
Ku Must Load Directly onto the Chromosome End in Order to Mediate Its Telomeric Functions
The Ku heterodimer associates with the Saccharomyces cerevisiae telomere, where it impacts several aspects of telomere structure and function. Although Ku avidly binds DNA ends via a preformed channel, its ability to associate with telomeres via this mechanism could be challenged by factors known to bind directly to the chromosome terminus. This has led to uncertainty as to whether Ku itself binds directly to telomeric ends and whether end association is crucial for Ku's telomeric functions. To address these questions, we constructed DNA end binding–defective Ku heterodimers by altering amino acid residues in Ku70 and Ku80 that were predicted to contact DNA. These mutants continued to associate with their known telomere-related partners, such as Sir4, a factor required for telomeric silencing, and TLC1, the RNA component of telomerase. Despite these interactions, we found that the Ku mutants had markedly reduced association with telomeric chromatin and null-like deficiencies for telomere end protection, length regulation, and silencing functions. In contrast to Ku null strains, the DNA end binding defective Ku mutants resulted in increased, rather than markedly decreased, imprecise end-joining proficiency at an induced double-strand break. This result further supports that it was the specific loss of Ku's telomere end binding that resulted in telomeric defects rather than global loss of Ku's functions. The extensive telomere defects observed in these mutants lead us to propose that Ku is an integral component of the terminal telomeric cap, where it promotes a specific architecture that is central to telomere function and maintenance.
Published in the journal:
. PLoS Genet 7(8): e32767. doi:10.1371/journal.pgen.1002233
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002233
Summary
The Ku heterodimer associates with the Saccharomyces cerevisiae telomere, where it impacts several aspects of telomere structure and function. Although Ku avidly binds DNA ends via a preformed channel, its ability to associate with telomeres via this mechanism could be challenged by factors known to bind directly to the chromosome terminus. This has led to uncertainty as to whether Ku itself binds directly to telomeric ends and whether end association is crucial for Ku's telomeric functions. To address these questions, we constructed DNA end binding–defective Ku heterodimers by altering amino acid residues in Ku70 and Ku80 that were predicted to contact DNA. These mutants continued to associate with their known telomere-related partners, such as Sir4, a factor required for telomeric silencing, and TLC1, the RNA component of telomerase. Despite these interactions, we found that the Ku mutants had markedly reduced association with telomeric chromatin and null-like deficiencies for telomere end protection, length regulation, and silencing functions. In contrast to Ku null strains, the DNA end binding defective Ku mutants resulted in increased, rather than markedly decreased, imprecise end-joining proficiency at an induced double-strand break. This result further supports that it was the specific loss of Ku's telomere end binding that resulted in telomeric defects rather than global loss of Ku's functions. The extensive telomere defects observed in these mutants lead us to propose that Ku is an integral component of the terminal telomeric cap, where it promotes a specific architecture that is central to telomere function and maintenance.
Introduction
Eukaryotic telomeres are comprised of repetitive G-rich sequence arrays and a host of proteins that associate with the repeats directly or as part of a complex [1], [2]. These proteins maintain telomeres by facilitating the replication of telomeric DNA and by protecting the natural chromosomal ends from end-joining and resection activities associated with the repair of DNA double-strand breaks (DSBs). Perturbation of the normal structure and function of the telomere, through the loss of telomere sequence or telomere-associated proteins, can result in genomic instability, checkpoint activation, and cellular senescence or apoptosis [3].
Understanding the structural relationship of telomere-associated proteins with telomeric DNA is integral to our comprehension of the maintenance, structure, and function of telomeres. To date, proteins have been shown to associate with telomeric DNA via at least one of three mechanisms. Some telomere-associated proteins, such as Rap1 in Saccharomyces cerevisiae [4] and TRF1 and TRF2 in mammalian cells [5], utilize a myb/homeodomain to bind directly and with high affinity to double-stranded (ds) telomeric DNA sequences. Other proteins, such as Cdc13 in S. cerevisiae [6] and the TEBPα/β heterodimer in Oxytricha nova [7], utilize one or more oligonucleotide-oligosaccharide binding (OB) folds to associate avidly and specifically with single-stranded (ss) G-rich telomeric DNA. Because telomeres terminate with G-rich overhangs, ss telomeric DNA binding proteins are localized to the chromosome end. The third mechanism by which proteins associate with telomeres is via protein-protein interactions. For example, in S. cerevisiae, Sir3 and Sir4 are recruited to telomeric repeat-containing chromatin via Rap1 [8], and in mammalian cells, TIN2 localizes to ds telomeric repeats via its interaction with TRF1 and TRF2 [9], [10]. Proteins capable of binding telomeric DNA directly also can employ this mode of association. For example, POT1, a protein that binds directly to the G-overhang through its OB folds [11], also associates with ds telomeric repeats through a series of protein-protein interactions terminating with TRF1 [12]. Thus, the ability to bind to telomeric DNA directly does not preclude telomeric association via protein-protein interactions.
In contrast to the above examples, the mechanism for the evolutionarily conserved Ku heterodimer's association to the telomere is uncertain. Comprised of the Ku70 and Ku80 subunits, Ku loads onto DNA ends via a preformed ring consisting of an expansive base and narrow bridge that encircles the DNA [13], [14]. Ku binds DNA ends in a sequence-independent manner through a limited number of contacts with the sugar phosphate backbone [14]. Even though Ku binds DNA in a sequence independent manner, it does load onto DNA ends in a specific orientation. The loading face is comprised predominantly of Ku80, while the lagging face consists mostly of Ku70. This orientation is possibly dictated by steric and electrostatic features present on the lagging face that impede the DNA's access to this end of the DNA binding channel.
Given its high affinity for DNA ends, Ku could localize to telomeric chromatin through direct DNA end binding [15]. In vitro assays have shown that Ku does associate with a telomeric DNA substrate in this way, as long as it is allowed access to the DNA before Cdc13 is added [16]. However, Cdc13 is associated with telomeres throughout the cell cycle in vivo [17]. Thus, the means by which telomeric end binding by Ku might occur in vivo in the context of Cdc13 and other telomeric binding proteins that avidly bind either ds or ss telomeric DNA remains unclear. In addition, the telomeres of many species are thought to exist in a higher order structure known as a t-loop, where the terminal G-strand 3′ overhang invades proximal ds telomeric DNA to form a d-loop, thereby concealing the DNA end [18]. In fact, one proposed function of this structure is to prevent Ku's association with the chromosome's terminus, in turn, preventing telomeres from engaging in Ku-dependent, nonhomologous end-joining (NHEJ) [19].
In addition to binding DNA, Ku70 and Ku80 each have N terminal α/β domains which lie laterally to the DNA binding channel and are thought to mediate Ku's interaction with other factors [14]. Ku has been shown to bind or co-purify with telomere-associated factors such as Sir4 in budding yeast and RAP1, TRF1, and TRF2 in mammalian cells [20]–[23]. In the case of Sir4, this has been shown to require residues in the yeast Ku80 α/β domain. Also, Ku's interaction with TLC1, the RNA component of telomerase, is disrupted by a mutation (yku80-135i) that maps to the Ku80 α/β domain [24]. Therefore, Ku might also associate with telomeres via its interaction with telomeric factors. Consistent with this, the association of budding yeast Ku80 with subtelomeric chromatin is reduced in the absence of Sir4 [25]. Thus, the possibility exists that Ku is recruited to telomeric repeats through these interactions, independent of its DNA end binding activity.
Defining how Ku associates with telomeres is crucial for understanding its non-overlapping roles in a myriad of telomeric processes. For example, in S. cerevisiae, Ku is required for the localization of Est2, the catalytic subunit of yeast telomerase, to telomeres in G1 [17]. This recruitment depends upon a direct interaction between Ku and a 48-nt stem-loop in TLC1 [24]. Ku also protects the telomeric 5′ strand from resection by Exo1 [26], [27], a 5′-3′ exonuclease and flap-endonuclease involved in a variety of DNA repair processes including resection of DSBs [28], [29]. Studies with the Yku80 separation-of-function mutant yku80-135i, however, indicate that Ku can regulate telomere length independently of its end protection function [24].
In addition to, and distinct from, its role in telomere replication, Ku aids in the formation of silent telomeric chromatin [30], which results in transcriptional repression of adjacent genes, a phenomenon referred to as telomeric silencing or telomere position effect [31]. Ku promotes the recruitment of Sir4 to sub-telomeric DNA, [25], [32], [33] and separation-of-function mutants have demonstrated that Ku's interaction with Sir4, while required for Ku's role in telomeric silencing, is dispensable for Ku's role in telomere end protection and length homeostasis [34].
Yeast Ku also is required for the normal localization of telomeres at the nuclear periphery via two pathways that are independent of Sir4 and silent chromatin [35], [36]. One of these pathways is S-phase specific and relies on Ku's interaction with TLC1 and telomerase's interaction with the integral nuclear membrane protein Mps3 [37]. Although perinuclear tethering can promote telomeric silencing, Ku's influence on silencing can occur, at least at some telomeres, independently of its effects on tethering. This is supported by the finding that the perinuclear localization of truncated versions of Tel VI-R and Tel VII-L is maintained in a yku70-Δ background (i.e., the tethering of these telomeres is Ku-independent), whereas the telomeric silencing of Tel VI-R ADE2 and Tel VII-L URA3 reporters is lost (i.e., the silencing of these telomeres is Ku-dependent) [35], [38].
In addition to telomeres, Ku localizes to DSBs, where it plays a crucial role in NHEJ. Separation-of-function mutations that result in isolated NHEJ or telomeric defects suggest that Ku plays distinct roles at telomeric versus broken ends [20], [24], [34], [36], [39], [40]. For example, mutations in helix 5 of the yeast Ku70 α/β domain impact selectively on NHEJ, whereas mutations in helix 5 of the yeast Ku80 α/β domain impact selectively on telomeric silencing [40]. The structural position of NHEJ versus telomeric function-specific residues and the polarity with which the heterodimer loads onto DNA ends led us to propose a ‘two-face’ model for how Ku's NHEJ and telomeric functions are spatially organized [40]. In this model, we proposed that Ku's outward face, juxtaposed with the DNA terminus when bound to a broken end, is dedicated to its NHEJ functions, whereas its inward face, which would be oriented toward telomeric chromatin when bound to a telomeric end, is dedicated to Ku's telomeric functions. This model assumes, however, that Ku loads directly onto telomeric ends and that Ku's telomeric functions are dependent upon this mode of binding.
To determine whether Ku associates with telomeres and mediates one or more of its telomeric roles via end binding, we generated DNA end binding defective S. cerevisiae Ku heterodimers and examined their association with telomeric chromatin and their impact on telomere function. We found that the loss of DNA end binding activity greatly reduced Ku's association with telomeres and rendered Ku deficient in its ability to protect telomeres from nucleolytic processing, to maintain telomere length, and to contribute to the formation of silent telomeric chromatin. Therefore, Ku must access and directly associate with telomeric ends in order for telomeres to adopt their proper architecture and perform their genome-protective functions.
Results
Construction of DNA end binding–defective Ku heterodimers
To determine whether DNA end binding is required for Ku's association with telomeres and its telomeric functions, we sought to create S. cerevisiae Ku heterodimers that were specifically defective in their ability to bind DNA ends by mutagenizing residues that interact with DNA. We used the crystal structure of human Ku bound to DNA (PDB 1JEY) [14] and multiple sequence alignments containing yeast and human Ku70 (Yku70 or hKu70) and Ku80 (Yku80 or hKu80) subunits to identify potential DNA contacting residues in yeast Ku. Twenty-four residues in human Ku, distributed between the two subunits, contact DNA in the crystal structure. Because of poor sequence conservation between most of these residues and the aligning residues in yeast, we limited single amino acid substitutions to a subset of five residues that ranked in the top 20th percentile of our previous analyses of evolutionarily conserved amino acid residues of Ku70 and Ku80 [40] (Figure 1). We also targeted Yku70-R456, the yeast residue that aligned with hKu70-R444, which was in close proximity to DNA in the human Ku-DNA co-crystal and highly conserved throughout evolution. In addition to these single amino acid substitutions, we targeted residues that corresponded to small runs of DNA contacting residues in each of the human subunits (Yku70-IMFQ266-269 and Yku80-SKKDS400-404 which correspond to hKu70-ALSR255-258 and hKu80-YDKRA397-401).

The impact of the mutations on DNA end binding was determined first by individually expressing each mutant allele in yeast lacking the corresponding wild type genomic copy and assaying DNA end binding activity in whole cell extracts (WCE) using an in vitro electrophoretic mobility shift assay (EMSA) [41]. In this assay, a 1000-fold molar excess of non-labeled closed circular DNA inhibits nonspecific binding to the 198 bp radiolabeled DNA fragment, resulting in an electrophoretic mobility shift of the radiolabeled fragment that is dependent on Ku binding to the DNA ends [41] (see also Figure 2A). As sought after, we saw significantly reduced end binding activity in two of the single missense mutant strains examined, yku70-K422E and yku70-R456E. Although Ku is known to bind DNA ends independently of other factors, we also examined purified recombinant WT and Yku70-R456E/Yku80 (Figure S1) heterodimers. We observed a 50 - to 100-fold reduction in the DNA end binding affinity of the mutant (Figure 2B), confirming that the targeting indeed affected Ku's inherent ability to bind DNA ends.

As shown in Figure 1B, the yku70-IMFQ266-269QDEY and yku80-SKKDS400-404DEEDD mutations (referred to hereafter as yku70-p1 and yku80-p1, respectively) targeted amino acids on opposing sides of the DNA binding channel. Whereas expression of yku70-p1 with YKU80 or yku80-p1 with YKU70 did not result in a severe deficiency in DNA end binding, expression of both panel mutant alleles resulted in undetectable DNA end binding activity in the WCE EMSA (Figure 2A and Figure S2). These results suggest that there are synergistic contributions to Ku's stable association with DNA ends mediated by amino acids on each side (i.e., leading and lagging) of the DNA binding channel.
Prior to utilizing the alleles identified as being DNA end binding defective for in vivo analyses, we needed to determine whether the loss of DNA end binding was the indirect result of markedly reduced steady state protein levels or loss of heterodimer formation. Using epitope tagged versions of Yku70 and Yku80, we found that the mutations did not substantially alter steady state protein levels (Figure 2C, input) or the ability of the mutant subunits to form heterodimers (Figure 2C, α-FLAG IP). Anti-FLAG immunoprecipitates from yku70-R456EFLAG, yku70-p1FLAG, and yku80-p1myc18 single mutant WCE contained WT levels of both subunits, despite the presence of the mutation. The levels of Yku70-K422EFLAG and Yku80myc18, and Yku70-p1FLAG and Yku80-p1myc18 were only slightly reduced in the yku70-K422EFLAG YKU80myc18 single mutant, and yku70-p1FLAG yku80-p1myc18 double mutant strains' input and anti-FLAG IPs. Thus, the yku70-K422E and yku70-R456E single mutants and the combination of the yku70-p1 and yku80-p1 panel mutant alleles appeared to directly impact Ku's ability to bind DNA ends.
As with heterodimerization, the interpretation of our in vivo findings would be confounded if the mutations conferring a DNA end binding defect also disrupted Ku's ability to associate with the telomeric factor Sir4 or the telomerase subunit TLC1. Yeast Ku interacts with Sir4 via the Yku80 subunit [40]; therefore, we examined whether the Yku80-Sir4 interaction was retained in the DNA end binding defective mutants using a previously reported Yku80-Sir4 yeast two-hybrid system with a LEU2 reporter [20]. Consistent with the prior observation that the Yku80-Sir4 yeast two-hybrid interaction was independent of Yku70 [20], we found that the presence of yku70-R456E, yku70-p1 or yku70-K422E instead of YKU70 did not impair the association between Yku80 and Sir4, as evidenced by growth of strains expressing these alleles, the YKU80 BAIT, and the SIR4 PREY on media lacking leucine (Figure 3A, compare strains 3 and 6 with 1, and 11 with 9). Significantly, the substitution of yku80-p1 for YKU80 on the BAIT plasmid in YKU70 or yku70-p1 cells did not inhibit growth in the absence of leucine (Figure 3A, compare strains 7 and 8 with 1). These results suggest that the mutations did not hinder the association between Yku80 and Sir4.

We also assayed the in vivo association of TLC1 with the Yku70-R456E Yku80 and Yku70-p1 Yku80-p1 Ku heterodimers. To do so, we immunoprecipitated mutant or WT Ku from asynchronous cultures expressing myc or FLAG tagged-Yku80 and performed real-time, reverse transcriptase polymerase chain reaction (RT-PCR) to detect TLC1 RNA. The yku70-R456E strain exhibited a level of TLC1 enrichment comparable to the YKU70 strain in both myc - and FLAG-tagged YKU80 backgrounds (Figure 3B; P = 0.51 and 0.20, respectively). Enrichment for TLC1 was not seen in the ‘no tag’ control or in a tlc1-Δ48 strain, which is defective for interaction with Yku80 [17](P = 0.007). Likewise, actin RNA was not enriched in any of the reactions, demonstrating specificity of the co-immunoprecipitation. TLC1 was enriched similarly in yku70-p1 yku80-p1FLAG anti-FLAG IPs (P = 0.17). Thus, both of these DNA end binding defective Ku heterodimers were capable of binding TLC1 in vivo.
DNA end binding–defective Ku heterodimers show decreased association with telomeric chromatin
The above analyses indicated that we had succeeded in introducing mutations into Ku that markedly impacted on DNA end binding without substantially altering protein stability, heterodimerization or interactions with known telomeric factors [40]. Therefore, we proceeded to address whether selective loss of Ku's DNA end binding activity altered its association with telomeric chromatin in vivo by performing chromatin immunoprecipitation (ChIP) assays. We found an 80–86% decrease in the amount of telomeric DNA that immunoprecipitated with Yku80myc in the yku70-R456E strain relative to the amount immunoprecipitated in the YKU70 strain (Figure 4A and Figure S3A). This decrease was not due to decreased levels of Yku80myc or decreased immunoprecipitation efficiency, as protein levels appeared equal in both the input and α-myc IP samples (Figure 4B and Figure S3B). We also performed ChIP experiments with the yku70-p1 yku80-p1 double panel mutant strain (Figure S3A), which showed undetectable DNA end binding in the WCE EMSA (Figure 2A). The immunoprecipitates isolated from the double panel mutant contained less than 1% of the telomeric DNA isolated from WT immunoprecipitates. However, we could not rule out the possibility that this was due to the reduced immunoprecipitation efficiency that was observed in this mutant under these conditions (Figure S3B). Thus, we conclude that, at the very least, the vast majority of Ku's localization to telomeres requires that it bind directly to telomeric ends.

Loss of Ku's DNA end binding activity results in the loss of telomere end protection
An important role of Ku at telomeres is to protect the C-rich telomeric strand from exonucleolytic degradation by Exo1. The mechanism by which this is accomplished has not been directly studied; however, the simplest model posits that Ku sterically hinders Exo1's access and/or progression by loading directly onto telomeric ends. By examining the telomeric G-overhangs in the DNA end binding defective strains, we could now test this model directly. To do this, XhoI-digested genomic DNA prepared from asynchronous cultures was hybridized with a C-rich radiolabeled telomeric probe and subjected to native gel electrophoresis as previously described [34] (Figure 5A, native). The signal was quantified and normalized to the total telomeric G-strand DNA signal, which was obtained by in-gel DNA denaturation followed by hybridization with the same probe (Figure 5A, denatured, and 5B). Two of the DNA end binding defective Ku mutants, yku70-R456E and yku70-p1 yku80-p1, exhibited readily detectable ss G-rich DNA signals similar to a yku70-Δ strain. Thus, a loss of DNA end binding correlated with a loss of telomere end protection.

In apparent contradiction to this correlation, the yku70-K422E mutant strain, which was also defective in DNA end binding in the WCE EMSA, exhibited only a slight increase in ss G-rich DNA, comparable to strains expressing yku70-p1 or yku80-p1 (Figure 5). This could indicate that Ku's end binding activity is, in fact, not required for Ku's telomere end protection function. However, yku70-K422E single mutants also appeared similar to WT in all additional assays performed (telomere length, telomeric silencing, synthetic lethality with tlc1-Δ, temperature sensitivity, and imprecise NHEJ; Figure S4 and data not shown). It was possible that this mutant protein was unfolded in vitro or differentially sensitive to the WCE EMSA conditions. An alternative explanation was that the Yku70-K422E mutant protein, with its mutated residue on the lagging face of the DNA binding channel (Figure 1), was stabilized at telomeric ends in vivo. We reasoned that if this was the case then the combination of the yku70-K422E allele with the yku80-p1 allele, whose mutated residues are on the loading face of the DNA binding channel (Figure 1), might destabilize the mutant Yku70-K422E-containing Ku's association with DNA ends in vivo and give rise to defective telomere end protection. Indeed, we found a synergistic increase in the ss G-rich DNA signal in the yku70-K422E yku80-p1 double mutant strain (Figure 5). An increase was not observed, however, when yku70-K422E was combined with the yku80-8 allele, which contains a mutated residue on Ku's loading face. This mutation impairs Ku's ability to bind Sir4 [40], but it does not affect Ku's ability to bind DNA ends (Figure S5). These findings suggest that the ability of Ku to protect telomeres from exonucleolytic resection requires that Ku bind directly to the telomeric end.
Loss of Ku's DNA end binding activity results in short telomeres
As described earlier, a second important telomeric function of Ku is in the maintenance of telomere length. Whereas it is known that this requires Ku's interaction with TLC1, it is unknown whether it requires Ku's direct association with telomeric ends. To address this question, we determined the length of telomeres present in yku70-R456E and yku70-p1 yku80-p1 mutant strains. We found that the telomeres in these mutants were nearly as short as those in yku70-Δ and yku80-Δ null strains (Figure 6A). In contrast, the telomeres in the yku70-p1 and yku80-p1 single mutants were of WT length or only slightly shortened. Additionally, as illustrated in the yku70-R456E mutant, the degree of shortening was much greater than in a tlc1-Δ48 mutant, in which Ku can no longer bind TLC1. Moreover, the telomeres were no shorter in a yku70-R456E tlc1-Δ48 double mutant than in a yku70-R456E single mutant (Figure 6B). Therefore, and consistent with the TLC1 co-immunoprecipitation results (Figure 3B), the shortening observed in these DNA end binding defective Ku mutants was due to a defect downstream of TLC1 association.
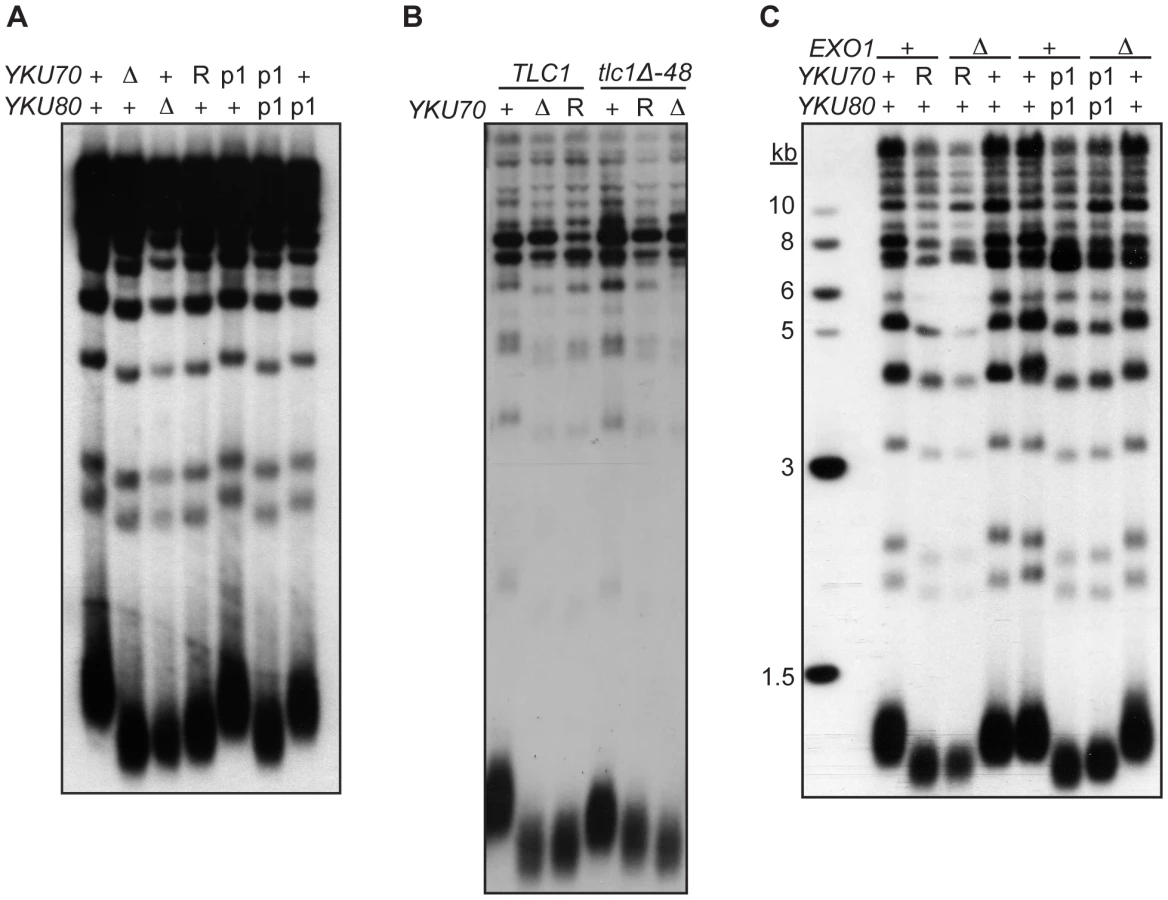
These data suggest that Ku must load onto and stably associate with the telomere end in order to mediate its role in telomere length regulation, yet it was possible that the telomere length defect in the DNA end binding defective strains was due to the increased single-strandedness of the G-rich telomeric strand. If the failure to protect telomeric ends was the sole contributing factor to the shortened telomeres present in the end binding defective Ku mutants, then the deletion of EXO1 should fully restore the length of the telomeres. We found that, although the deletion of EXO1 fully complemented the defect in end protection of DNA end binding defective mutants (Figure S6), it did not fully restore telomeres to a WT length (Figure 6C). Thus, Ku's ability to bind telomeric ends contributes directly to the maintenance of telomere length beyond simple telomeric end protection.
Loss of Ku's DNA end binding activity results in the loss of telomeric silencing
Whereas we predicted Ku's association with the telomeric end would be required for its role in telomere end protection and length regulation, a role for Ku's DNA end binding in the formation of subtelomeric heterochromatin was not as clearly anticipated [30], [42], [43]. In the formation of subtelomeric chromatin, Ku is recruited to the centromere-proximal region of subtelomeric DNA in a Sir4-dependent manner [25]. Therefore, the DNA end binding activity of Ku may not be required for Ku's ability to promote the silencing of subtelomeric reporters. To address this question, we utilized a strain containing a URA3 telomeric reporter gene, which is subject to Ku-dependent silencing [43], to examine whether the DNA end binding mutations resulted in telomeric silencing defects. WT strains grow poorly on medium lacking uracil (and robustly on 5-FOA medium) because of the spread of telomeric heterochromatin and secondary transcriptional repression of the adjacent URA3 gene. Cells with a silencing deficiency, such as yku70-Δ, display the opposite growth pattern – robust growth in the absence of uracil and minimal growth in the presence of 5-FOA. As in the previous assays of Ku's telomere end protection and length maintenance functions, the yku70-p1 yku80-p1 double mutant strain exhibited a markedly defective phenotype, whereas the single panel mutant strains, yku70-p1 and yku80-p1, displayed a phenotype similar to that of WT (Figure 7A). The silencing defect in the yku70-R456E mutant was not as severe as the defect observed in the yku70-Δ or double panel mutant strain, but was clearly evident by the larger colony size and greater colony number on the plate lacking uracil (Figure 7A). This partial silencing defect was also evident when we examined the expression of a second telomeric reporter gene in this strain, ADE2. The reversible nature of telomeric silencing results in variegation, such that WT colonies are mostly red (due to ADE2 repression) with some white sectors (due to ADE2 expression). However, colonies from strains that are deficient in telomeric silencing are white in appearance. Utilizing this reporter, we found that the double-panel mutant and yku70-Δ strains were white, hence, defective for telomeric silencing (Figure 7A). The yku70-R456E strain exhibited a very slight variegated appearance, suggesting that it was not completely defective in silencing; however, it was clearly dysfunctional when compared to WT, yku70-p1, and yku80-p1 strains.

Rap1 plays a central role in the establishment of telomeric silencing by binding ds telomeric repeats and recruiting the essential silencing proteins Sir3 and Sir4 to chromatin [8]. Because excess degradation of the telomeric sequence would result in the loss of Rap1 binding sites, the telomeric silencing defect of the DNA end binding defective Ku mutant strains could be an indirect consequence of their defect in telomere end protection. To determine if such was the case, we repeated the silencing assay in an exo1-Δ background. Deletion of EXO1 restored the level of ss, G-rich DNA in the DNA end binding defective mutants to near WT levels (Figure S6), which was further reflected by suppression of the temperature sensitive phenotype (Figure 7B). Despite restoring ds telomeric DNA, we found that deleting EXO1 only slightly restored silencing, as evidenced by the slightly decreased growth of yku70-R456E exo1-Δ and yku70-p1 yku80-p1 exo1-Δ strains on medium lacking uracil relative to their corresponding EXO1 strains (compare Figure 7B to 7A). Similarly, the deletion of EXO1 led to the appearance of very faint pink colonies in the DNA end binding defective mutants (Figure 7B). Altogether, these results indicate that the DNA end binding activity of Ku (and, hence, its direct association with telomeric DNA) is required for its role in telomeric silencing.
Loss of Ku's DNA end binding activity results in increased imprecise NHEJ
In each of the telomeric function assays performed, the yku70-R456E and yku70-p1 yku80-p1 mutants exhibited loss of function similar to that of yku70-Δ or yku80-Δ strains; however, the interaction assays (e.g., for heterodimerization, TLC1 interaction, and Yku80-Sir4 yeast two-hybrid interaction) argued against a global null phenotype. This was further revealed in an assay for imprecise NHEJ in which the DNA end binding defective mutants could be distinguished clearly from Ku-deficient strains. The yeast strain used in this assay contains an HO endonuclease cleavage site at the mating type (MAT) locus but lacks HML and HMR donor sequences, which are required for the normal homology directed repair of HO-cleaved MAT [44]. The expression of HO endonuclease, and, hence, the induction of a DSB, is under the control of a galactose-inducible promoter. Because of the absence of a homologous donor sequence, only NHEJ can repair a DSB created by HO at MAT in this strain. When cells are plated on galactose-containing medium, HO is constitutively expressed and available to re-cleave any accurately repaired cleavage sites, precluding cell growth. However, when the HO cleavage site is altered by an imprecise end-joining event, a colony can form. This imprecise NHEJ occurs in approximately 0.1% of plated WT cells [44].
Previous studies have demonstrated that colony survival in the presence of constitutively expressed HO (i.e., imprecise NHEJ) is reduced to 0.0001% of cells plated in the absence of Ku [44]; therefore, we expected to observe little colony formation when the DNA end binding defective Ku mutations were examined in this assay. However, unlike the yku70-Δ strain, the yku70-R456E strain gave rise to ample colonies when grown on galactose (Figure 8A, +HO). This strain also was engineered to test whether the yku70-R456E mutation impacted cell viability in the absence of TLC1 (and, therefore, telomerase), which requires Ku's telomere end protection function [40]. In contrast to the result of the imprecise NHEJ assay, the effect of yku70-R456E mutation on survival in the absence of telomerase was similar to that of a yku70-Δ mutation because both resulted in synthetic lethality when combined with a tlc1-Δ mutation (Figure 8A, tlc1-Δ). Thus, the seemingly WT behavior of the yku70-R456E mutant in the HO assay (versus the more null-like behavior in the telomere function assays mentioned above) was not due to a difference in the general requirement for Ku in the different strain backgrounds.

We noted that the yku70-R456E mutants appeared to grow more robustly than WT when plated as serial dilutions on galactose-containing medium (Figure 8A, +HO). To confirm this, as well as to test the yku70-p1 yku80-p1 double panel mutant for this phenotype, we repeated the assay in a strain containing an intact chromosomal copy of TLC1. We consistently found that both DNA end binding defective mutants displayed greater than WT number of colonies on galactose-containing medium (Figure 8B). The yku70-Δ mutant, in contrast, showed minimal growth on galactose. Additionally, as with the telomeric assays, yku70-p1 and yku80-p1 single mutant strains behaved like WT with comparable levels of survival (Figure 8B). Therefore, the phenotype of the DNA end binding defective mutants was different not only from the yku70-Δ strain, but also from WT. Taken together, these results provide phenotypic evidence that argue against the telomeric phenotypes simply being a reflection of a generalized Ku deficiency.
Discussion
We found that the singular loss of Ku's ability to bind DNA ends had dramatic effects on its association with telomeric chromatin and its telomeric functions. These results demonstrate that Ku predominantly associates with telomeres via direct DNA end binding and that DNA end binding is required for Ku to carry out its various telomeric functions. Thus, Ku is an integral component of the telomere cap.
So, what is involved in this integral association between Ku and telomeric DNA? The phenotypes seen with the DNA end binding defective mutants and the position of these mutations within the Ku heterodimer provide clues that address this question. There appear to be at least two distinct steps involved: the initial loading of Ku onto the telomeric end, and the stabilization of Ku at telomeres. For example, the yku70-R456E mutant contains a single amino acid substitution on the loading side of the central channel (Figure 1). More telomeric chromatin is associated with Yku70-R456E Yku80 heterodimers (Figure 4) than one would expect based upon in vitro DNA end binding assays with recombinant protein (Figure 2B). This is likely due to the differences in substrates in the two assays – naked DNA fragments versus telomeres, and these data suggest that, once this mutant Ku heterodimer is able to load onto telomeric ends in vivo, a fraction can be stabilized. In vitro, this mutant protein is not as stably bound to DNA as WT, as suggested by the excessive smearing seen in the EMSA (Figure 2B). The synergistic loss of telomeric function and synergistic increase in imprecise NHEJ observed with the yku70-K422E yku80-p1 double versus the yku70-K422E and yku80-p1 single mutants (Figure 5 and Figure S4) also points to factors influencing the stabilization of Ku's binding to DNA ends in vivo relative to the in vitro assays.
The loss of Ku's ability to bind DNA ends affected each of the telomeric functions examined in this study. Least surprisingly, loss of DNA end binding resulted in a telomere end protection defect, with an EXO1-dependent increase in G-overhang DNA as observed in yku-Δ strains (Figure 5 and Figure S6). These data are consistent with Ku's DNA end binding contributing to end protection through direct steric hindrance of Exo1. However, the mechanism of Ku-mediated telomere end protection is likely to be more complex. Despite the association of Yku80 (and presumably Yku70) with telomeres throughout the cell cycle [17], its role in the protection of de novo telomeres is limited to G1 [45], a time when the Cdc13-Stn1-Ten1 (CST) complex is dispensable [46]. This may be the case at native telomeres as well [45]. Thus, Ku or telomeres may be altered as cells progress through S phase, after which telomeres no longer require Ku, but rather depend on the CST complex, as well as Rap1, Rif1 and Rif2, for their end protection [45], [46]. That end binding is required for Ku's role in telomere end protection is also consistent with the observation that Ku is required to prevent the initiation of telomere resection [45].
Ku has been shown to repress recombination near telomeres. It has been proposed that this repression is mediated through Ku's promotion of a telomere fold-back architecture via interactions with the subtelomeric core X element [47], [48]. Such an architecture may also serve to protect telomeres from end resection. Such a structure is likely to unfold with progression through S phase and telomere replication, perhaps limiting its role to G1. This opening of the fold-back structure is consistent with the greater amount of sub-telomeric DNA found in Yku80 ChIPs from cells in G1 compared to the amount seen after cells have entered S phase [17]. Determining whether the DNA end binding Ku mutants retain their interaction with core X and/or repress recombination near telomeres will be important toward our understanding of the significance of Ku – core X interactions.
In contrast to a defect in telomere end protection, the defect in telomeric silencing observed in the DNA end binding defective mutants (Figure 7A) was not necessarily expected for two reasons. First, Ku does not impact on Sir4′s association with telomeric repeat-associated chromatin, but rather on its association with subtelomeric nucleosomal chromatin [33]. Second, the association of Ku with subtelomeric chromatin is diminished in the absence of Sir4, suggesting that Ku is recruited to the subtelomere via its association with Sir4 [25]. How, then, might Ku's association with the very end affect subtelomeric chromatin? The answer to this question may also relate to a role for Ku in mediating a fold-back structure at telomeres and the importance of this structure for the formation or maintenance of silent telomeric chromatin [47]–[50]. We note that, although, the truncated Tel V-R and Tel VII-L telomeres used in our silencing assays lack core X elements, Ku has been shown to associate with non-core X sequences near telomeres and has core X-independent effects on recombination repression [47], [48].
Ku also contributes to the tethering of some telomeres to the nuclear periphery, a phenomenon that contributes to telomeric silencing [43], [51]. Thus, it is possible (and indeed likely) that the Ku DNA end binding mutations, which alter Ku's association with telomeres, also impact on the localization of some telomeres to the nuclear envelope. Therefore, might the effects these mutations have on telomeric silencing be a consequence of loss of telomere perinuclear localization? Although we can not eliminate this possibility at those native telomeres that are subjected to both Ku-mediated localization and silencing, the perinuclear anchoring of the truncated Tel V-R and Tel VII-L telomeres used in our telomeric silencing assays is not Ku-dependent [38], [52]. Therefore, the results from this assay reveal that the silencing defects observed in DNA end binding defective mutants are independent of Ku's role in nuclear tethering of telomeres.
The short telomere phenotype seen in Ku DNA end binding defective mutants suggests that the association of Ku with telomeric ends is also essential for Ku's ability to positively regulate telomere length. Ku-TLC1 association was not significantly diminished in the DNA end binding defective mutants (Figure 3B), arguing against this simple explanation for the short telomeres in these mutants (Figure 6A). Moreover, yku70-R456E mutants had telomeres that were significantly shorter than the telomeres present in tlc1-Δ48 mutants (Figure 6B). These results indicate that binding TLC1 (and presumably telomerase) alone is insufficient and that end binding is essential for Ku-mediated telomere length regulation. Deletion of EXO1, while suppressing the increased ss G-strand telomeric DNA, had only minor effects on the short telomere phenotype, arguing against the loss of end protection in the end binding defective mutants as the primary contributor to the loss of telomere length (Figure 6C). Therefore, we conclude that the direct association of Ku with the very end is required for Ku to contribute to telomere length maintenance.
By targeting residues in Ku that were predicted to contact DNA, we generated Ku heterodimers that were defective for DNA end binding yet proficient for TLC1 association in vivo. These mutants are the reverse complement of the yku80-135i mutant, which is defective for its ability to bind TLC1 but fully competent for binding DNA ends [24]. These data suggest that Ku's association with TLC1 does not involve the central channel and that Ku has separate determinants for binding DNA ends and TLC1. Whether Ku can simultaneously bind telomeric DNA and TLC1 RNA has yet to be determined and would have implications for molecular models of how Ku influences telomere length regulation.
We observed that strains that were defective in DNA end binding did not phenocopy Ku-deficient strains in an assay for imprecise end-joining (Figure 8). This result supports the conclusion that the telomere defects in these strains were not due to a generalized deficiency in function. Quantification of survival under constant DSB-inducing conditions, and subsequent sequencing of the repaired junctions, revealed that the DNA end binding defective mutants exhibited an increase in survival relative to WT and that the repaired junctions were distinct from junctions observed in WT and null strains (Figure 8 and data not shown). Thus, these mutants exhibit a novel phenotype. This likely relates to the fact that, in contrast to yku70-Δ or yku80-Δ strains, Ku is still present in these mutants; in contrast to WT strains, in which Ku is also present, the Ku that is present exhibits markedly reduced binding to DSBs. How the presence of these mutant proteins might result in more efficient re-ligation through an error-prone end-joining pathway is unknown. One possibility is that they have retained not only their interactions with telomeric factors (such as Sir4 and TLC1 as we have shown), but also with NHEJ factors. Consequently, DNA end binding defective Ku heterodimers might titrate one or more factors away from DSBs, which results in an increase in Ku-independent error-prone NHEJ.
Our findings show that Ku must access and load onto telomeric ends in order for it to participate in its various telomeric functions in S. cerevisiae. It will be interesting to determine whether Ku's end binding property is also required at human telomeres. Ku in human cells has been shown to prevent the rapid loss of telomeric repeats and the generation of so-called t-circles (extrachromosomal circular DNA containing telomeric repeats) [53]. Perhaps in human cells, Ku plays a role in stabilizing the t-loop structure, similar to its proposed role in maintaining the protective fold-back architecture of yeast telomeres.
Materials and Methods
Strains and plasmids
The strains and plasmids used in this study are described in Table S1 and Table S2, respectively. Strains lacking YKU70 and/or YKU80 were constructed using one-step allele replacement with the indicated selection marker. YAB285, YAB438 and YAB439 were constructed by integrating yku70-R456E, yku70-p1 and yku80-p1 into YTSF79 [17] by pop-in/pop-out technique. Mutations were introduced into YKU70 and YKU80 containing plasmids using oligonucleotide single-stranded mutagenesis as described [40].
Electrophoretic mobility shift assays (EMSA)
For EMSA using yeast WCEs, cell lysates were prepared, and EMSA was performed using a modification of a previously published protocol [41]. Five ml cultures were lysed with glass beads in lysis buffer [420 mM KCl, 42 mM HEPES, 4.2 mM EDTA, 0.1 mM dithiothreitol (DTT)]. Approximately 10–15 µg of protein from each cell lysate was incubated with (1.5 ng) 32P end-labeled Bgl II – Nru I fragment (198 bp) of pcDNA3.1 (Invitrogen) for 15 minutes at room temperature, along with 1 µg unlabeled closed circular pcDNA3.1. The samples were loaded onto a 5% polyacrylamide gel in Tris-glycine buffer and run at 30 mA for approximately 45 minutes. The gel was dried and exposed to film or a phosphorimager screen and analyzed using a Storm865 imaging system (Molecular Dynamics). Quantitation of the phosphorimager image was done using ImageQuant software.
For EMSA using recombinant Ku, binding reactions were prepared containing 20 mM Tris-HCl pH 8, 50 mM NaCl, 1 mM DTT, 5 mM MgCl2, 0.5 mM EDTA, 100 µg/ml BSA, 11% glycerol, 1.5 ng of 32P - labeled DNA (as used for the yeast WCE EMSA), and the indicated amounts of purified recombinant Ku. Reactions were incubated for 15 minutes at room temperature. The reactions were loaded onto a 5% polyacrylamide gel, which was prerun at 40 mA for 30 minutes. Electrophoresis was carried out in Tris-glycine buffer at 40 mA for 100 minutes. The gel was dried and analyzed as above.
Expression and purification of recombinant yeast Ku
Ku70, Ku70-R456E and Ku80 were individually subcloned into pFastBac HT A (Invitrogen) opened with NcoI and NotI. An N-terminal 6xHis tag followed by a linker region and a TEV protease recognition sequence precedes the multiple cloning site.
Individual recombinant baculoviruses expressing either Ku70, Ku70-R456E or Ku80 were made from the bacmids and used to infect Sf9 cells by the Baylor Baculovirus Core. Sf9 cell stocks (Invitrogen) were maintained as suspension cultures in spinner vessels prior to infection. Sf9 cells were co-infected at a MOI of 1 for each recombinant virus for 48 hours in a 1 liter spinner vessel in 500 ml of Grace's Insect Medium (Invitrogen) + 10% FBS at a density of 1.0×106 cells/ml. At 48 hours post-infection, the cells were centrifuged at 800× g for 10 minutes, washed one time in PBS, and the cell pellets were immediately placed in a −80°C freezer for storage.
Cells were disrupted by the addition of lysis buffer (50 mM phosphate buffer pH 8.0, 300 mM NaCl, 0.05% Tween 20, 5 mM β-mercaptoethanol, 10 mM imidazole). The lysates were incubated on ice for 30–60 minutes and then sonicated on ice using a Misonix sonicator probe. The lysate was cleared by ultracentrifugation at 26K rpm, 4°C for 15 minutes and then incubated with Ni-NTA agarose (Qiagen) rotating for 2 hours at 4°C. The resin was then washed twice in buffer (50 mM phosphate buffer pH 8.0, 300 mM NaCl, 5 mM β-mercaptoethanol, 20 mM imidazole). Recombinant Ku was eluted in buffer (50 mM phosphate buffer pH 8.0, 300 mM NaCl, 5 mM β-mercaptoethanol, 250 mM imidazole). The Ku containing eluates were identified by SDS-polyacrylamide gel electrophoresis (PAGE) and Coomassie stain, then combined and concentrated on an 50NMDL Amicon filter (Millipore). The Ku retentate was then put over a Superdex 200 16/60 sizing column and Ku dimer containing eluates were concentrated on a 50NMDL Amicon filter (Millipore). The purity of the sample was verified by SDS-PAGE and silver staining. To remove the N-terminal 6xHis tag, Ku was incubated at 16°C overnight with AcTEV protease (Invitrogen) in buffer [50 mM Tris-HCl pH 8.0, 300 mM NaCl, 0.5 mM EDTA, 1 mM DTT]. The digested sample was diluted 10-fold with buffer (50 mM Tris pH 8 and 300 mM NaCl) and then loaded again onto a Ni-NTA column. The Ku containing fractions were combined and concentrated via a 50NMDL Amicon filter (Millipore) in 50 mM Tris-HCl pH 8.0, 300 mM NaCl. Storage buffer (0.5 mM EDTA, 1 mM DTT, 40% glycerol) was added and Ku was then aliquoted, flash frozen and stored at −80°C.
Heterodimerization assays
YAB226 (yku70-Δ yku80-Δ) was co-transformed with FLAG-tagged versions of YKU70, yku70-p1, yku70-R456E, or yku70-K422E and myc-tagged versions of YKU80 or yku80-p1. Fifty ml asynchronous cultures were lysed with glass beads in TMG-200 (10 mM Tris-HCl, pH 8.0, 1 mM MgCl2, 10% [v/v] glycerol, 0.1 mM DTT, 0.1 mM EDTA plus 200 mM NaCl). Forty µl of anti-FLAG agarose (Sigma A220) was added to 150 µg of protein extract. After 3 hours of incubation at 4°C, the beads were washed in TMG-200 plus 0.5% Tween and resuspended in 40 µl TMG. Input and IP samples were subjected to SDS-PAGE on 10% gels and analyzed by western blotting with anti-myc (9E10, Sigma M4439) and anti-PGK (Molecular Probes A6457) primary antibodies and IRDye 800CW conjugated goat anti-mouse secondary antibody (LiCor). Fluorescence was visualized using the LiCor Odyssey® Infared Imaging System. After quantitation was performed using ImageQuant software (Molecular Dynamics), the membrane was stripped and re-probed with anti-FLAG (M2, Sigma F3165).
Yeast two-hybrid assays
YAB327 is a yku70-Δ derivative of EGY48 [20], which was grown on 5-FOA-containing media to select against the presence of the URA3-containing LacZ reporter plasmid. YAB327 was transformed with YKU70 (pVL1874), yku70-R456E (pAB558), yku70-p1 (pAB608), yku70-K422E (pAB710) or pRS416 and then a pEG202-based BAIT plasmid containing YKU80 (pEGku80) or yku80-p1 (pAB599) and a pJG4-5-based PREY vector containing full-length Sir4 (pB42AD::SIR4). Interaction between the BAIT and PREY constructs was determined by growth on Gal –Leu plates.
TLC1 co-immunoprecipitation, RNA isolation, and quantitative RT-PCR
Procedures were performed using modified versions of described protocols [17], [54], [55]. Fifty mL cultures (A600 = 0.8–1.0) were lysed in TMG-50 using silica beads, and extracts containing approximately 4 mg protein were prepared. For samples immunoprecipitated with anti-myc antibody (9E10, Sigma M4439), Protein G beads (EMD Chemicals) were added after they were incubated overnight at 4°C. Both FLAG conjugated beads (Sigma A220) and Protein G beads were washed in TMG-50 plus 0.5% Tween before being resuspended in 50 µl TMG-50. The resuspended beads (and 50 µl aliquots of the inputs) were added to 350 çl of Proteinase K solution and incubated at 37°C for 30 minutes. Following phenol:chloroform extraction and ethanol precipitation, nucleic acids were resuspended in 100 µl RNase free water. The RNA was subjected to RNA cleanup and on-column DNase I treatment using the RNeasy kit (Qiagen). Two µg of input RNA (11 µl of IP RNA) was used as a template for cDNA synthesis using the Flex cDNA synthesis kit (Quanta Biosciences). Quantitative PCR was performed on 5 µl of the cDNA synthesis reaction using the Perfecta SYBR Green FastMix, ROX (Quanta Biosciences) on an ABI7300 real time PCR system (Applied Biosystems). Actin and TLC1 primers used were the same as described [54].
ABI7300 software was used to determine threshold and CT values. CT values were averaged and plotted using MS Excel. Percent input was determined by multiplying 100 by 2 (Adjusted input CT – IP CT), where adjusted input CT = Input CT - log2 (dilution factor).
Chromatin immunoprecipitation (ChIP) assays
Fifty ml cultures of YTSF23 (YKU70 YKU80) [17], YTSF79 (YKU70 YKU80myc18) [17], YAB285 (yku70-R456E YKU80myc18), YAB438 (yku70-p1 YKU80myc18) and YAB439 (yku70-p1 yku80-p1myc18) were grown at 28°C to an A600 = 0.8–1.0. Formaldehyde crosslinking was carried out, as described by Aparicio [56]. Cell pellets were washed with 1 M sorbitol, resuspended in approximately 5 ml zymolyase buffer (1 M sorbitol, 50 mM Tris, 10 mM β-ME), and incubated with 125 µl zymolyase (50 mg/ml 20T zymolyase) for 1 hour at 28°C, as described [57]. Spheroplasts were pelleted and resuspended in approximately 500 µl NP-S buffer [57] before brief (2 minutes) glass bead lysis to further disrupt cells. The pellets were re-suspended in FA-lysis buffer [56] and sonicated with a Misonix Sonicator 3000 (power setting 5, 90 sec, 3 cycles). Total protein levels were adjusted so that equal amounts of protein were present in each sample. Five µl of 9E10 (Sigma) were added to IP samples and incubated overnight at 4°C. Protein G agarose beads (Calbiochem IP04) were equilibrated with lysis buffer, added to IP samples, and incubated at 4°C for 3 hours. Beads were washed at room temperature according to Aparacio, et al. [56]. Crosslinks were reversed, and the DNA was ethanol precipitated. The DNA fragments (∼500 bp) were re-suspended in 50 µl of TE and brought up to 300 µl with a 0.4N NaOH, 10 mM EDTA solution. The samples were heated to 100°C for 10 minutes before being blotted onto a Hybond XL nylon membrane (Amersham) using a dot blot manifold (Schleicher & Schuell). The blot was probed with a telomere-specific, end-labeled oligonucleotide (AB766) or a randomly labeled 3.2 kb DNA fragment containing TyB sequence excised from plasmid pAB126. After excess probe was washed away, the blot was exposed to phosphorimager screen for quantitation.
To monitor for efficiency of immunoprecipitation, 50 µl aliquots of the Input and IP samples were run on a 10% polyacrylamide gel, transferred to a nitrocellulose membrane, and probed with anti-myc (9E10, Sigma M4439) and anti-PGK (Molecular Probes A6457) primary antibodies and IRDye 800CW conjugated goat anti-mouse secondary antibody (LiCor).
Telomere Southern blots and telomeric G-strand overhang assays
Southern blot analysis of telomeres and telomeric G-strand overhang assays were done as described previously [34], [58].
Telomeric silencing assays
Plasmids containing WT or mutant yku70 and yku80 alleles or empty vector controls were co-transformed into YAB219 (yku70-Δ yku80-Δ) and YAB353 (yku70-Δ yku80-Δ exo1-Δ). Cultures were grown to mid-log phase in –Trp –Leu media. Five-fold serial dilutions were spotted onto –Trp –Leu, –Trp –Leu –Ura, and –Trp –Leu low ade plates, to monitor the expression of URA3 and ADE2. Growth was determined after 2 to 4 days at 28°C. Temperature sensitivity was determined by plating the serial dilutions onto –Trp –Leu plates and incubating at 37°C for 2 to 4 days.
Error-prone NHEJ assay
Plasmids containing yku70 and/or yku80 alleles or empty vector controls were transformed into YAB199 (yku70-Δ), YAB198 (yku70-Δ) or YAB273 (yku70-Δ yku80-Δ). Five-fold serial dilutions of asynchronous cultures were spotted onto –Trp and –Trp Gal plates (for YAB198 and YAB199) –Trp –Leu and –Trp –Leu Gal plates (YAB273). For single colony experiments, asynchronous cultures were diluted and 100 µl were spread onto these sets of plates. Colonies were counted after 2 to 4 days of incubation at 28°C.
Statistical analyses
A student's T-test was used to determine whether the differences in values shown on bar graphs were statistically significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. LingerBRPriceCM 2009 Conservation of telomere protein complexes: shuffling through evolution. Crit Rev Biochem Mol Biol 44 434 446
2. PalmWde LangeT 2008 How shelterin protects mammalian telomeres. Annu Rev Genet 42 301 334
3. De LangeT 2005 Telomere-related genome instability in cancer. Cold Spring Harb Symp Quant Biol 70 197 204
4. KonigPGiraldoRChapmanLRhodesD 1996 The crystal structure of the DNA-binding domain of yeast RAP1 in complex with telomeric DNA. Cell 85 125 136
5. CourtRChapmanLFairallLRhodesD 2005 How the human telomeric proteins TRF1 and TRF2 recognize telomeric DNA: a view from high-resolution crystal structures. EMBO Rep 6 39 45
6. Mitton-FryRMAndersonEMHughesTRLundbladVWuttkeDS 2002 Conserved structure for single-stranded telomeric DNA recognition. Science 296 145 147
7. HorvathMPSchweikerVLBevilacquaJMRugglesJASchultzSC 1998 Crystal structure of the Oxytricha nova telomere end binding protein complexed with single strand DNA. Cell 95 963 974
8. CockellMPalladinoFLarocheTKyrionGLiuC 1995 The carboxy termini of Sir4 and Rap1 affect Sir3 localization: evidence for a multicomponent complex required for yeast telomeric silencing. J Cell Biol 129 909 924
9. YeJZDonigianJRvan OverbeekMLoayzaDLuoY 2004 TIN2 binds TRF1 and TRF2 simultaneously and stabilizes the TRF2 complex on telomeres. J Biol Chem 279 47264 47271
10. KimSHBeausejourCDavalosARKaminkerPHeoSJ 2004 TIN2 mediates functions of TRF2 at human telomeres. J Biol Chem 279 43799 43804
11. LeiMPodellERCechTR 2004 Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection. Nat Struct Mol Biol 11 1223 1229
12. LiuDSafariAO'ConnorMSChanDWLaegelerA 2004 PTOP interacts with POT1 and regulates its localization to telomeres. Nat Cell Biol 6 673 680
13. DynanWSYooS 1998 Interaction of Ku protein and DNA-dependent protein kinase subunits with nucleic acids. Nucleic Acids Res 26 1551 1559
14. WalkerJRCorpinaRAGoldbergJ 2001 Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 412 607 614
15. GravelSLarriveeMLabrecquePWellingerRJ 1998 Yeast Ku as a regulator of chromosomal DNA end structure. Science 280 741 745
16. WuTJChiangYHLinYCTsaiCRYuTY 2009 Sequential loading of Saccharomyces cerevisiae Ku and Cdc13p to telomeres. J Biol Chem 284 12801 12808
17. FisherTSTaggartAKZakianVA 2004 Cell cycle-dependent regulation of yeast telomerase by Ku. Nat Struct Mol Biol 11 1198 1205
18. GriffithJDComeauLRosenfieldSStanselRMBianchiA 1999 Mammalian telomeres end in a large duplex loop. Cell 97 503 514
19. de LangeT 2009 How telomeres solve the end-protection problem. Science 326 948 952
20. RoyRMeierBMcAinshADFeldmannHMJacksonSP 2004 Separation-of-function mutants of yeast Ku80 reveal a Yku80p-Sir4p interaction involved in telomeric silencing. J Biol Chem 279 86 94
21. O'ConnorMSSafariALiuDQinJSongyangZ 2004 The human Rap1 protein complex and modulation of telomere length. J Biol Chem 279 28585 28591
22. HsuHLGilleyDGalandeSAHandeMPAllenB 2000 Ku acts in a unique way at the mammalian telomere to prevent end joining. Genes Dev 14 2807 2812
23. SongKJungDJungYLeeSGLeeI 2000 Interaction of human Ku70 with TRF2. FEBS Lett 481 81 85
24. StellwagenAEHaimbergerZWVeatchJRGottschlingDE 2003 Ku interacts with telomerase RNA to promote telomere addition at native and broken chromosome ends. Genes Dev 17 2384 2395
25. MartinSGLarocheTSukaNGrunsteinMGasserSM 1999 Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell 97 621 633
26. MaringeleLLydallD 2002 ExoI-dependent single-stranded DNA at telomeres activates subsets of DNA damage and spindle checkpoint pathways in budding yeast yku70Δ mutants. Genes Dev 16 1919 1933
27. BertuchAALundbladV 2004 EXO1 contributes to telomere maintenance in both telomerase-proficient and telomerase-deficient Saccharomyces cerevisiae. Genetics 166 1651 1659
28. ZhuZChungWHShimEYLeeSEIraG 2008 Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134 981 994
29. MimitouEPSymingtonLS 2008 Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455 770 774
30. BoultonSJJacksonSP 1998 Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. EMBO J 17 1819 1828
31. GottschlingDEAparicioOMBillingtonBLZakianVA 1990 Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell 63 751 762
32. MishraKShoreD 1999 Yeast Ku protein plays a direct role in telomeric silencing and counteracts inhibition by rif proteins. Curr Biol 9 1123 1126
33. LuoKVega-PalasMAGrunsteinM 2002 Rap1-Sir4 binding independent of other Sir, yKu, or histone interactions initiates the assembly of telomeric heterochromatin in yeast. Genes Dev 16 1528 1539
34. BertuchAALundbladV 2003 The Ku heterodimer performs separable activities at double strand breaks and chromosome termini. Mol Cell Biol 23 8202 8215
35. HedigerFNeumannFRVan HouweGDubranaKGasserSM 2002 Live imaging of telomeres: yKu and Sir proteins define redundant telomere-anchoring pathways in yeast. Curr Biol 12 2076 2089
36. TaddeiAHedigerFNeumannFRBauerCGasserSM 2004 Separation of silencing from perinuclear anchoring functions in yeast Ku80, Sir4 and Esc1 proteins. EMBO J 23 1301 1312
37. SchoberHFerreiraHKalckVGehlenLRGasserSM 2009 Yeast telomerase and the SUN domain protein Mps3 anchor telomeres and repress subtelomeric recombination. Genes Dev 23 928 938
38. ThamWHWyitheJSFerrignoPKSilverPAZakianVA 2001 Localization of yeast telomeres to the nuclear periphery is separable from transcriptional repression and telomere stability functions. Mol Cell 8 189 199
39. DrillerLWellingerRJLarriveeMKremmerEJaklinS 2000 A short C-terminal domain of Yku70p is essential for telomere maintenance. J Biol Chem 275 24921 24927
40. Ribes-ZamoraAMihalekILichtargeOBertuchAA 2007 Distinct faces of the Ku heterodimer mediate DNA repair and telomeric functions. Nat Struct Mol Biol 14 301 307
41. MilneGTJinSShannonKBWeaverDT 1996 Mutations in two Ku homologs define a DNA end-joining repair pathway in Saccharomyces cerevisiae. Mol Cell Biol 16 4189 4198
42. NugentCIBoscoGRossLOEvansSKSalingerAP 1998 Telomere maintenance is dependent on activities required for end repair of double-strand breaks. Curr Biol 8 657 660
43. LarocheTMartinSGGottaMGorhamHCPrydeFE 1998 Mutation of yeast Ku genes disrupts the subnuclear organization of telomeres. Curr Biol 8 653 656
44. LeeSEPaquesFSylvanJHaberJE 1999 Role of yeast SIR genes and mating type in directing DNA double-strand breaks to homologous and non-homologous repair paths. Curr Biol 9 767 770
45. BonettiDClericiMAnbalaganSMartinaMLucchiniG 2010 Shelterin-like proteins and Yku inhibit nucleolytic processing of Saccharomyces cerevisiae telomeres. PLoS Genet 6 e1000966 doi:10.1371/journal.pgen.1000966
46. VodenicharovMDWellingerRJ 2006 DNA degradation at unprotected telomeres in yeast is regulated by the CDK1 (Cdc28/Clb) cell-cycle kinase. Mol Cell 24 127 137
47. MarvinMEBeckerMMNoelPHardySBertuchAA 2009 The association of yKu with subtelomeric core X sequences prevents recombination involving telomeric sequences. Genetics 183 453 467, 451SI-413SI
48. MarvinMEGriffinCDEyreDEBartonDBLouisEJ 2009 In Saccharomyces cerevisiae, yKu and subtelomeric core X sequences repress homologous recombination near telomeres as part of the same pathway. Genetics 183 441 451, 441SI-412SI
49. Strahl-BolsingerSHechtALuoKGrunsteinM 1997 SIR2 and SIR4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes Dev 11 83 93
50. PrydeFELouisEJ 1999 Limitations of silencing at native yeast telomeres. EMBO J 18 2538 2550
51. TaddeiAGasserSM 2004 Multiple pathways for telomere tethering: functional implications of subnuclear position for heterochromatin formation. Biochim Biophys Acta 1677 120 128
52. HedigerFBerthiauASvan HouweGGilsonEGasserSM 2006 Subtelomeric factors antagonize telomere anchoring and Tel1-independent telomere length regulation. Embo J 25 857 867
53. WangYGhoshGHendricksonEA 2009 Ku86 represses lethal telomere deletion events in human somatic cells. Proc Natl Acad Sci U S A 106 12430 12435
54. LiSMakovetsSMatsuguchiTBlethrowJDShokatKM 2009 Cdk1-dependent phosphorylation of Cdc13 coordinates telomere elongation during cell-cycle progression. Cell 136 50 61
55. LivengoodAJZaugAJCechTR 2002 Essential regions of Saccharomyces cerevisiae telomerase RNA: separate elements for Est1p and Est2p interaction. Mol Cell Biol 22 2366 2374
56. AparicioOMGeisbergJVSekingerEYangAMoqtaderiZ 2005 Chromatin immunoprecipitation for determining the association of proteins with specific genomic regions in vivo. AusubelFMBrentRKingstonREMooreDDSeidman JG etal Current Protocols in Molecular Biology: John Wiley & Sons, Inc 21.23.21 21.23.17
57. DionMFKaplanTKimMBuratowskiSFriedmanN 2007 Dynamics of replication-independent histone turnover in budding yeast. Science 315 1405 1408
58. LendvayTSMorrisDKSahJBalasubramanianBLundbladV 1996 Senescence mutants of Saccharomyces cerevisiae with a defect in telomere replication identify three additional EST genes. Genetics 144 1399 1412
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 8
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- An EMT–Driven Alternative Splicing Program Occurs in Human Breast Cancer and Modulates Cellular Phenotype
- Chromosome Painting Reveals Asynaptic Full Alignment of Homologs and HIM-8–Dependent Remodeling of Chromosome Territories during Meiosis
- A Pre-mRNA–Associating Factor Links Endogenous siRNAs to Chromatin Regulation
- Discovery of Sexual Dimorphisms in Metabolic and Genetic Biomarkers
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy