a role in RSZ breakage in the absence of spindle tension, we assessed the impact of inactivating relevant gene products. These included; (i) Scc1/Mcd1 (hereon referred to as Scc1), a component of the cohesion complex that holds sister chromatids together until their disjunction at the onset of anaphase [47], [50], (ii) Esp1, a caspase-like cysteine protease that promotes sister chromatid separation by mediating the cleavage of Scc1 [51], (iii) Ycg1 and Ysc4, two non SMC components of the condensin complex, required for mitotic chromosome compaction [52], [53], and (iv) Top2, a type II topoisomerase that catalyzes decatetation of DNA strands between the sister chromatids to allow their resolution and facilitate chromosome condensation [48], [49], [54].
A set of mec1-4 mad2Δ strains, each expressing temperature sensitive scc1, esp1, ycg1, ysc4, or top2 alleles were released from alpha-factor arrest into YPD + MBC media at 37°C, a restrictive temperature for all of the conditional alleles utilized. Samples were collected 3 hours after the release and analysed for RSZ breakage (Figure 2C). As expected, RSZ breakage was observed in the mec1-4 mad2Δ control strain. The breakage was also observed in strains expressing a temperature sensitive scc1 or esp1 allele, suggesting that RSZ breakage occurred independently of the status of the cohesins. In contrast, inactivation of Top2, Ycg1, or Ysc4 suppressed chromosome breakage, suggesting that mitotic chromosome condensation might be involved in RSZ breakage in the absence of spindle tension.
To rule out the possibility that the top2, ycg1, or ysc4 suppression was mediated by their impact on S phase progression, either by allowing replication forks to progress through RSZs [2] or by committing cells to inviability before forks reach a RSZ [19], we assessed their impact on the status of S phase progression. A WT, mec1-4 mad2Δ, or mec1-4 mad2Δ strain expressing a temperature sensitive allele of either top2 or ycg1 was released from G1 arrest into YPD+MBC media at 37°C. Samples were collected at various time points after the release and subjected to fluorescent activated cell scan (FACS) analysis (Materials and Methods). In a WT control, cells proceeded through S phase and completed bulk genome duplication by 80 minutes following alpha-factor arrest/release (Figure 2F). In contrast, S phase progression in a mec1-4 mad2Δ culture, like that in a mec1-4 culture [2] was delayed (Figure 2F), consistent with earlier observations that the status of MAD2 did not confer any effects on DNA replication [55]. Thermal inactivation of top2 or ycg1 did not exert a notable effect on S phase progression, suggesting that the top2, ycg1, or ysc4 suppression was unlikely to be due to their impact on DNA replication. Taken together, we conclude that Top2/condensin-mediated mitotic chromosome condensation triggers RSZ breakage in the absence of mitotic spindles.
RSZ breakage in the presence of spindle tension also requires Top2 and condensin function
Results thus far showed that breakage of RSZ, like that of mammalian CFSs, can occur in the absence of spindle tension. Importantly however, breakage of RSZs or CFSs occurs during normal cell proliferation in the presence of spindle tension. Thus, it was formally possible that the Top2/condensin dependent RSZ breakage was a mechanism operating specifically in the absence of mitotic spindles. To address this, we assessed the effects of inactivating Top2, Ycg1, or Ysc4 in the presence of spindle tension. A set of mec1-4 MAD2 BUB2 strains expressing temperature sensitive alleles of top2, ycg1, or ysc4 was released from G1 arrest into a fresh YPD media in the absence of spindle poison. Samples were collected 3 hours after the release and analysed for RSZ breakage. The results showed that top2, ycg1, or ysc4 suppressed RSZ breakage (Figure 3A). In contrast, thermal inactivation of Scc1 or Esp1 did not prevent the breakage (Figure 3B). These results suggest that the genetic requirement (and the mechanism, by extension) of RSZ breakage in the presence or absence of spindle tension is likely to be the same. We conclude that Top2/condensin mediated mitotic chromosome condensation is required for RSZ breakage during normal cell proliferation irrespective of the status of spindle tension.
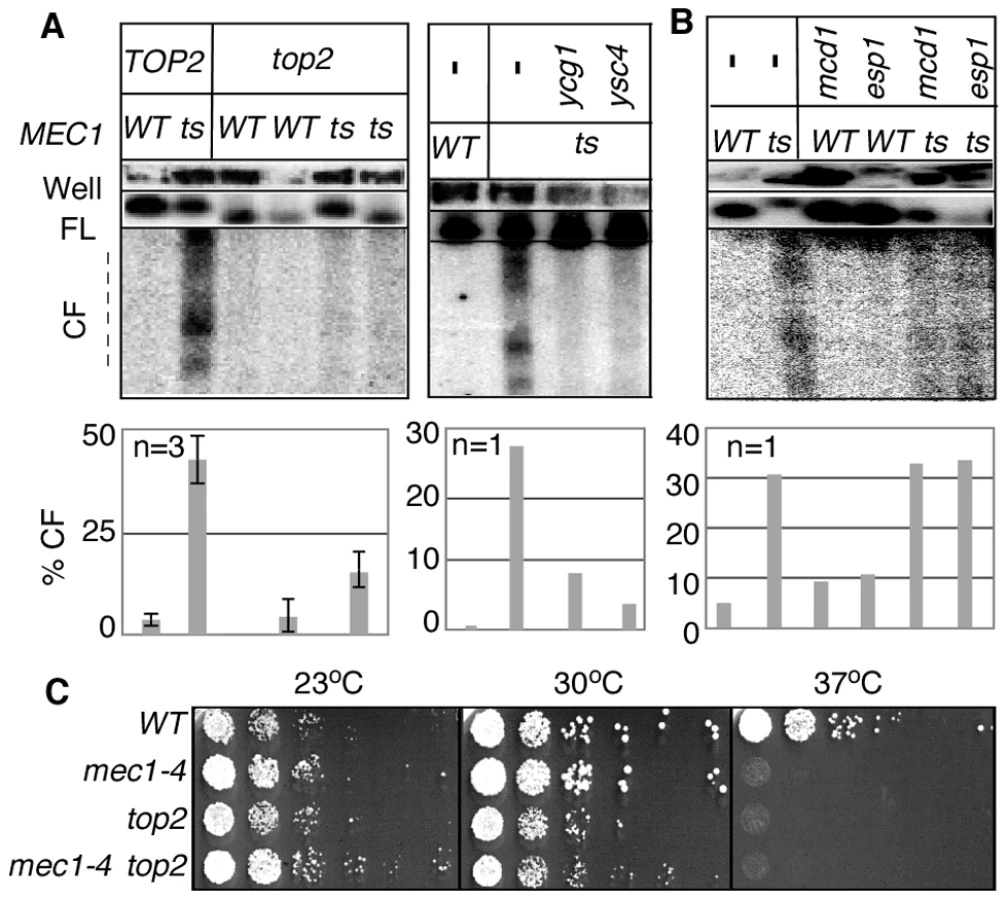
As expected from the essential nature of TOP2 and condensin, inactivation of these gene products did not rescue the lethality of mec1-4 at non-permissive temperature (Figure 3C; data not shown).
RSZ breakage occurs independently of anaphase or cytokinesis
If RSZ breakage is independent of spindle tension, then a prediction might be that it should also be independent of the events downstream of the SAC execution point, such as anaphase, mitotic exit, and cytokinesis. We tested this by monitoring the occurrence of these events in mec1-4 and MEC1 cultures as they proceeded through a synchronous cell cycle in the presence of spindle tension. Samples were collected at various time points following alpha-factor arrest/release and assayed for RSZ-expression, Scc1-cleavage, a readout for the onset of anaphase [43], and Clb2-degradation, a readout for exit from mitosis [47] (Figure 4).

In the MEC1 culture, cells completed bulk genome duplication between 60–75 minutes following alpha-factor release (Figure 4A panels i and v). An Scc1 cleavage product was observed starting from 75 minutes after release (Figure 4A panels ii and v). Levels of Clb2 peaked at 45 and 60 minutes following the release and decreased rapidly thereafter (Figure 4A, panels iii and v). These results indicate that, in the MEC1 culture, the completion of bulk genome duplication (60–75′), onset of anaphase (75–90′), and exit from mitosis (90′) occurred in a temporally ordered manner. PFGE/Southern analysis of ChrIII showed that chromosome breakage in this culture remained at background levels (Figure 4A panel iv).
In the mec1-4 culture, the cells remained stuck in mid-S phase from about 60 minutes following the release, suggesting that anaphase did not take place (Figure 3B panels i and v). Scc1 cleavage was not observed (Figure 3B panels ii and v). A modest reduction in Clb2 levels was observed starting 90 min after the release, although the extent of reduction was notably less than that observed in the MEC1 culture (Figure 3B, panels iii and v). In this mec1-4 culture, DSBs began to accumulate starting at t = 90–105 minutes (Figure 3B panels iv and v). These results demonstrate that RSZ-expression in the presence of spindle tension occurs in the absence of Scc1 cleavage, Clb2 degradation, or, by extension, the onset of anaphase or exit from mitosis, respectively.
Next, the occurrence of cytokinesis was assessed. To this end, we generated MEC1 and mec1-4 strains expressing an endogenous copy of MYO1-GFP, and monitored the appearance of binucleate cells with or without the Myo1-GFP signal (Figure 4C panel iv versus panel v). MYO1 encodes a component of the actomyosin ring that localizes to the bud neck from early S phase (e.g. Figure 4C panel ii). The Myo1 ring remains at the neck until cytokinesis, during which the ring constricts and Myo1 disappears from the bud neck [56] (Figure 4C panel v). MEC1 MYO1-GFP and mec1-4 MYO1-GFP cells were released from alpha-factor arrest into fresh YPD media at a restrictive temperature in the absence of spindle poison. Samples were collected at various time points and assessed for the morphology of DNA (via a DAPI stain) and the status of Myo1-GFP ring. In both cultures, the Myo1-GFP ring appeared by 45 minutes after release (Figure 4C panel vi). In MEC1 cells, the Myo1-GFP ring remained at the bud neck until 75 minutes and disappeared by 105 minutes, indicating that cytokinesis had occurred. In contrast, the Myo1-GFP ring in mec1-4 cells remained at the bud neck throughout the duration of the experiment, indicating that cytokinesis did not take place (Figure 4C panel vi). In this culture, the fraction of cells that had undergone anaphase – i.e. those containing two DAPI staining bodies that were separated by MyoI-GFP ring (e.g. Figure 4C panel iv) remained low, in agreement with the lack of Scc1 cleavage (Figure 4Bii). In the MEC1 control, the fraction of binucleate cells reached about 50% by t = 75 minutes; thereafter, the fraction decreased as the cells underwent cytokinesis. RSZ expression in the mec1-4 culture was observed at t = 120 minutes (data not shown).
Taken together, these results show that RSZ breakage in the presence of spindle tension occurs independently of anaphase, mitotic exit, or cytokinesis. The simplest interpretation would be that the breakage occurs before the onset of anaphase.
Discussion
The aim of this study was to examine the mechanism of chromosome breakage at RSZ, a MEC1-sensitive fragile site and a model for mammalian CFSs. Specifically, we tested involvement of the following possibilities, each of which had been implicated in mammalian fragile site expression: (i) errors in replication fork restart,; (ii) premature mitotic chromosome condensation; (iii) spindle tension; (iv) anaphase; or (v) cytokinesis. Evidence revealed that Top2 and condensin proteins were required for RSZ breakage; in contrast, the key proteins involved in replication fork restart, spindle tension, anaphase, or cytokinesis were dispensable.
In all eukaryotes examined to date, an essential function of Top2 and condensins is chromosome compaction [48], [49], [53], [54], [57], [58]. Although the extent of mitotic chromosome condensation in budding yeast is about two orders of magnitude less than that observed in metazoan cells (the compaction ratio is 160 in yeast versus 10,000–20,000 in metazoans; 57,58), inactivation of budding yeast Top2 or condensin subunits also results in a chromosome compaction defect [52], [59], [60], in agreement with the notion that the mechanism is evolutionarily conserved.
Taken together, we propose a model whereby two temporally and genetically distinguishable events mediate chromosome breakage at RSZs (Figure 5). In WT cells, the genome duplication is complete by the end of S phase (Figure 5Ai). During mitotic prophase, the duplicated genome undergoes Top2 - and condensin - dependent chromosome compaction (Figure 5Aii) in preparation for its disjunction during anaphase (Figure 5Aiii). In the absence of Mec1 function, replication forks stall at RSZs (Figure 5Bi); despite this, the cells exit S phase and proceed through the cell cycle. During prophase, the incompletely duplicated genome of mec1-4 cells becomes subjected to Top2 - and condensin - dependent chromosome compaction (Figure 5Bii). We propose that internal stress generated during this process promotes the conversion of stalled forks to a DSB. The molecular mechanism underlying the catalysis of breakage is unknown, but may involve a nuclease that is yet to be identified (below). Above evidence indicates that chromosome breakage is independent of spindle tension or tension-dependent events such as anaphase or cytokinesis. This breakage is also independent of the status of sister chromatid cohesins, consistent with the report that cohesion removal, while essential for sister chromatid resolution, is dispensable for mitotic chromosome compaction [61]. In the absence of Top2 or condensins (Figure 5C), chromosome condensation does not take place; therefore, the incompletely duplicated genome of mec1-4 cells is not subjected to the internal stress that triggers the conversion of stalled forks to DSBs. Nevertheless, the cells die, likely due to the lack of an essential Top2 or condensin function(s) [52], [62].

With regard to the dispensability of the replication fork restart process, it is important to note that the list of candidate genes examined is not exhaustive, and therefore, we cannot rigorously eliminate its involvement based on this line of evidence. Nevertheless, our results unequivocally rule out the involvement of some of the key players in replication fork restart that had previously been implicated in breakage at different types of fragile sites (see below); the RAD52 epistasis group proteins, the Sgs1BLM-Top3 complex, the Srs2 helicase, and the Mus81-Mms4 endonuclease [38]–[42].
The dispensability of spindle tension is not surprising in the light of the fact that the distribution pattern of RSZs is different from that of the spindle tension mediated breaks. Specifically, RSZs are found between active replication origins along the entire length of the chromosome except for the centromeric region ([2]; N. Hashash and R. Cha, unpublished data). In contrast, spindle tension-dependent DSBs tend to occur around the centromere, the locus of greatest spindle tension [22], [63]. Mammalian CFSs, like RSZs, are found along the chromosome arms. Furthermore, the fact that mammalian fragile sites are defined as loci of recurrent breaks or gaps on metaphase chromosome spreads, obtained from cultures treated with spindle poisons such as colchicines [3], [4], strongly suggest that expression of mammalian fragile sites, like that of RSZs, occurs independently of spindle tension.
The amount of force exerted by a pair of microtubules at the centromere (i.e. 20 piconewton [pN]) is estimated to be at least an order of magnitude smaller than that required to break the chromosome (i.e. 480 pN) [64], [65]. Assuming that the intra-chromosomal stress generated during mitotic chromosome compaction is less than that generated by the spindles, it is likely that the Top2/condensin-dependent RSZ breakage is mediated by an endonuclease. As a means to test whether Top2 was the responsible enzyme, we performed Top2 ChIP-on-CHIP analysis in MEC1 and mec1-4 cells, reasoning that if Top2 catalyzed the cleavage, we might observe its enrichment at RSZs. Analysis thus far has failed to show any such enrichment, suggesting that its direct involvement was unlikely (N Hashash, R Cha, Y Katou, K Shirahege; unpublished data). Nevertheless, this observation alone does not eliminate the possibility, because Top2 may dissociate from the ends of the DSB after DNA cleavage, and therefore would not normally remain enriched at RSZs. Alternatively, the cleavage might be mediated by a different protein, for example, Yen1, an evolutionarily conserved Holiday junction resolvase that is activated during M phase of the cell division cycle [66] or proteins involved in post replication repair [67]. It is also possible that the DSBs at RSZs result from cleavage of single stranded DNA associated with stalled forks [68].
A positive role for Top2 and condensin in chromosome breakage is unexpected in light of the observations that their inactivation caused, rather than prevented, DSB formation [e.g. [22], [23], [69], [70]. Also surprising is the dispensability of anaphase or cytokinesis in RSZ breakage. Upon a closer examination, however, it becomes apparent that the chromosome breakage examined in each study was at different types of fragile loci in the genome, in that they differed with respect to their structure (e.g. a hairpin or a specific protein-DNA complex), distribution (e.g. chromosome arms versus the centromeres) and/or the timing of their expression (e.g. during S phase, before anaphase, or during cytokinesis) [2], [16], [18]–[20], [22], [23], [41], [69], [71]. These observations provide further support for the notion that both the stability and the expression of each type of fragile sites is under a specific genetic and regulatory control [19].
Among the different types of fragile sites identified and characterized to date, the RSZ appears to be the closest structural and functional homolog of mammalian fragile sites. Furthermore, among the currently proposed mechanisms of mammalian fragile site expression, the mechanism of RSZ breakage inferred in the current study seems to be most consistent with the original definition of a mammalian fragile site, that it is a heritable locus of recurrent breaks or gaps on metaphase chromosome spreads [3]. Taken together, it is tempting to speculate that the mechanism of RSZ breakage and that of CFS expression, at least for those that are sensitive to the loss of ATR or ATM functions [12], [13], might be conserved and that the mammalian Top2 and condensin may similarly play a role in promoting fragile site expression.
Materials and Methods
Yeast strains and media
All strains were of the SK1 background except those noted. Relevant genotypes of the strains are listed in Table S1. Unless specified otherwise, cells were grown in YPD (1% [w/v] yeast extract, 2% [w/v] bacto-peptone, 2% [w/v] glucose) media. To obtain a synchronous culture for cell cycle analysis, mid-log cultures were arrested with 5 µg/ml alpha-factor for 3 hours before being released to fresh YPD media. For temperature-sensitive strains, cells were arrested with alpha-factor at 23°C before releasing to YPD media prewarmed to a restrictive temperature. To induce microtubule deploymerization, cells were grown in the presence of either 15 µg/ml nocodazole (Sigma-Aldrich) or 40 µg/ml carbendazim (MBC; Sigma-Aldrich).
Fluorescence-activated cell scan (FACS)
Cells from 1 ml of relevant samples were fixed (40% [v/v] ethanol, 0.1 M sorbitol) for at least 3 hours before being pelleted, resuspended in RNase solution (50 mM Tris-HCl pH 7.5, 100 µg/ml RNaseA) and incubated overnight at 37°C. The next day, the cells were treated with 500 µl of pepsin solution (50 mM HCl, 5 mg/ml pepsin) for a minimum of 5 minutes at room temperature, before being resuspended in 1 ml SYTOX solution (50 mM Tris-HCl pH 7.5, 1 µM SYTOX Green; Invitrogen Molecular Probes). The samples were incubated overnight at 4°C. The next day, they were analyzed on a Becton Dickinson FACSan using Cell Quest software (Becton Dickinson).
Chromosome breakage analysis by pulse field gel electrophoresis (PFGE)/Southern blot analysis
Chromosome-sized DNA in agarose plugs for PFGE was prepared as described [72]. Electrophoresis was performed at 14°C in a Bio-Rad CHEF Mapper under the following condition: a voltage gradient of 6 V/cm, switch times of 5–30 sec, a switch angle of 115°, in a 1% agarose gel in 0.5× TBE for 24 hours. The DNA in gels was transferred to nylon membranes and hybridized with 32P-labeled CHA1 probe, a 757 bp HindIII-BamHI fragment (−156 to +601 of the ORF) restricted from a pUC19 based plasmid (pRSC38). The image was visualized and signals quantified using a Storm 860 PhosphorImager and ImageJ software, respectively.
Fluorescence microscopy
900 µl from appropriate samples was incubated with 100 µl 37% (w/v) formaldehyde (Fisher Scientific) for 10 minutes at room temperature. The cells were pelleted and washed twice with 1 ml PBS, and then resuspended in 200 µl PBS. A 10 µl sample of the cell suspension was spread onto a glass microscope slide and left to dry. Before application of the glass coverslip, 2 µl of 4,6-diamino-2-phenylindole (DAPI; Sigma) solution (1.5 µg/ml in Vectashield mounting medium [Vector Lab]) was dotted onto the dried cells. Fluorescence microscopy was performed on a Deltavision Spectris system.
Western blots
Whole-cell extracts were prepared from cell suspension in 20% trichloroacetic acid by agitation with glass beads. Precipitated proteins were solubilized in SDS-PAGE sample buffer and appropriate dilutions were subjected to SDS-PAGE and Western blotting. Antibodies utilized for Western blotting were, rabbit polyclonal anti-Clb2 (Santa Cruz Biotechnology Inc), mouse monoclonal anti-HA (12CA5; NIMR, London), mouse monoclonal anti-MYC (9E10; NIMR, London), and rat monoclonal anti-tubulin (YL1/2; Abcam). For each antibody a 1∶1000 dilution was used for Western Blotting except for anti-tubulin, which was used at a 1∶5000 dilution.
Supporting Information
Zdroje
1. BidnenkoV, EhrlichS, MichelB (2002) Replication fork collapse at replication terminator sequences. EMBO J 21 : 3898–3907.
2. ChaRS, KlecknerN (2002) ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 297 : 602–606.
3. MagenisR, HechtR, LovrienE (1970) Heritable fragile site on chromosome 16: probable localization of haptoglobin locus in man. Science 170 : 85–87.
4. Sutherland G, Hecht F (1985) Fragile sites on human chromosomes. New York: Oxford University Press.
5. Hill T (1996) Features of the chromosome terminus region. In: Neidhardt F, editor. Escherichia coli and Salmonella: Cellular and Molecular Biology. Washington DC: ASM Press. pp. 1602–1615.
6. DurkinS, GloverT (2007) Chromosome fragile sites. Ann Rev Gen 41 : 169–192.
7. GloverT, SteinC (1987) Induction of sister chromatid exchanges at common fragile sites. Am J Hum Genet 41 : 882–890.
8. SmeetsD, van de KlundertF (1990) Common fragile sites in man and three closely related primate species. Cytogenet Cell Genet 53 : 8–14.
9. ElderF, RobinsonT (1989) Rodent common fragile sites: are they conserved? Evidence from mouse and rat. Chromosoma 97 : 459–464.
10. LairdC, JaffeE, KarpenG, LambM, NelsonR (1987) Fragile sites in human chromosomes as regions of late-replicating DNA. TIB 3 : 274–281.
11. GloverT, BergerC, CoyleJ, EchoB (1984) DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum Genet 67 : 136–142.
12. CasperA, NghiemP, ArltM, GloverT (2002) ATR regulates fragile site stability. Cell 111 : 779–789.
13. Ozer-GalaiE, SchwartzM, RahatA, KeremB (2008) Interplay between ATM and ATR in the regulation of common fragile site stability. Oncogene 27 : 2109–2117.
14. SchwartzM, ZlotorynskiE, GoldbergM, OzeriE, RahatA, et al. (2005) Homologous recombination and nonhomologous end-joining repair pathways regulate fragile site stability. Genes Dev 19 : 2715–2726.
15. El AchkarE, Gerbault-SeureauM, MulerisM, DutrillauxB, DebatisseM (2005) Premature condensation induces breaks at the interface of early and late replicating chromosome bands bearing common fragile sites. Proc Natl Acad Sci U S A 102 : 18069–18074.
16. NaimV, RosselliF (2009) The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat Cell Biol 11 : 761–768.
17. ChanK, Palmai-PallagT, YingS, HicksonI (2009) Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol 11 : 753–760.
18. AdmireA, ShanksL, DanzlN, WangM, WeierU, et al. (2006) Cycles of chromosome instability are associated with a fragile site and are increased by defects in DNA replication and checkpoint controls in yeast. Genes Dev 20 : 159–173.
19. HashashN, JohnsonAL, ChaRS (2011) Regulation of fragile sites expression in budding yeast by MEC1, RRM3 and hydroxyurea. J Cell Sci 124 : 181–185.
20. LemoineF, DegtyarevaN, LobachevK, PetesT (2005) Chromosomal translocations in yeast induced by low levels of DNA polymerase: A model for chromosome fragile sites. Cell 120 : 587–598.
21. TorresJ, BesslerJ, ZakianV (2004) Local chromatin structure at the ribosomal DNA causes replication fork pausing and genome instability in the absence of the S. cerevisiae DNA helicase Rrm3p. Genes Dev 18 : 498–503.
22. SpellRM, HolmC (1994) Nature and distribution of chromosomal intertwinings in Saccharomyces cerevisiae. Mol Cell Biol 14 : 1465–1476.
23. BermejoR, CarpraT, Gonzalez-HuiciV, FachinettiD, CocitoA, et al. (2009) Genome-organizing factors Top2 and Hmo1 prevent chromosome fragility at sites of S phase transcription. Cell 138 : 870–884.
24. KatoR, OgawaH (1994) An essential gene, ESR1, is required for mitotic cell growth, DNA repair and meiotic recombination in Saccharomyces cerevisiae. Nucleic Acids Res 22 : 3104–3112.
25. CarballoJA, ChaRS (2007) Meiotic roles of Mec1, a budding yeast homolog of mammalian ATR/ATM. Chromosome Res 15 : 539–550.
26. CarballoJA, JohnsonAL, SedgwickSG, ChaRS (2008) Phosphorylation of the axial element protein Hop1 by Mec1/Tel1 ensures meiotic interhomolog recombination. Cell 132 : 758–770.
27. WeinertTA, KiserGL, HartwellLH (1994) Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev 8 : 652–665.
28. ZhaoX, MullerEGD, RothsteinR (1998) A suppressor of two essential checkpoint genes identifies novel protein that negatively affects dNTP pools. Mol Cell 2 : 329–340.
29. ZhaoX, ChabesA, DomkinV, ThelanderL, RothsteinR (2001) The ribonucleotide reductase inhibitor Sml1 is a new target of the Mec1/Rad53 kinase cascade during growth and in response to DNA damage. EMBO J 20 : 3544–3553.
30. DesanyB, AlcasabasAA, BachantJB, ElledgeSJ (1998) Recovery from DNA replicational stress is the essential function of the S phase checkpoint pathway. Genes Dev 12 : 2956–2970.
31. AndresonB, GuptaA, GeorgievaB, RothsteinR (2010) The ribonucleotide reductase inhibitor, Sml1, is sequentially phosphorylated, ubiquitylated and degraded in response to DNA damage. Nucleic Acids Res 38 : 6490–6501.
32. LopesM, Cotta-RamusinoC, PellicioliA, LiberiG, PlevaniP, et al. (2001) The DNA replication checkpoint response stablizes stalled replication forks. Nature 412 : 557–561.
33. TerceroJ, DiffleyJ (2001) Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 412 : 553–556.
34. MishmarD, RahatA, SchererS, NyakaturaG, HinzmannB, et al. (1998) Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site. Proc Natl Acad Sci U S A 95 : 8141–8146.
35. ZhangH, FreudenreichC (2007) An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol Cell 27 : 367–379.
36. BrewerBJ, FangmanWL (1988) A replication fork barrier at the 3′ end of yeast ribosomal RNA genes. Cell 55 : 637–643.
37. DalgaardJ, KlarA (2000) swi1 and swi3 perform imprinting, pausing, and termination of DNA replication in S. pombe. Cell 102 : 745–751.
38. CoxM, GoodmanM, KreuzerK, SherrattD, SandlerS, et al. (2000) The importance of repairing stalled replication forks. Nature 404 : 37–41.
39. CollinsS, MillerK, MaasN, RoguevA, FillinghamJ, et al. (2007) Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature 446 : 806–810.
40. LambertS, MizunoK, BlaisonneauJ, MartineauS, ChanetR, et al. (2010) Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol Cell 346–359.
41. LobachevK, GordeninD, ResnickM (2002) The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell 108 : 183–193.
42. CotéA, LewisS (2008) Mus81-dependent double-strand DNA breaks at in vivo-generated cruciform structures in S. cerevisiae. Mol Cell 31 : 800–812.
43. LewD, BurkeD (2003) The spindle assembly and spindle position checkpoints. Annu Rev Genet 37 : 251–282.
44. KimE, BurkeD (2008) DNA damage activates the SAC in an ATM/ATR dependent manner, independently of the kinetochore. PLoS Genetics 4: e1000015 doi:10.1371/journal.pgen.1000015.
45. KrishnanV, NirantarS, CrastaK, ChengA, SuranaU (2004) DNA replication checkpoint prevents precocious chromosome segregation by regulating spindle behavior. Mol Cell 16 : 687–700.
46. BachantJ, JessenS, KavanaughS, FieldingC (2005) The yeast S phase checkpoint enables replicating chromosomes to bi-orient and restrain spindle extension during S phase distress. J Cell Biol 168 : 999–1012.
47. UhlmannF, WernicD, PoupartM-A, KooninE, NasmythK (2000) Cleavage of cohesin by the CD clan protease separin triggers anaphase in yeast. Cell 103 : 375–386.
48. UemuraT, OhkuraH, AdachiY, MorinoK, ShiozakiK, et al. (1987) DNA topoisomerase II is required for condensation and separation of mitotic chromosomes in S. pombe. Cell 50 : 917–925.
49. Gimenez-AbianJ, ClarkeD, DevlinJ, Gimenez-AbianM, De la TorreC, et al. (2000) Premitotic chromosome individualization in mammalian cells depends on topoisomerase II activity. Chromosoma 109 : 235–244.
50. GuacciV, KoshlandD, StrunnikovA (1997) A direct link between sister chromtid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell 91 : 47–57.
51. CioskR, ZachariaeW, MichaelisC, ShevchenkoA, MannM, et al. (1998) An ESP1/PDS1 complex regulates loss of sister chromatid cohesion at the metaphase to anaphase transition in yeast. Cell 93 : 1067–1076.
52. LavoieB, TuffoK, OhS, KoshlandD, HolmC (2000) Mitotic chromosome condensation requires Brn1p, the yeast homologue of Barren. Mol Biol Cell 11 : 1293–1304.
53. LosadaA, HiranoT (2005) Dynamic molecular linkers of the genome: the first decade of SMC proteins. Genes Dev 19 : 1269–1287.
54. Gimenez-AbianJ, ClarkeD, MullingerA, DownesC, JohnstonR (1995) A postprophase topoisomerase II-dependent chromatid core separation step in the formation of metaphase chromosomes. J Cell Biol 131 : 7–17.
55. AlexandruG, ZachariaeW, SchleifferA, NasmythK (1999) Sister chromatid separation and chromosome re-duplication are regulated by different mechanisms in response to spindle damage. EMBO J 18 : 2707–2721.
56. BiE, MaddoxP, LewD, SalmonE, McMillanJ, et al. (1998) Involvement of an actomyosin contractile ring in Saccharomyces cerevisiae cytokinesis. J Cell Biol 142 : 1301–1312.
57. LavoieB, HoganE, KoshlandD (2002) In vivo dissection of the chromosome condensation machinery: reversibility of condensation distinguishes contributions of condensin and cohesin. J Cell Biol 156 : 805–815.
58. GuacciV, HoganE, KoshlandD (1994) Chromosome condensation and sister chromatid pairing in budding yeast. J Cell Biol 125 : 517–530.
59. StrunnikovA, HoganE, KoshlandD (1995) SMC2, a Saccharomyces cerevisiae gene essential for chromosome segregation and condensation, defines a subgroup within the SMC family. Genes Dev 9 : 587–599.
60. VasA, AndrewsC, Kirkland MateskyK, ClarkeD (2007) In vivo analysis of chromosome condensation in Saccharomyces cerevisiae. Mol Biol Cell 18 : 557–568.
61. LosadaA, HiranoM, HiranoT (2002) Cohesin release is required for sister chromatid resolution, but not for condensin-mediated compaction, at the onset of mitosis. Genes Dev 16 : 3004–3016.
62. DiNardoS, VoelkelK, SternglanzR (1984) DNA topoisomearse II mutant of Saccharomyces cerevisiae: Topoisomerase II is required for segregation of daughter molecules at the termination of DNA replication. Proc Natl Acad Sci U S A 81 : 2616–2620.
63. FengW, BachantJ, CollingwoodD, RaghuramanM, BrewerB (2009) Centromere replication timing determines different forms of genomic instability in Saccharomyces cerevisiae checkpoint mutants during replication stress. Genetics 183 : 1249–1260.
64. BloomK (2008) Beyond the code: the mechanical properties of DNA as they relate to mitosis. Chromosoma 117 : 103–110.
65. BensimonD, SimonA, CroquetteV, BensimonA (1995) Stretching DNA with a receding meniscus: Experiments and models. Phys Rev Lett 74 : 4754–4757.
66. MatosJ, BlancoM, MaslenS, SkehelJ, SCW (2011) Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell 147 : 158–172.
67. BroomfieldS, HryciwT, XiaoW (2001) DNA postreplication repair and mutagenesis in Saccharomyces cerevisiae.. Mutat Res 486 : 167–184.
68. FengW, Di RienziS, RaghuramanM, BrewerB (2011) Replication stress-induced chromosome breakage is correlated with replication fork progression and is preceded by single-stranded DNA formation. G3 1 : 327–335.
69. BaxterJ, DiffleyJ (2008) Topoisomerase II inactivation prevents the completion of DNA replication in budding yeast. Mol Cell 30 : 790–802.
70. LosadaA, HiranoM, HiranoT (1998) Identification of Xenopus SMC protein complexes required for sister chromatid cohesion. Gene Dev 12 : 1986–1997.
71. LambertS, WatsonD, SheedyD, MartinB, CarrA (2005) Gross chromosomal rearrangements and elevated recombination at an inducible site-specific recombination fork barrier. Cell 121 : 689–702.
72. BordeV, GoldmanAS, LichtenM (2000) Direct coupling between meiotic DNA replication and recombination initiation. Science 290 : 806–809.
73. ChaRS, WeinerBM, KeeneyS, DekkerJ, KlecknerN (2000) Progression of meiotic DNA replication is modulated by interchromosomal interaction proteins, negatively by Spo11p and positively by Rec8p. Genes Dev 14 : 493–503.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- A Mutation in the Gene Causes Alternative Splicing Defects and Deafness in the Bronx Waltzer Mouse
- Mutations in (Hhat) Perturb Hedgehog Signaling, Resulting in Severe Acrania-Holoprosencephaly-Agnathia Craniofacial Defects
- Classical Genetics Meets Next-Generation Sequencing: Uncovering a Genome-Wide Recombination Map in
- Regulation of ATG4B Stability by RNF5 Limits Basal Levels of Autophagy and Influences Susceptibility to Bacterial Infection
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy