
Viral Evasion of a Bacterial Suicide System by RNA–Based Molecular Mimicry Enables Infectious Altruism
Abortive infection, during which an infected bacterial cell commits altruistic suicide to destroy the replicating bacteriophage and protect the clonal population, can be mediated by toxin-antitoxin systems such as the Type III protein–RNA toxin-antitoxin system, ToxIN. A flagellum-dependent bacteriophage of the Myoviridae, ΦTE, evolved rare mutants that “escaped” ToxIN-mediated abortive infection within Pectobacterium atrosepticum. Wild-type ΦTE encoded a short sequence similar to the repetitive nucleotide sequence of the RNA antitoxin, ToxI, from ToxIN. The ΦTE escape mutants had expanded the number of these “pseudo-ToxI” genetic repeats and, in one case, an escape phage had “hijacked” ToxI from the plasmid-borne toxIN locus, through recombination. Expression of the pseudo-ToxI repeats during ΦTE infection allowed the phage to replicate, unaffected by ToxIN, through RNA–based molecular mimicry. This is the first example of a non-coding RNA encoded by a phage that evolves by selective expansion and recombination to enable viral suppression of a defensive bacterial suicide system. Furthermore, the ΦTE escape phages had evolved enhanced capacity to transduce replicons expressing ToxIN, demonstrating virus-mediated horizontal transfer of genetic altruism.
Published in the journal:
. PLoS Genet 8(10): e32767. doi:10.1371/journal.pgen.1003023
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003023
Summary
Abortive infection, during which an infected bacterial cell commits altruistic suicide to destroy the replicating bacteriophage and protect the clonal population, can be mediated by toxin-antitoxin systems such as the Type III protein–RNA toxin-antitoxin system, ToxIN. A flagellum-dependent bacteriophage of the Myoviridae, ΦTE, evolved rare mutants that “escaped” ToxIN-mediated abortive infection within Pectobacterium atrosepticum. Wild-type ΦTE encoded a short sequence similar to the repetitive nucleotide sequence of the RNA antitoxin, ToxI, from ToxIN. The ΦTE escape mutants had expanded the number of these “pseudo-ToxI” genetic repeats and, in one case, an escape phage had “hijacked” ToxI from the plasmid-borne toxIN locus, through recombination. Expression of the pseudo-ToxI repeats during ΦTE infection allowed the phage to replicate, unaffected by ToxIN, through RNA–based molecular mimicry. This is the first example of a non-coding RNA encoded by a phage that evolves by selective expansion and recombination to enable viral suppression of a defensive bacterial suicide system. Furthermore, the ΦTE escape phages had evolved enhanced capacity to transduce replicons expressing ToxIN, demonstrating virus-mediated horizontal transfer of genetic altruism.
Introduction
Toxin-antitoxin (TA) systems are ubiquitously distributed in plasmids and chromosomes of prokaryotes [1]–[3]. TA systems are divided into three Types, depending upon the nature of the interacting partners [4]. These small, bicistronic, genetic loci were originally identified as plasmid maintenance systems [5] though they are also involved in stress responses [6] and formation of persister cells [7] amongst other roles [8]. All three Types have been shown to protect from bacteriophage (phage) infection. The Type I hok/sok locus excludes T4 [9], the Type II mazEF locus can prevent spread of phage P1 infection [10] and the Type III toxIN locus can inhibit multiple phages in multiple host backgrounds [11], [12].
ToxIN was the first characterised Type III protein-RNA TA system, encoded by plasmid pECA1039 of the phytopathogen Pectobacterium atrosepticum [11]. Recent crystallographic studies showed that ToxN is a 19.7 kDa endoribonuclease of the Kid family, which is held inactive as a hetero-hexameric triangular structure, with three ToxN monomers in complex with three, 36-nucleotide, pseudoknots of antitoxic ToxI RNA [4], [13]. ToxIN was originally discovered through shared sequence similarity with an abortive infection (Abi) system, AbiQ, from Lactococcus lactis [14].
Due to constant selection pressure from viral predation, bacteria have developed multiple routes of defence; these include altering the cell surface to avoid phage adsorption, cleavage of viral nucleic acids by restriction-modification or CRISPR/Cas systems, preventing viral DNA injection, and through Abi systems [15]. Abi systems are lethal to their hosts when activated in a phage-infected cell. This prevents productive phage propagation – and thus protects the clonal bacterial population through ‘altruistic’ suicide [16]. By sequencing mutants of phages that had spontaneously ‘escaped’ an Abi system, it has been possible to examine the activity of the Abi systems and also the components of each phage required to activate these systems [17], [18].
To investigate how phages interact with the ToxIN dual functions (Abi and TA), we sought to characterise phage mutants resistant to ToxIN. A ToxIN-sensitive phage, ΦTE, which infects Pectobacterium atrosepticum 1043 (Pba) [19], evolved low-frequency spontaneous escape mutants that were ToxIN-insensitive. In this study, we have sequenced the genomes of both wild type and escape ΦTE phages in order to identify the factors involved in phage-ToxIN recognition.
Results
ΦTE is a flagellum-dependent rv5-like virus
Mutant Pba strain SCC34 has a mucoid morphology that provides resistance to infection by lipopolysaccharide (LPS)-dependent phages, such as the generalised transducing phage ΦM1 [20], [21]. ΦTE was initially isolated from treated sewage effluent enriched for phages using SCC34 as host, as part of a screen for lipopolysaccharide (LPS)-independent phages. Using transmission electron microscopy, ΦTE was seen to have an isometric, icosahedral, head measuring 98 ±4 nm from flat face to flat face and a tail measuring 124±3 nm when extended (Figure 1A), or 65±7 nm when contracted (Figure 1B). ΦTE was classified as a member of the Myoviridae of the order Caudovirales [22]. A bank of ΦTE-resistant transposon-mutants of Pba was generated in order to identify the host receptor for ΦTE. These mutants of Pba remained sensitive to ΦM1 but were resistant to the flagellum-dependent phage, ΦAT1 [23]. Seven of the transposon insertions conferring ΦTE-resistance disrupted genes involved in flagellum biosynthesis (Table S1), suggesting that the host receptor for ΦTE was the flagellum. Restriction enzyme digestion analysis of the extracted ΦTE genome showed that it was terminally redundant and circularly permuted (Figure S1).

ΦTE can evolve to escape ToxIN-mediated abortive infection
Serial dilutions of ΦTE were plated into top-lawns of Pba containing ToxIN plasmid pTA46 [11]. The Efficiency of Plating (EOP) of ΦTE was ∼1×10−8 compared with the ToxIN-frameshift (ToxIN-FS) negative controls. Phages were then isolated from six individual ΦTE plaques that formed on Pba (pTA46). These phages were plaque purified and passaged twice by growth on Pba without the ToxIN plasmid, and re-tested for sensitivity to ToxIN. The six mutant phage strains, denoted ΦTE-A to -F, plated on Pba (pTA46) with EOPs of 1.01, 1.07, 1.23, 0.90, 1.08 and 1.30, respectively, indicating that they had heritable mutations allowing them to ‘escape’ Abi by ToxIN. To identify the escape mutation, the genomes of ΦTE wild type (wt) and three escape ΦTE strains were sequenced.
Genomic sequencing of wild-type and escape strains of ΦTE
Genomic DNAs were extracted from ΦTE, ΦTE-A, ΦTE-C and ΦTE-E and subjected to 454 sequencing. ΦTE had a dsDNA genome of 142,349 bp encoding 242 putative ORFs and two tRNAs (Figure 1C and Table S2). The coding regions of the phage were divided into four gene clusters, carried on both strands (Figure 1C).
Whilst 44% of ΦTE gene products had no detectable similarity with any protein in the NCBI database, a further 27% shared significant amino acid sequence identity with proteins encoded by the Salmonella phage PVP-SE1 [24] (Table S2). In many of these cases, the second most significant hit came from Escherichia coli phage rv5 (GenBank Accession no. DQ832317), which is the closest relative to PVP-SE1 [24]. PVP-SE1 has a similarly-sized genome to ΦTE, a similar morphology and is also terminally redundant and circularly permuted [24]. The tBLASTx results comparing ΦTE with PVP-SE1 and rv5 are shown (Figure 1C). Altogether, this suggests that ΦTE should be included within the “rv5-like virus” genus of the Myoviridae [24].
ΦTE escape phages expand a repetitive antitoxin sequence
Escape phage ΦTE-A had a final genome size of 142,457 bp, whilst ΦTE-C and ΦTE-E were siblings with an identical genome of 142,497 bp. By comparing the finished sequences of ΦTE and the escape phages, a single difference was identified; the escape genomes had expanded in size within the intergenic locus between phiTE_202 and phiTE_203. This ‘escape locus’ between 106,714–106,772 bp of ΦTE wt (Figure 1C), contained a nucleotide sequence closely resembling that of the ToxI antitoxin from ToxIN (Figure 2). Within the escape phages, the number of DNA repeats had been expanded from 1.5 repeats to 4.5 (ΦTE-A) or 5.5 (ΦTE-C and -E; Figure 3, Table S3). The remaining escape phages were analysed by specifically sequencing amplicons generated from these escape loci. In phages ΦTE-B and ΦTE-D, the locus had also expanded to 5.5 repeats. The nature of each repeat varied in one position, in that a trinucleotide ‘TTT’ (denoted 3T) sometimes appeared in a repeat as a dinucleotide ‘TT’ (denoted 2T) (Figure 2 and Table S3). This difference separated ΦTE-B and -D from ΦTE-C and -E.
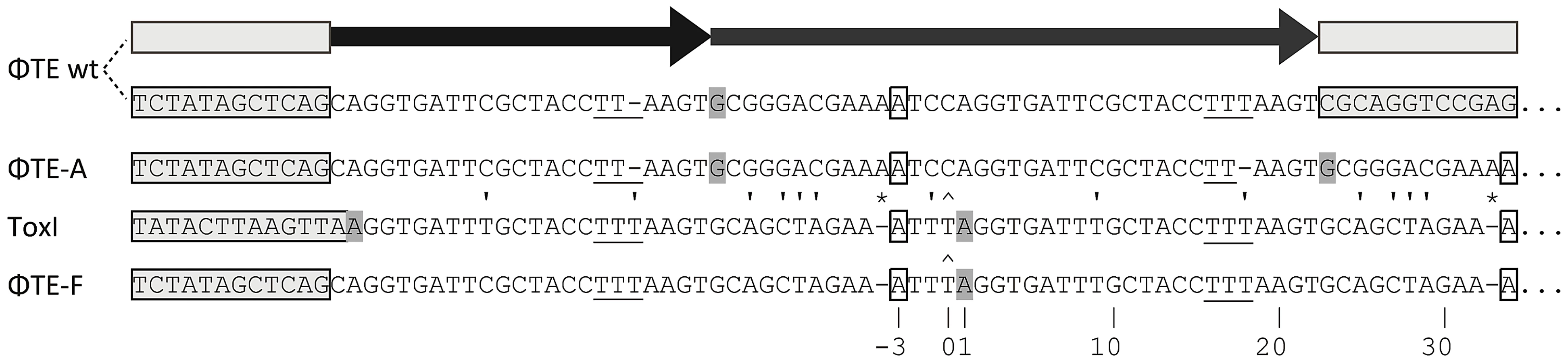

To ensure this escape response was not an isolated phenomenon, a second round of escape phage selection was performed. The escape phenotype of four new isolates, ΦTE-G to -J, was checked by plating the phages on Pba (pTA46); they plated with EOPs of 1.38, 1.67, 1.43 and 1.62, respectively. The escape loci of these four new escape phages were sequenced. Though isolated four months apart, we found that ΦTE-G had the same mutation as ΦTE-B and -D, whilst ΦTE-H had the same mutation as ΦTE-A. This suggests that these phages, though genetically identical, are not true siblings from a single escape event, and that an identical escape route can be taken in successive rounds of selection. This second batch also provided a new expansion, for phages ΦTE-I and -J (Figure 3 and Table S3).
One interesting outlier from the first isolation, ΦTE-F, appeared to have recombined with the ToxIN plasmid used during selection and incorporated ToxI into its genome, partially losing the ΦTE ‘pseudo-ToxI’ sequence in the process (Figure 2 and Figure 3). This recombination event generated mosaic repeats; the initial and final ‘repeats’ contained sequences from both pseudo-ToxI and ToxI itself (Figure 3 and Table S3).
Functional analysis of the escape locus from ΦTE
An alignment of the ToxI RNA sequence with the predicted pseudo-ToxI RNA showed that the majority of nucleotide positions were conserved (Figure 4A). When the mutated positions were mapped onto the structure of antitoxic ToxI RNA in complex with the ToxN protein [13], they formed five groups as defined by their proximity within the structure (Figure 4B and 4C). To ascertain whether the aligned pseudo-ToxI was in fact a functional antitoxin, a single repeat was cloned into an expression vector, pTA100 [11] and tested for the ability to inhibit ToxN over-expressed from a second, pBAD30-based [25] vector, within an E. coli model (Figure 4D). In this assay, pseudo-ToxI could not suppress ToxN toxicity (Figure 4D). Each mutation group was therefore examined in isolation to compare the contribution each made to rendering pseudo-ToxI inactive within this assay (Figure 4E). Groups 1, 2, 3 and 5 had no impact on the antitoxic activity of ToxI, alone or in concert, whilst group 4 was solely responsible for knocking-out ToxI function (Figure 4E). Group 4 comprised three contiguous nucleotides. The contribution of each individual group 4 nucleotide substitution was assessed. Mutations C27G and U28A were each independently sufficient to knock-out ToxI, either in the pseudo-ToxI (ie. with mutations 1, 2, 3 and 5) or ToxI backgrounds (Figure 4E). As pseudo-ToxI was not active as a single, aligned, repeat (Figure 4D), it was also decided to clone and test the whole escape loci from ΦTE wt, ΦTE-A and ΦTE-F (Figure 4F). Only the sequence from ΦTE-F (which had recombined with ToxI) was antitoxic (Figure 4F).

Finally, in attempts to generate a more native context, the escape loci from ΦTE wt, ΦTE-A and ΦTE-F were cloned to replace ToxI within the native toxIN operon (Figure 5A). This cloning required the use of a transposon-marked derivative of the original ToxIN plasmid, pECA1039-Km3 [11]. In this case, only recombinants with the insert from ΦTE-F could be obtained, presumably due to toxic effects preventing cloning of ΦTE wt and ΦTE-A. The ΦTE-F plasmid retained Abi activity (Figure 5B), as tested with ΦS61 [12], which was used in order to prevent any possible interaction of an infecting ΦTE with the regions of the ΦTE genome cloned onto the test plasmid construct.

Expression of pseudo-ToxI antitoxin can inhibit abortive infection
As the protection assays above (Figure 4D and 4E) relied on over-expression of the toxic and antitoxic components, it was considered that they may act as poor approximations to the relative stoichiometries of ToxN, ToxI and pseudo-ToxI during ΦTE infections. An assay was therefore designed to focus on whether Abi could be perturbed by excess antitoxin (Figure 6A). An excess was generated by first cloning both control and ΦTE-derived antitoxin sequences into the multiple cloning site of the high copy-number plasmid pBluescript II SK - (Fermentas), oriented so that they would be transcribed from the lacZα promoter. This constitutive promoter would ensure an over-abundance of our test RNA. These constructs were used in attempts to alter the Abi phenotype of an independent, pACYC184-based [26] ToxIN plasmid (pTRB101) within Pba.

In the presence of the positive control ToxI plasmid, Abi against ΦTE was greatly suppressed, compared to the vector control (Figure 6B). Escape locus sequences from ΦTE-A and -F were also able to suppress Abi to similar levels as ToxI, whilst the same locus from ΦTE wt did not have an effect (Figure 6B). To confirm that this result was dependent on the specific sequence cloned into pBluescript, further negative controls were tested; a section of the ΦTE genome upstream of the escape locus (269 bp, from 99,267 to 99,536 bp), the E. coli coding sequence for ompA (a known substrate of ToxN) [13] and a non-cognate antitoxic ToxI from the ToxIN system of Bacillus thuringiensis. None of these three negative controls made any significant impact on the Abi phenotype of ToxIN against ΦTE (Figure 6B). To confirm that this was not a phage-specific effect, the experiments were repeated with phages ΦM1 and ΦS61 [12]. The same trend was observed, with protection from Abi only observed with plasmids containing either the escape loci of ΦTE-A and -F, or cognate ToxI (Figure 6B). The effect of ToxIN upon ΦS61 was so pronounced that, when no protection was provided from the test construct, no phage plaques could be observed. This produced an EOP of <1×10−8 (Figure 6B) and provided a stark indication of which constructs acted as antitoxins. Collectively, these data show that Abi can be suppressed by an expanded pseudo-ToxI sequence, or cognate ToxI, within the infected cell.
ΦTE expresses antitoxin mimics during infection
Having ascertained that expression of pseudo-ToxI during infection could alter the Abi outcome, we studied the levels of antitoxic RNA and ToxN protein during ΦTE infections of Pba containing FLAG-tagged ToxIN plasmid pMJ4 [12]. When infecting Pba with ΦTE wt, there were no detectable differences in ToxI or ToxN levels between the infected and uninfected controls, as measured by S1 nuclease assays and Western blotting of the ToxN-FLAG protein, respectively (Figure 7A). To see if an ‘escape’ ΦTE phage caused an alteration in the ToxI:ToxN ratio, the same experiment was performed with phage ΦTE-F. No detectable differences were observed following infection relative to the uninfected controls (Figure 7B).
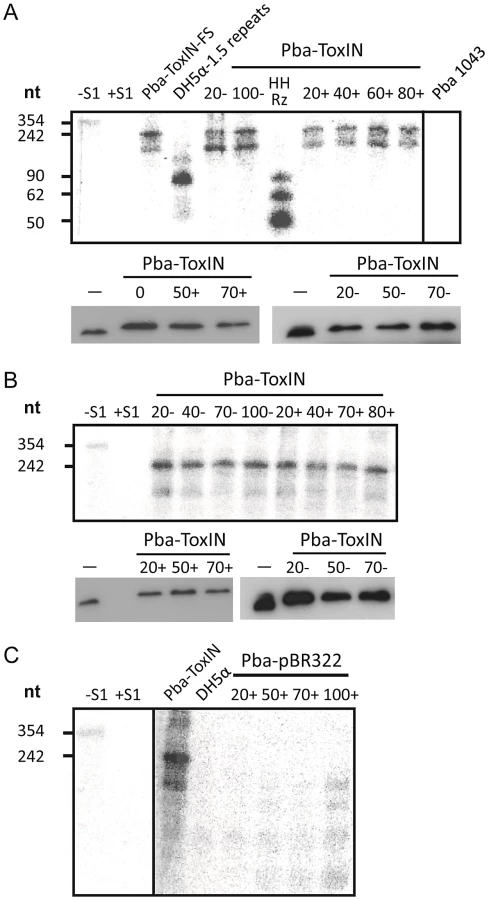
Using the same probe, it was then possible to examine whether ToxI was expressed from the recombinant escape phage ΦTE-F, during infection of Pba carrying a vector-only pBR322 (NEB) control rather than a ToxIN plasmid. Again, this was monitored by taking regular total cellular RNA samples during an infection cycle and detecting the escape transcripts of phage ΦTE-F by S1 nuclease assay (Figure 7C). These assays showed specific detectable transcripts from the ΦTE-F escape locus that increased during phage infection. The transcripts from the ΦTE-F escape locus were shorter, and expressed at a lower level, than observed for the Pba-ToxIN (pMJ4) control (Figure 7C). The shorter length was consistent with the differences between the ΦTE-F escape locus and ToxI.
Generalised transduction of ToxIN by ΦTE
ΦTE was tested for the ability to transduce selectable markers. ΦTE was able to transduce chromosomal markers from Pba strains SCC34 and SCC14 [27] along with plasmids pKD46 [28] and pBluescript II KS+ (Table 1). ΦTE wt and escape phages ΦTE-A and -F were then used in attempts to transduce plasmids pTRB101 (ToxIN) and pTRB102 (ToxIN-FS). Phages ΦTE-A and ΦTE-F plated on pTRB101 with an EOP of ∼1, whereas ΦTE wt had an EOP of 1×10−7. It was therefore necessary to plate ∼107 more ΦTE wt phages to obtain a pTRB101 transducing lysate, than with the two isolated escape phage strains. Once generated, however, the ΦTE wt pTRB101 lysate then transduced approximately as efficiently as the ΦTE-A and -F lysates (Table 1). The resulting pTRB101 transductant strains were then confirmed as having gained Abi activity. Finally, ΦTE-F was used to transduce a marked derivative of the native ToxIN plasmid, pECA1039, proving that phage-mediated horizontal transmission of the naturally-occurring phage-resistance replicon was also possible (Table 1).

Discussion
We isolated and characterised phage ΦTE, a flagellum-dependent generalised transducing Myoviridae member of the “rv5-like virus” genus (Figure 1 and Figure S1). The genome of ΦTE did not contain any evidence of a lysogeny module or remnant integrases, confirming ΦTE to be a lytic phage. ΦTE was shown to undergo Abi by the Type III TA system, ToxIN. Abi resulted in the selection of ToxIN-insensitive mutant ΦTE ‘escape’ phages. Genome sequencing identified a ΦTE ‘escape locus’ containing a repetitive DNA sequence, which was similar to the repetitive 36 nucleotide sequence of each unit from the ToxI antitoxic non-coding RNA (Figure 2 and Figure 3; Table S3). ΦTE wt contained 1.5 repeats of this ‘pseudo-ToxI’ sequence, whilst the escape phages had expanded this number (Figure 3). In one case, ΦTE-F, a recombination event had occurred between the phage genome and the plasmid carrying toxIN (Figure 2 and Figure 3). Alignment with, and consequent mutagenesis of, ToxI, identified those mutations of pseudo-ToxI which impaired antitoxic activity within over-expression assays (Figure 4). Next, we developed a novel Abi inhibition assay, in which a cell is pre-loaded with potentially antitoxic RNAs and then tested to see whether this abundance of RNA would be sufficient to inhibit the ToxN protein and thereby Abi (Figure 6A). This assay showed that pseudo-ToxI adequately mimicked ToxI activity and suppressed ToxN anti-phage activity in vivo (Figure 6B). Biochemical study of ToxI RNA levels during ΦTE-F infection indicated that pseudo-ToxI would indeed be expressed during the replication cycle of ΦTE phages (Figure 7). ΦTE was also shown to be able to transduce the native ToxIN plasmid (Table 1).
By comparing the nucleotide sequence from a single repeat of pseudo-ToxI with that of ToxI (Figure 4A) and considering how the mutations may alter the shape of the resulting RNA pseudoknot (Figure 4B and 4C), it became clear how pseudo-ToxI may act to inhibit ToxN. The Group 1 mutation is U-1C (Figure 4B and 4C). This change is in the initial ssRNA tail, with the base clearly extending away from the complex (Figure 4C). The next alteration (denoted by, ∧, in Figure 2 and Figure 3) can be discounted as it only appears as a mutation against the first ToxI repeat; this has a non-consensus T rather than the consensus C, which would otherwise match pseudo-ToxI. Group 2 can be considered a compensatory set of mutations, as U9C is matched by A25G; together, they form the end duplex interaction of the pseudoknot central stack [13]. Variability in each pseudo-ToxI arises from whether the group 3 mutation is present, deleting the sequence ‘UUU’ to ‘UU’ at U16-U18. This region forms the second loop of the ToxI pseudoknot and is not vital for interaction with ToxN, so is likely to allow for some flexibility in length. Jumping ahead to group 5, this is an additional A at the 3′ end of each pseudo-ToxI, which conserves the sequence around the defined cleavage site. Mutations in groups 1, 2, 3 and 5 were all shown not to effect pseudo-ToxI antitoxicity (Figure 4E). This highlights the innate plasticity in pseudoknot formation and the interaction of a Type III antitoxin with its cognate toxic protein. There are limits to this plasticity, however, as group 4 contains knock-out mutations; positions 27–29 are CUA in ToxI, but GAC in pseudo-ToxI. Mutation A29C has no effect, but both C27G and U28A are individually sufficient to knock-out ToxI antitoxicity in over-expression assays (Figure 4E). This can be readily explained when examining the ToxIN structure, as both C27 and U28 are involved in extensive hydrogen-bond networks at the previously defined major interface between ToxI and ToxN [13].
From these analyses it is clear why it is necessary to expand the ΦTE escape locus in order to obtain an escape phenotype. Though the ΦTE wt locus has 1.5 DNA repeats, it does not encode one full pseudo-ToxI RNA (Figure 2). The phasing of the repetitive DNA and active RNA sequences differ, so that within the ΦTE wt locus, it is not possible to generate a pseudo-ToxI RNA that could readily form a pseudoknot. Only by expanding this locus can a phage then express active pseudo-ToxI RNAs that can fold properly to mimic cognate ToxI and suppress ToxN. We suggest that these expansions of pseudo-ToxI may have arisen through strand slippage during replication. When we previously cloned these regions, we often observed a range of PCR products containing 2.5 to 5.5 copies of the repeat sequences [11]. Strand slippage would help to generate the observed diversity in the number of repeats and mosaic nature of each expanded escape locus. This could also reflect the generation of ToxI in nature, and may help to explain the diversity in the number of repeats shown within the family of ToxI sequences [3]. This expansion to either 4.5 or 5.5 repeats also highlights how the levels of expression must be exquisitely tuned, in order to inhibit Abi within the native infection setting.
This observed mode of escape from Abi differs from either the specific point mutations observed in the sak and sav genes, (which allow lactococcal phages to escape AbiK and AbiV, respectively [17], [18]), or the extremely large-scale genomic re-arrangements observed in phages escaping AbiK and AbiT [29]. Here, ΦTE carries an inactive antitoxin which can be selectively upregulated through expansions in genomic DNA content. In the case of ΦTE-F, the phage actively recombined with the ToxIN Abi system to hijack the antitoxic component and protect itself from the host defences. A similar case was previously observed when a virulent phage transferred a host methylase gene into its genome, providing protection from the host restriction-modification system [30]. Within ΦTE-F, however, it is an RNA that provides the advantage. Large non-coding RNAs have been identified within prophages from a restricted number of bacterial species, and are thought to be involved in host lysis and virion production [31]. Selected temperate phages use non-coding RNAs to mediate superinfection immunity [32]. To the best of our knowledge this is the first example, however, where an infecting phage evolves a non-coding RNA to suppress host defensive mechanisms and ensure phage replication.
As the observed transduction efficiencies for ΦTE, though low, are similar to values for transducing phages of Citrobacter rodentium and Pseudomonas aeruginosa [33], [34], ΦTE would make a suitable tool for functional genomics. In the environment, the apparently low efficiency of transduction will be counter-acted by the abundant microbiological populations and high frequency of infection. This identification of ΦTE as a transducing phage also provided us with the opportunity to investigate transfer of ToxIN. Whilst conjugation has been used to transfer both chromosomally encoded AbiV [35] and other plasmid-based Abi systems [36], pECA1039 does not contain any obvious components for conjugal transfer [11]. Instead, we were able to show transduction of ToxIN plasmids by ΦTE phages, making this a key example of phage-mediated transfer of phage-resistance mechanisms. Given the mode of action of ToxIN, this particular case may demonstrate a de facto example of “infectious altruism”. This finding may also help to explain the widespread horizontal transfer of both chromosomal and plasmid-borne Type III TA systems observed in a recent bioinformatic study [3].
We can consider this three-way, phage-host-plasmid, interaction, from the point of view that ToxIN, as a toxin-antitoxin system, is an addictive and selfish element [8]. Selection of an escape phage would indeed have the negative effect of allowing more productive infection within host cells containing ToxIN, but it may also lead to increased dissemination of the ToxIN plasmid. Considering the selfish phage, this would then allow ΦTE escape phages to select against other, ToxIN-sensitive, phages competing for the same bacterial host. The interactivity between these three elements shapes the evolution of further methods of defence and counter-attack. It will be interesting to identify how other phages escape Abi by ToxIN and indeed whether these escape mechanisms are as widespread as the resistance mechanisms themselves.
Materials and Methods
Bacterial strains, bacteriophages, and growth conditions
Bacterial strains and bacteriophages are listed in Table S4. All experiments were performed with E. coli strain DH5α (Gibco/BRL) or Pectobacterium atrosepticum SCRI1043 [19] and derivatives thereof. Bacteriophage ΦTE was isolated from sewage effluent collected from the outlet at Milton sewage treatment plant, near Cambridge, UK. E. coli strains were grown at 37°C and Pba was grown either at 25°C on plates or at 25, 28, or 30°C as required for liquid culture, in Luria broth (LB) at 250 rpm or on LB-agar (LBA). LBA contained 1.5% w v−1 or 0.35% w v−1 agar, to make LBA plates or top-LBA, respectively. Growth (OD600) was measured using a spectrophotometer set to 600 nm. When required, media was supplemented with ampicillin (Ap) at 100 µg ml−1, chloramphenicol (Cm) at 50 µg ml−1, kanamycin (Km) at 50 µg ml−1 and spectinomycin (Sp) at 50 µg ml−1. Bacteriophages were isolated as described [23] though using SCC34 as the host strain. Phage lysates were made as described [37]. Phages were stored at 4°C in phage buffer; 10 mM Tris-HCl pH 7.4, 10 mM MgSO4, 0.01% w v−1 gelatin. Efficiency of Plating (EOP) was calculated after overnight incubation of phage lysate serial dilutions in a top-LBA lawn of each bacterial host, using (pfu on test strain/pfu on control strain).
Electron microscopy
Electron microscopy was performed at the Multi-Imaging Centre, University of Cambridge, using a Tecnai G2 series transmission electron microscope. Samples were prepared by first placing charge-discharged copper grids onto 50 µl drops of high titre (≥1×1010 pfu ml−1) lysates of ΦTE for 1 min. Each grid was then washed briefly with three drops of distilled water, followed by staining with 1% phosphotungstic acid for 1 min. The accelerating voltage was 120.0 kV and the direct magnification was 25,000×.
ΦTE genomic sequencing
Bacteriophage DNA was extracted with phenol/chloroform, using phase-lock gel tubes (Eppendorf) and following the manufacturer's instructions as for bacteriophage λ. The extracted DNA was subjected to pyrosequencing on a Roche 454 Genome Sequencer FLX at the DNA sequencing facility, Department of Biochemistry, University of Cambridge. Contigs were assembled using Newbler (Roche).
ORFs were identified in the ΦTE genome using Glimmer and LongORFS [38]. Homologues of predicted proteins were identified using BLASTp [39]. ΦTE tRNAs were identified using tRNAScan-SE [40]. RBSfinder [41] was used to identify ribosome binding sites (Table S2). The ΦTE genome was viewed and annotated using Artemis [42] and the GenBank submission file was generated using Sequin (NCBI). Figure 1C was generated by using Adobe Illustrator to adapt an output from the CGView Server [43].
Plasmid construction
Molecular biology techniques were performed as described previously [44]. All primers were obtained from Sigma-Genosys and Invitrogen and are listed in Table S5. All plasmids constructed and/or used in this study are listed in Table S6, along with the primers used for their construction. All recombinant plasmid sequences were verified by DNA sequencing.
Protection assays
When required, media was supplemented with Ap, Sp, D-glucose (glu) at 0.2% w v−1, L-arabinose (L-ara) at 0.1% w v−1 and isopropyl-β-D-thiogalactopyranoside (IPTG) at 1 mM. Double-plasmid carrying DH5α strains were grown as 10 ml overnight cultures, then used to inoculate 25 ml of LB, Ap and glu in 250 ml flasks, and grown at 37°C and 250 rpm, from a starting OD600 of ∼0.04, until exponential phase (∼1×108 colony forming units (cfu) ml−1). Samples were removed, washed with phosphate buffered saline (PBS) serially diluted and plated for viable counts at 37°C on LBA, Ap, Sp plates containing either i) glu, so neither toxin or antitoxin were expressed; ii) glu and IPTG, to express the antitoxin; iii) L-ara, to express the toxin; or iv) L-ara and IPTG to express both the toxin and antitoxin.
Measuring ToxI and ToxN levels during phage infection
Two cultures of 180 ml of LB containing Ap were inoculated with 2 ml of overnight cultures of Pba (pBR322) or Pba (pMJ4), respectively. Cultures were grown at 25°C and 180 rpm to an OD600 of 1 and each split into 2×80 ml; one of which was infected with phage to a multiplicity of infection (MOI) of 1 while the other served as a negative control without infection. Cultures were left 10 min without shaking for phage adsorption, then shaken at 25°C and 180 rpm. Samples for OD600 measurement, RNA preparation and protein analysis were taken regularly during infection. Total RNA was isolated using the TRIZOL method and subsequently DNase treated. Cell pellets for Western blot analysis were resuspended in 1× PBS according to OD600 measurement.
Western blotting
One ml cell samples were taken, pelleted and resuspended in 1× PBS according to OD600. The protein was quantified using Nanodrop (ThermoScientific) and equal amounts of protein (150 µg) were resolved by 12% PAGE. Proteins were transferred to a PVDF-membrane and blocked for 1 hour in 1× PBS containing 5% milk powder. Immunodetection of FLAG-tagged ToxN was performed overnight at 4°C in 1× PBS using anti-FLAG M2 antibody (Sigma). Goat anti-mouse IgG-HRP (Santa Cruz) was used as secondary antibody. Bands were visualised on X-Ray film using the SuperSignal West Pico Chemiluminescent Substrate Kit (Pierce).
Construction of size markers for S1-nuclease assays
Plasmid pTA96 was generated to act as a size marker in the S1-nuclease assays, using overlap extension PCR. Briefly, the 5′ flanking fragment encoding the toxIN promoter was amplified using pTA47 as template with PF221 and PF162. The 3′ flanking fragment containing 1.5 toxI repeats and toxN-FS was amplified using PF168 and PF222 and pTA47 as template. The overlap PCR used PF221 and PF222 and the 5′ and 3′ flanking sequences as templates, the product was digested with EcoRI and HindIII and cloned into EcoRI/HindIII digested pBR322. A second size marker, HHRz, was prepared as described [45].
S1-nuclease protection assays
An antisense probe covering the complete ToxI sequence was made by amplification of the ToxI locus from plasmid pTA110, using primers PF217 and PF218, and subsequent in vitro transcription and gel extraction of the probe as described [46] generating a uniformly 32P-UTP labeled antisense transcript. Ten g of DNase-treated total RNA was hybridised to the antisense probe overnight at 68°C in a total volume of 30 µl containing 38% or 23% formamide for the ΦTE and ΦTE-F total RNA, respectively, 40 mM PIPES/KOH (pH 6.4), 1 mM EDTA and 400 mM NaCl. Reactions were treated with S1-nuclease (Invitrogen) (1 U µl−1) for 1.5 hr at 37°C in a total volume of 300 µl of 1× S1-nuclease buffer, to degrade any single-stranded nucleic acids. Double-stranded hybridisation products were precipitated, resuspended and resolved by 10% PAGE. Bands were visualised by phosphorimaging (BioRad Personal FX phosphorimager).
Generalised transduction using ΦTE
High titre (≥1×1010 pfu ml−1) lysates of ΦTE were prepared on the bacterial donor strain. An appropriate volume of the transducing lysate (either 10 or 100 µl) was added to 5 ml overnight cultures of the recipient strain. This was incubated on a roller wheel at 30°C for 45–60 min. The cells were then pelleted and the supernatant was removed. The remaining pellet was resuspended in 250 µl of LB and 100 µl of this suspension was plated onto LBA containing the appropriate selection. Controls for contamination and spontaneous antibiotic resistance were performed by also plating samples of the phage lysates and samples of the recipient strains inoculated with phage buffer, respectively. Any transductants obtained were streaked out twice prior to use, to reduce any bacteriophage carry-over. Transductants receiving ToxIN plasmids pTRB101 or pECA1039-Km12 were also confirmed as having Abi activity, using phage ΦTE wt for pTRB101 and both ΦTE wt and ΦM1 for pECA1039-Km12.
Additional methods
Further detailed methods for characterisation of the ΦTE genome are available in Text S1.
Supporting Information
{kind=link}
Zdroje
1. FozoEM, MakarovaKS, ShabalinaSA, YutinN, KooninEV, et al. (2010) Abundance of type I toxin-antitoxin systems in bacteria: searches for new candidates and discovery of novel families. Nucl Acids Res 38 : 3743–3759.
2. LeplaeR, GeeraertsD, HallezR, GuglielminiJ, DrezeP, et al. (2011) Diversity of bacterial type II toxin-antitoxin systems: a comprehensive search and functional analysis of novel families. Nucl Acids Res 39 : 5513–5525.
3. BlowerTR, ShortFL, RaoF, MizuguchiK, PeiXY, et al. (2012) Identification and classification of bacterial Type III toxin-antitoxin systems encoded in chromosomal and plasmid genomes. Nucl Acids Res doi:10.1093/nar/gks231
4. BlowerTR, SalmondGP, LuisiBF (2011) Balancing at survival's edge: the structure and adaptive benefits of prokaryotic toxin-antitoxin partners. Curr Opin Struc Biol 21 : 109–118.
5. OguraT, HiragaS (1983) Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc Natl Acad Sci U S A 80 : 4784–4788.
6. GerdesK, ChristensenSK, Lobner-OlesenA (2005) Prokaryotic toxin-antitoxin stress response loci. Nat Rev Microbiol 3 : 371–382.
7. MaisonneuveE, ShakespeareLJ, JorgensenMG, GerdesK (2011) Bacterial persistence by RNA endonucleases. Proc Natl Acad Sci U S A 108 : 13206–13211.
8. MagnusonRD (2007) Hypothetical functions of toxin-antitoxin systems. J Bacteriol 189 : 6089–6092.
9. PecotaDC, WoodTK (1996) Exclusion of T4 phage by the hok/sok killer locus from plasmid R1. J Bacteriol 178 : 2044–2050.
10. HazanR, Engelberg-KulkaH (2004) Escherichia coli mazEF-mediated cell death as a defense mechanism that inhibits the spread of phage P1. Mol Genet Genomics 272 : 227–234.
11. FineranPC, BlowerTR, FouldsIJ, HumphreysDP, LilleyKS, et al. (2009) The phage abortive infection system, ToxIN, functions as a protein-RNA toxin-antitoxin pair. Proc Natl Acad Sci U S A 106 : 894–899.
12. BlowerTR, FineranPC, JohnsonMJ, TothIK, HumphreysDP, et al. (2009) Mutagenesis and functional characterisation of the RNA and protein components of the toxIN abortive infection and toxin-antitoxin locus of Erwinia. J Bacteriol 191 : 6029–6039.
13. BlowerTR, PeiXY, ShortFL, FineranPC, HumphreysDP, et al. (2011) A processed non-coding RNA regulates an altruistic bacterial antiviral system. Nat Struc Mol Biol 18 : 185–190.
14. EmondE, HollerBJ, BoucherI, VandenberghPA, VedamuthuER, et al. (1998) AbiQ, an abortive infection mechanism from Lactococcus lactis. Appl Environ Microbiol 64 : 4748–4756.
15. LabrieSJ, SamsonJE, MoineauS (2010) Bacteriophage resistance mechanisms. Nat Rev Microbiol 8 : 317–327.
16. ChopinMC, ChopinA, BidnenkoE (2005) Phage abortive infection in lactococci: variations on a theme. Curr Opin Microbiol 8 : 473–479.
17. BouchardJD, MoineauS (2004) Lactococcal phage genes involved in sensitivity to AbiK and their relation to single-strand annealing proteins. J Bacteriol 186 : 3649–3652.
18. HaaberJ, RousseauGM, HammerK, MoineauS (2009) Identification and characterization of the phage gene sav, involved in sensitivity to the lactococcal abortive infection mechanism AbiV. Appl Environ Microbiol 75 : 2484–2494.
19. BellKS, SebaihiaM, PritchardL, HoldenMT, HymanLJ, et al. (2004) Genome sequence of the enterobacterial phytopathogen Erwinia carotovora subsp. atroseptica and characterization of virulence factors. Proc Natl Acad Sci U S A 101 : 11105–11110.
20. TothIK, MulhollandV, CooperV, BentleyS, ShihY-L, et al. (1997) Generalized transduction in the potato blackleg pathogen Erwinia carotovora subsp. atroseptica by bacteriophage ΦM1. Microbiology 143 : 2433–2438.
21. Evans TJ (2009) Investigation of bacteriophages and their use in the analysis of enterobacterial virulence. Ph.D Thesis, Department of Biochemistry, University of Cambridge, UK.
22. AckermannHW, DuBowMS, GershmanM, Karska-WysockiB, KasatiyaSS, et al. (1997) Taxonomic changes in tailed phages of enterobacteria. Arch Virol 142 : 1381–1390.
23. EvansTJ, TraunerA, KomitopoulouE, SalmondGP (2010) Exploitation of a new flagellatropic phage of Erwinia for positive selection of bacterial mutants attenuated in plant virulence: towards phage therapy. J Appl Microbiol 108 : 676–685.
24. SantosSB, KropinskiAM, CeyssensPJ, AckermannHW, VillegasA, et al. (2011) Genomic and Proteomic Characterization of the Broad-Host-Range Salmonella Phage PVP-SE1: Creation of a New Phage Genus. J Virol 85 : 11265–11273.
25. GuzmanLM, BelinD, CarsonMJ, BeckwithJ (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177 : 4121–4130.
26. ChangAC, CohenSN (1978) Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol 134 : 1141–1156.
27. LiuH, CoulthurstSJ, PritchardL, HedleyPE, RavensdaleM, et al. (2008) Quorum sensing coordinates brute force and stealth modes of infection in the plant pathogen Pectobacterium atrosepticum. PLoS Pathog 4: e1000093 doi:10.1371/journal.ppat.1000093
28. DatsenkoKA, WannerBL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97 : 6640–6645.
29. LabrieSJ, MoineauS (2007) Abortive infection mechanisms and prophage sequences significantly influence the genetic makeup of emerging lytic lactococcal phages. J Bacteriol 189 : 1482–1487.
30. HillC, MillerLA, KlaenhammerTR (1991) In vivo genetic exchange of a functional domain from a type II A methylase between lactococcal plasmid pTR2030 and a virulent bacteriophage. J Bacteriol 173 : 4363–4370.
31. WeinbergZ, PerreaultJ, MeyerMM, BreakerRR (2009) Exceptional structured noncoding RNAs revealed by bacterial metagenome analysis. Nature 462 : 656–659.
32. RobertsF, AllisonGE, VermaNK (2007) Transcription-termination-mediated immunity and its prevention in bacteriophage SfV of Shigella flexneri. J Gen Virol 88 : 3187–3197.
33. PettyNK, ToribioAL, GouldingD, FouldsI, ThomsonN, DouganG, SalmondGPC (2007) A generalized transducing phage for the murine pathogen Citrobacter rodentium. Microbiology 153 : 2984–2988.
34. MonsonR, FouldsI, FowerakerJ, WelchM, SalmondGPC (2011) The Pseudomonas aeruginosa generalized transducing phage phiPA3 is a new member of the phiKZ-like group of ‘jumbo’ phages, and infects model laboratory strains and clinical isolates from cystic fibrosis patients. Microbiology 157 : 859–867.
35. HaaberJ, MoineauS, HammerK (2009) Activation and transfer of the chromosomal phage resistance mechanism AbiV in Lactococcus lactis. Appl Environ Microbiol 75 : 3358–3361.
36. KlaenhammerTR, SanozkyRB (1985) Conjugal transfer from Streptococcus lactis ME2 of plasmids encoding phage resistance, nisin resistance and lactose-fermenting ability: evidence for a high-frequency conjugative plasmid responsible for abortive infection of virulent bacteriophage. J Gen Microbiol 131 : 1531–1541.
37. PettyNK, FouldsIJ, PradelE, EwbankJJ, SalmondGP (2006) A generalized transducing phage (ΦIF3) for the genomically sequenced Serratia marcescens strain Db11: a tool for functional genomics of an opportunistic human pathogen. Microbiology 152 : 1701–1708.
38. DelcherAL, BratkeKA, PowersEC, SalzbergSL (2007) Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23 : 673–679.
39. AltschulSF, GishW, MillerW, MyersEW, LipmanDJ (1990) Basic local alignment search tool. J Mol Biol 215 : 403–410.
40. LoweTM, EddySR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucl Acids Res 25 : 955–964.
41. SuzekBE, ErmolaevaMD, SchreiberM, SalzbergSL (2001) A probabilistic method for identifying start codons in bacterial genomes. Bioinformatics 17 : 1123–1130.
42. RutherfordK, ParkhillJ, CrookJ, HorsnellT, RiceP, et al. (2000) Artemis: sequence visualization and annotation. Bioinformatics 16 : 944–945.
43. GrantJR, StothardP (2008) The CGView Server: a comparative genomics tool for circular genomes. Nucl Acids Res 36: W181–184.
44. FineranPC, EversonL, SlaterH, SalmondGP (2005) A GntR family transcriptional regulator (PigT) controls gluconate-mediated repression and defines a new, independent pathway for regulation of the tripyrrole antibiotic, prodigiosin, in Serratia. Microbiology 151 : 3833–3845.
45. PrzybilskiR, GrafS, LescouteA, NellenW, WesthofE, et al. (2005) Functional hammerhead ribozymes naturally encoded in the genome of Arabidopsis thaliana. Plant Cell 17 : 1877–1885.
46. PrzybilskiR, RichterC, GristwoodT, ClulowJS, VercoeRB, et al. (2011) Csy4 is responsible for CRISPR RNA processing in Pectobacterium atrosepticum. RNA Biol 8 : 517–528.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- A Mutation in the Gene Causes Alternative Splicing Defects and Deafness in the Bronx Waltzer Mouse
- Mutations in (Hhat) Perturb Hedgehog Signaling, Resulting in Severe Acrania-Holoprosencephaly-Agnathia Craniofacial Defects
- Classical Genetics Meets Next-Generation Sequencing: Uncovering a Genome-Wide Recombination Map in
- Regulation of ATG4B Stability by RNF5 Limits Basal Levels of Autophagy and Influences Susceptibility to Bacterial Infection
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy