
Increasing Signal Specificity of the TOL Network of mt-2 by Rewiring the Connectivity of the Master Regulator XylR
Prokaryotic transcription factors (TFs) that bind small xenobiotic molecules (e.g., TFs that drive genes that respond to environmental pollutants) often display a promiscuous effector profile for analogs of the bona fide chemical signals. XylR, the master TF for expression of the m-xylene biodegradation operons encoded in the TOL plasmid pWW0 of Pseudomonas putida, responds not only to the aromatic compound but also, albeit to a lesser extent, to many other aromatic compounds, such as 3-methylbenzylalcohol (3MBA). We have examined whether such a relaxed regulatory scenario can be reshaped into a high-capacity/high-specificity regime by changing the connectivity of this effector-sensing TF within the rest of the circuit rather than modifying XylR structure itself. To this end, the natural negative feedback loop that operates on xylR transcription was modified with a translational attenuator that brings down the response to 3MBA while maintaining the transcriptional output induced by m-xylene (as measured with a luxCDABE reporter system). XylR expression was then subject to a positive feedback loop in which the TF was transcribed from its own target promoters, each known to hold different input/output transfer functions. In the first case (xylR under the strong promoter of the upper TOL operon, Pu), the reporter system displayed an increased transcriptional capacity in the resulting network for both the optimal and the suboptimal XylR effectors. In contrast, when xylR was expressed under the weaker Ps promoter, the resulting circuit unmistakably discriminated m-xylene from 3MBA. The non-natural connectivity engineered in the network resulted both in a higher promoter activity and also in a much-increased signal-to-background ratio. These results indicate that the working regimes of given genetic circuits can be dramatically altered through simple changes in the way upstream transcription factors are self-regulated by positive or negative feedback loops.
Published in the journal:
. PLoS Genet 8(10): e32767. doi:10.1371/journal.pgen.1002963
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002963
Summary
Prokaryotic transcription factors (TFs) that bind small xenobiotic molecules (e.g., TFs that drive genes that respond to environmental pollutants) often display a promiscuous effector profile for analogs of the bona fide chemical signals. XylR, the master TF for expression of the m-xylene biodegradation operons encoded in the TOL plasmid pWW0 of Pseudomonas putida, responds not only to the aromatic compound but also, albeit to a lesser extent, to many other aromatic compounds, such as 3-methylbenzylalcohol (3MBA). We have examined whether such a relaxed regulatory scenario can be reshaped into a high-capacity/high-specificity regime by changing the connectivity of this effector-sensing TF within the rest of the circuit rather than modifying XylR structure itself. To this end, the natural negative feedback loop that operates on xylR transcription was modified with a translational attenuator that brings down the response to 3MBA while maintaining the transcriptional output induced by m-xylene (as measured with a luxCDABE reporter system). XylR expression was then subject to a positive feedback loop in which the TF was transcribed from its own target promoters, each known to hold different input/output transfer functions. In the first case (xylR under the strong promoter of the upper TOL operon, Pu), the reporter system displayed an increased transcriptional capacity in the resulting network for both the optimal and the suboptimal XylR effectors. In contrast, when xylR was expressed under the weaker Ps promoter, the resulting circuit unmistakably discriminated m-xylene from 3MBA. The non-natural connectivity engineered in the network resulted both in a higher promoter activity and also in a much-increased signal-to-background ratio. These results indicate that the working regimes of given genetic circuits can be dramatically altered through simple changes in the way upstream transcription factors are self-regulated by positive or negative feedback loops.
Introduction
The mechanisms that bacteria use to transduce external stimuli into specific responses rely on connected transcriptional factors that shape circuit-like input/output devices [1]. Such networks are comprised of interacting molecular components and can adopt different topologies [2]. The responses of a specific regulatory network to given stimuli are then fine-tuned by the dynamics of its interacting constituents [3], [4]. Free-living bacteria have evolved to respond and adapt to the perturbations derived from a fluctuating environment by increasing the complexity of their regulatory circuits [5]. The TOL plasmid pWW0 of the soil bacterium Pseudomonas putida mt-2 is a good example. This plasmid encodes two catabolic operons for biodegradation of m-xylene [6] that are subject to an intricate regulatory control involving the interplay among various transcription factors (TFs) [7], [8]. The master regulatory element of the system is the σ54-dependent regulator XylR, which, in the presence of its natural inducers, acts on the Pu promoter of the upper TOL operon. In addition, XylR triggers the expression of the gene that encodes a second regulator, XylS, via the Ps promoter [9]. Due to the divergent character of the Ps and the Pr promoters (driving expression of xylR, Figure 1), the activation of the Ps promoter not only triggers the expression of xylS but also leads to the down-regulation of xylR transcription [10]. XylR is optimally activated by the primary substrates of the TOL system, such as m-xylene or toluene. However, this TF is not entirely specific for these effectors, as it also responds to a large number of structural analogs. These analogs include both non-substrates as well as metabolic intermediates of m-xylene biodegradation, e.g., 3-methylbenzylalcohol (3MBA) [6], [11], resulting in a degree of naturally occurring effector promiscuity. The transcriptional output produced by XylR on the target promoters Pu and Ps is in turn limited by intracellular concentrations of the TF [12], [13] and σ54 [14]. This extant configuration of the system not only leads to a quick response to XylR effectors when cells enter the stationary phase, but it also restricts the Pu promoter to low capacity i.e. poor maximum output. The existing characteristics of the XylR-based regulatory network have likely evolved for adjusting the tradeoff between transcriptional efficiency and physiological burden in the natural context, constraining the output of the system. This natural control of the xylR expression loop in the context of the TOL plasmid limits the value of the system as the primary component of whole-cell biosensors [15], [16]. Previous attempts to increase the performance of XylR/Pu-based biosensing devices have included in vitro evolution of the TF [17], [18], construction of regulatory cascades [19] and improvement of the ribosome binding sequence (RBS) of the reporter genes [20]. None of these approaches, however, solve the problem of effector promiscuity. The issue at stake is, therefore, whether we can artificially change such an effector-relaxed/low-output circuit regime into a high-signal specificity/high-capacity counterpart without modifying the XylR protein.

In this work, we report one strategy to overcome the constraints imposed by the natural architecture of the TOL network on the function of the XylR/Pu regulatory node of the plasmid. To this end, we adopted a Pu-luxCDABE reporter integrated into the chromosome of P. putida for accurately measuring system performance. In this genetic background, we then designed and tested various combinations of translation signals, promoter strengths and regulatory loops aimed at [i] suppressing the effect of effector promiscuity on XylR/Pu output and [ii] enhancing the response to optimal inducers (e.g., m-xylene). The results herein demonstrate that the working regimes of regulatory nodes, including their signal specificity, can be dramatically altered by changing the upstream connectivity of the TFs involved in the network instead of mutating the structure of the corresponding proteins.
Results
Monitoring activity of the Pr/xylR/Pu regulatory loop with a formatted P. putida reporter strain
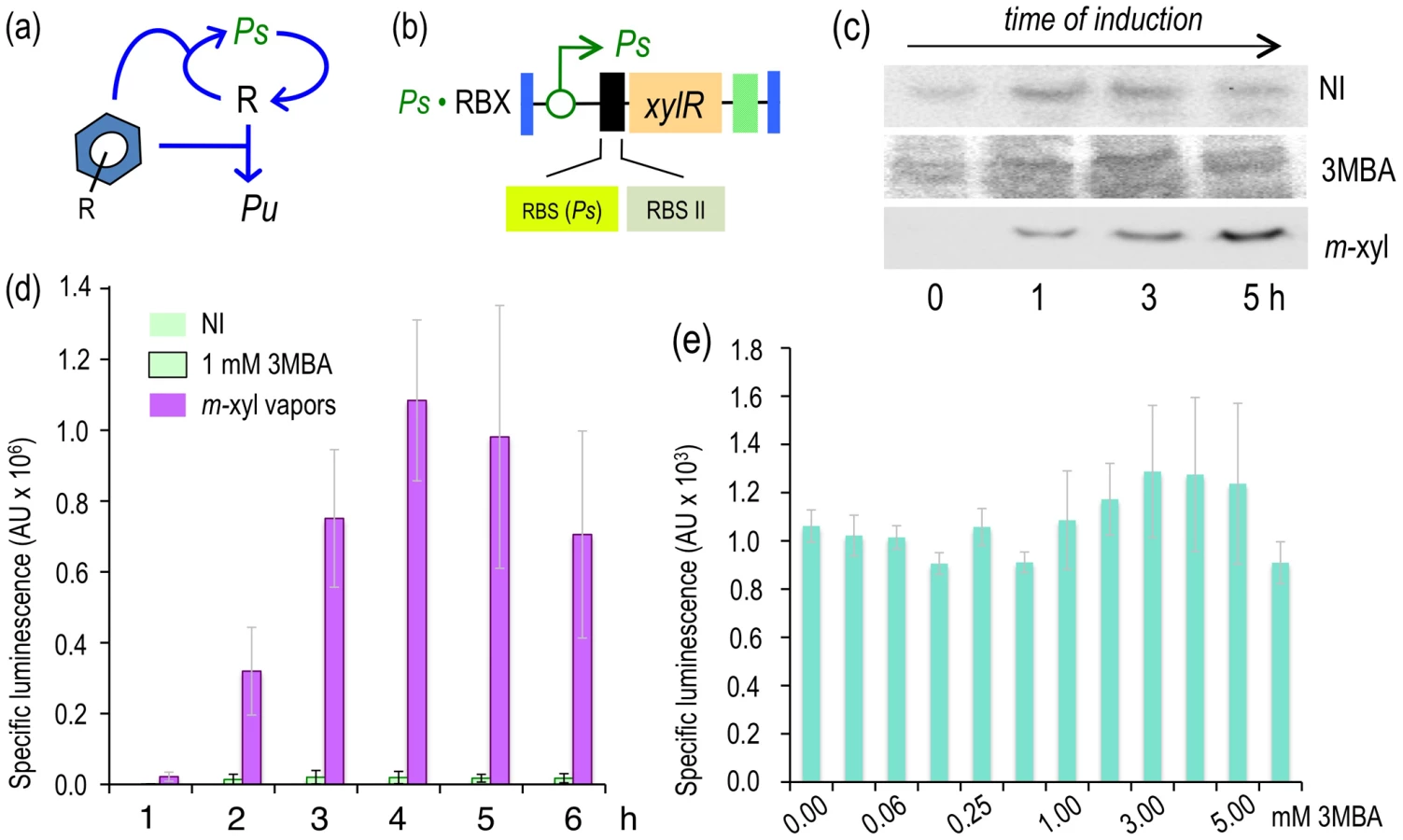
As shown in Figure 1, the event that triggers the regulatory and metabolic program encoded in the TOL plasmid is the binding of the pathway substrate to the XylR protein [7] and the ensuing activation of the Pu promoter for expression of the upper operon [6]. This results from the interplay of four components: [i] the aromatic effector, [ii] the Pr promoter that transcribes xylR, [iii] the XylR protein itself and [iv] the Pu promoter that is targeted by XylR. Although many other host factors influence the activity of the system in vivo [14], [21], [22], the set comprising the inducer/Pr/XylR/Pu forms the master regulatory device that determines signal specificity, i.e., the responsiveness of Pu to different aromatic effectors [11], [23]. The relational map of this node is depicted in Figure 2a. Exposure to aromatic effectors gives rise to a form of the XylR protein that both activates Pu and represses Pr, i.e., downregulates its own transcription. To have a reliable test system for comparing the inputs and outputs associated with this node, we engineered these components in a strain of P. putida bearing a transcriptional Pu-luxCDABE fusion inserted into its chromosome via a mini-transposon vector (P. putida Pu·LUX, Figure 2c). To ensure a faithful comparison of the input (i.e., inducer) and output (light emission) transfer function for each of the configurations tested, we assembled xylR expression in a specialized plasmid called pTn7 Gm FRT [24]. This vector targets any DNA segment inserted therein to a natural attTn7 site present in the genome of P. putida KT2440 [25] in a specific orientation. Furthermore, once inserted, the Gm resistance marker can be excised through site-specific recombination between two flanking FTR sequences, thereby leaving the cells free of antibiotic resistances. To set a benchmark for the subsequent studies, we first produced a strain with the Pr/XylR/Pu regulatory parts connected in the same configuration as the natural TOL plasmid (Figure 2a). To this end, a DNA segment encoding the xylR gene placed under its native promoter was assembled in the aforementioned Tn7 vector to yield pTn7-BX (Figure 2b). The insert was then delivered to the P. putida Pu·LUX chromosome and the Gm marker was deleted as shown in Figure 2c and 2d, thereby generating P. putida BX, which was thereafter the reference reporter strain. Note that all subsequent strains handled below carry the Pu-luxCDABE already described.

To quantify the response of the regulatory node of Figure 2a implemented in P. putida BX, the strain was grown in liquid medium and exposed to either optimal inducer vapor (m-xylene) or to 1.0 mM of a suboptimal effector (3MBA) and the resulting bioluminescence was recorded 5–6 h post-addition. This timing does not significantly affect luminescence (e.g. see Figure 3 below). While the background reading of the Pu output was in the range of 103 luminescence units/OD600, the m-xylene-induced levels were close to 106 (Figure 2e). These results confirmed the inducibility and strength of the Pu promoter and set a minimum and maximum window of activity for the rest of the work. The addition of 3MBA in the assay produced a luminescence readout that was only approximately 15% of that obtained with the optimal XylR effector but still very high relative to the background, non-induced levels. Such a difference between one inducer and the other is not understood mechanistically, as the apparent binding affinities of both good and bad XylR inducers are similar [26]. In either case, it is plausible that the output of the sensing device as a result of induction by either aromatic compound is limited by the intracellular concentrations of XylR [13], which curbs the robustness and sensitivity of the system [3], [4]. With this background, we wondered whether we could exacerbate the difference between optimal and non-optimal inducers (and thus increase signal specificity) by artificially increasing some of the parameters of the existing node (Figure 2a), by rewiring its connections or by both methods. Various approaches to this goal are explained below.

Translational attenuation of XylR decreases the response of Pu to 3MBA
One theoretical way to modify the sensitivity of a signaling route involves changing the levels of the proteins involved in the process [27]. With this in mind, we entertained the possibility that lowering XylR concentrations could suppress the response of the regulatory device (Figure 2a) to 3MBA while preserving the induction of the same system by m-xylene. To test this, we constructed a variant of the node that kept the same relational organization but caused a drop in the levels of XylR by attenuating the protein's translation with tandem, repeated non-overlapping RBSs (Figure 3b) [28]. This modification was expected to lower translation of the downstream ORF without any effect on mRNA stability [29]. To implement this change, the Tn7 plasmid pTn7-Pr·RBX was built as explained in the Materials and Methods section and delivered into attTn7 of P. putida Pu·LUX, as explained previously. The resulting strain, P. putida Pr·RBX, was identical to the reference strain P. putida BX except for a translational attenuator at the 5′-upstream untranslated region (5′-UTR) of the xylR gene (Figure 3b).
We next verified that the changes in the (UTR) of xylR lowered the net expression levels of the regulator without affecting its production kinetics. Thus, in parallel, we grew strains P. putida BX and P. putida Pr·RBX in LB medium and exposed them to 1.0 mM of the suboptimal effector 3MBA. Then, we performed Western blot analyses of the cell extracts with an anti-XylR antibody at various times after induction (Figure 3a and 3b). The pattern of induction in the strain with the wild-type 5′-UTR (P. putida BX, Figure 3a) was such that expression of XylR reached a maximum during the period 1–2 hours after exposure to the inducer, followed by a decrease at longer times, which was expected from the negative feedback loop that governs xylR expression (Figure 2a). The evolution of XylR in the counterpart strain bearing the modified 5′-UTR (P. putida Pr·RBX, Figure 3b) developed similarly, but the net concentration of XylR per cell was clearly lower. To examine the consequences of the different levels of the regulator in the response of the Pu-luxCDABE reporter to the suboptimal inducer, 3MBA was added to cultures of P. putida BX and P. putida Pr·RBX as before and their luminescence measured over time (Figure 3c). The results indicate that the overall output of the strain that expresses lower amount of XylR (P. putida Pr·RBX) was 4–7-fold lower than the strain carrying xylR controlled by its natural upstream region (P. putida BX). As a control, we also measured the response of the two strains with the different xylR 5′-UTRs to the optimal effector, m-xylene. As shown in Figure 3d, Pu output in the xylR 5′-UTR-modified strain P. putida Pr·RBX displayed a similar trend (although at somewhat lower levels) than the reference counterpart P. putida BX. However, we observed that the response of the cells to each inducer was more divergent in strain P. putida Pr·RBX than in strain P. putida BX. These results suggested that decreasing concentrations of XylR had the effect of widening the relative gap between the induction caused by 3MBA and m-xylene. Yet, the change in the xylR 5′-UTR was insufficient to entirely suppress the response of Pu to the suboptimal inducer. Moreover, lower XylR levels also caused a low-capacity regime with the optimal effector. Therefore, the next question was how to keep and even enhance Pu readout in response to m-xylene while removing the effect of 3MBA.
Engineering a positive feedback loop (PFL) for augmenting the output of the Pu/XylR regulatory node
As shown in Figure 3d, the data indicate that decreasing intracellular XylR by changing the xylR 5′-UTR sequence caused a reduction of Pu response to m-xylene by approximately 50%. It is thus plausible that the intracellular levels of the regulator determine the capacity (i.e., the maximum output) of the promoter. As the intracellular level of XylR under its native transcriptional control [13] is small and tends to decrease upon induction with aromatic effectors ([10] and Figure 3a and 3b), we wondered how making xylR transcription subject to a PFL (instead of the extant negative auto-regulation, Figure 2a) could affect the sensitivity and the capacity of the regulatory node to m-xylene and 3MBA. Positive auto-regulatory loops are prone to off/on expression patterns [30], [31] in a very TF concentration-sensitive fashion [32], [33]. We reasoned it would be possible to find a window of xylR expression that could trigger the on state with the optimal effector and leave the loop with 3MBA in the off state.
The first attempt in this direction involved the replacement of the native xylR promoter (Pr) by Pu, the promoter that is triggered by effector-bound XylR (Figure 4a). To this end, the same Pu sequence employed to construct the Pu-luxCDABE reporter was amplified with PCR primers and placed in front of a promoterless xylR gene preceded by the modified 5′-UTR [28] discussed above. The resulting expression device was then inserted at the attTn7 site of the reporter P. putida chromosome (Figure 4b) [25], as described in the Materials and Methods to generate P. putida Pu·RBX. Note that this strain is entirely isogenic to P. putida Pr·RBX except that the xylR gene is expressed through Pu and not through Pr. This configuration changes the connectivity of the Pu/XylR node from a negative auto-inhibition device (Figure 2a) to a PFL (Figure 4a). To verify that such a modification in fact transforms the expression pattern of XylR in vivo, we used a Western blot to assay the accumulation of the protein in the reference strain (P. putida BX) and in P. putida Pu·RBX in the presence and absence of m-xylene. As shown in Figure 4c, the non-induced P. putida BX expressed XylR at low levels with a tendency to accumulate at later growth stages [34]. As expected, exposure to m-xylene under the same conditions resulted in lower XylR levels that appeared to decrease over time. The situation with the strain engineered with a forward loop (P. putida Pu·RBX) was similar under non-induced conditions but entirely different when cells were exposed to m-xylene. As shown in Figure 4c (bottom), the intracellular concentration of XylR quickly increased after one hour of induction, reaching very high levels at later growth stages. This experiment demonstrated not only that the positive loop engineered for expression of xylR worked as predicted but also that the effect of m-xylene on Pu was enough to switch the state of the loop to from low to high activity (see 0 h vs. 5 h of Figure 4c).

To examine whether expressing xylR through such an artificial regulatory device was translated into a high-capacity XylR/Pu node regime, we quantified the luminescence emitted by cultures of P. putida Pu·RBX induced with m-xylene vapors. As shown in Figure 4d, despite sustaining an attenuated translation of xylR because of the modified 5′-UTR introduced into this strain, the readout of Pu activity in P. putida Pu·RBX was as high as in the strain with the wild-type node (P. putida BX, Figure 2e) and more than twofold greater than the strain with the modified 5′-UTR but with wild-type regulatory connectivity (P. putida Pr·RBX, Figure 3d).
Response of P. putida Pu·RBX to 3MBA
The result of the creation of the construct discussed above was a circuit (Figure 4a) that responded to the bona fide XylR inducer (m-xylene) with a transcriptional strength comparable to the wild-type (Figure 2a) because the lower level of XylR caused by translational attenuation had been compensated for by a PFL. However, what is the effect of such a change on effector specificity? To examine this question, we monitored the luminescent response of a culture of P. putida Pu·RBX to 3MBA over time (Figure 5a) as well as the sensitivity of the same cells to increasing concentrations of this suboptimal inducer (Figure 5b). As a control, we employed the strain bearing the wild-type architecture of the regulatory node (P. putida BX). The results of our experiments (Figure 5) indicate that the response in the PFL-engineered strain to 3MBA was twofold greater than the response of the wild-type construct. This magnification is expected in such PFL regulatory motifs, which are prone to amplify the response to the trigger signal once it reaches a given threshold [30], [32]. This scenario was confirmed by the results shown in Figure 5b in which the responses of P. putida Pu·RBX and P. putida BX to 3MBA were followed along with moderate incremental increases of inducer concentrations. While Pu activity derived from the wild-type regulatory motif was only gradually increasing at 3MBA concentrations beyond 0.12 mM, the equivalent PFL strain displayed an abrupt change of Pu activity in cultures with 0.12 mM inducer (Pu very low) vs. those with 0.25 mM (Pu high to very high) and above. This phenomenon likely reflects the switch-on typically caused by the passing of a threshold in auto-inducing regulatory loops [32], [35].

While the results noted in Figure 5 did not by themselves elucidate the fundamental mechanism underlying our primary question of interest (discrimination of two chemically related inducers of Pu, see above), they demonstrated that the response of the XylR/Pu node to inducer could be made more digital in a fashion dependent on the concentration of the TF involved. These results suggested that one could keep the node in an entirely off state when XylR levels are below a certain threshold, while triggering a high activity regime once the threshold has been surpassed. On this basis, we recreated the same PFL but pursued a higher limit for XylR auto-induction in a way such that optimal and suboptimal effectors could trigger or not, respectively, a high-activity of the downstream Pu promoter.
A Ps promoter-based PFL enhances activity and specificity of the XylR/Pu regulatory node
The results above indicate that making XylR expression subject to a PFL increases the amplitude of the XylR/Pu response to both optimal (m-xylene) and suboptimal effectors (3MBA), which means that both effectors cause XylR (controlled by the attenuated Pu-based PFL) to reach the TF threshold imposed by this auto-inducing architecture (Figure 4a) [30]. The subsequent question was whether suppression of any response to 3MBA could be brought about by moving the window of effector-induced xylR transcription in the PLF to a range that could still trigger full response to m-xylene but remain impervious to the suboptimal inducer. To check this, we simply replaced the Pu promoter of the P. putida Pu·RBX (Figure 4a) with a weaker but still XylR-responding promoter, Ps of the TOL plasmid [36]. As XylR activates Ps in response to aromatic effectors at a lower level than Pu [37], we hypothesized that a Ps-based PFL would make the switch-on threshold more difficult to reach for a suboptimal inducer. We constructed a P. putida strain (Figure 6a) placing xylR and the RBS II [28] downstream of the regulatory region of the xylS gene including its own RBS(Ps), using the same methods employed for construction of other strains (see Materials and Methods). This new strain, which was engineered with a Ps-based PFL (Figure 6a), was named P. putida Ps·RBX (Figure 6b). To examine the response of the new regulatory loop of this strain to either effector, P. putida Ps·RBX was grown in the absence or presence of each aromatic inducer and the intracellular levels of XylR recorded over time along with light emission. Figure 6c reveals that 3MBA failed to trigger the auto-activation loop for XylR expression, suggesting that the levels of the TF were insufficient to switch on the PFL. Consistent with those results, 3MBA also failed to cause any significant activation of the downstream Pu-luxCDABE reporter (Figure 6d). This situation did not change when more inducer was added to the culture (Figure 6e), thereby confirming that the silencing of the PFL could be traced to nothing else but XylR. In contrast, when the same P. putida Ps·RBX cells were induced with m-xylene, the cells exhibited a noticeable accumulation of the XylR protein over time (Figure 6c) as well as a strong emission of light (Figure 6d). In fact, the output of the Pu-luxCDABE reporter was twofold higher than that of the wild-type regulatory node of P. putida BX (Figure 2e). These results indicated that, unlike the native effector-responding device of the TOL plasmid, the regulatory architecture implemented in P. putida Ps·RBX could discriminate between optimal and suboptimal inducers in a fashion that was not dependent on their concentration but on their chemical structure alone. Unfortunately, the very low levels of expression of XylR under this PFL made detection of intracellular XylR concentrations difficult in cells exposed to 3MBA (Figure 6c). The mechanistic basis of effector discrimination could therefore be inferred but not really proven. To overcome this uncertainty, we resorted to a further perturbation of the system as explained below.
Increasing the basal expression of the Ps-based PFL restores innate promiscuity of the XylR/Pu node
The increase in the signal specificity of the XylR/Pu node reported above could be attributed to a change in the threshold necessary to trigger the response produced by the new regulatory loop of xylR. Should this be the case, any resetting of such a threshold back to its former sensitivity range is predicted to restore the response to the suboptimal effector, 3MBA. How can this be accomplished without varying the architecture of the node yet again? To solve this conundrum, we decided to replace the wild-type xylR sequence of the Ps-based PFL device with the variant xylRv17. This mutant encodes a XylR derivative that is responsive to all aromatic effectors of the wild-type protein, but it is also able to trigger low-level activity of target promoters in the absence of any inducer [17], [26]. The expected result of having xylRv17 expressed under the control of a Ps-based PFL is therefore to downshift the threshold of active TF that is necessary for switching on the auto-inducing device. To test these predictions, we first constructed a control strain P. putida BX17, which was identical to P. putida BX except that the encoded TF sequence is xylRv17 [26] instead of wild-type xylR. As shown in Figure 7a, P. putida BX17 displayed a basal Pu activity level in the absence of effectors ≥7-fold higher than that of the strain bearing the wild-type xylR gene. The test strain, in contrast, was the same as the P. putida Ps·RBX examined above, but xylR had been similarly replaced by xylRv17, thus giving rise to P. putida Ps·RBX17 (see the Materials and Methods for construction details). The only difference between the two is the minor semi-constitutive expression of xylRv17 compared with the original TF. This disparity has, however, dramatic consequences in the sensitivity of the regulatory device as a whole to 3MBA. Figure 7b and 7c compares the emission of light of the P. putida BX17 (xylRv17 under TOL plasmid Pr promoter), P. putida Ps·RBX (wild-type xylR expressed through the Ps-based PFL) and P. putida Ps·RBX17 (same but xylRv17) strains with and without 3MBA. The results indicate that P. putida Ps·RBX17 was nearly as responsive to this effector as the strain bearing the native configuration of the regulatory system. Consistent with the results reported above (Figure 6), no light emission above background levels could be detected from P. putida Ps·RBX under the same conditions. Taken together, these data strengthened the notion that up - or downshifting of the auto-activation threshold of the PFL by adjusting the concentration or activity of the TF resulted in a regulatory device whose specificity to given effectors could be drastically changed.

Discussion
The regulatory networks that control gene expression in cells and organisms have evolved to accurately adjust their reactions to specific stimuli [38]. Traditionally, the well-characterized pieces of these regulatory networks haven been exploited to engineer cells with new responses. Although this approach was useful for constructing a plethora of strains presenting new phenotypes [15], [20], [39]–[41], it was not until the onset of systems and synthetic biology that we began to understand how the output of a specific circuit was conditioned by the shape of the network in which the parts are interconnected. Thus, bottom-up approaches shed light on the intrinsic properties of regulatory networks, allowing for the rational design of newly engineered genetic circuits [3], [42]. Prokaryotic regulatory systems have been used in the construction of bacterial strains with biotechnological applications, such as whole-cell biosensors to detect environmental pollutants [15], [41], [43], [44]. Such biosensors are generally based on the association of input/output components that usually include one bacterial transcriptional regulator that acts as a sensor module and a reporter gene coupled to its cognate promoter [39], [45]. The specificity of engineered regulatory networks primarily relies on the responsiveness of the transcriptional factor to the signal of interest [41]. Based on this understanding, the quest for new signal specificities has been generally based on in vitro modification of the sensor module [17], [18], [26], [46]. In this work, we have demonstrated that, by rational rewiring of the architecture of a specific regulatory network, it is possible to modify the input-output function to increase the amplitude or the specificity of the response without modifying the core sensor part of the circuit. To this end, we took advantage of the well-characterized TOL network of P. putida pWW0 [7], where XylR controls the expression of several genes by binding two target promoters, Pu and Ps [6]. As shown in Figure 2, XylR responds strongly to m-xylene and, to a lesser extent but still significantly, to 3MBA. As shown above, by modifying the connectivity of the components of the regulatory system, we could [i] increase the general amplitude of the output and [iii] generate a super-specific response to the optimal inducer by filtering the response to the less favorable XylR effector. To accomplish this, we first considered simply lowering the concentration of some of the components of the regulatory system, with the aim of increasing the activation threshold and thus increasing specificity [47]. This approach is not without precedent, as previous studies indicate that controlling the expression levels of MAP kinases in regulatory cascades, through gene expression or post-translational modifications, make it possible to change the activation profile of the system [27]. However, this did not suffice for discrimination between optimal and suboptimal effectors (Figure 3c and 3d). In the natural and translationally attenuated context, the levels of XylR are maintained within limits through a negative feedback loop mediated by the Pr promoter (Figure 1; [10], [34]). We demonstrated above that replacing this negative auto-regulation by a PFL leads to an amplification of the system output in a fashion typical of bistable switches [30], [31], [47]. Furthermore, the combination of a translational attenuator with PFLs endowed with different auto-induction parameters resulted in regulatory devices with activation thresholds far enough apart to discriminate between the two XylR effectors tested. Although other approaches have been used to increase signal sensitivity [19], [48], this is, to the best of our knowledge, the first instance that modifies the specificity of a sensor system by simply rewiring the connectivity of the parts involved. We argue this approach is extraordinarily promising for improving the performance of whole-cell biosensors [49], more so when combined with modification of the core TF [17] or the output modules [28], [50] in the design of optimized devices.
Finally, we have not failed to observe that a large number of regulatory nodes for biodegradative and detoxification operons [51] follow a general architecture that we have designated the master control loop (MCL, Figure 8). This theme, which is also implicit in many metabolic and regulatory networks [52] consists of an upstream signal (i.e., the metabolic substrate or effector) that both influences expression of the cognate regulator as well as the interaction of the same TF with the downstream target promoter. The 3 components of the motif can interact at 4 sites of the related object and present up to 16 theoretically possible configurations. The native arrangement of the TOL regulatory network, as well as those that have been engineered for the sake of this work, are simply variants of such a general layout. The work above suggests that this motif is endowed with extraordinary plasticity for responding to the specifications of any given regulatory need in terms of capacity, inducibility and signal specificity. We propose such an MCL motif as a frame of reference for the further development of regulatory devices á la carte, as required in contemporary metabolic engineering and other fundamental and biotechnological applications.

Materials and Methods
Strains, culture conditions, and general procedures
P. putida KT2440 [53], its derivatives and the E. coli strains used in this study were grown in Luria-Bertani (LB) medium and handled with standard procedures. E. coli CC118λpir was used as the host for propagating plasmids based on an R6K origin of replication [54]. When required, the media was amended with the specified concentrations of 3-methylbenzylalcohol (3MBA) or saturating vapors of m-xylene. Antibiotics were used at the following concentrations: piperacillin (Pip) 40 µg/ml, chloramphenicol (Cm) 30 µg/ml, gentamycin (Gm) 10 µg/ml, tetracycline (Tc) 10 µg/ml and potassium tellurite (Tel) at 80 µg/ml. PCR reactions were performed as follows: 50–100 ng of the template indicated in each case was mixed in a 100-µl reaction mixture with 50 pmol of each of the primers specified and 2.5 units of Pfu DNA polymerase (Stratagene). The samples were then subjected to 30 cycles of 1 min at 95°C, 1 min at 58°C and 3 min at 72°C. Clones were first verified by colony PCR [55] using 1.25 units of Taq DNA polymerase (Roche) and later confirmed with DNA sequencing. Other gene cloning techniques and standard molecular biology procedures were performed according to [55].
Construction of hybrid Tn7 delivery plasmids
DNA segments containing the xylR gene under the control of different promoter architectures were cloned in vector pTn7-Gm FRT [24] for their eventual insertion at the native attTn7 site of the P. putida chromosome [25]. Such insertions occur always at the same site and in the same orientation, thus generating entirely equivalent strains [24]. To construct the corresponding mini-Tn7 delivery vectors, we first engineered a series of pUC18Not derivatives [54] carrying the DNA segments at stake as follows. A 2.8-kb KpnI-SacI fragment of pBBXylR [56] containing the construct Pr → xylR (i.e., the xylR gene expressed through its native promoter of the TOL plasmid) was inserted in a pUC18Not variant that lacked the EcoRI site, thereby generating pBXe. This plasmid was used as the frame for replacing the native promoter region of xylR with the 3 alternative 5′-upstream sequences employed in this work. In the first case, P. putida mt-2 genomic DNA was amplified with the primersPR 1F (5′-CGctcgagGTTAACATAATCGGAGACTGC-3′) and PRrbs 2R (5′-CCGgaattcCATGCTTAATTTCTCCTCTTTTTGTTTTCCTCTTGTTTTTAT-3′). The resulting 545-bp product contained the native Pr promoter and the adjacent sequence down to the natural RBS (bold) but with an added RBSII (underlined in the sequence [28] between the original RBS and the cognate ATG codon in italics). Furthermore, the amplified segment was flanked by the EcoRI and XhoI restriction sites introduced in the primers (lower case in the sequences above). In the second case, primers PU 1F (5′-ACGCctcgagCCCGGGAAAGCGCGATGA-3′) and PUrbs 2R (5′-CGgaattcCATGCTTAATTTCTCCTCT TTTGAAGGGTCACCACTATTTTT-3′) amplified a 464-bp segment containing Pu all the way down to the RBS of the xylU gene (bold), which was then followed by the native RBSII (underlined) and ATG (italics) of the xylR gene, flanked by EcoRI and XhoI sites (lower case). Finally, the primers PS 1F (5′-CGctcgagTTGTTTTCCTCTTGTTTTTATCG-3′) and PSrbs 2R (5′-CGgaattcCATGCTTAATTT CTCCTCTTTAGTTCACGGTTCTCTTATT-3′) resulted in a EcoRI-XhoI 256-bp fragment containing the second XylR-responsive promoter Ps of the TOL plasmid (Figure 1) down to the RBS of the xylS gene (bold) and followed by the RBSII (underlined) and ATG (italics) of the xylR gene as before. Each of these 3 EcoRI-XhoI restriction products were cloned into the corresponding sites of the pBXe plasmid, thereby replacing the original xylR upstream region with refactored counterparts and originating pPrRBX, pPuRBX and pPsRBX. These plasmids were separately digested with NotI, which excised DNA segments carrying Pr → xylR, Pr-(RBSPr RBSII) → xylR, Pu-(RBSPu RBSII) → xylR and Ps-(RBSPs RBSII) → xylR. These were cloned in the same orientation into the NotI site of pTn7-Gm FRT [24] generating pTn7-BX, pTn7-Pr·RBX, pTn7-Pu·RBX and pTn7-Ps·RBX. For the constructs bearing the semi-constitutive XylR variant named XylRv17 (which carries mutations F48I and L222R; [17]), a 713-bp EcoRI-AvrII fragment-spanning DNA sequence corresponding to the A domain of xylRv17 was excised from plasmid pBBxylRv17 [17] and recloned into the corresponding sites of pBXe or pPs·RBX, yielding pBX17 and pPs·RBX17, respectively. These plasmids were then digested with NotI and the fragments encoding Pr → xylRv17 and Ps-(RBSPs RBSII) → xylRv17 were cloned, as before, in vector pTn7-Gm FRT [24], thereby generating pTn7-BX17 and pTn7-Ps·RBX17.
Strain construction
Standardization of the various regulatory devices for xylR expression and measurement of network output required the engineering of a reference Pu-luxCDABE reporter P. putida strain. To this end, we first digested plasmid pattPuLUX [24] with EcoRI/XmaI to delete an internal 86-bp fragment containing the E. coli attTn7 insertion site [57]. The ends of the digested plasmid were blunted with T4 DNA polymerase and relegated to generate the plasmid pPu·LUX. This construct was then digested with NotI, and the fragment containing a Pu-luxCDABE fusion ligated to pJMT6, a mini-Tn5 delivery vector with a potassium tellurite (Tel) resistance cassette [58], producing pTn5 Tel-Pu·LUX. This plasmid was conjugally transferred to P. putida KT2440 (see below) and TelR exconjugants tested for insertion of the hybrid mini-Tn5 Tel element carrying the Pu-luxCDABE fusion. One of these clones was called P. putida Pu·LUX and retained for further use as receptor of the different variants of the mini-Tn7 transposons borne by the plasmids pTn7-BX, pTn7-Pr·RBX, pTn7-Pu·RBX, pTn7-Ps·RBX (see above). Insertion of the corresponding segments in the naturally occurring attTn7 site of P. putida Pu·LUX was confirmed through colony PCR using a primer that anneals within the sequence of the B domain of xylR (NDoCAvrII: 5′-GCGAATGGCCTAGG CCGTAATACTG-3′) and one at the attTn7 insertion site within the glmS gene (PpuglmS 2R: 5′-GTGCGTGCCCGTGGTGG-3′). The ensuing collection of GmR strains were deleted of this antibiotic marker by transient expression of yeast flippase encoded by the plasmid pBBFLP, which brings about site-specific recombination of the FRT sequences that flank the resistance gene [56]. The same strategy was followed in the case of pTn7-BX17 and pTn7-Ps·RBX17, although the GmR marker was not removed in these cases. The final outcome of all these manipulations was the isogenic strain collection P. putida BX, P. putida Pr·RBX, P. putida Pu·RBX, P. putida Ps·RBX, P. putida BX17 and P. putida Ps·RBX17.
Plasmid transfer and mini-transposon delivery into P. putida
pTn5Tel-Pu·LUX and mini-Tn7 derivatives (pTn7-BX, pTn7-Pr·RBX, pTn7-Pu·RBX, pTn7-Ps·RBX, pTn7-BX17 and pTn7-PsRBX17) and pBBFLP were conjugally passed from the donor E. coli strain indicated in each case into the different P. putida recipients with a filter mating technique [54]. To this end, a mixture of donor, recipient and helper strain E. coli HB101 (pRK600) was deposited on 0.45-µm filters in a 1∶1∶3 ratio and incubated for 8 h at 30°C on the surface of LB-agar plates. Mini-Tn7 derivatives were co-mobilized along with the transposase-encoding genes tnsABCD into the recipient strains by including E. coli CC118λpir (pTNS1) in the mating mixture [59]. After incubation, the cells were resuspended in 10 mM MgSO4, and the appropriate dilutions plated on M9/succinate amended with suitable antibiotics for counter-selection of the donor and helper strains and growth of the P. putida clones that had acquired the desired insertions. Bona fide transposition was verified in every case by checking the sensitivity of individual exconjugants to the delivery vector marker, piperacillin.
Bioluminescence assays
To measure bioluminescence production of P. putida cells carrying luxCDABE fusions, 2-ml cultures of the strains under study were first pre-grown in 10-ml test tubes overnight in LB medium at 30°C. The cultures were then diluted to an OD600 of 0.05 and grown up to an OD600 = 1.0 in 100-ml flasks. At this point, the cultures were exposed to m-xylene or 3MBA, as indicated for each case. When required, 200-µl aliquots of these cultures were placed in 96-well plates (NUNC), and light emission and OD600 were measured in a Victor II 1420 Multilabel Counter (Perkin Elmer). The specific bioluminescence values were calculated by dividing the obtained values of total light emission (in arbitrary units) by the optical density of the culture (OD600). The specific bioluminescence values shown represent the average of at least three biological replicates.
Protein techniques
Protein analyses were performed according to published protocols [55]. For detection of the XylR (wild-type and variants), 5 µg of whole protein extract of P. putida cells was denatured in a sample buffer containing 2% SDS and 5% ß-mercaptoethanol and run on 10% polyacrylamide gels. These were subsequently blotted onto a polyvinylidene difluoride (PVDF) membrane (Immobilon-P, Millipore) using a semi-dry electrophoresis transfer apparatus (BioRad). After protein transfer, the membranes were blocked for 2 h at room temperature with MBT buffer (0.1% Tween and 5% skim milk in phosphate-buffered saline, PBS). For detection of XylR, the membranes were incubated with MBT buffer containing a dilution 1/2000 of anti-XylR Phab [13]. The membranes were subjected to 5-min washing steps in 40 ml of MBT buffer alone or MBT with 0.1% sodium deoxycholate in the case of the membranes hybridized with Phabs. To detect the anti-XylR Phab bound to the XylR bands, an anti-M13 peroxidase conjugate was utilized (1/5000 dilution in MBT). The membranes were incubated for 1 h at room temperature with a secondary antibody and washed 5 times in MBT buffer for 5 min each, as before. XylR was developed by reaction of the treated membrane with a chemiluminescent substrate (ECL, Amersham Pharmacia Biotech) and recorded on x-ray film.
Zdroje
1. PerezJC, GroismanEA (2009) Evolution of transcriptional regulatory circuits in bacteria. Cell 138 : 233–244.
2. WallME, HlavacekWS, SavageauMA (2004) Design of gene circuits: lessons from bacteria. Nat Rev Genet 5 : 34–42.
3. Silva-RochaR, de LorenzoV (2010) Noise and robustness in prokaryotic regulatory networks. Annu Rev Microbiol 64 : 257–275.
4. Van HijumSAFT, MedemaMH, KuipersOP (2009) Mechanisms and evolution of control logic in prokaryotic transcriptional regulation. Microbiol Mol Biol Rev 73 : 481–509.
5. Silva-RochaR, TamamesJ, dos SantosVM, de LorenzoV (2011) The logicome of environmental bacteria: merging catabolic and regulatory events with Boolean formalisms. Environ Microbiol 13 : 2389–2402.
6. RamosJL, MarquesS (1997) Transcriptional control of the Pseudomonas TOL plasmid catabolic operons is achieved through an interplay of host factors and plasmid-encoded regulators. Annu Rev Microbiol 51 : 341–373.
7. Silva-RochaR, de JongH, TamamesJ, de LorenzoV (2011) The logic layout of the TOL network of Pseudomonas putida pWW0 plasmid stems from a metabolic amplifier motif (MAM) that optimizes biodegradation of m-xylene. BMC Syst Biol 5 : 191.
8. Silva-RochaR, de LorenzoV (2011) A composite feed-forward loop I4-FFL involving IHF and Crc stabilizes expression of the XylR regulator of Pseudomonas putida mt-2 from growth phase perturbations. Mol Biosyst 7 : 2982–2990.
9. InouyeS, NakazawaA, NakazawaT (1987) Expression of the regulatory gene xylS on the TOL plasmid is positively controlled by the xylR gene product. Proc Natl Acad Sci USA 84 : 5182–5186.
10. MarquésS, GallegosMT, ManzaneraM, HoltelA, TimmisKN, et al. (1998) Activation and repression of transcription at the double tandem divergent promoters for the xylR and xylS genes of the TOL plasmid of Pseudomonas putida. J Bacteriol 180 : 2889–2894.
11. AbrilMA, MichanC, TimmisKN, RamosJL (1989) Regulator and enzyme specificities of the TOL plasmid-encoded upper pathway for degradation of aromatic hydrocarbons and expansion of the substrate range of the pathway. J Bacteriol 171 : 6782–6790.
12. Perez-MartinJ, de LorenzoV (1996) VTR expression cassettes for engineering conditional phenotypes in Pseudomonas: activity of the Pu promoter of the TOL plasmid under limiting concentrations of the XylR activator protein. Gene 172 : 81–86.
13. FraileS, RoncalF, FernandezLA, de LorenzoV (2001) Monitoring intracellular levels of XylR in Pseudomonas putida with a single-chain antibody specific for aromatic-responsive enhancer-binding proteins. J Bacteriol 183 : 5571–5579.
14. JuradoP, FernandezLA, de LorenzoV (2003) Sigma 54 levels and physiological control of the Pseudomonas putida Pu promoter. J Bacteriol 185 : 3379–3383.
15. BehzadianF, BarjesteH, HosseinkhaniS, ZareiAR (2011) Construction and characterization of Escherichia coli whole-cell biosensors for toluene and related compounds. Curr Microbiol 62 : 690–696.
16. GarmendiaJ, de las HerasA, GalvaoTC, de LorenzoV (2008) Tracing explosives in soil with transcriptional regulators of Pseudomonas putida evolved for responding to nitrotoluenes. Micro Biotech 1 : 236–246.
17. de Las HerasA, de LorenzoV (2011) Cooperative amino acid changes shift the response of the sigma-dependent regulator XylR from natural m-xylene towards xenobiotic 2,4-dinitrotoluene. Mol Microbiol 79 : 1248–1259.
18. GarmendiaJ, DevosD, ValenciaA, de LorenzoV (2001) A la carte transcriptional regulators: unlocking responses of the prokaryotic enhancer-binding protein XylR to non-natural effectors. Mol Microbiol 42 : 47–59.
19. de Las HerasA, de LorenzoV (2012) Engineering whole-cell biosensors with no antibiotic markers for monitoring aromatic compounds in the environment. Methods Mol Biol 834 : 261–281.
20. YuQ, LiY, MaA, LiuW, WangH, et al. (2011) An efficient design strategy for a whole-cell biosensor based on engineered ribosome binding sequences. Anal Bioanal Chem 401 : 2891–2898.
21. de LorenzoV, HerreroM, MetzkeM, TimmisKN (1991) An upstream XylR - and IHF-induced nucleoprotein complex regulates the sigma 54-dependent Pu promoter of TOL plasmid. EMBO J 10 : 1159–1167.
22. RescalliE, SainiS, BartocciC, RychlewskiL, De LorenzoV, et al. (2004) Novel physiological modulation of the Pu promoter of TOL plasmid: negative regulatory role of the TurA protein of Pseudomonas putida in the response to suboptimal growth temperatures. J Biol Chem 279 : 7777–7784.
23. DelgadoA, RamosJL (1994) Genetic evidence for activation of the positive transcriptional regulator Xy1R, a member of the NtrC family of regulators, by effector binding. J Biol Chem 269 : 8059–8062.
24. de Las HerasA, ChavarriaM, de LorenzoV (2011) Association of dnt genes of Burkholderia sp. DNT with the substrate-blind regulator DntR draws the evolutionary itinerary of 2,4-dinitrotoluene biodegradation. Mol Microbiol 82 : 287–299.
25. LambertsenL, SternbergC, MolinS (2004) Mini-Tn7 transposons for site-specific tagging of bacteria with fluorescent proteins. Environ Microbiol 6 : 726–732.
26. GalvãoTC, MencíaM, de LorenzoV (2007) Emergence of novel functions in transcriptional regulators by regression to stem protein types. Mol Microbiol 65 : 907–919.
27. O'ShaughnessyEC, PalaniS, CollinsJJ, SarkarCA (2011) Tunable signal processing in synthetic MAP kinase cascades. Cell 144 : 119–131.
28. AndersenJB, SternbergC, PoulsenLK, BjornSP, GivskovM, et al. (1998) New unstable variants of green fluorescent protein for studies of transient gene expression in bacteria. Appl Environ Microbiol 64 : 2240–2246.
29. AlexcievK, UschevaA, PavlovaM, YavachevL, IvanovI (1989) Expression of synthetic calcitonin genes in plasmid vectors containing tandemly repeated non-overlapping ribosome binding sites. Int J Biochem 21 : 987–996.
30. SneppenK, KrishnaS, SemseyS (2010) Simplified models of biological networks. Annu Rev Biophys 39 : 43–59.
31. HermsenR, EricksonDW, HwaT (2011) Speed, sensitivity, and bistability in auto-activating signaling circuits. PLoS Comput Biol 7: e1002265 doi:10.1371/journal.pcbi.1002265.
32. ShahNA, SarkarCA (2011) Robust network topologies for generating switch-like cellular responses. PLoS Comput Biol 7: e1002085 doi:10.1371/journal.pcbi.1002085.
33. RaiN, AnandR, RamkumarK, SreenivasanV, DabholkarS, et al. (2012) Prediction by promoter logic in bacterial quorum sensing. PLoS Comput Biol 8: e1002361 doi:10.1371/journal.pcbi.1002361.
34. BertoniG, Perez-MartinJ, de LorenzoV (1997) Genetic evidence of separate repressor and activator activities of the XylR regulator of the TOL plasmid, pWW0, of Pseudomonas putida. Mol Microbiol 23 : 1221–1227.
35. MitrophanovAY, GroismanEA (2008) Positive feedback in cellular control systems. Bioessays 30 : 542–555.
36. HoltelA, TimmisKN, RamosJL (1992) Upstream binding sequences of the XylR activator protein and integration host factor in the xylS gene promoter region of the Pseudomonas TOL plasmid. Nucl Acids Res 20 : 1755–1762.
37. MarquésS, HoltelA, TimmisKN, RamosJL (1994) Transcriptional induction kinetics from the promoters of the catabolic pathways of TOL plasmid pWW0 of Pseudomonas putida for metabolism of aromatics. J Bacteriol 176 : 2517–2524.
38. LimWA (2010) Designing customized cell signalling circuits. Nat Rev Mol Cell Biol 11 : 393–403.
39. de Las HerasA, CarrenoCA, Martinez-GarciaE, de LorenzoV (2010) Engineering input/output nodes in prokaryotic regulatory circuits. FEMS Microbiol Rev 34 : 842–865.
40. WackettLP (2008) Biosensors. Microbial Biotechnology 1 : 331–332.
41. van der Meer JR (2011) Bacterial sensors: Synthetic design and application principles; Amos M, editor. New Jersey: Morgan & Claypool.
42. NandagopalN, ElowitzMB (2011) Synthetic Biology: Integrated gene circuits. Science 333 : 1244–1248.
43. OlaniranA, MotebejaneR, PillayB (2008) Bacterial biosensors for rapid and effective monitoring of biodegradation of organic pollutants in wastewater effluents. J Environ Monit 10 : 889.
44. RonEZ (2007) Biosensing environmental pollution. Curr Opin Biotechnol 18 : 252–256.
45. van der MeerJR, BelkinS (2010) Where microbiology meets microengineering: design and applications of reporter bacteria. Nat Rev Microbiol 8 : 511–522.
46. LoogerLL, DwyerMA, SmithJJ, HellingaHW (2003) Computational design of receptor and sensor proteins with novel functions. Nature 423 : 185–190.
47. DubnauD, LosickR (2006) Bistability in bacteria. Mol Microbiol 61 : 564–572.
48. KarigD, WeissR (2005) Signal-amplifying genetic circuit enables in vivo observation of weak promoter activation in the Rhl quorum sensing system. Biotechnol Bioeng 89 : 709–718.
49. de Las Heras A, de Lorenzo V (2010) Genetic constructs: Molecular tools for the assembly of environmental bacterial biosensors. In: Timmis KN, editor. Handbook of Hydrocarbon and Lipid Microbiology. Berlin: Springer-Verlag pp. 2651–2676.
50. PedelacqJD, CabantousS, TranT, TerwilligerTC (2006) Engineering and characterization of a superfolder green fluorescent protein. Nature Biotech 24 : 79–88.
51. TropelD, van der MeerJR (2004) Bacterial transcriptional regulators for degradation pathways of aromatic compounds. Microbiol Mol Biol Rev 68 : 474–500.
52. CamasFM, PoyatosJF (2008) What determines the assembly of transcriptional network motifs in Escherichia coli? PLoS ONE 3: e3657 doi:10.1371/journal.pone.0003657.
53. NelsonKE, WeinelC, PaulsenIT, DodsonRJ, HilbertH, et al. (2002) Complete genome sequence and comparative analysis of the metabolically versatile Pseudomonas putida KT2440. Environ Microbiol 4 : 799–808.
54. de LorenzoV, TimmisKN (1994) Analysis and construction of stable phenotypes in gram-negative bacteria with Tn5 - and Tn10-derived minitransposons. Methods Enzymol 235 : 386–405.
55. Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: A laboratory manual. New York: Cold Spring Harbor.
56. de Las HerasA, CarreñoCA, de LorenzoV (2008) Stable implantation of orthogonal sensor circuits in Gram-negative bacteria for environmental release. Env Microbiol 10 : 3305–3316.
57. McKownRL, OrleKA, ChenT, CraigNL (1988) Sequence requirements of Escherichia coli attTn7, a specific site of transposon Tn7 insertion. J Bacteriol 170 : 352–358.
58. Sanchez-RomeroJM, Diaz-OrejasR, de LorenzoV (1998) Resistance to tellurite as a selection marker for genetic manipulations of Pseudomonas strains. Appl Env Microbiol 64 : 4040–4046.
59. ChoiKH, GaynorJB, WhiteKG, LopezC, BosioCM, et al. (2005) A Tn7-based broad-range bacterial cloning and expression system. Nat Methods 2 : 443–448.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- A Mutation in the Gene Causes Alternative Splicing Defects and Deafness in the Bronx Waltzer Mouse
- Mutations in (Hhat) Perturb Hedgehog Signaling, Resulting in Severe Acrania-Holoprosencephaly-Agnathia Craniofacial Defects
- Classical Genetics Meets Next-Generation Sequencing: Uncovering a Genome-Wide Recombination Map in
- Regulation of ATG4B Stability by RNF5 Limits Basal Levels of Autophagy and Influences Susceptibility to Bacterial Infection
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy