
Mutations in and Reveal That Cartilage Matrix Controls Timing of Endochondral Ossification by Inhibiting Chondrocyte Maturation
Differentiating cells interact with their extracellular environment over time. Chondrocytes embed themselves in a proteoglycan (PG)-rich matrix, then undergo a developmental transition, termed “maturation,” when they express ihh to induce bone in the overlying tissue, the perichondrium. Here, we ask whether PGs regulate interactions between chondrocytes and perichondrium, using zebrafish mutants to reveal that cartilage PGs inhibit chondrocyte maturation, which ultimately dictates the timing of perichondral bone development. In a mutagenesis screen, we isolated a class of mutants with decreased cartilage matrix and increased perichondral bone. Positional cloning identified lesions in two genes, fam20b and xylosyltransferase1 (xylt1), both of which encode PG synthesis enzymes. Mutants failed to produce wild-type levels of chondroitin sulfate PGs, which are normally abundant in cartilage matrix, and initiated perichondral bone formation earlier than their wild-type siblings. Primary chondrocyte defects might induce the bone phenotype secondarily, because mutant chondrocytes precociously initiated maturation, showing increased and early expression of such markers as runx2b, collagen type 10a1, and ihh co-orthologs, and ihha mutation suppressed early perichondral bone in PG mutants. Ultrastructural analyses demonstrated aberrant matrix organization and also early cellular features of chondrocyte hypertrophy in mutants. Refining previous in vitro reports, which demonstrated that fam20b and xylt1 were involved in PG synthesis, our in vivo analyses reveal that these genes function in cartilage matrix production and ultimately regulate the timing of skeletal development.
Published in the journal:
. PLoS Genet 7(8): e32767. doi:10.1371/journal.pgen.1002246
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002246
Summary
Differentiating cells interact with their extracellular environment over time. Chondrocytes embed themselves in a proteoglycan (PG)-rich matrix, then undergo a developmental transition, termed “maturation,” when they express ihh to induce bone in the overlying tissue, the perichondrium. Here, we ask whether PGs regulate interactions between chondrocytes and perichondrium, using zebrafish mutants to reveal that cartilage PGs inhibit chondrocyte maturation, which ultimately dictates the timing of perichondral bone development. In a mutagenesis screen, we isolated a class of mutants with decreased cartilage matrix and increased perichondral bone. Positional cloning identified lesions in two genes, fam20b and xylosyltransferase1 (xylt1), both of which encode PG synthesis enzymes. Mutants failed to produce wild-type levels of chondroitin sulfate PGs, which are normally abundant in cartilage matrix, and initiated perichondral bone formation earlier than their wild-type siblings. Primary chondrocyte defects might induce the bone phenotype secondarily, because mutant chondrocytes precociously initiated maturation, showing increased and early expression of such markers as runx2b, collagen type 10a1, and ihh co-orthologs, and ihha mutation suppressed early perichondral bone in PG mutants. Ultrastructural analyses demonstrated aberrant matrix organization and also early cellular features of chondrocyte hypertrophy in mutants. Refining previous in vitro reports, which demonstrated that fam20b and xylt1 were involved in PG synthesis, our in vivo analyses reveal that these genes function in cartilage matrix production and ultimately regulate the timing of skeletal development.
Introduction
Vertebrate bone is produced by two major developmental processes, intramembranous ossification (forming dermal bones) and endochondral ossification (forming chondral bones). The latter process has many stages that must be coordinated in space and time. First, some cells in the mesenchymal condensation differentiate as chondrocytes, which secrete a cartilage extracellular matrix rich in proteoglycans (PGs), such as chondroitin sulfate PGs [1], [2]. A thin layer of cells, the perichondrium, surrounds the developing cartilage. Later, a subset of chondrocytes undergoes a developmental transition, expressing markers of chondrocyte maturation, including indian hedgehog (ihh) and collagen type 10a1 (col10; [3]). Meanwhile, cells of the perichondrium that overlie the maturing chondrocytes differentiate as bone-forming osteoblasts.
Tissue interactions between cartilage and perichondrium ensure that these early events in endochondral ossification are coordinated in space and time [4]–[6]. Critically, the growth factor Ihh produced by maturing chondrocytes induces perichondral bone, and both mouse Ihh and zebrafish ihha mutants have delayed perichondral ossification [7], [8]. Although specific signaling pathways and transcription factors are involved [9]–[11], the full ontology of genes that regulate the timing of endochondral ossification is still unknown, but may include genes involved in extracellular matrix production.
Mutations disrupting PG synthesis commonly affect skeletal tissues and can change the timing of skeletal development. Hereditary Multiple Exostoses is caused by mutations in Exostosin (Ext) genes, which encode enzymes that synthesize heparan sulfate PGs [12], [13]. Mouse and zebrafish models of Ext loss-of-function reveal defects in endochondral ossification associated with delays in chondrocyte maturation, which were attributed to altered Ihh signaling [14]–[16]. Mutations affecting various components of PG synthesis, from sugar precursor production enzymes to sulfation enzymes, affect dramatically the shape and composition of skeletal tissues, and also can delay endochondral ossification [14], [17]–[20]. Interestingly, acceleration or inhibition of developmental timing may be a function of the class of PG. Evidence above indicates that mutations in HSPG synthesis delay endochondral ossification, while mutations in chondroitin sulfate PG (CSPG) synthesis may accelerate this bone-forming process. Aggrecan is the major chondroitin sulfate PG (CSPG) in cartilage, and Aggrecan (Acan) mutant chicks exhibit increased levels of Col10 expression [21], which these authors interpreted as evidence that endochondral ossification had initiated earlier. Here we perform direct investigation of developmental timing in order to test the hypothesis that loss of CSPGs accelerates endochondral ossification.
In a forward genetic screen of skeletal tissues in developing zebrafish larvae, we isolated a class of mutants with increased chondral bone and decreased cartilage matrix at 6 days post-fertilization (dpf). Positional cloning identified mutations in fam20b and xylosyltransferase1 (xylt1), two critical genes for PG synthesis that are associated with human disease [22]–[27]. Biochemical and histochemical analyses revealed that mutants in both genes have defects in CSPG production. Rescue experiments, gene expression studies, ultrastructural, and mutational analyses all argue that PG mutant chondrocytes undergo precocious maturation, thus inducing early perichondral bone. Our analyses of in vivo models for loss of function in fam20b or xylt1 refine previous in vitro reports [23], [28] by demonstrating an important role for these PG synthesis genes in cartilage matrix production in intact, developing animals. In closing, we discuss the phenotype of fam20b mutant zebrafish with respect to excessive bone formation in Raine syndrome patients, who contain mutations in the paralogous gene FAM20C [26], [27].
Results
Two mutant loci with increased bone matrix and decreased cartilage matrix phenotypes
Screening of larval skeletal phenotypes in mutagenized zebrafish revealed a class of mutant with normal gross anatomy at 6 dpf, but with specific defects in the degree of cartilage and bone tissue formation (Figure 1A, 1B). We identified four independently derived lines (b1125, b1127, b1128, and b1189), in which homozygous mutants showed increased ossification of many bones, as judged by Alizarin red staining (Figure 1C–1H). In addition, these mutants had decreased Alcian blue staining of cartilage matrix in all cartilaginous elements (Figure 1C–1H), which reflected defects in the production or secretion of cartilage extracellular matrix. Cartilage and bone phenotypes were both present in 100% of mutants examined, although there was variation in the degree to which Alizarin red staining increased. Complementation crosses, scored for these skeletal phenotypes, suggested mutations at two genetic loci among these four mutants. The b1125 and b1127 mutations failed a genetic complementation test, producing 51/212 (24%) mutant larvae when heterozygotes were crossed to one another. Likewise, b1128 and b1189 failed to complement, producing 60/208 (29%) mutant larvae when heterozygotes were crossed. All other pair-wise complementation crosses produced larvae with wild-type skeletal development. The b1127;b1128 double mutants showed Alizarin red staining that was similar to each single mutant, although the loss of Alcian blue staining seemed more severe (Figure 1F, quantified below), suggesting that both of these loci drive production of cartilage matrix. Other than these changes to skeletal tissues, overall mutant cranial morphology appeared smaller at 6 dpf (Figure 1A, 1B). In particular, mutant chondral bones appeared shorter than those in wild-type siblings. All homozygous mutants can grow to viable adults, allowing us to investigate whether morphological phenotypes were more apparent at later stages. Compared to wild-type siblings, mutant adults displayed foreshortened upper and lower jaws, hypoplastic midface, and bulging eyes (Figure 1I, 1J). Imaging Alizarin red-stained head skeletons with optical projection tomography (OPT) revealed that morphological defects in adult mutant heads were accompanied by altered craniofacial skeletal morphology (Figure 1K, 1L), predominantly an apparent loss of anterior neurocranial growth.

Identification of fam20b and xylt1 lesions in skeletal matrix mutants
Genetic mapping confirmed that the two complementation groups of skeletal mutants mapped to independent loci, and sequencing nearby candidates revealed molecular lesions in fam20b and xylt1 that underlie the skeletal phenotype. RAD mapping and subsequent simple sequence repeat (SSR) mapping identified a genetic interval of 0.2 cM on LG20 containing the b1127 mutation (Figure 2A; [29]; see Materials and Methods). The lesion in fam20bb1127 mutants (991T>C) disrupted a highly conserved cysteine residue (C331R; Figure 2B, 2C; [30]). For the b1125 allele, cDNA sequencing revealed a 1162C>T mutation in the seventh coding exon of fam20bb1125, which would alter amino acid 388 from Gln to STOP, thereby truncating the last 22 amino acids, including a highly conserved Cys residue at aa389 (Figure 2B, 2C; [30]). fam20bb1125 transcripts were down-regulated at 55 hours post-fertilization (hpf), while fam20bb1127 transcripts were present at wild-type levels (data not shown), which is consistent with nonsense-mediated RNA degradation [31]. Therefore, mapping and sequence data suggested that mutations within fam20b caused the b1125 and b1127 phenotypes.

As the first steps in identifying molecular lesions in xylt1, RAD mapping and subsequent SSR mapping defined a genetic interval of 0.7 cM on LG3 containing the b1128 mutation (Figure 2D; [29]). Sequencing of xylt1 from b1128 cDNA and gDNA revealed a splice donor mutation (2103G>A) in exon 9 that would produce a frameshifted and truncated C-terminal portion of the zebrafish Xylt1 protein from at least amino acid 702 (out of 919, Figure 2E). For the b1189 allele, cDNA sequencing revealed a 1600T>G mutation in the seventh coding exon of xylt1b1189, which altered highly-conserved amino acid 534 from Ser to Ala (Figure 2E, 2F). xylt1b1128 transcripts were down-regulated at 55 hpf, while xylt1b1189 transcripts were present at wild-type levels (data not shown), which is in agreement with nonsense-mediated RNA degradation [31]. These data suggested that the b1128 and b1189 phenotypes were caused by mutations in xylt1.
Expressing wild-type fam20b rescues b1125 and b1127
We used rescue experiments to test our conclusion that lesions in fam20b caused the skeletal phenotypes of b1125 and b1127. Mutant embryos were injected with Tol2 expression plasmids driving expression of wild-type fam20b, and then were assayed for skeletal phenotypes. Wild-type fam20b cDNA rescued fam20bb1127 (n = 10/13) and fam20bb1125 (n = 3/8) skeletal phenotypes when driven by the ubiquitous beta-actin2 promoter (Figure 3A–3D, data not shown; [32]). There was variation in the degree to which the entire skeleton, or even an entire skeletal element, was rescued, which would be expected from mosaicism inherent to the transient injection protocol. We observed in these rescues a correlation between chondrocytes surrounded by faint Alcian blue staining and overlying bone (n = 11 skeletal elements). That is, in adjacent patches of chondrocytes, lighter stained Alcian blue matrix was covered with abundant Alizarin red staining, whereas darker stained Alcian blue matrix was not surrounded by heavy Alizarin red staining (Figure 3D, dashed lines), a finding that will be of significance below. No differences in skeletal phenotypes at 6 dpf were observed in control wild-type embryos that were injected with wild-type fam20b expression constructs (Figure S1A). Also, the mutant skeletal phenotype was not rescued when fam20bb1127 mutant embryos were injected with Tol2 expression plasmids containing mutant fam20bb1127 (n = 0/23) or fam20bb1125 (n = 0/12) cDNA (Figure 3E, data not shown). To address whether over-expression of fam20b was having non-specific effects, we subjected xylt1 mutants to Tol2-mediated fam20b over-expression. Wild-type fam20b was not sufficient to rescue skeletal defects in xylt1b1128 mutants (n = 0/11; Figure S1B). Experiments injecting xylt1b1128 mutants with wild-type xylt1 expression constructs did not lead to rescue of the skeletal phenotype (data not shown), which may be due to the fact that our xylt1 cDNA constructs contained a transmembrane domain that was removed from similar mis-expression studies [28], [33].

As measured by Alcian blue staining, PG secretion by developing chondrocytes in the anterior pharyngeal arches was not detected at 48 hours post-fertilization (hpf), but was abundant at 60 hpf (Figure S2). To understand when fam20b plays a role during development, wild-type fam20b expression was induced under the control of a heat-shock-inducible promoter (hsp70l; [32]). A single induction of wild-type fam20b at 55 hpf proved sufficient to rescue skeletal phenotypes of fam20bb1125 mutant larvae at 6 dpf (n = 6/7; Figure S1C–S1E), although again there was variation in the ability of a transient injection to rescue completely even a given skeletal element. In summary, we conclude that fam20b is the mutated gene in b1125 and b1127, and our data argue that the timing of fam20b action in producing the craniofacial skeleton correlates with the onset of overt chondrogenesis.
fam20b and xylt1 are expressed in developing chondrocytes
Transcripts for fam20b and xylt1 in wild-type larvae were detected by RT-PCR from 3 hpf through 54 hpf (Figure 4A, 4F), demonstrating that fam20b and xylt1 were expressed during the mid-blastula transition and early skeletogenic time points. In situ hybridization revealed fam20b and xylt1 expression in chondrocytes of developing skeletal elements at 53 hpf, 63 hpf, and 72 hpf, but these levels decreased by 4 dpf (Figure 4B–4E, 4G–4J, data not shown). xylt1 transcripts were readily apparent in osteoblasts of newly-forming dermal bone at 3 dpf (Figure 4K, 4L), which is interesting to consider under the proposition that dermal bones progress through a transient chondrogenic phase [34]. However, neither fam20b nor xylt1 transcripts were detected in developing perichondrium at 3 dpf, 4 dpf, or 6 dpf, which spanned the time perichondral osteoblasts appear (Figure 4D, 4E, 4I, 4J, and data not shown). Apart from their up-regulated expression in skeletal tissues, fam20b showed diffuse ubiquitous expression in brain and craniofacial mesenchyme, and xylt1 transcripts were expressed in discrete domains of the brain, such as the developing forebrain (Figure 4B, 4C, 4G, 4H). In summary, fam20b and xylt1 are expressed in developing chondrocytes, but not in cells of the perichondrium, even at stages when we know bone-forming cells have differentiated within this tissue. Our findings are thus consistent with the notion that mutations in these genes act directly in chondrocytes to produce the mutant cartilage phenotype, but only indirectly in causing elevated perichondral bone.

fam20b and xylt1 mutants exhibit partial loss of cartilage PGs
Quantifying spectrometrically lysates from at least three Alcian blue-stained clutches for fam20b or xylt1 single mutants, or fam20b;xylt1 double mutants (see Materials and Methods; [35]), we found statistically significant changes in Alcian blue levels between all genotypes tested (Figure 5A–5D, 5F; ANOVA p<0.0001). Levels of Alcian blue in fam20b and xylt1 mutants were 50±3.0% and 57±2.0% of their wild-type siblings, respectively, while fam20b;xylt1 double mutants further reduced Alcian blue levels to 39±2.6% of their wild-type siblings (Figure 5F). Recently, we showed that a null mutation in UDP-xylose synthase1 (uxs1) abolished zygotic production of UDP-xylose [18], a sugar that ultimately is the substrate for both Fam20b and Xylt1 during PG synthesis (Figure 5G; [22], [23]). We used the uxs1 mutant to estimate whether fam20b and xylt1 mutants were null alleles. Relative to fam20b and xylt1 single and double mutants, cartilages from uxs1 mutants showed even less Alcian blue staining (Figure 5E), reducing levels to 22±1.5% of those in wild-type siblings (Figure 5F). Therefore, quantitiative comparisons with uxs1 mutants suggested that the fam20bb1127 and xylt1b1128 alleles did not completely eliminate cartilage PG production; even fam20b;xylt1 double mutants were less severe than uxs1 mutants.

To show by an independent method that fam20b and xylt1 mutants had cartilage PG defects that were less severe than uxs1 mutants, we analyzed GAG disaccharide levels using biochemical and immunohistochemical methods. Both heparan sulfate PGs (HSPGs) and chondroitin sulfate PGs (CSPGs) are xylose-dependent, so both of these classes of PG may be affected in fam20b and xylt1 mutants. HPLC analyses on lysates of whole 5 dpf larvae demonstrated that fam20b and xylt1 mutants had decreased levels of CSPG disaccharides, which are predominant in cartilage [36], and again, the losses were less severe than seen in uxs1 mutants (Figure S3). HSPG disaccharide levels were not affected consistently among these mutants; fam20b and uxs1 mutants showed decreases, whereas xylt1 mutants did not (Figure S3). Consistent with the biochemical data, immunodetection assays revealed losses of both CSPGs and HSPGs specifically around developing chondrocytes in fam20b mutants (Figure 6A–6F). No loss of CSPGs in xylt1 mutant larvae were detected (data not shown), suggesting that the CSPG antibody may recognize epitopes still present in xylt1 mutants. Together, these biochemical and immunohistochemical data revealed cartilage PG defects in fam20b and xylt1 mutants. Furthermore, the quantitative comparisons with uxs1 mutant embryos suggested either that fam20bb1127 and xylt1b1128 are not null alleles, or that redundant genes compensate for their loss of function (see Discussion).

Since both Fam20b and Xylt1 depend upon UDP-xylose, and hence uxs1 function, in order to promote GAG synthesis (Figure 5G), loss of uxs1 function should mask the skeletal phenotypes of fam20b and xylt1 embryos. Consistent with fam20b and xylt1 being downstream of uxs1, skeletal phenotypes of fam20b;uxs1 and xylt1;uxs1 larvae appeared identical to uxs1 single mutants, including severe loss of Alcian blue staining and undetectable levels of perichondral bone, as predicted (Figure 7A–7H). Despite the strong uxs1 mutant phenotype, elimination of just one copy of uxs1 did not sensitize zebrafish skeletons to the loss of fam20b or xylt1. Trans-heterozygous larvae displayed normal skeletal phenotypes, and no enhancement of the homozygous fam20b and xylt1 mutant phenotypes was observed when larvae also were heterozygous for the uxs1 mutation (data not shown). These epistasis experiments provided genetic support that fam20b and xylt1 function in the PG synthesis pathway in vivo.

Mutants have increased bone due to precocious differentiation of perichondral osteoblasts
We find that two parameters of bone matrix production along the endochondral ossification pathway are increased in both fam20b and xylt1 mutants, which is in contrast to decreased production of cartilage matrix. First, the frequency of chondral bone Alizarin red staining at 6 dpf was increased (see Figure 1C–1H for example of material scored), whereas dermal bone staining did not consistently show statistically significant effects (Figure 8A, 8B). Second, the amount of staining in a given chondral bone (e.g., the ceratohyal) was also increased significantly in fam20b and xylt1 mutants (Figure 8C–8G). The increased bone resided on the inner surface of the perichondrium (see Figure 9H, 9I), as expected for chondral, rather than dermal, bones. Therefore, fam20b and xylt1 mutants showed specific increases in the amount of perichondral bone.


The observed increase in perichondral bone might result from three possible scenarios: 1) osteoblasts differentiate at the correct time, but exhibit enhanced secretory activity; 2) osteoblasts differentiate at the correct time, but a larger pool of pre-osteoblasts exists in mutant perichondria; or 3) osteoblasts differentiate early. Examination of skeletal phenotypes and cellular and molecular markers of osteoblasts at time points prior to 6 dpf demonstrated that the increase in mutant perichondral bone resulted from early initiation of osteogenesis. While neither mutants nor wild types showed signs of perichondral bone at 3 dpf (Figure 9A–9C), Alizarin red staining was observed in chondral bones of fam20b and xylt1 mutants, but not wild types, at 4.5 dpf (Figure 9D–9F, arrows). Bone formation was not accelerated in time further in fam20b;xylt1 double mutants, as perichondral bone was not detected at 3 dpf, but was prominent by 4.5 dpf (data not shown). To address the cellular basis for early perichondral bone, fam20b and xylt1 mutants were bred into transgenic zebrafish expressing EGFP under a promoter that is restricted to osteoblasts (Tg(sp7:EGFP)b1212; [37]). Transgenic mutants showed GFP expression and Alizarin red fluorescence in the perichondrium of chondral bones by 4.5 dpf, whereas their wild-type siblings showed neither of these markers at this time point (Figure 9G, 9H, arrows). Further support for the temporal shift in bone development came from similar analyses at 6 dpf, when wild types demonstrated levels of GFP and Alizarin red staining that were comparable to those observed in the perichondrium of mutant chondral bones at 4.5 dpf (data not shown). Expression of molecular markers of osteoblasts, such as col10a1 and runx2b, was increased in the perichondrium of mutant chondral bones at 4 dpf, compared to that seen in their wild-type siblings (Figure 9J–9L, arrows, data not shown). In total, these data argue that the cellular basis of excessive bone in fam20b and xylt1 mutants is the precocious differentiation of secretory osteoblasts in mutant perichondria.
Early chondrocyte maturation promotes precocious perichondral bone in PG mutants
Our expression data (Figure 4C, 4F), biochemical data (Figure 5F, Figure S3), and rescue experiments (Figure 3C, 3D) suggest that the premature perichondral bone in fam20b and xylt1 mutants is caused by chondrocyte defects. Indian hedgehog (Ihh) is an osteo-inductive signal expressed by chondrocytes as they mature, and is required for perichondral bone formation [7], [8], [38]. Therefore, we hypothesized that Ihh and other markers of chondrocyte maturation would appear earlier in fam20b and xylt1 mutants. The transcription factor Runx2 positively regulates chondrocyte maturation through transcriptional activation of such genes as col10a1 and ihh [39]–[41], so our hypothesis predicted premature up-regulation of runx2 genes and their downstream targets. In support, transcript levels for runx2b were up-regulated in mutant chondrocytes at 3 dpf compared to wild-type chondrocytes (Figure 10A–10C), and col10a1, ihha, and ihhb expression was up-regulated in mutant chondrocytes earlier than in chondrocytes of wild-type siblings (Figure 10D–10L). Expression of sox9a and col2a1a was high in both xylt1 mutant and wild-type chondrocytes at 3 dpf (data not shown). On the other hand, both transcripts were down-regulated in mature chondrocytes by 6 dpf (data not shown), suggesting that some markers of chondrocyte gene expression were not affected in PG mutants. Importantly, the initiation of chondrocyte differentiation was not accelerated overall in mutants, as Alcian blue staining of pharyngeal cartilages was absent at 48 hpf and present at 60 hpf both in wild types and in PG mutants (Figure S2). Therefore, the timing of chondrocyte differentiation was accelerated only after mutant cells had begun to immerse themselves in PG-rich extracellular matrix. The relative timing of these events is consistent with our ability to rescue the fam20b mutant phenotype with induction of wild-type fam20b at 55 hpf (see Figure S1).
Given that PG mutant chondrocytes expressed molecular markers of maturation prematurely, we performed ultrastructural analyses to assay whether PG mutant chondrocytes showed early cellular features of chondrocyte hypertrophy. Examined by transmission electron microscopy at 84 hpf, wild-type chondrocytes in the central portion of the ceratohyal contained abundant rough ER, Golgi complexes, and mitochondria, indicative of their high biosynthetic and secretory activity (Figure 11A; [42]). By comparison, xylt1 mutant chondrocytes in the central portion of the ceratohyal at 84 hpf displayed ultrastructural hallmarks of hypertrophic differentiation, including increased cell size, reduction in number of biosynthetic organelles, and cytoplasmic clearing (Figure 11B). Although chondrocytes along almost the entire ceratohyal showed premature hypertrophy in mutants, the most pronounced changes in cellular morphology were in the center of this cartilage element, consistent with the timing and location of molecular changes in gene expression (see Figure 10). Near the end of the xytl1 mutant ceratohyal, we observed chondrocytes that appeared like those in the center of the wild-type ceratohyal (data not shown). Furthermore, we observed differences between wild type and mutants in cartilage ECM ultrastructure. Wild-type cartilage ECM contained well-dispersed matrix proteins within fibrillar collagen, forming a well-defined ECM network (Figure 11C). Both territorial (pericellular) and interterritorial (outer) matrix showed uniform organization. In contrast, xylt1 mutant cartilage ECM was electron dense, tightly packed, and highly fibrillar (Figure 11D). As a result, xylt1 mutant chondrocytes were closer to each other than the cells in WT cartilage (Figure 11B). Furthermore, territorial matrix lateral to xylt1 mutant chondrocytes contained more poorly defined, electron dense precipitates 20–50 nm in diameter than seen in wild-type matrix (* in Figure 11C, 11D). In addition to verifying matrix defects, our ultrastructural analyses demonstrated that xylt1 mutant chondrocytes displayed cellular hallmarks of hypertrophic differentiation prior to those of wild-type siblings.

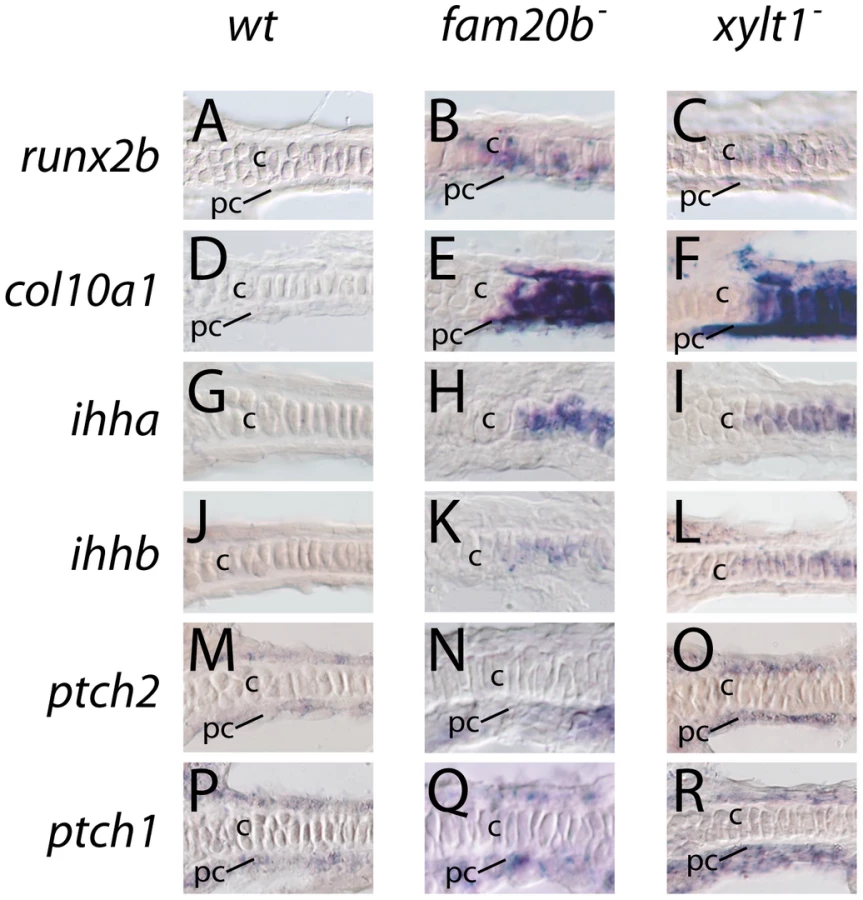
Given these accelerated molecular and cellular features of chondrocyte maturation in PG mutants, we used molecular and genetic means to demonstrate that chondrocyte Ihh expression signaled prematurely to induce perichondral bone. Expression of ptch2, a downstream marker of Hh signaling, was increased in ceratohyal perichondrium of fam20b and xylt1 mutants at 3 dpf (Figure 10M–10O), suggesting increased Ihh signaling in PG mutants. No differences in perichondral ptch1, gli2, and gli3 expression were observed between xylt1 or fam20b mutants and wild types (Figure 10P–10R, data not shown), perhaps reflecting the notion that potential transcriptional targets of Hh signaling are not employed in every cell type [43]. Because ihha mutant zebrafish have delayed perichondral ossification [7], we could test the functional significance of early ihha expression by creating fam20b;ihha and xylt1;ihha double mutants. Perichondral bone was suppressed in fam20b;ihha and xylt1;ihha double mutants (Figure 12A–12H), showing that ihha is required for early perichondral bone in fam20b and xylt1 mutants. While epistatic to the bone phenotypes, the ihha mutation did not alter cartilage matrix reduction of fam20b and xylt1 mutants, consistent with the interpretation that ihha acts downstream of the cartilage matrix defects. In summary, these data argue that cartilage PG defects in fam20b and xylt1 mutants primarily accelerated the timing of chondrocyte maturation and ihha expression, which then secondarily triggered early perichondral bone formation.

Discussion
Investigation of the vertebrate skeletal system has revealed substantial insight into the structural and functional roles of proteoglycans (PGs; [14]–[16], [18]–[20]). Here, we provide experimental data linking functions of long-studied (Xylt1) and recently-identified (Fam20b) members of the PG synthesis pathway to skeletal development in vivo. Xylosyltransferases (Xylts) have been known for 40 years to initiate glycosaminoglycan side chain outgrowth onto protein cores of PGs by transferring xylose to serine residues (Figure 5G; [22], [44]). Based on tissue culture and biochemical assays, Xylts were shown to function in the synthesis of both heparan sulfate PGs (HSPGs) and chondroitin sulfate PGs (CSPGs; [28], [45]–[47]). Xylt1 expression increased during the course of chondrogenic differentiation in vitro [48], and high Xylt serum activity has been linked to osteoarthritis [49]. Our expression, biochemical, and mutational analyses argue strongly for a predominant role of Xylt1 in CSPG production, specifically in cartilage matrix, thus providing an in vivo context for decades of in vitro studies.
In contrast to Xylts, Fam20 molecules were only recently identified, and our studies reveal in vivo functions of Fam20b [30]. Knock-down and over-expression studies in cell lines indicated that mouse Fam20c could promote odontoblast differentiation from mesenchymal stem cells [50], although the molecular mechanism for this role of Fam20c remains unclear. Renewed focus on Fam20 molecules arose from the revelation that humans with a skeletal disease called Raine syndrome have mutations in FAM20C [26], [27]. Subsequently, the paralogous protein Fam20b was shown in vitro to phosphorylate xylose on a nascent glycosaminoglycan side chain (Figure 5G), which appears to increase the likelihood that disaccharide repeats will be added by Exostosins and Chondroitin synthases [23]. Our expression and biochemical data show that fam20b functions similar to xylt1 in cartilage PG production in vivo, and our analyses of xylt1, fam20b, and uxs1 zebrafish mutants highlight the importance of xylosylation in skeletal development. Furthermore, all of these fam20b and xylt1 mutants are homozygous viable, providing the only current vertebrate models in which to study how Fam20 and Xylt molecules affect the variety of human disease-related physiological processes for which they have been implicated [24]–[27], [49]. In particular, we note that humans with Raine syndrome display similar facial dysmorphies and osteosclerosis as seen in fam20b mutant zebrafish, potentiating these fish as an informative model by which to understand the etiology of Raine syndrome.
The mutations in fam20b and xylt1 reported here help identify functional domains of the enzymes these genes encode. While Fam20b domains previously have not been probed experimentally, the conservation of amino acid residues among Fam20 family members suggests functional sites [30]. Both fam20bb1125 and fam20bb1127 disrupt cysteine residues that are highly conserved among vertebrates and may play a role in Fam20b molecular structure. Future in vitro enzyme assays will confirm whether such mutations abolish Fam20b kinase activity. Functional analyses of Xylt1 domains [28], [33], [51] did not target residues affected in our mutants, so xylt1b1128 and xylt1b1189 will provide new insights into Xylt1 function. The replacement of serine with alanine in xylt1b1189 may alter minimally protein biochemical properties. As we show, this serine residue is conserved in Xylt1's across vertebrates, suggesting that it might serve as a target for phosphorylation that ultimately impacts enzyme performance. The xylt1b1128 mutation alters some amino acids and then truncates a domain that is associated with protein-protein interactions (88645 superfamily; www.ensembl.org); thus, this alteration might change substrate recognition or inhibit molecular interactions with other glycosyltransferases in the proposed multienzyme complex [22], [52]. Future experiments designed to investigate whether skeletal defects of PG synthesis mutants arose merely from quantitative reduction in PG levels or rather also qualitative differences in protein substrate recognition will provide unique insight into the functional roles of PGs in vivo.
Our biochemical analyses demonstrate that the mutations in fam20b and xylt1 reported here do not abolish cartilage PG synthesis completely. Mutations in both fam20b and xylt1 reduce cartilage PG synthesis to similar extents; fam20b;xylt1 double mutants are quantitatively more severe than either mutant alone; and uxs1 mutants, which represent a complete loss of zygotic xylose-dependent PG production [18], exhibit an even more dramatic reduction in cartilage PG synthesis than fam20b;xylt1 double mutants. Therefore, the alleles reported here might not be null, and/or redundant genes partially mask their loss. Five total zebrafish fam20 genes have been reported (fam20a, fam20b, fam20c1, fam20c2, and fam20c3; [30]), and zebrafish have two xylt genes (xylt1, orthologous to human XYLT1, and xylt2, orthologous to human XYLT2; http://uswest.ensembl.org/Danio_rerio/). Expression and functional analyses of the full set of zebrafish fam20 and xylt genes would test for redundant activities that might compensate for the loss of fam20b and xylt1. We do not expect xylt2 to compensate for a loss of xylt1 function during zebrafish skeletogenesis, because mice deficient for Xylt2 exhibit polycystic kidney disease, but do not have skeletal defects [53].
Our finding that mutations in genes encoding CSPG synthesis enzymes accelerate endochondral ossification reveals that CSPGs can negatively regulate skeletogenic timing. This finding suggests to us that CSPGs might serve as therapeutics for skeletal defects that result from precocious developmental timing, such as craniosynostoses [54], [55]. Since various mutants affecting HSPG synthesis do not have increased perichondral bone [14]–[16], the gain in perichondral bone we describe here must be a CSPG-specific effect. In addition to this qualitative difference between HSPGs and CSPGs, PG levels may be interpreted quantitatively by developing skeletal cells. Homozygous uxs1 mutants demonstrate loss of Alcian blue staining in cartilage in the absence of zygotic CSPGs, but uxs1 mutants have delayed, rather than accelerated, bone formation [17], [18]. These data suggest that intermediate levels of CSPGs are required for the unique osteogenic acceleration observed in fam20b and xylt1 mutants. In fact, degradation of CSPGs may be a normal, required stage of endochondral ossification, for lighter Alcian blue staining is associated with more mature cartilage matrix (see Figure 5; [56], [57]). Another explanation for the lack of accelerated bone formation in uxs1 mutants is that these fish fail to produce both CSPGs and HSPGs. The gain in bone formation that loss of CSPGs imparts during endochondral ossification may depend upon HSPG function. This assertion is not supported by our data, however, as fam20b mutants exhibit increased perichondral bone while suffering loss of HSPG production in chondrocytes.
Together, our data argue that defective cartilage PG synthesis alters expression of transcription factors that determine the rates of chondrocyte maturation, thus changing the timing of perichondral bone formation. Increased expression of runx2b in fam20b and xylt1 mutant chondrocytes likely drives early expression of chondrocyte maturation markers [39]–[41], such as ihh co-orthologs, which induce early perichondral bone [7], [8], [38]. As such, fam20b mutants may provide a completely new etiology for Raine syndrome: defects in chondrocyte differentiation underlie the increased perichondral bone (i.e., osteosclerosis) and skeletal dysmorphies observed in these humans [26], [27]. Premature cellular hallmarks of chondrocyte hypertrophy, including cytoplasmic clearing and loss of rough ER, Golgi complex, and mitochondria, accompany molecular features of early chondrocyte maturation in xylt1 mutants. This premature terminal differentiation is also accompanied by changes in cartilage ECM, which contains well-defined, but more tightly packed collagen fibrils, and that might result from failure of GAGs to properly incorporate into the matrix.
The general hypothesis emerging from our work that CSPGs negatively regulate chondrocyte maturation is consistent with a recent, detailed study of the chick Aggrecan mutant [21]. Future work will aim to decipher the mechanism by which mutations in the cartilage PG synthesis pathway impact the timing of chondrocyte differentiation. Recent studies, for example, highlight a novel role for CSPGs in modulating growth factor signaling in developing cartilage [21], [58]. Here, we demonstrate that mutations in the CSPG synthesis pathway can accelerate developmental timing, thus expanding the ontology of genes regulating the rate of skeletogenesis.
Materials and Methods
Zebrafish lines
All fish lines were maintained and embryos raised according to established protocols [59] with IACUC approval. We obtained the b1125, b1127, b1128, and b1189 mutant alleles through mutagenesis with N-ethyl-N-nitrosourea (ENU) in an AB background [59]. ihhahu2131 fish were obtained from P. Ingham.
Histological stains
Embryos were fixed in 2% PFA in PBS for 1 hr., washed in 100 mM Tris pH 7.5/10 mM MgCl2 for 10 min., stained in 0.04% Alcian blue/10 mM MgCl2/70% EtOH pH 7.5 overnight, taken through graded EtOH series (80% EtOH/100 mM Tris pH 7.5/10 mM MgCl2; 50% EtOH/100 mM Tris pH 7.5; 25% EtOH/100 mM Tris pH 7.5), bleached in 3% H2O2/0.5% KOH for 10 min. with lids open, washed twice in 25% glycerol/0.1% KOH for 10 min. each, stained in 0.01% Alizarin red/25% glycerol/0.1% KOH pH 7.5 for 30 min., and de-stained with two washes of 50% glycerol/0.1% KOH. Larvae were also incubated in 0.003% Alizarin red in Embryo Medium to visualize live mineralized bone.
Mapping, cloning, and genotyping PG mutants
All oligonucleotide sequences appear in Figure S4. Sequence alignments were made using MultAlin [60]. RAD mapping localized b1127 to LG20 [29]. Subsequent simple sequence repeat (SSR) mapping identified a genetic interval of 0.2 cM between SSRs defined by primers A+B and C+D on scaffold 2914 of Zv7 (http://uswest.ensembl.org/Danio_rerio/Info/Index) containing the b1127 mutation (Figure 2A). In this interval, the SSR marker z20582 showed zero cross-overs with the mutant phenotype in 862 meioses. RNA isolated (TRI Reagent; Ambion Inc.) from live 5 dpf Alizarin red-stained zebrafish larvae that were screened for increased perichondral bone were made into cDNAs (First Strand Synthesis kit; Invitrogen Corp.). Full sequence of fam20b cDNA was determined by overlapping PCR fragments, generated from primers I+J and K+L. Four genes (angptl1, ralgps2, blactl, and fam20b) around z20582 were sequenced, and of these, mutant-specific coding sequence changes were identified only in the sixth coding exon of fam20b (Figure 2B, data not shown). PCR-based genotyping assays showed perfect correspondence between the fam20bb1127 skeletal phenotype and this mutation (0 cross-overs/614 meioses; Figure 2B). The second allele in the fam20b complementation group, b1125, also mapped tightly to fam20b. The map cross contained three cross-overs between fam20bb1125 and z20582 among 602 meioses (0.5 cM). b1127 was genotyped by digesting PCR product from primers V+W with HaeII, which only cuts mutant sequence. b1125 was genotyped with the marker z10805.
RAD mapping localized b1128 to LG3 [29]. Subsequent SSR mapping defined a genetic interval of 0.7 cM between SSRs defined by primers E+F and G+H on scaffold 380 of Zv7 containing the mutation (Figure 2D). We focused on a predicted gene (LOC560951; www.ensembl.org) within this interval with homology to xylosyltransferase1 (xylt1). Comparison of the Ensembl-predicted protein to Xylt1 of other vertebrates suggested that the annotated version of Xylt1 for zebrafish was lacking about 50 amino acids at the N-terminal portion of the protein. Therefore, we used 5′RACE on a 3 dpf cDNA library, using the nested primers M and N, along with universal primers (Clontech Laboratories, Inc.), to reveal an unannotated xylt1 exon in zebrafish over 50 kb upstream of the annotated version. PCR and sequencing analyses confirmed the gene structure of zebrafish xylt1, which consists of 11 exons, similar to human XYLT1 (Figure 2E). Full sequence of xylt1 cDNA generated from primers O+N, P+Q, R+S, and T+U was submitted to GenBank (Accession: HQ692884). Sequencing of xylt1 from b1128 cDNA and gDNA revealed a splice donor mutation (G2103A) in exon 9 (Figure 2E). PCR-based genotype assays showed perfect correspondence between the xylt1b1128 phenotype and this splice site mutation (0 cross-overs/738 meioses; Figure 2E). Sequencing from 5 dpf xylt1b1128 mutant cDNAs showed that in the absence of the wild-type splice donor site, cryptic sites were used (6/6 clones). Although the most common of these (5/6 clones) resulted in a tetranucleotide insertion (Figure 2E, 2F), all mutant cDNAs would produce a frameshifted and truncated C-terminal portion of the zebrafish Xylt1 protein from at least amino acid 702 (out of 919). The second allele in the xylt1 complementation group, b1189, also had a unique mutation in xylt1. cDNA sequencing revealed a T1600G mutation in the seventh coding exon of xylt1b1189, which altered highly-conserved amino acid 534 from Ser to Ala (Figure 2E, 2F). PCR-based genotype assays showed perfect correspondence between this mutation and the xylt1b1189 phenotype (0 cross-overs/632 meioses; Figure 2E). b1128 was genotyped by digesting PCR product from primers X+Y with DdeI, which only cuts wild-type sequence. b1189 was genotyped by digesting PCR product from primers Z+AA with HhaI, which only cuts mutant sequence.
fam20b rescue
Full-length fam20b was amplified from mutant and wild-type cDNAs with primers BB+CC and inserted into pDONR211 by BP recombination (Invitrogen Corp.). LR recombination reactions inserted fam20b under the control of beta-actin2 or hsp70l promoters with GFP fused in-frame in the destination vector pDestTol2CG2, which also contained cmlc2-GFP [32]. mRNA from pCS2FA-transposase (http://chien.neuro.utah.edu/tol2kitwiki) was made (mMessage mMachine; Ambion Inc.). Approximately 3 nl of 100 ng/µl of fam20b expression plasmid, 70 ng/µl transposase RNA, and 0.2% phenol red were injected into one to four-cell embryos. Embryos containing green hearts were screened, and for heat shock activation, 55 hpf embryos were incubated at 40C for 20 min.
Expression analyses
Whole-mount and section RNA in situ hybridization were carried out as described [61], [62]. Probes used were runx2b, ihha, ihhb, ptch1, ptch2, col10a1 [18]. Nomenclature for ptch genes reflects a recent update, whereby previously termed ptc1 is now ptch2 and ptc2 is now ptch1 (www.zfin.org). Probes for fam20b and xylt1 were created by cloning PCR amplicons from primers K+L and R+S, respectively, into pCR4-TOPO (Invitrogen Corp), digesting with NotI, and transcribing with T3 Polymerase. Primers DD+L and P+EE were used for fam20b and xylt1 RT-PCR analyses, respectively. Immunostaining was carried out as previously reported [18].
Alcian blue quantitation
Since Alcian blue binds sulfated glycosaminoglycans (GAGs; [63]), defects in cartilage PG production should be reflected by Alcian blue levels. As an indication of the levels of sulfated glycosaminoglycans in cell and tissue lysates, we modified the protocol of Bjornsson et al. (1998) as follows: larvae underwent all the stages except the Alizarin red staining of the Alcian blue, Alizarin red procedure outlined above, and tails were removed for genotyping. Samples were lysed (DNeasy kit; QIAGEN Inc.), spun briefly, and read spectrometrically at 620 nm. All analyses were performed in triplicate. Error bars represent standard error of the means.
HPLC analyses
Glycosaminoglycan (GAG) biochemistry was carried out by the UCSD Glycotechnology Core Resource, according to protocols Pre006, Mis002, Mod004, Pro006, and Pro007 (http://glycotech.ucsd.edu/desc.html). Some disaccharide species did not appear in the majority of samples, due to their overall low levels, and were not included in analyses.
Quantitation of perichondral bone
Digital images of flat-mounted, ventral views of ceratohyals in live Alizarin red-stained 6 dpf larvae were obtained under equivalent microscope settings, and subsequently analyzed with ImageJ software (http://rsbweb.nih.gov/ij/). Standard threshold values were applied to each image to define mineralized portions of ceratohyal perichondria. From eight samples each for genotyped mutants and wild types, mineralized area from the two ceratohyals was determined by the Analyze Particles function. Error bars represent standard error of the means.
OPT imaging
Fish were fixed overnight in 4% PFA, washed for an hour in 1% KOH, bleached in 3% H2O2/0.5% KOH for 40 min. with lids open, washed in 1% KOH, stained overnight in 0.003% Alizarin red in 1% KOH, and de-stained in 1% KOH. After eyes were removed, heads were embedded in agarose, washed twice in methanol, and cleared in benzyl alcohol∶benzyl benzoate (2∶1). Images were captured using Bioptonics OPT Scanner 3001 M (MRC Technology).
Transmission electron microscopy
Samples were processed as previously described [42], with the following modification. Zebrafish embryos were fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate at room temperature for 1 hr and then at 4°C for at least 24 hr. They were then rinsed in 0.1 M sodium cacodylate and post-fixed with 1% osmium tetroxide in 0.1 M sodium cacodylate for 1 hr. Following additional rinsing, specimens were dehydrated step-wise in ethanol and then propylene oxide, infiltrated with resin step-wise, and then embedded in resin for 48 hr at 60°C. Ultra-thin 50 nm sections were collected on a Leica Ultracut Microtome and analyzed on a Phillips CM-12 Transmission Electron Microscope provided by VUMC Cell Imaging Shared Resource.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HamAWCormackDH 1987 Ham's histology Philadelphia Lippincott xiv 732
2. CaplanABoyanB 1994 Endochondral bone formation: the lineage cascade. HallB Bone Boca Raton CRC Press 1 46
3. EamesBFde la FuenteLHelmsJA 2003 Molecular Ontogeny of the Skeleton. Birth Defects Research (Part C) 69 93 101
4. KronenbergHM 2003 Developmental regulation of the growth plate. Nature 423 332 336
5. YoonBSPogueROvchinnikovDAYoshiiIMishinaY 2006 BMPs regulate multiple aspects of growth-plate chondrogenesis through opposing actions on FGF pathways. Development 133 4667 4678
6. VortkampA 2001 Interaction of growth factors regulating chondrocyte differentiation in the developing embryo. Osteoarthritis Cartilage 9 Suppl A S109 117
7. HammondCLSchulte-MerkerS 2009 Two populations of endochondral osteoblasts with differential sensitivity to Hedgehog signalling. Development 136 3991 4000
8. St-JacquesBHammerschmidtMMcMahonAP 1999 Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev 13 2072 2086
9. VortkampALeeKLanskeBSegreGVKronenbergHM 1996 Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein [see comments]. Science 273 613 622
10. LanskeBKaraplisACLeeKLuzAVortkampA 1996 PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth [see comments]. Science 273 663 666
11. BiWHuangWWhitworthDJDengJMZhangZ 2001 Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc Natl Acad Sci U S A 98 6698 6703
12. ZakBMCrawfordBEEskoJD 2002 Hereditary multiple exostoses and heparan sulfate polymerization. Biochim Biophys Acta 1573 346 355
13. DuncanGMcCormickCTufaroF 2001 The link between heparan sulfate and hereditary bone disease: finding a function for the EXT family of putative tumor suppressor proteins. J Clin Invest 108 511 516
14. ClementAWiwegerMvon der HardtSRuschMASelleckSB 2008 Regulation of zebrafish skeletogenesis by ext2/dackel and papst1/pinscher. PLoS Genet 4 e1000136 doi:10.1371/journal.pgen.1000136
15. HiltonMJGutierrezLMartinezDAWellsDE 2005 EXT1 regulates chondrocyte proliferation and differentiation during endochondral bone development. Bone 36 379 386
16. KozielLKunathMKellyOGVortkampA 2004 Ext1-dependent heparan sulfate regulates the range of Ihh signaling during endochondral ossification. Dev Cell 6 801 813
17. WiwegerMIAvramutCMde AndreaCEPrinsFAKosterAJ 2011 Cartilage ultrastructure in proteoglycan-deficient zebrafish mutants brings to light new candidate genes for human skeletal disorders. J Pathol 223 531 542
18. EamesBFSingerASmithGAWoodZAYanYL 2010 UDP xylose synthase 1 is required for morphogenesis and histogenesis of the craniofacial skeleton. Dev Biol 341 400 415
19. SohaskeyMLYuJDiazMAPlaasAHHarlandRM 2008 JAWS coordinates chondrogenesis and synovial joint positioning. Development 135 2215 2220
20. MatsumotoKLiYJakubaCSugiyamaYSayoT 2009 Conditional inactivation of Has2 reveals a crucial role for hyaluronan in skeletal growth, patterning, chondrocyte maturation and joint formation in the developing limb. Development 136 2825 2835
21. DomowiczMSCortesMHenryJGSchwartzNB 2009 Aggrecan modulation of growth plate morphogenesis. Dev Biol 329 242 257
22. StoolmillerACHorwitzALDorfmanA 1972 Biosynthesis of the chondroitin sulfate proteoglycan. Purification and properties of xylosyltransferase. J Biol Chem 247 3525 3532
23. KoikeTIzumikawaTTamuraJKitagawaH 2009 FAM20B is a kinase that phosphorylates xylose in the glycosaminoglycan-protein linkage region. Biochem J 421 157 162
24. SchonSSchulzVPranteCHendigDSzliskaC 2006 Polymorphisms in the xylosyltransferase genes cause higher serum XT-I activity in patients with pseudoxanthoma elasticum (PXE) and are involved in a severe disease course. J Med Genet 43 745 749
25. SchonSPranteCBahrCTarnowLKuhnJ 2006 The xylosyltransferase I gene polymorphism c.343G>T (p.A125S) is a risk factor for diabetic nephropathy in type 1 diabetes. Diabetes Care 29 2295 2299
26. SimpsonMAScheuerleAHurstJPattonMAStewartH 2009 Mutations in FAM20C also identified in non-lethal osteosclerotic bone dysplasia. Clin Genet 75 271 276
27. SimpsonMAHsuRKeirLSHaoJSivapalanG 2007 Mutations in FAM20C are associated with lethal osteosclerotic bone dysplasia (Raine syndrome), highlighting a crucial molecule in bone development. Am J Hum Genet 81 906 912
28. MullerSDisseJSchottlerMSchonSPranteC 2006 Human xylosyltransferase I and N-terminal truncated forms: functional characterization of the core enzyme. Biochem J 394 163 171
29. MillerMRAtwoodTSEamesBFEberhartJKYanYL 2007 RAD marker microarrays enable rapid mapping of zebrafish mutations. Genome Biol 8 R105
30. NalbantDYounHNalbantSISharmaSCobosE 2005 FAM20: an evolutionarily conserved family of secreted proteins expressed in hematopoietic cells. BMC Genomics 6 11
31. ChangYFImamJSWilkinsonMF 2007 The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem 76 51 74
32. KwanKMFujimotoEGrabherCMangumBDHardyME 2007 The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn 236 3088 3099
33. GottingCMullerSSchottlerMSchonSPranteC 2004 Analysis of the DXD motifs in human xylosyltransferase I required for enzyme activity. J Biol Chem 279 42566 42573
34. AbzhanovARoddaSJMcMahonAPTabinCJ 2007 Regulation of skeletogenic differentiation in cranial dermal bone. Development 134 3133 3144
35. BjornssonS 1998 Quantitation of proteoglycans as glycosaminoglycans in biological fluids using an alcian blue dot blot analysis. Anal Biochem 256 229 237
36. WatanabeHYamadaYKimataK 1998 Roles of aggrecan, a large chondroitin sulfate proteoglycan, in cartilage structure and function. J Biochem 124 687 693
37. DeLaurierAEamesBFBlanco-SanchezBPengGHeX 2010 Zebrafish sp7:EGFP: a transgenic for studying otic vesicle formation, skeletogenesis, and bone regeneration. Genesis 48 505 511
38. LongFChungUIOhbaSMcMahonJKronenbergHM 2004 Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development 131 1309 1318
39. InadaMYasuiTNomuraSMiyakeSDeguchiK 1999 Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dev Dyn 214 279 290
40. EamesBFSharpePTHelmsJA 2004 Hierarchy revealed in the specification of three skeletal fates by Sox9 and Runx2. Dev Biol 274 188 200
41. StrickerSFundeleRVortkampAMundlosS 2002 Role of Runx genes in chondrocyte differentiation. Dev Biol 245 95 108
42. SarmahSBarrallo-GimenoAMelvilleDBTopczewskiJSolnica-KrezelL 2010 Sec24D-dependent transport of extracellular matrix proteins is required for zebrafish skeletal morphogenesis. PLoS ONE 5 e10367 doi:10.1371/journal.pone.0010367
43. SteccaBRuizIAA 2010 Context-dependent regulation of the GLI code in cancer by HEDGEHOG and non-HEDGEHOG signals. J Mol Cell Biol 2 84 95
44. GrebnerEEHallCWNeufeldEF 1966 Glycosylation of serine residues by a uridine diphosphate-xylose: protein xylosyltransferase from mouse mastocytoma. Arch Biochem Biophys 116 391 398
45. EskoJDStewartTETaylorWH 1985 Animal cell mutants defective in glycosaminoglycan biosynthesis. Proc Natl Acad Sci U S A 82 3197 3201
46. CuellarKChuongHHubbellSMHinsdaleME 2007 Biosynthesis of chondroitin and heparan sulfate in chinese hamster ovary cells depends on xylosyltransferase II. J Biol Chem 282 5195 5200
47. PonighausCAmbrosiusMCasanovaJCPranteCKuhnJ 2007 Human xylosyltransferase II is involved in the biosynthesis of the uniform tetrasaccharide linkage region in chondroitin sulfate and heparan sulfate proteoglycans. J Biol Chem 282 5201 5206
48. PranteCBiebackKFunkeCSchonSKernS 2006 The formation of extracellular matrix during chondrogenic differentiation of mesenchymal stem cells correlates with increased levels of xylosyltransferase I. Stem Cells 24 2252 2261
49. SchonSHuepGPranteCMullerSChristR 2006 Mutational and functional analyses of xylosyltransferases and their implication in osteoarthritis. Osteoarthritis Cartilage 14 442 448
50. HaoJNarayananKMuniTRamachandranAGeorgeA 2007 Dentin matrix protein 4, a novel secretory calcium-binding protein that modulates odontoblast differentiation. J Biol Chem 282 15357 15365
51. MullerSSchottlerMSchonSPranteCBrinkmannT 2005 Human xylosyltransferase I: functional and biochemical characterization of cysteine residues required for enzymic activity. Biochem J 386 227 236
52. SchwartzNBRodenLDorfmanA 1974 Biosynthesis of chondroitin sulfate: interaction between xylosyltransferase and galactosyltransferase. Biochem Biophys Res Commun 56 717 724
53. CondacESilasi-MansatRKosankeSSchoebTTownerR 2007 Polycystic disease caused by deficiency in xylosyltransferase 2, an initiating enzyme of glycosaminoglycan biosynthesis. Proc Natl Acad Sci U S A 104 9416 9421
54. WilkieAO 1997 Craniosynostosis: genes and mechanisms. Hum Mol Genet 6 1647 1656
55. MelvilleHWangYTaubPJJabsEW 2010 Genetic basis of potential therapeutic strategies for craniosynostosis. Am J Med Genet A
56. IshizekiKNawaT 2000 Further evidence for secretion of matrix metalloproteinase-1 by Meckel's chondrocytes during degradation of the extracellular matrix. Tissue Cell 32 207 215
57. FarquharsonCWhiteheadCCLoveridgeN 1994 Alterations in glycosaminoglycan concentration and sulfation during chondrocyte maturation. Calcif Tissue Int 54 296 303
58. CortesMBariaATSchwartzNB 2009 Sulfation of chondroitin sulfate proteoglycans is necessary for proper Indian hedgehog signaling in the developing growth plate. Development 136 1697 1706
59. WesterfieldM 2007 The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio) Eugene Univ. of Oregon Press
60. CorpetF 1988 Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res 16 10881 10890
61. YanYLWilloughbyJLiuDCrumpJGWilsonC 2005 A pair of Sox: distinct and overlapping functions of zebrafish sox9 co-orthologs in craniofacial and pectoral fin development. Development 132 1069 1083
62. Rodriguez-MariAYanYLBremillerRAWilsonCCanestroC 2005 Characterization and expression pattern of zebrafish Anti-Mullerian hormone (Amh) relative to sox9a, sox9b, and cyp19a1a, during gonad development. Gene Expr Patterns 5 655 667
63. LevRSpicerSS 1964 Specific Staining of Sulphate Groups with Alcian Blue at Low Ph. J Histochem Cytochem 12 309
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 8
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- An EMT–Driven Alternative Splicing Program Occurs in Human Breast Cancer and Modulates Cellular Phenotype
- Chromosome Painting Reveals Asynaptic Full Alignment of Homologs and HIM-8–Dependent Remodeling of Chromosome Territories during Meiosis
- A Pre-mRNA–Associating Factor Links Endogenous siRNAs to Chromatin Regulation
- Discovery of Sexual Dimorphisms in Metabolic and Genetic Biomarkers
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy