
A Mutation in the Gene Causes Alternative Splicing Defects and Deafness in the Bronx Waltzer Mouse
Sensory hair cells are essential for hearing and balance. Their development from epithelial precursors has been extensively characterized with respect to transcriptional regulation, but not in terms of posttranscriptional influences. Here we report on the identification and functional characterization of an alternative-splicing regulator whose inactivation is responsible for defective hair-cell development, deafness, and impaired balance in the spontaneous mutant Bronx waltzer (bv) mouse. We used positional cloning and transgenic rescue to locate the bv mutation to the splicing factor-encoding gene Ser/Arg repetitive matrix 4 (Srrm4). Transcriptome-wide analysis of pre–mRNA splicing in the sensory patches of embryonic inner ears revealed that specific alternative exons were skipped at abnormally high rates in the bv mice. Minigene experiments in a heterologous expression system confirmed that these skipped exons require Srrm4 for inclusion into the mature mRNA. Sequence analysis and mutagenesis experiments showed that the affected transcripts share a novel motif that is necessary for the Srrm4-dependent alternative splicing. Functional annotations and protein–protein interaction data indicated that the encoded proteins cluster in the secretion and neurotransmission pathways. In addition, the splicing of a few transcriptional regulators was found to be Srrm4 dependent, and several of the genes known to be targeted by these regulators were expressed at reduced levels in the bv mice. Although Srrm4 expression was detected in neural tissues as well as hair cells, analyses of the bv mouse cerebellum and neocortex failed to detect splicing defects. Our data suggest that Srrm4 function is critical in the hearing and balance organs, but not in all neural tissues. Srrm4 is the first alternative-splicing regulator to be associated with hearing, and the analysis of bv mice provides exon-level insights into hair-cell development.
Published in the journal:
. PLoS Genet 8(10): e32767. doi:10.1371/journal.pgen.1002966
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002966
Summary
Sensory hair cells are essential for hearing and balance. Their development from epithelial precursors has been extensively characterized with respect to transcriptional regulation, but not in terms of posttranscriptional influences. Here we report on the identification and functional characterization of an alternative-splicing regulator whose inactivation is responsible for defective hair-cell development, deafness, and impaired balance in the spontaneous mutant Bronx waltzer (bv) mouse. We used positional cloning and transgenic rescue to locate the bv mutation to the splicing factor-encoding gene Ser/Arg repetitive matrix 4 (Srrm4). Transcriptome-wide analysis of pre–mRNA splicing in the sensory patches of embryonic inner ears revealed that specific alternative exons were skipped at abnormally high rates in the bv mice. Minigene experiments in a heterologous expression system confirmed that these skipped exons require Srrm4 for inclusion into the mature mRNA. Sequence analysis and mutagenesis experiments showed that the affected transcripts share a novel motif that is necessary for the Srrm4-dependent alternative splicing. Functional annotations and protein–protein interaction data indicated that the encoded proteins cluster in the secretion and neurotransmission pathways. In addition, the splicing of a few transcriptional regulators was found to be Srrm4 dependent, and several of the genes known to be targeted by these regulators were expressed at reduced levels in the bv mice. Although Srrm4 expression was detected in neural tissues as well as hair cells, analyses of the bv mouse cerebellum and neocortex failed to detect splicing defects. Our data suggest that Srrm4 function is critical in the hearing and balance organs, but not in all neural tissues. Srrm4 is the first alternative-splicing regulator to be associated with hearing, and the analysis of bv mice provides exon-level insights into hair-cell development.
Introduction
Hair cells of the hearing and balance organs are specialized mechanoreceptors that convert mechanical stimuli to electrical signals. These signals are transmitted to the central nervous system via connecting afferent neurons. Hair cells of the hearing organ are specialized further as inner hair cells (IHCs) and outer hair cells (OHCs). IHCs are the primary auditory receptors, whereas the electromotile OHCs (and their neural feedback loops) are amplifiers of the mechanical stimulus [1]. In mice, hair cells become responsive to mechanical stimuli between embryonic day (E)17 and postnatal day (P)4 [2], [3]. During this period, the mechanosensing stereociliary bundles of the hair cells grow and become organized into rows of increasing height [4]. Defects in either stereocilium formation or afferent synaptogenesis lead to deafness and impaired balance [5]–[8].
The development of sensory hair cells is governed by several known transcription factors. For example, Sox2, Eya1, and the Notch effectors Hes1 and Hey1/2 are key transcriptional regulators of the specification process that guides the undifferentiated otocyst cells towards a prosensory fate [9]. The prosensory cells can then differentiate into either hair cells or supporting cells, depending on the presence or absence of the basic helix-loop-helix transcription factor Atoh1 within the cell. Genetic deletion of Atoh1 leads to the complete absence of hair cells [10], whereas the ectopic expression of Atoh1 in supporting cells can induce the formation of stereociliary bundles and the expression of hair-cell markers [11]–[13]. Atoh1 also induces the expression of at least 2 other transcription factors (i.e. Pou4f3 and Gfi1) that are required for the terminal differentiation of hair cells [14]–[17].
Proper hair-cell differentiation has also been shown to depend on microRNA-96 (miR-96), a post-transcriptional regulator of gene expression. Mutations in the miR-96 genes of both humans and mice have been associated with deafness [18], [19], and the analysis of mice harboring such mutations has demonstrated that this miR is required for the maturation of stereociliary bundles, as well as for the establishment of auditory nerve connections [20]. In addition, the analysis of knockout mice lacking the miR-processing protein Dicer1 in the inner ear supports the notion that miR-dependent regulation of gene expression plays a critical role in hair-cell differentiation [21], [22].
In an effort to identify additional regulatory mechanisms that are necessary for hair-cell development, we analyzed the bv mouse line, whose inner ear pathology suggested that the bv mutation disrupts a gene that is key to the differentiation of most hair-cell types [23]–[26]. Although the hair cells of homozygous bv (bv/bv) mice are morphologically intact until E17.5, neither the IHCs in the hearing organ nor the vestibular hair cells (VHCs) in the balance organs develop normally beyond this point. Specifically, the IHCs and VHCs fail to form synapses with afferent neurons, exhibit delayed stereociliary-bundle growth, and tend to degenerate by P3–5 [25]–[27]. These hair-cell defects are associated with the deafness and impaired balance observed in these animals. The bv/bv mouse is unique amongst the deaf mouse models in that IHC degeneration is not accompanied by the loss of OHCs [23], [27].
In this study, we localize the deafness-causing gene defect of the bv mouse line to the splicing factor-encoding gene Srrm4 (also known as nSR100 [28]). Because Srrm4 is expressed broadly in neural tissues, we used tissue-selective transgenic rescue to examine the biological importance of Srrm4 in and outside of the inner ear. The results of these rescue experiments indicated that defective Srrm4 function specifically in hair cells is the main, if not the only, cause of the bv phenotype. We evaluated the molecular function of Srrm4 using a transcriptome-wide approach, and found that it was required for neuron-like alternative splicing in the developing sensory patches of the inner ear but not in other Srrm4-expressing tissues that we examined. The majority of the affected pre-mRNAs encoded proteins with functions related to neurotransmission and secretion, confirming the notion that alternative splicing factors can selectively alter specific functional modules in the cell. Moreover, Srrm4-dependent splicing in hair cells affected transcriptional regulators that are known to control cell differentiation and presynaptic vesicle processing in neural tissues. Thus, our analysis of the bv mouse line suggests that Srrm4-dependent regulation of alternative exon choice has a profound effect on the differentiation program of sensory hair cells.
Results
The Bronx waltzer mouse line harbors a mutation in the Srrm4 gene
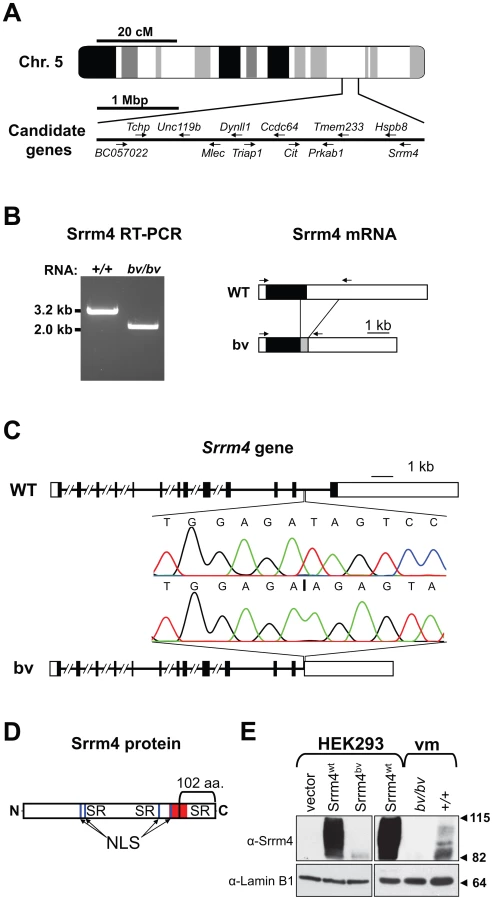
Although the gene affected by the bv mutation was not identified in previous studies, it had been mapped to a 4-mega base pair (bp) interval on chromosome 5 [29]. We examined the tissue expression profiles and putative functions of all 63 genes localized within this interval and selected 12 for further analysis (Figure 1A). In amplifying the transcripts of the 12 genes, we found that one, which encodes the splicing factor Srrm4, was abnormally short in bv/bv mice (Figure 1B). Sequence analysis showed that the shortened Srrm4 transcript lacked hundreds of nucleotides and retained an intronic sequence (Figure 1B), whereas the other amplified transcripts did not contain any mutations (data not shown). Sequencing of the 3′ end of the Srrm4 gene in bv/bv mice revealed a 2,710-bp deletion that removed a portion of the last intron and the entire coding region of the last exon but left the polyadenylation site intact (Figure 1C and Table S1). The affected last exon of Srrm4 encodes potentially important domains of the Srrm4 protein, including the C-terminal SR repeats and a region that is highly conserved between Srrm4 and its closest paralogue, Srrm3 (Figure 1D). We used Western blotting to examine expression of the wild-type Srrm4 (Srrm4wt) protein and that of the mutant form encoded by the bv mouse genome (Srrm4bv). In nuclear pellets generated from Srrm4wt-transfected HEK293 cells and from the sensory regions of the balance organs (i.e. vestibular maculas) in wild-type mice, the protein was detected as multiple bands between 82 and 115 kDa (Figure 1E). In contrast, in nuclear pellets generated from Srrm4bv-transfected HEK293 cells and the vestibular maculas of bv/bv mice, only a single, faint band was detected at ∼82 kDa (Figure 1E). Thus, the bv mutation not only truncates the Srrm4 protein but also interferes with either the stability or synthesis of the truncated protein.
The Bronx waltzer phenotype is caused by defective Srrm4 function in hair cells
Next, we used in situ hybridization to examine the expression pattern of wild-type Srrm4 in the inner ear, and found that the Srrm4 mRNA was detected in all sensory regions of hearing and balance organs (Figure 2A). In the cochlea, the antisense Srrm4 probe labeled the IHCs, OHCs, and spiral ganglion (Figure 2B). In the utricle, the most intensive staining was found at the periphery of the sensory macula (Figure 2C), where the density of VHCs is highest [30]. In the crista ampullaris, the VHC-containing regions were strongly positive, whereas the non-sensory septum cruciatum (present in the anterior but not the lateral crista [31]), was not labeled (Figure 2D, asterisk). The negative-control, Srrm4 sense probe did not hybridize with any of the sensory regions in the inner ear (Figure S1). These data indicate that Srrm4 is expressed in the sensory hair cells and the spiral ganglion. RT-PCR experiments showed that the Srrm4 mRNA was also present in the brain but not in the kidney, liver, or spleen (data not shown), consistent with the previously reported neural expression pattern of Srrm4 [28].

Although Srrm4 is expressed broadly in neural tissues, we hypothesized that the Srrm4 defect in hair cells is the cause of the bv phenotype. We tested this possibility by transgenic rescue. Specifically, a Myo7a-Srrm4 transgene (Figure 3A) was constructed in which the hair-cell specific promoter of Myo7a [32] controlled the transcription of wild-type Srrm4. Transgenic founder mice were generated via pronuclear injection of the Myo7a-Srrm4 transgene, and bred into the bv/bv line. In situ hybridizations of inner ear samples with a probe corresponding to the coding region in Srrm4 exon 13 demonstrated that the Myo7a-Srrm4 transgene was expressed in the balance organs and IHCs of the transgenic bv/bv mice (Figure S2). In contrast, the inner ears of bv/bv mice did not hybridize with the Srrm4 exon 13 probe (Figure S2), confirming that exon 13 is missing from the Srrm4 transcript in the bv/bv mice. We evaluated the hearing of Myo7a-Srrm4 transgenic bv/bv mice and non-transgenic bv/bv littermates by measuring the auditory brainstem response (ABR) of these animals on P21–28, using broadband sounds. The ABR measurements confirmed that the bv/bv mice were severely hearing impaired (Figure 3B), whereas the hearing thresholds of several Myo7a-Srrm4 transgenic bv/bv mice were at the wild-type level (Figure 3B and 3C). Nevertheless, the extent to which hearing was restored in individual Myo7a-Srrm4 transgenic bv/bv mice varied (Figure 3C) by transgenic lineage (Figure S3), and probably reflected differences in transgene insertion site or copy number.
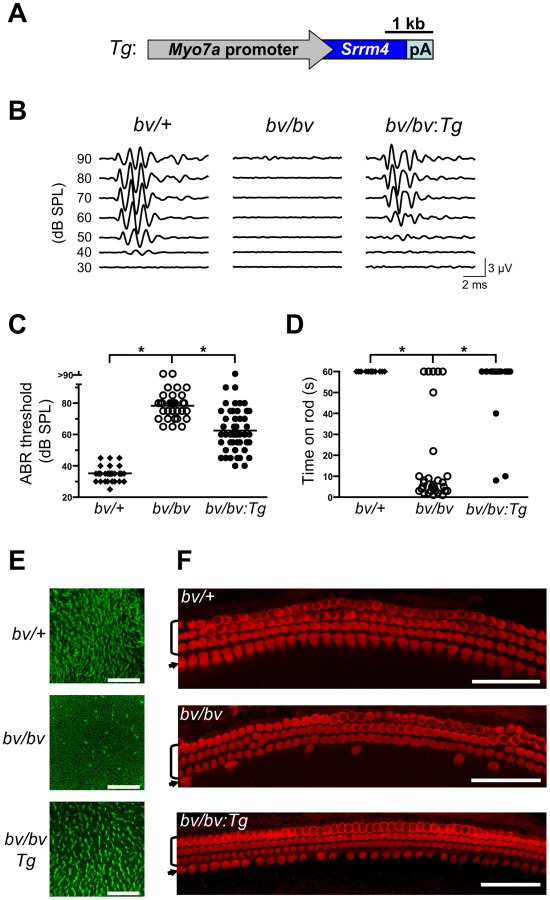
We also assessed balance in the transgenic bv/bv animals, by measuring the length of time they could remain on a horizontal rod. The performance of the Myo7a-Srrm4 transgenic bv/bv mice was similar to that of bv/+ animals, which were able to remain on the rod for the duration of the assay (60 s); in contrast, most bv/bv mice fell within 10 s (Figure 3D).
We next analyzed the effects of the Myo7a-Srrm4 transgene on hair-cell survival in bv/bv mice. The sensory regions were dissected from the balance organs of bv/+, bv/bv, and Myo7a-Srrm4 transgenic bv/bv mice on P5, and the actin-rich stereociliary bundles of VHCs were visualized using fluorescently labeled phalloidin. Looking at the balance organs of bv/bv and Myo7a-Srrm4 transgenic bv/bv mice, we found that the vast majority of stereociliary bundles were absent in the former but present at nearly normal density in the latter (see utricle in Figure 3E and quantitative analysis in Figure S4A). In the cochleas of bv/bv mice, both actin staining of stereocilia and immunofluorescence-based visualization of the hair-cell protein Myo7a indicated that 71% of the IHCs were absent on P5. In contrast, in the Myo7a-Srrm4 transgenic bv/bv mice 63% of IHCs were present at this time (Figure 3F and Figure S4). These results indicate that the Srrm4 mutation is responsible for the hair-cell loss, deafness, and balance defect in the bv mouse line. Our data also support the notion that the inner ear pathology in bv/bv mice is caused by defects in the hair cells rather than in the neurons.
The Bronx waltzer mice are subject to alternative splicing defects in the inner ear but not in the cerebellum
Srrm4 belongs to the family of SR-related proteins, which act as regulators of alternative pre-mRNA splicing [33], [34]. Therefore, we examined whether alternative splicing was altered in the embryonic hair cells of bv/bv mice, using a transcriptome-wide approach. Specifically, embryonic hair cells (and the adjacent supporting cells) were acquired from the vestibular maculas of bv/bv and bv/+ mice, by laser-capture microdissection, on E16.5, i.e. ∼1 day before the onset of hair-cell degeneration. RNA from the captured tissue was analyzed using the new Affymatrix chip ‘Mouse Exon Junction Array’ (MJAY, Figure 4A). MJAY contains more than half a million exon and exon-exon junction probe sets (see probe-set design in Figure 4B), and interrogates all of the splicing events supported by mouse EST/mRNA evidence within the UCSC/Ensembl databases. Processing of the MJAY data was carried out largely in the Partek Genomics Suite (see details in Methods), based on concepts that were previously described for the analysis of Human Exon Junction Array data [35]. The frequency of an alternative splicing event was considered to differ significantly between the bv/bv and bv/+ samples if the difference in normalized intensities for at least two probe sets per splicing event (Figure 4C) resulted in P-values less than 0.05. Seventy-six candidate alternative splicing events were found and tested further by RT-PCR, using primers that annealed with the constitutive exons upstream and downstream of the alternative exons. These reactions validated 24 alternative splicing events in the vestibular maculas of bv/bv mice (Figure 4D and Figure S5A). Notably, examination of these splicing defects indicated that, in the bv/bv cells, certain alternative exons were either spliced into the mature mRNA at reduced frequency or completely skipped. Common features of the affected exons included conservation among vertebrates (data not shown) and – with the exception of Add1 exon 15 – a neuron-specific inclusion pattern (Figure S5C). Therefore, we used ‘conservation’ and ‘neuron-specific splicing’ (based on EST evidence) as new criteria with which to scrutinize the list of exons for which a single probe set suggested abnormal splicing in the bv/bv mice. RT-PCR revealed that, among the 283 new candidate exons, 30 were incorrectly spliced in the bv/bv sample (Figure S5B). Thus, overall, RT-PCR verified 54 changes in splicing in the bv/bv mouse (see Table S2). We used the DAVID software [36] to analyze the gene ontology (GO) annotations of the encoded proteins, and found that the lowest P values were for those associated with the ‘transmission of nerve impulse’ (Benjamini-Hochberg corrected P-value = 0.00047), ‘secretion by cell’ (P-value = 0.0064), and other closely related GO terms (e.g. ‘cell-cell signaling’).

The majority of splicing defects we found in the vestibular macula of bv/bv mice (81%) had not been reported in an earlier study that examined Srrm4 function in the Neuro2A cell line [28]. Conversely, although nPTB (exon 10) was found to be a key target of Srrm4 in the Neuro2A cells [28], it was spliced normally in the vestibular maculas of bv/bv mice (Figure S5D). Nevertheless, there were striking instances of overlap as well. For example, the RE1 silencing transcription factor (Rest, exon 4) was reported as a target of Srrm4 in the Neuro2A cells [37], and RT-PCR showed that the same Rest exon was differently spliced in the inner ears of bv/bv and bv/+ mice (Figure S5E). Furthermore, the Srrm4-dependent splicing of Rest had been shown to affect the expression of numerous Rest-regulated genes in Neuro2A cells [37], and our analysis of the MJAY data suggested that Rest-regulated genes [38] were overrepresented among those whose expression was reduced in the vestibular macula of bv/bv mice (χ2 test P<0.0001, Figure S6). Notably, our data showed that the Phf21a/Bhc80 mRNA, which encodes a negative modulator of Rest-dependent transcriptional regulation [39]–[41], was also differentially spliced in the vestibular maculas of bv/bv and bv/+ mice (Figure 4D). These results support the notion that Srrm4 modifies gene expression in hair cells, probably through the alternative splicing of specific transcriptional regulators.
We also wanted to test whether the bv mutation led to splicing alterations in Srrm4-expressing tissues other than the inner ear. We focused on the cerebellum, based on our RT-PCR analysis showing that the Srrm4 transcript is highly expressed in this tissue (Figure S7A), and in situ hybridization data in the Allen Brain Atlas [42] indicating that the neuron-rich layers of the cerebellum contain large amounts of Srrm4 mRNA. Notably, although the analysis of MJAY data identified 18 alternative exons as potentially differently spliced in the cerebellums of bv/bv and bv/+ mice on P15, anlaysis by RT-PCR failed to validate such an outcome (Figure S7C and S7D). Furthermore, both the MJAY and RT-PCR data showed that, in the bv mouse line, inclusion rates for alternative exons that were abnormally spliced in the vestibular macula were unaltered in the cerebellum (Figure 4E and 4F and Figure S7E). Given that the Srrm4 mRNA is highly expressed in the neocortex [28], we used RT-PCR to test the inclusion rates of 10 Srrm4-regulated exons in this tissue. Again, we found no alterations in the inclusion rates of tested alternative exons in the investigated brain region of bv/bv mice (Figure S8). These findings are supported by the lack of obvious histological alterations in the cerebellum and neocortex of bv/bv mice (Figure S7B and Figure S9). In sum, the bv mutation does not lead to apparent defects in these Srrm4-expressing tissues.
Srrm4-dependent alternative splicing requires the C-terminal region of Srrm4 and a novel sequence motif in the target pre–mRNA
We next used a reconstituted system to evaluate whether the Srrm4bv protein retains molecular function. Specifically, HEK293 cells were transfected with Srrm4bv, Srrm4wt, or empty vector (control) alongside various minigenes consisting of exons and introns. Each minigene construct contained an exon that was incorrectly spliced in the vestibular macula of bv/bv mice, the flanking intronic sequences (∼300 bp), and two constitutive exons (Figure 5A). Of the 54 alternative exons whose inclusion rates were found to be altered in the vestibular maculas of bv/bv mice, 12 were randomly selected for these minigene experiments. RT-PCR-based evaluation of pre-mRNA splicing demonstrated that all 12 exons required Srrm4wt for alternative splicing in the transfected cells, and that Srrm4bv was unable to promote such splicing (Figure 5A and Figure S10A). When the minigenes were co-transfected with a construct encoding an SR protein other than Srrm4 (i.e. Srsf1), the inclusion rates of the alternative exons did not increase above background levels (Figure S10A). Thus, the minigene experiments confirmed that splicing of the tested exons is dependent on Srrm4wt, and also indicated that the bv truncation prevents the expression of functional Srrm4 protein in transfected HEK293 cells.

We also tested the functional status of Srrm4bv in vivo, using zebrafish as an animal model. The endogenous Srrm4 mRNA of zebrafish (zSrrm4) was knocked down by injecting a previously described zSrrm4 morpholino (MO) [28] into fish eggs. Some of these eggs were also injected with either an mRNA encoding a MO-insensitive wild-type zSrrm4 (zSrrm4wt) or the zebrafish version of a MO-insensitive Srrm4bv (zSrrm4bv). Three days later, the hair cells were visualized in the lateral line of zebrafish larvae using the fluorescent dye FM1–43 [43], [44]. We found that in the zSrrm4 MO-injected fish, the body axis was abnormally curved (Figure 5C versus Figure 5B, upper panel). This deformity in the body axis has previously been described for Srrm4 knock-down zebrafish, and has been attributed to neuronal defects [28]. In addition, we found that the number of hair cells was dramatically reduced in the zSrrm4 MO-injected fish (Figure 5C versus Figure 5B, lower panel, and quantitative analyses in Figure S10B and S10C). Co-injection of the zSrrm4wt mRNA with the MO rescued both the body axis deformity and the hair-cell loss (Figure 5D), whereas co-injection of the zSrrm4bv mRNA did not (Figure 5E, and statistical analyses in Figure S5B and S5C). These data suggest that Srrm4bv is not functional, regardless of the expression system. Furthermore, our data show that although the loss of Srrm4 function has a broader phenotypic impact in zebrafish than in mice, Srrm4 is essential for hair-cell development in both species.
We hypothesized that a unique sequence motif may mark the Srrm4-regulated exons for splicing. Initially, we focused on exon sequences, testing a 9-nucleotide long exon whose splicing we had found to be Srrm4 regulated (i.e. Dtna exon 11). However, random mutation of 5 consecutive nucleotides in the 9-nucleotide exon did not affect its Srrm4-dependent splicing (). Next, we used the MEME software [45] to search for consensus motifs in both the Srrm4-regulated exons (n = 54) and the 50-nucleotide long portions of introns that are directly adjacent to these exons. MEME identified 3 motifs with P-values lower than 0.05, including a novel UGC motif and the known binding sequences of 2 splicing factors (i.e. U2af1 and the U1 small nuclear ribonucleoprotein). Alignment of the intron sequences upstream and downstream of the UGC motif showed that it is located near the 3′ end of the polypyrimidine tract (Figure 5F). To test whether UGC commonly occurs upstream of exons (i.e., regardless of Srrm4-dependent alternative splicing), we assessed all 50-bp regions that lie directly upstream of an exon in the Cacna1d and Ergic3 pre-mRNAs (n = 60). MEME did not detect UGC as a frequent motif in these sequences. Thus, UGC might be important for specifically Srrm4-dependent splicing.
Next, we used G-to-A point mutations to disrupt the selected UGC motifs in 6 minigenes that were randomly chosen from those that require Srrm4 for alternative splicing. In all cases, the mutations inhibited Srrm4-dependent exon inclusion (Figure 5G, Figure S11B–S11C). Mutagenesis experiments were also carried out to test the importance of the other two nucleotides in the UGC motif. We found that C-to-U substitutions (Figure 5G and Figure S11C), but not U-to-C/G/A mutations (data not shown) inhibited Srrm4-dependent exon inclusion. Thus, although a U nucleotide most often precedes the functionally relevant GC motif, only the GC nucleotides are required for Srrm4-dependent alternative splicing.
Blast searches revealed that the functionally relevant GC motifs are conserved among vertebrate species (Figure S11D). To test whether these motifs interact with Srrm4, we carried out streptavidin pull-down assays using two types of biotin-labeled RNA oligos. The ‘wild-type’ RNA oligo corresponded exactly to a 40-nucleotide long sequence around the splice acceptor site of the Srrm4-regulated exon in the Ergic3 pre-mRNA, whereas the ‘mutated’ RNA oligo contained a GC-to-AU substitution (Figure 5H). Western blot analysis of cell lysates prepared from flag-Srrm4-transfected HEK293 cells showed that only the ‘wild-type’ RNA efficiently pulled down flag-tagged Srrm4 (Figure 5H). These results suggest that conserved GC motifs upstream of the splice acceptor sites of Srrm4-regulated exons are necessary for the interaction between Srrm4 and the pre-mRNA.
Discussion
In the present study, we show that the hearing and balance defects of bv/bv mice are caused by a mutation in the Srrm4 gene. Using the bv mouse line and a genome-wide screening method to analyze the molecular function of Srrm4 in vivo, we identified Srrm4 as a key regulator of pre-mRNA splicing in the inner ear. Moreover, we found that Srrm4 was required for the alternative splicing of a specific set of exons that are marked by GC motifs near the 3′ ends of the polypyrimidine tracts. The Srrm4 mutation in bv/bv mice also affected gene expression in the sensory patches of the inner ear, suggesting that Srrm4 controls a cascade of transcriptome-modifying events. Based on this analysis of the bv mouse line, we propose that Srrm4-regulated alternative splicing is critical for the differentiation of all sensory hair-cell types except the OHCs.
Although Srrm4 is expressed broadly in neural tissues, we did not detect splicing defects in the cerebellum and neocortex of P15 bv/bv mice. Moreover, if neurogenesis is impaired in bv/bv mouse embryos, the consequences of this defect are not readily detectable by Nissl staining at P15. Nevertheless, we cannot rule out the possibility that pre-mRNA splicing is affected at other time points or in other brain regions in these animals. Notably, a recent study showed that neurogenesis was impaired in E13/14 wild-type mice after neural progenitors in the ventricular zone were electroporated with vectors encoding an Srrm4-targeting shRNA [37]. Also, in examining the brains of bv mice, Matsuda and colleagues observed that immunofluorescence-based visualization of the parvalbumin-expressing GABAergic interneurons [46] detected abnormally few parvalbumin-expressing cells in the auditory cortex, somatosensory cortex, and anterior cingulate, whereas the visual cortex and the amygdala complex were unaffected. A possible interpretation of these data is that the Srrm4 defect in bv/bv mice directly affects the differentiation of interneurons in certain brain regions. Alternatively, some of the observed changes in the number of parvalbumin-expressing cells could be secondary to the hearing and balance defects in bv/bv mice. This possibility is consistent with the fact that congenital deafness has been shown to prevent the maturation of GABAergic transmission in the auditory cortex [47], [48], and sensory hearing loss has been associated with a decrease in the number of parvalbumin-positive cells in the superior olivary complex [49]. In addition, the lack of vestibular input has been reported to cause a reduction in the expression of various calcium-binding proteins, including parvalbumin, in the medial vestibular nucleus [50]. Thus, additional studies will be necessary to establish the etiology of the altered GABAergic interneuron density in certain brain regions of bv/bv mice.
In zebrafish, MO-mediated knock-down of Srrm4 has an obvious effect on both neural differentiation [28] and hair-cell development (Figure 5). Why does Srrm4 deficiency have a much greater impact on neural differentiation in zebrafish than in mice? One possible explanation is that splicing proteins other than Srrm4 have Srrm4-like functions in the mouse brain, but not in that of zebrafish. However, a more complex explanation is suggested by two findings. Firstly, whereas approximately 70% of the IHCs die between E18 and P5 in the bv mouse, this trend does not continue after P5 [27]. Secondly, the surviving IHCs are most likely functional because the bv/bv mice are not completely deaf. Together these data suggest that Srrm4 is not needed in the inner ear after a critical phase in development. This “critical phase” hypothesis is supported by the gene expression profile of the splicing suppressor polypyrimidine tract binding protein 1 (PTBP1), which has been shown to inhibit the constitutive inclusion of at least some Srrm4-regulated exons in Neuro2A cells [28]. PTBP1 is expressed in neural cells only during the early phases of differentiation [51]. Thus, Srrm4 may not be needed during the later phases of development when neural cells no longer contain PTBP1. We speculate that although Srrm4 deficiency could possibly lead to splicing defects in the neurons of both mice and zebrafish during early development, Srrm4-independent regulatory mechanisms are sufficient to support neuron differentiation until the end of the critical phase in mice but not in zebrafish.
Our finding that Srrm4-dependent exon inclusion requires the presence of a GC motif near the 3′ end of the polypyrimidine tract suggests that this motif serves as a cis-regulatory element for Srrm4-dependent splicing. Cis-regulatory elements are short sequence motifs that recruit RNA-binding proteins [52]; they can either enhance or suppress exon inclusion depending on which splicing factors are recruited and – in some cases – the position of the cis-regulatory element relative to the exon [53], [54]. Pre-mRNAs co-regulated by the same RNA-binding protein usually contain the same cis-regulatory element. Thus, the presence of the same motif next to almost every affected exon in the Srrm4 mutant mouse suggests that the inclusion of these exons into the mRNA is regulated by the same Srrm4-dependent mechanism.
Our RNA pull-down experiment suggested that the GC motif is necessary for the interaction between Srrm4 and the RNA. Whether this interaction is direct or mediated through other proteins remains to be determined. Notably, the GC motif is not the only sequence in the pre-mRNA that is important for the regulation of Srrm4-dependent splicing events. A previous study showed that pyrimidine-rich motifs are often present in introns that flank Srrm4-regulated exons [28], and that these pyrimidine-rich motifs are binding sites for PTBP1 [28]. Because the GC motifs are located near the 3′ end of the polypyrimidine sequences, it is tempting to speculate that the recruitment of either Srrm4 or Srrm4-binding proteins to the pre-mRNA may interfere with the binding of PTBP1.
The fact that the Srrm4-regulated exons were found more frequently in the transcripts of proteins that are annotated with the GO terms ‘transmission of nerve impulse’ and ‘secretion by cell’ than in the transcripts of a random set of proteins suggests that the protein products of the Srrm4-regulated pre-mRNAs are functionally linked. We explored this possibility by collecting PubMed data on the subcellular localizations of, and interactions among, the affected proteins; given that we wanted to maximize the amount of information gathered, we did not restrict these PubMed searches to hair cell-related publications. Based on the information collected, we charted the likely subcellular localization of the affected proteins on a schematic model of the basolateral portion of a hair cell (Figure 6). This model illustrates that the majority of the proteins encoded by Srrm4-regulated transcripts may be associated with synaptic vesicles and the presynaptic plasma membrane. Notably, 42% of the proteins in this model that are encoded by Srrm4-regulated pre-mRNAs and have known protein-protein binding partners in the PubMed database interact with each other (see reference list in Table S3). Thus, both GO annotation analysis and the protein-protein interaction patterns suggest that the Srrm4-dependent modifications cluster predominantly in a single functional module of the proteome, and that this module is responsible for secretion and neurotransmission at the presynaptic side of the synapse. This analysis also suggests that the Srrm4-regulated proteins with uncharacterized molecular functions (e.g. Plekha6, 6330403A02Rik, and C230096C10Rik) are more likely involved in secretion or neurotransmission than in other biological processes.

Sustained high rates of neurotransmission from the IHCs and VHCs to their respective neural afferents require specialized presynaptic structures termed synaptic ribbons. Most OHCs contain synaptic ribbons only temporarily during differentiation; the only OHCs in which synaptic ribbons persist are those that are most apical [55]. Interestingly, in the bv/bv mice the OHCs are the only hair cells to remain intact, and many of the proteins with splicing defects are localized to synaptic ribbons (i.e. Cacna1d, Cask, Erc2, Rims2, Snap91, and Synj1 [56]). Thus, it seems plausible that the cell-type specificity of the synaptogenesis defect is due to an absence of protein isoforms that are specifically required for the formation of synaptic ribbons. Alternatively, it is possible that the mechanism that supports the inclusion of Srrm4-regulated exons in the cerebellum and neocortex of bv/bv mice also protects the OHCs from degeneration. These hypotheses could be tested by analyzing pre-mRNA splicing in the embryonic OHCs of bv/bv and control mice. However, RNA collection selectively from embryonic OHCs is technically challenging because of the physical proximity of OHCs and IHCs in the developing inner ear. Therefore, the analysis of pre-mRNA splicing in the OHCs of bv/bv mice is yet to be carried out.
Although we found that the majority of the splicing defects in the bv mouse line were associated with the secretory and synaptic apparatuses, the alternative splicing of at least two ciliary protein-encoding mRNAs (i.e. Bbs9 and Wdr35) were also altered. In addition, the Srrm4 mutation led to reduced expression of the receptor-like inositol phosphatase Ptprq, which is required for the development of stereociliary bundles in the cochlea [57]. Srrm4-dependent splicing also affected at least 3 mRNAs that encode nuclear proteins (i.e. Rest, Bhc80, and Mef2d). Two of these (i.e. Rest and Bhc80) have been shown to have opposing effects on gene expression and have been reported to control vesicle processing and exocytosis through translational regulation [41], [58], [59]. We found that the genes regulated by Rest - and Bhc80 – but not those regulated by Mef2d – were overrepresented among the 44 whose expression was most reduced in the vestibular macula of bv/bv mice. Thus, the Srrm4-dependent splicing of the Rest and Bhc80 pre-mRNAs supports our hypothesis that Srrm4 plays a role in maturation of the regulated secretory apparatus in hair cells. Altered splicing of the Rest mRNA and reduced expression of the Rest target genes in the context of reduced Srrm4 function were described previously in Neuro2A cells subjected to RNA interference [37]. Thus, both in vivo and in vitro data suggest that the loss of Srrm4 function leads to a cascade of transcriptome alterations that affect both pre-mRNA splicing and gene expression. Further studies defining the importance of individual Srrm4-regulated exons in hair-cell development will enable us to elucidate the detailed pathogenesis of hair-cell degeneration in bv/bv mice.
In summary, in analyzing the bv mouse line we have identified Srrm4 as a regulator of alternative splicing that is required for the differentiation of hair cells in the hearing and balance organs. We propose that a Srrm4-regulated cascade of transcriptome modifying events adjusts the proteome of differentiating hair cells such that they take on neuron-like functions. Our study adds alternative splicing to the list of mechanisms that are critical for hair-cell differentiation. Given that some deafness-causing mutations are known to be localized to alternative exons (i.e. R643X in PCDH15 [60] and R500X in TRIC [61]), understanding the regulation of alternative exon choice in the inner ear is expected to create therapeutic opportunities for the prevention of deafness.
Methods
Genetic analysis of the bv mouse line
The bv mouse strain was recovered from cryopreserved sperm samples (obtained from the European Mutant Mouse Archive), by intracytoplasmic sperm injection. All experiments and procedures were approved by the Animal Care and Use Committee of the University of Iowa. For mutation analysis, RNA was isolated from the inner ear of bv/bv and wild-type mice (E16.5), using the Trizol reagent. The coding regions of candidate mRNAs were amplified from the RNA samples using RT-PCR (see primers in Table S4), and the PCR products were sequenced. To amplify and sequence the genome adjacent to the deletion site in bv/bv mice, “genome walks” were carried out using the PCR-based Genome Walker Universal kit (Clontech Laboratories, Inc.) and a gene specific primer that anneals to the penultimate exon in Srrm4 (5′-ACGGGACCTAAAGTATGGTGAGAAAG-3′). For genotyping, the presence or absence of the bv mutation was detected by PCR using tail DNA extracts, and 2 sets of primer pairs (wild-type allele: 5′-GGGAAGAGGTGGAGTATGTTG-3′ and 5′-CCTCGTGCTGGCATAGCTTTC-3′; bv allele: 5′-GAAAGAACCACAGCCCCGAGAA-3′ and 5′-CTGGGCAGGAGGGTACTTCTATAC-3).
Generation and analysis of Srrm4 transgenic mice
The Myo7a-Srrm4 transgene was constructed by subcloning the mouse Srrm4-encoding cDNA downstream of the mouse Myo7a promoter and upstream of the SV40 polyadenylation site in the pSTEC-1 vector, using standard PCR and subcloning methods (see PCR primers in Table S5). The Myo7a-Srrm4 expression cassette was isolated from pSTEC-1 by restriction digestion, and sent to Xenogen Corp. for the production of transgenic mice. The ABR thresholds of mice were measured at P21–28, using a previously described open-field system and broadband click stimuli [62]. The ability of mice to balance (P70–80) was evaluated by measuring the time each mouse could remain on a fixed horizontal rod (1.8 cm in diameter) following two training trials. Actin and Myo7a staining of whole-mount preparations of PFA-fixed cochlear and vestibular tissues was carried out as previously described [63], using the following reagent and antibodies: Alexa-488 labeled phalloidin (Invitrogen Corp.), rabbit anti-Myo7a antibody (Proteus Biosciences, Inc.), and Alexa-594 labeled anti-rabbit IgG (Invitrogen Corp.).
In situ hybridization
Digoxigenin-labeled antisense exon 13 probe (coding nucleotides 1521–1827 in Srrm4), sense and antisense Srrm4 riboprobes (coding nucleotides 23–188) were generated using the DIG RNA Labeling Mix (Roche), and hybridized to inner ear samples of mice of various genotypes as described previously [17].
Laser-capture microdissection and RNA extraction for microarray analysis
Inner ears of mouse embryos (E16.5) were embedded in Tissue-Tek O.C.T. Compound (Sakura Finetech, Inc.), frozen in liquid nitrogen, and cryosectioned. Sections were further processed for laser-capture microdissection using the Arcturus Histogene Frozen Section Staining kit (Applied Biosystems). The manufacturer's staining protocol was modified in that RNAse inhibitor (ProtectRNA, Sigma) was added to every solution in the kit that contains more than 5% water. The vestibular macula was captured from the inner ear sections using the Laser Capture Microdissection system (Pixcell II, Arcturus, Mountain View, CA). RNA was isolated from the captured tissue using the PicoPure RNA isolation kit. RNA was also extracted from the cerebellums of mice at P15, using the Trizol Reagent (Invitrogen). The cerebellar RNA was treated with DNase and further purified using the RNeasy mini kit (Qiagen).
Microarray analysis
RNA samples for microarray analysis were processed using the NuGEN WT-Ovation Pico RNA Amplification System, NuGEN WT-Ovation Exon Module, and NuGEN FL-Ovation cDNA Biotin Module. Samples were hybridized to Mouse Exon Junction Microarrays (MJAY, Affymetrix Inc.). MJAY were scanned with an Affymetrix Model 7G upgraded scanner, and data were collected using GeneChip Operating Software. Raw microarray CEL files were imported into Partek Genomics Suite (Partek, Inc.). Signal intensities for the probe sets were quantile normalized and median polished using Robust Multichip Average background correction. The signal intensities of exon probe sets were used to calculate the overall expression level of each gene represented in MJAY. Normalized probe-set intensities (Inorm) were calculated by dividing the background-corrected signal intensities of exon and exon-junction probe sets by the background-corrected gene-expression signal of the corresponding gene. The Inorm in the bv/bv and bv/+ samples was analyzed by two-tailed Student t-test. Probe sets with significantly different Inorm (P<0.05) were queried against the Affymetrix annotation map file (which contains alternative/constitutive annotations for each measured splicing event) using simple Visual Basic for Application scripts, and probe sets that measure constitutive events were filtered out. The remaining probe sets were queried against the “SIB Alt-Splicing track” in the UCSC Genome Browser to identify and eliminate those that show either more than 50% identity with more than one gene or measure alternative promoter activity. The sequences of the remaining probe sets were queried against the mouse genome to identify those that measure the same splicing events. We required that probe sets targeting competing isoforms have opposite Inorm trends.
RT–PCR and minigene-based validation of MJAY data
RT-PCR was carried out essentially as described previously [64]. We defined an alternative exon as ‘differently spliced’ in the bv/bv and bv/+ samples if the RT-PCR data indicated that the inclusion rates for the exon were at least 1.5-fold different between the compared samples (Table S6 contains the inclusion rates calculated based on the RT-PCR data shown in Figure 3D and 3F, Figures S4, S7, and S8). Table S7 lists all primers that were used to generate the data shown in Figure 3D and 3F, Figures S4, S7, and S8.
For minigene-based validation of Srrm4-dependent splicing events, alternative exons and adjacent ∼300 bp intronic sequences were PCR amplified and subcloned into the exon trap pET-01 vector (Mobitec, see primers in Table S5). Mouse Srrm4wt and Srrm4bv were amplified by RT-PCR (see primers in Table S5) from inner ear RNA and subcloned into the pcDNA3.1 expression vector. The Srsf1 expression construct (Addgene plasmid 17990) has been described previously [65]. The minigines, Srrm4-encoding constructs, and the Srsf1-encoding plasmid were transfected into HEK293 cells using the Lipofectamine LTX and PLUS reagents (Invitrogen), and RNA was extracted from the cells 24 hours later using the RNeasy mini kit (Qiagen). RNA was reverse transcribed with Superscript III, and analyzed by RT-PCR using primers that annealed to the constitutive exons (primers: 5′-CACTTGGTGGAAGCTCTCTACC-3′ and 5′-CCACCTCCAGTGCCAAGGTC-3′). Site-directed mutagenesis of minigenes was carried out using overlap-extension PCR.
Srrm4 knock-down and rescue experiments in zebrafish
The Srrm4 knock-down experiments were carried out in a transgenic zebrafish line developed by Haas and Gilmour [66]. In the neuromasts of these transgenic zebrafish, the claudin B promoter drives the expression of a membrane-tethered GFP (Tg[CldnB-mGFP]). zSrrm4 expression was knocked down by injecting the transgenic zebrafish (2-cell stage) with a previously described zSrrm4 MO [28] (5′-TTCTCCCAAAAGTACGCCAGCCATG-3′ from Gene Tools, Philomath, OR; 5 ng zSrrm4 morpholino/embryo). Since injection of 5 ng of the zSrrm4 MO led to non-specific toxicity, a p53-targeting MO (5′-GCGCCATTGCTTTGCAAGAATTG-3′ from Gene Tools; 5 ng/embryo) was co-injected. 3 days after injection, zebrafish larvae were incubated with 3 µM FM1–43 dye for 30 s to label the mechanosensing hair cells in the neuromasts. The FM1–43 staining led to a bright green signal that was much more intense than the GFP signal of the CldnB-mGFP transgene. After staining with FM1–43, the zebrafish were rinsed, anesthetized (0.02% 3-aminobenzoic acid ethyl ester), mounted in 3% methylcellulose, and photographed. The zSrrm4wt and zSrrm4bv mRNAs used for rescue experiments were generated using the mMessage mMachine kit (Ambion) and CS2+ plasmids that contained the zSrrm4wt and zSrrm4bv cDNAs (see cloning primers in Table S5). The zSrrm4wt and zSrrm4bv mRNAs were injected into zebrafish embryos (4-cell stage, 10 ng mRNA/embryo) that had previously been injected with MOs targeting zSrrm4 and p53. After the embryos had been maintained for 3 days, the mechanosensing hair cells were stained using the FM1–43 dye as described above.
Western blotting
Nuclear fractions were isolated from the vestibular maculas of 44 wild-type and 44 bv/bv mice, on E16.5, using the Nuclear Complex Co-IP kit (Active Motif) according to the manufacturer's instructions. The obtained nuclear fractions were treated with Enzymatic Shearing Cocktail (Active Motif) and centrifuged at 16,000 g for 15 min at 4°C. The pellets were dissolved in SDS sample buffer, boiled for 3 min, resolved by SDS-PAGE, and electroblotted onto nitrocellulose membranes. Following a blocking incubation step, goat anti-Srrm4 antibody (sc-139291 from Santa Cruz Biotechnology Inc.) diluted 1∶200 or rabbit anti-Lamin B1 antibody (ab16048 from Abcam) diluted 1∶5,000 was added to the membranes for 14 hours. After multiple washing steps, membranes were incubated with HRP-conjugated secondary antibodies (anti-goat IgG and anti-rabbit IgG). Immunoblot signals were visualized using an Enhanced Chemiluminescence Detection System (Pierce Biotechnology).
RNA pull-down assays
Flag-tagged Srrm4 was subcloned into the pcDNA3.1 expression vector and transfected into HEK293 cells. 24 hours after transfection, the cells were harvested and resuspended in buffer DG (containing 80 mM Potassium Glutamate, 0.1 mM EDTA, 10% glycerol, 0.01% NP40, 0.1 mM PMSF, 1 mM DTT, 16 µg/ml chymostatin, 10 µl/ml protease inhibitor cocktail [from Sigma], and 20 mM Hepes-KOH, pH 7.9). The cells were then sonicated, incubated on ice for 15 min, and centrifuged at 16,000 g for 15 min at 4°C. The supernatant was collected and diluted ∼5-fold in buffer DG supplemented with 2.2 mM MgCl2, 0.1 mg/ml tRNA (Invitrogen) and 1 U/ml RNase OUT. Mixtures of biotinylated RNA oligos (4 µg) and NutrAvidin agarose resin (∼45 µl from Pierce Biotechnology) were added to the cell lysates. Following 1.5-h incubation at 4°C, the resin was washed 6 times with buffer DG (supplemented with 2.2 mM MgCl2, 0.1 mg/ml tRNA, and 1 U/ml RNase OUT), resuspended in 45 µl of 2×SDS sample buffer, and boiled for 5 min. After a brief centrifugation, the supernatant fraction was resolved by SDS-PAGE, and protein was electroblotted onto nitrocellulose membranes. The membranes were blocked and incubated with 1∶1,000 dilution of a monoclonal anti-flag antibody (Sigma) for 14 hours. Following multiple washing steps, membranes were incubated with a HRP-conjugated secondary antibody (anti-mouse IgG). Signal was visualized with an Enhanced Chemiluminescence Detection System.
Accession codes
Complete microarray datasets have been deposited at Gene Expression Omnibus under SuperSeries accession number GSE33591.
Supporting Information
Zdroje
1. DallosP (2008) Cochlear amplification, outer hair cells and prestin. Curr Opin Neurobiol 18 : 370–376.
2. GéléocGS, HoltJR (2003) Developmental acquisition of sensory transduction in hair cells of the mouse inner ear. Nat Neurosci 6 : 1019–1020.
3. LelliA, AsaiY, ForgeA, HoltJR, GéléocGS (2009) Tonotopic gradient in the developmental acquisition of sensory transduction in outer hair cells of the mouse cochlea. J Neurophysiol 101 : 2961–2973.
4. FrolenkovGI, BelyantsevaIA, FriedmanTB, GriffithAJ (2004) Genetic insights into the morphogenesis of inner ear hair cells. Nat Rev Genet 5 : 489–498.
5. RichardsonGP, de MonvelJB, PetitC (2011) How the genetics of deafness illuminates auditory physiology. Annu Rev Physiol 73 : 311–334.
6. GillespiePG, MüllerU (2009) Mechanotransduction by hair cells: models, molecules, and mechanisms. Cell 139 : 33–44.
7. DrorAA, AvrahamKB (2010) Hearing impairment: a panoply of genes and functions. Neuron 68 : 293–308.
8. MeyerAC, MoserT (2010) Structure and function of cochlear afferent innervation. Curr Opin Otolaryngol Head Neck Surg 18 : 441–446.
9. DriverEC, KelleyMW (2009) Specification of cell fate in the mammalian cochlea. Birth Defects Res C Embryo Today 87 : 212–221.
10. BerminghamNA, HassanBA, PriceSD, VollrathMA, Ben-ArieN, et al. (1999) Math1: an essential gene for the generation of inner ear hair cells. Science 284 : 1837–1841.
11. ZhengJL, GaoWQ (2000) Overexpression of Math1 induces robust production of extra hair cells in postnatal rat inner ears. Nat Neurosci 3 : 580–586.
12. WoodsC, MontcouquiolM, KelleyMW (2004) Math1 regulates development of the sensory epithelium in the mammalian cochlea. Nat Neurosci 7 : 1310–1318.
13. IzumikawaM, MinodaR, KawamotoK, AbrashkinKA, SwiderskiDL, et al. (2005) Auditory hair cell replacement and hearing improvement by Atoh1 gene therapy in deaf mammals. Nat Med 11 : 271–276.
14. XiangM, GanL, LiD, ChenZY, ZhouL, et al. (1997) Essential role of POU-domain factor Brn-3c in auditory and vestibular hair cell development. Proc Natl Acad Sci U S A 94 : 9445–9450.
15. VahavaO, MorellR, LynchED, WeissS, KaganME, et al. (1998) Mutation in transcription factor POU4F3 associated with inherited progressive hearing loss in humans. Science 279 : 1950–1954.
16. WallisD, HamblenM, ZhouY, VenkenKJ, SchumacherA, et al. (2003) The zinc finger transcription factor Gfi1, implicated in lymphomagenesis, is required for inner ear hair cell differentiation and survival. Development 130 : 221–232.
17. HertzanoR, MontcouquiolM, Rashi-ElkelesS, ElkonR, YücelR, et al. (2004) Transcription profiling of inner ears from Pou4f3(ddl/ddl) identifies Gfi1 as a target of the Pou4f3 deafness gene. Hum Mol Genet 13 : 2143–2153.
18. MencíaA, Modamio-HøybjørS, RedshawN, MorínM, Mayo-MerinoF, et al. (2009) Mutations in the seed region of human miR-96 are responsible for nonsyndromic progressive hearing loss. Nat Genet 41 : 609–613.
19. LewisMA, QuintE, GlazierAM, FuchsH, De AngelisMH, et al. (2009) An ENU-induced mutation of miR-96 associated with progressive hearing loss in mice. Nat Genet 41 : 614–618.
20. KuhnS, JohnsonSL, FurnessDN, ChenJ, InghamN, et al. (2011) miR-96 regulates the progression of differentiation in mammalian cochlear inner and outer hair cells. Proc Natl Acad Sci U S A 108 : 2355–2360.
21. FriedmanLM, DrorAA, MorE, TenneT, TorenG, et al. (2009) MicroRNAs are essential for development and function of inner ear hair cells in vertebrates. Proc Natl Acad Sci U S A 106 : 7915–7920.
22. SoukupGA, FritzschB, PierceML, WestonMD, JahanI, et al. (2009) Residual microRNA expression dictates the extent of inner ear development in conditional Dicer knockout mice. Dev Biol 328 : 328–341.
23. DeolMS, Gluecksohn-WaelschS (1979) The role of inner hair cells in hearing. Nature 278 : 250–252.
24. TuckerJB, MackieJB, BussoliTJ, SteelKP (1999) Cytoskeletal integration in a highly ordered sensory epithelium in the organ of Corti: reponse to loss of cell partners in the Bronx waltzer mouse. J Neurocytol 28 : 1017–1034.
25. CheongMA, SteelKP (2002) Early development and degeneration of vestibular hair cells in bronx waltzer mutant mice. Hear Res 164 : 179–189.
26. SobkowiczHM, InagakiM, AugustBK, SlapnickSM (1999) Abortive synaptogenesis as a factor in the inner hair cell degeneration in the Bronx Waltzer (bv) mutant mouse. J Neurocytol 28 : 17–38.
27. WhitlonDS, GabelC, ZhangX (1996) Cochlear inner hair cells exist transiently in the fetal Bronx Waltzer (bv/bv) mouse. J Comp Neurol 364 : 515–522.
28. CalarcoJA, SuperinaS, O'HanlonD, GabutM, RajB, et al. (2009) Regulation of vertebrate nervous system alternative splicing and development by an SR-related protein. Cell 138 : 898–910.
29. BussoliTJ, KellyA, SteelKP (1997) Localization of the bronx waltzer (bv) deafness gene to mouse chromosome 5. Mamm Genome 8 : 714–717.
30. LiA, XueJ, PetersonEH (2008) Architecture of the mouse utricle: macular organization and hair bundle heights. J Neurophysiol 99 : 718–733.
31. DesaiSS, AliH, LysakowskiA (2005) Comparative morphology of rodent vestibular periphery. II. Cristae ampullares. J Neurophysiol 93 : 267–280.
32. BoëdaB, WeilD, PetitC (2001) A specific promoter of the sensory cells of the inner ear defined by transgenesis. Hum Mol Genet 10 : 1581–1589.
33. LongJC, CaceresJF (2009) The SR protein family of splicing factors: master regulators of gene expression. Biochem J 417 : 15–27.
34. ShepardPJ, HertelKJ (2009) The SR protein family. Genome Biol 10 : 242.
35. XingY, StoilovP, KapurK, HanA, JiangH, et al. (2008) MADS: a new and improved method for analysis of differential alternative splicing by exon-tiling microarrays. RNA 14 : 1470–1479.
36. HuangDW, ShermanBT, LempickiRA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4 : 44–57.
37. RajB, O'HanlonD, VesseyJP, PanQ, RayD, et al. (2011) Cross-regulation between an alternative splicing activator and a transcription repressor controls neurogenesis. Mol Cell 43 : 843–850.
38. AbrajanoJJ, QureshiIA, GokhanS, MoleroAE, ZhengD, et al. (2010) Corepressor for element-1-silencing transcription factor preferentially mediates gene networks underlying neural stem cell fate decisions. Proc Natl Acad Sci USA 107 : 16685–16690.
39. HakimiMA, BocharDA, ChenowethJ, LaneWS, MandelG, et al. (2002) A core-BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc Natl Acad Sci USA 99 : 7420–7425.
40. IwaseS, JanumaA, MiyamotoK, ShonoN, HondaA, et al. (2004) Characterization of BHC80 in BRAF-HDAC complex, involved in neuron-specific gene repression. Biochem Biophys Res Commun 322 : 601–608.
41. KlajnA, FerraiC, StucchiL, PradaI, PodiniP, et al. (2009) The rest repression of the neurosecretory phenotype is negatively modulated by BHC80, a protein of the BRAF/HDAC complex. J Neurosci 29 : 6296–62307.
42. JonesAR, OverlyCC, SunkinSM (2009) The Allen Brain Atlas: 5 years and beyond. Nat Rev Neurosci 10 : 821–828.
43. GaleJE, MarcottiW, KennedyHJ, KrosCJ, RichardsonGP (2001) FM1–43 dye behaves as a permeant blocker of the hair-cell mechanotransducer channel. J Neurosci 21 : 7013–7025.
44. MeyersJR, MacDonaldRB, DugganA, LenziD, StandaertDG, et al. (2003) Lighting up the senses: FM1–43 loading of sensory cells through nonselective ion channels. J Neurosci 23 : 4054–4065.
45. BaileyTL, WilliamsN, MislehC, LiWW (2006) MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res 34: W369–373.
46. MatsudaY, InoueY, IzumiH, KagaM, InagakiM, et al. (2011) Fewer GABAergic interneurons, heightened anxiety and decreased high-frequency electroencephalogram components in Bronx waltzer mice, a model of hereditary deafness. Brain Res 1373 : 202–210.
47. KotakVC, TakesianAE, SanesDH (2008) Hearing loss prevents the maturation of GABAergic transmission in the auditory cortex. Cereb Cortex 18 : 2098–2108.
48. SarroEC, KotakVC, SanesDH, AokiC (2008) Hearing loss alters the subcellular distribution of presynaptic GAD and postsynaptic GABAA receptors in the auditory cortex. Cereb Cortex 18 : 2855–2867.
49. HatanoM, FurukawaM, ItoM (2009) Changes in calbindin-D28k and parvalbumin expression in the superior olivary complex following unilateral cochlear ablation in neonatal rats. Acta Otolaryngol 129 : 839–845.
50. EugèneD, DeforgesS, GuimontF, IdouxE, VidalPP, et al. (2007) Developmental regulation of the membrane properties of central vestibular neurons by sensory vestibular information in the mouse. J Physiol 583 : 923–943.
51. SuckaleJ, WendlingO, MasjkurJ, JägerM, MünsterC, et al. (2011) PTBP1 is required for embryonic development before gastrulation. PLoS ONE 6: e16992 doi:10.1371/journal.pone.0016992.
52. LiQ, LeeJA, BlackDL (2007) Neuronal regulation of alternative pre-mRNA splicing. Nat Rev Neurosci 8 : 819–831.
53. WangZ, BurgeCB (2008) Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA 14 : 802–813.
54. WittenJT, UleJ (2011) Understanding splicing regulation through RNA splicing maps. Trends Genet 27 : 89–97.
55. RouxI, SafieddineS, NouvianR, GratiM, SimmlerMC, et al. (2006) Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell 127 : 277–289.
56. ZanazziG, MatthewsG (2009) The molecular architecture of ribbon presynaptic terminals. Mol Neurobiol 39 : 130–148.
57. GoodyearRJ, LeganPK, WrightMB, MarcottiW, OganesianA, et al. (2003) A receptor-like inositol lipid phosphatase is required for the maturation of developing cochlear hair bundles. J Neurosci 23 : 9208–9219.
58. PanceA, LiveseyFJ, JacksonAP (2006) A role for the transcriptional repressor REST in maintaining the phenotype of neurosecretory-deficient PC12 cells. J Neurochem 99 : 1435–1444.
59. D'AlessandroR, KlajnA, StucchiL, PodiniP, MalosioML, et al. (2008) Expression of the neurosecretory process in PC12 cells is governed by REST. J Neurochem 105 : 1369–1383.
60. AhmedZM, RiazuddinS, AhmadJ, BernsteinSL, GuoY, et al. (2003) PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum Mol Genet 12 : 3215–3223.
61. RiazuddinS, AhmedZM, FanningAS, LagzielA, KitajiriS, et al. (2006) Tricellulin is a tight-junction protein necessary for hearing. Am J Hum Genet 79 : 1040–1051.
62. WangemannP, NakayaK, WuT, MagantiRJ, ItzaEM, et al. (2007) Loss of cochlear HCO3 − secretion causes deafness via endolymphatic acidification and inhibition of Ca2+ reabsorption in a Pendred syndrome mouse model. Am J Physiol Renal Physiol 292: F1345–F1353.
63. NakanoY, KimSH, KimHM, SannemanJD, ZhangY, et al. (2009) A claudin-9-based ion permeability barrier is essential for hearing. PLoS Genet 5: e1000610 doi:10.1371/journal.pgen.1000610.
64. UleJ, UleA, SpencerJ, WilliamsA, HuJS, et al. (2005) Nova regulates brain-specific splicing to shape the synapse. Nat Genet 37 : 844–852.
65. PhairRD, MisteliT (2000) High mobility of proteins in the mammalian cell nucleus. Nature 404 : 604–609.
66. HaasP, GilmourD (2006) Chemokine signaling mediates self-organizing tissue migration in the zebrafish lateral line. Dev Cell 10 : 673–680.
67. Siepel A, Pollard KS, Haussler D (2006) New methods for detecting lineage-specific selection. Proceedings of the 10th International Conference on Research in Computational Molecular Biology (RECOMB). pp.190–205.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- A Mutation in the Gene Causes Alternative Splicing Defects and Deafness in the Bronx Waltzer Mouse
- Mutations in (Hhat) Perturb Hedgehog Signaling, Resulting in Severe Acrania-Holoprosencephaly-Agnathia Craniofacial Defects
- Classical Genetics Meets Next-Generation Sequencing: Uncovering a Genome-Wide Recombination Map in
- Regulation of ATG4B Stability by RNF5 Limits Basal Levels of Autophagy and Influences Susceptibility to Bacterial Infection
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy