
Distinct Cdk1 Requirements during Single-Strand Annealing, Noncrossover, and Crossover Recombination
Repair of DNA double-strand breaks (DSBs) by homologous recombination (HR) in haploid cells is generally restricted to S/G2 cell cycle phases, when DNA has been replicated and a sister chromatid is available as a repair template. This cell cycle specificity depends on cyclin-dependent protein kinases (Cdk1 in Saccharomyces cerevisiae), which initiate HR by promoting 5′–3′ nucleolytic degradation of the DSB ends. Whether Cdk1 regulates other HR steps is unknown. Here we show that yku70Δ cells, which accumulate single-stranded DNA (ssDNA) at the DSB ends independently of Cdk1 activity, are able to repair a DSB by single-strand annealing (SSA) in the G1 cell cycle phase, when Cdk1 activity is low. This ability to perform SSA depends on DSB resection, because both resection and SSA are enhanced by the lack of Rad9 in yku70Δ G1 cells. Furthermore, we found that interchromosomal noncrossover recombinants are generated in yku70Δ and yku70Δ rad9Δ G1 cells, indicating that DSB resection bypasses Cdk1 requirement also for carrying out these recombination events. By contrast, yku70Δ and yku70Δ rad9Δ cells are specifically defective in interchromosomal crossover recombination when Cdk1 activity is low. Thus, Cdk1 promotes DSB repair by single-strand annealing and noncrossover recombination by acting mostly at the resection level, whereas additional events require Cdk1-dependent regulation in order to generate crossover outcomes.
Published in the journal:
. PLoS Genet 7(8): e32767. doi:10.1371/journal.pgen.1002263
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002263
Summary
Repair of DNA double-strand breaks (DSBs) by homologous recombination (HR) in haploid cells is generally restricted to S/G2 cell cycle phases, when DNA has been replicated and a sister chromatid is available as a repair template. This cell cycle specificity depends on cyclin-dependent protein kinases (Cdk1 in Saccharomyces cerevisiae), which initiate HR by promoting 5′–3′ nucleolytic degradation of the DSB ends. Whether Cdk1 regulates other HR steps is unknown. Here we show that yku70Δ cells, which accumulate single-stranded DNA (ssDNA) at the DSB ends independently of Cdk1 activity, are able to repair a DSB by single-strand annealing (SSA) in the G1 cell cycle phase, when Cdk1 activity is low. This ability to perform SSA depends on DSB resection, because both resection and SSA are enhanced by the lack of Rad9 in yku70Δ G1 cells. Furthermore, we found that interchromosomal noncrossover recombinants are generated in yku70Δ and yku70Δ rad9Δ G1 cells, indicating that DSB resection bypasses Cdk1 requirement also for carrying out these recombination events. By contrast, yku70Δ and yku70Δ rad9Δ cells are specifically defective in interchromosomal crossover recombination when Cdk1 activity is low. Thus, Cdk1 promotes DSB repair by single-strand annealing and noncrossover recombination by acting mostly at the resection level, whereas additional events require Cdk1-dependent regulation in order to generate crossover outcomes.
Introduction
DNA double-strand breaks (DSBs) occur spontaneously during DNA replication and after exposure to certain genotoxic chemicals or ionizing radiation. Efficient repair of DSBs can be accomplished by nonhomologous end joining (NHEJ), which directly rejoins broken DNA ends, or by homologous recombination (HR), which utilizes a homologous DNA template to restore the genetic information lost at the break site (reviewed in [1]–[3]). Failure to repair DSBs can lead to genome instability and cell death.
HR is initiated by 5′-3′ nucleolytic degradation of the DSB ends to yield 3′-ended single-stranded DNA (ssDNA) tails. Replication protein A (RPA) binds to the ssDNA tails to remove their secondary DNA structures, but is then replaced by Rad51 aided by Rad52. Once formed, the Rad51 nucleofilaments search for homologous sequences and then promote invasion of the ssDNA into homologous donor double-stranded DNA to form a joint molecule with a displaced strand (D-loop) (reviewed in [1]–[3]). Following strand invasion, the 3′ end of the invading strand primes DNA synthesis using the donor sequence as a template, thus restoring those residues that were lost by resection [4].
According to the canonical double-strand break repair (DSBR) model [5], the displaced strand of the D-loop can anneal with the complementary sequence on the other side of the break (second end capture) to form a double Holliday junction (dHJ) intermediate. Random cleavage of the two HJs is expected to yield an equal number of noncrossover and crossover products. This DSBR model predicts that both crossover and noncrossover products derive from dHJ resolution. However, the finding that most DSB repair in somatic cells is not associated with crossovers [6] led to alternative models for noncrossover generation. In one of them, the action of helicases mediates the convergent branch migration of the two HJs, thus producing a hemicatenane structure that is decatenated to form exclusively noncrossover products [7]–[9]. A second mechanism, termed synthesis-dependent strand annealing (SDSA), leads to displacement of the invading strand that has been extended by DNA synthesis and that anneals with the complementary sequences exposed by 5′-3′ resection [10]–[12]. Because no HJ is formed, only noncrossover products are made. Interestingly, during meiotic recombination, where dHJ resolution into crossovers is essential to drive segregation of homologs to opposite poles, most crossovers are thought to arise via dHJ resolution, whereas noncrossovers form mostly by the SDSA pathway [13], [14].
When a DSB is flanked by direct repeats, its repair primarily occurs by single-strand annealing (SSA). Here, the resected DSB ends anneal with each other instead of invading a homologous DNA sequence (reviewed in [1]–[3]). Subsequent nucleolytic removal of the protruding single-stranded tails results in deletion of the intervening DNA sequence and one of the repeats. In principle, such a break can also be repaired by break-induced replication (BIR), where the repeat closer to the cut site can strand-invade the repeat that is further away and set up a recombination-dependent replication fork to copy all the distal sequences. However, SSA usually out-competes BIR, which is a kinetically slow process [15].
All the above HR pathways require 5′-3′ nucleolytic degradation of DNA ends and the strand-annealing activity of Rad52. In addition, DSBR, SDSA and BIR require the Rad51 protein, which is dispensable for SSA that does not involve strand invasion [16].
In Saccharomyces cerevisiae haploid cells, mitotic HR is generally restricted to the S and G2 phases of the cell cycle, when DNA has been replicated and a sister chromatid is available as an appropriate donor [17], [18]. This cell-cycle specificity depends on cyclin-dependent kinases (Cdks; Cdk1 in S. cerevisiae), which promote resection of the 5′ DSB ends to yield 3′-ended ssDNA tails that are necessary to initiate HR [17], [18]. End resection occurs through a biphasic mechanism: first the MRX complex and Sae2 clip 50–100 nucleotides from the 5′ DNA ends; then Exo1 or Sgs1-Top3-Rmi1 and Dna2 process the early intermediate to form extensive regions of ssDNA (reviewed in [19], [20]). The Sae2 protein has been shown to be a Cdk1 target in promoting ssDNA generation at DNA ends during both mitosis and meiosis [21], [22]. However, as Sae2 only resects a relatively small amount of DNA and other nucleases and helicases are required for efficient DSB resection, Cdk1 likely has additional targets in promoting this event.
Indeed, DSB end resection is also negatively regulated by the Yku heterodimer [23], [24] and by the checkpoint protein Rad9 [25], [26]. Interestingly, the ends of an endonuclease-induced DSB are resected in the G1 phase of the cell cycle (low Cdk1 activity) when Yku is lacking [24]. Moreover, RAD9 deletion allows DSB resection in G2 cells that display low Cdk1 activity due the overexpression of the Cdk1 inhibitor Sic1 [26]. These findings indicate that Cdk1 requirement for DSB resection is bypassed when the inhibitory function of either Yku or Rad9 is relieved.
Whether Cdk1 promotes other HR events is unknown. Some evidence suggests that HR steps other than DSB resection might be regulated by Cdk1 activity. For example, formation of Rad52 foci after ionizing radiation (IR) is less efficient in G1 than in G2, suggesting that Cdk1 might control Rad52 recruitment to DSBs [27]. Furthermore, Cdk1 targets the Srs2 helicase to dismantle D-loop structures, possibly by counteracting unscheduled Srs2 sumoylation [28]. Proteins implicated in late HR events have also been identified as potential Cdk substrates in other eukaryotes. In particular, human BRCA2 is phosphorylated by Cdks, and this phosphorylation has been proposed to negatively regulate Rad51 recombination activity [29]. Moreover, Cdk1-dependent phosphorylation of the fission yeast checkpoint protein Crb2 stimulates resolution of HR intermediates by the topoisomerase Top3 and the ReqQ helicase Rqh1 [30].
Here, we investigate the role of Cdk1 in homology-dependent repair of a DSB. We show that generation of 3′-ended ssDNA at the DSB ends bypasses Cdk1 requirement for the repair of a DSB by either SSA or noncrossover recombination, indicating that Cdk1 is dispensable for these repair events if DSB resection occurs. By contrast, resection is not sufficient to bypass Cdk1 requirement for generating crossover products. Thus, Cdk1 promotes SSA - and noncrossover-mediated recombination by regulating essentially the resection step, while Cdk1 controls further HR steps in order to allow crossover outcomes.
Results
The lack of Yku70 allows DSB repair by SSA in G1
HR is inhibited in G1 when Cdk1 activity is low, whereas it occurs during S and G2/M cell cycle phases when Cdk1 activity is high [17], [18]. Although it is well known that Cdk1 promotes resection of DSB ends [17], [18], [21], it is still unclear if other HR steps are regulated by Cdk1. To investigate whether DSB resection is the only step controlled by Cdk1 in HR-mediated DSB repair, we asked if generation of ssDNA at the DSB ends is sufficient to allow HR when Cdk1 activity is low. As DSB resection in G1 is inhibited by the Yku heterodimer and YKU70 deletion allows ssDNA generation at DSB ends in G1 cells [24], we asked if yku70Δ cells are capable to carry out HR in G1.
Homology-dependent repair of a DSB made between tandem DNA repeats occurs primarily by SSA [15], which requires DSB resection and re-annealing of RPA-covered ssDNA by the Rad52 protein [1], [31]. This process does not involve strand invasion and is therefore independent of Rad51 [16]. We deleted YKU70 in a strain where tandem repeats of the LEU2 gene are 0.7 kb apart and one of them (leu2::cs) is adjacent to a recognition site for the HO endonuclease (Figure 1A) [32]. The strain also harbors a GAL-HO construct that provides regulated HO expression. Since homology is restricted to only one DSB end (Figure 1A), the HO-induced break cannot be repaired by gene conversion, making SSA the predominant repair mode. HO was expressed by galactose addition to α-factor-arrested cells that were kept arrested in G1 with α-factor for the subsequent 4 hours. Galactose was maintained in the medium in order to permanently express HO, which can recurrently cleave the HO sites eventually reconstituted by NHEJ-mediated DSB repair. Kinetics of DSB repair was evaluated by Southern blot analysis with a LEU2 probe that also allowed following 5′-end resection on each side of the break by monitoring the disappearance of the HO-cut DNA bands. The quality and persistence of the cell cycle arrest was assessed by FACS analysis (Figure 1B) and by measuring Cdk1 kinase activity (Figure 1F). Consistent with the requirement of Cdk1 activity for DSB resection and repair, both the 1.8 kb and 3.2 kb HO-cut band signals remained high throughout the experiment in wild type G1 cells (Figure 1C and 1D), where the 2.9 kb SSA repair product was only barely detectable (Figure 1C and 1E). By contrast, the SSA repair product accumulated in yku70Δ G1 cells (Figure 1C and 1E), where both the 1.8 kb and 3.2 kb HO-cut band signals decreased (Figure 1C and 1D). The ability of yku70Δ cells to carry out SSA does not require Cdk1. In fact, Cdk1 activity, which was present in exponentially growing wild type and yku70Δ cells, dropped to undetectable levels after G1 arrest (time 0) and remained undetectable in both cultures throughout the experiment (Figure 1F). Thus, the lack of Yku allows DSB repair by SSA in G1, suggesting that ssDNA generation is sufficient to bypass Cdk1 requirement for SSA.

SSA-based DNA repair requires degradation of the 5′ DSB ends to reach the complementary DNA sequences that can then anneal. If SSA in yku70Δ G1 cells depends on generation of 3′-ended ssDNA at DSB ends, then failure of resection to reach the homologous distal leu2 sequence should prevent SSA. Interestingly, Cdk1-independent resection takes place in yku70Δ cells, but it is confined to DNA regions closed to the DSB site [24], suggesting that other proteins limit extensive DSB resection in the absence of Yku. We therefore asked whether increasing the distance between the complementary leu2 sequences prevented DSB repair by SSA in yku70Δ G1 cells. To this end, we monitored SSA-mediated repair of an HO-induced DSB in a strain where the donor leu2 sequence was positioned 4.6 kb away from the HO recognition site at leu2::cs (Figure 2A) [32]. HO expression was induced in α-factor-arrested cells that were kept blocked in G1 with α-factor in the presence of galactose (Figure 2B). Consistent with previous findings [24], resection in yku70Δ G1 cells was restricted to DNA regions closed to the break site. In fact, the 2.5 kb HO-cut signal decreased more efficiently in yku70Δ than in wild type G1 cells, whereas similar amounts of the 12 kb HO-cut signal were detectable in both wild type and yku70Δ G1 cells (Figure 2C and 2D). Thus, 5′-3′ nucleolytic degradation in yku70Δ G1 cells failed to proceed beyond the distal leu2 hybridization region. The inability of resection to uncover the homologous distal leu2 sequence prevented DSB repair by SSA in yku70Δ G1 cells. In fact, the 8 kb SSA repair product was only barely detectable in both wild type and yku70Δ G1 cells throughout the experiment (Figure 2C and 2E). By contrast, when a similar experiment was performed in G2-arrested cells (Figure 2B), where the inhibitory function of Yku on DSB resection is relieved [33], [34], the 8 kb SSA repair product was clearly detectable in wild type and yku70Δ cells (Figure 2C and 2E), which both showed also a decrease of the 12 kb HO-cut signals compared to the same strains arrested in G1 (Figure 2C and 2D). Thus, the ability of yku70Δ G1 cells to repair a DSB by SSA depends on the extent of resection.

The lack of Rad9 enhances resection in yku70Δ G1 cells
If ssDNA generation were the limiting step in SSA-mediated DSB repair in G1, then increasing the efficiency/extent of resection should enhance the ability of yku70Δ cells to carry out SSA in G1. The lack of the checkpoint protein Rad9 has been shown to allow DSB resection in G2 cells that displayed low Cdk1 activity due to high levels of the Cdk1 inhibitor Sic1 [26]. Thus, we asked whether the lack of Rad9 enhanced the efficiency of DSB resection in yku70Δ G1 cells. To compare resection efficiency independently of DSB repair, we monitored the appearance of the resection products at an HO-induced DSB generated at the MAT locus (Figure 3B) of G1-arrested (Figure 3A) cells, which were not able to repair this DSB because they lacked the homologous donor sequences HML and HMR [23]. As expected, wild type cells showed very low levels of the 3′-ended resection products (r1 to r5), which instead clearly accumulated in both yku70Δ and yku70Δ rad9Δ cells (Figure 3C and 3D). Moreover, the longest r4 and r5 resection products were detectable in yku70Δ rad9Δ cells 120 minutes earlier than in yku70Δ cells (Figure 3C and 3D), indicating that the lack of Rad9 enhances the resection efficiency of yku70Δ G1 cells. Interestingly, although RAD9 deletion was shown to allow MRX-dependent ssDNA generation in Sic1 overproducing G2 cells [26], rad9Δ G1 cells did not show increased efficiency of DSB resection compared to wild type cells (Figure 3C and 3D). Thus, Rad9 limits extensive resection in yku70Δ cells, but its lack is not sufficient, by itself, to escape the inhibitory effect of Yku on DSB resection in G1.

The lack of Rad9 enhances SSA in yku70Δ G1 cells
Because DSB resection in G1 was more efficient in yku70Δ rad9Δ cells than in yku70Δ cells, we asked whether the lack of Rad9 allows efficient SSA-mediated DSB repair in yku70Δ G1 cells carrying tandem repeats of the LEU2 gene 4.6 kb apart. Indeed, the amount of SSA repair products in G1 was much higher in yku70Δ rad9Δ cells than in wild type, yku70Δ or rad9Δ cells (Figure 4A–4C). Consistent with DSB resection being more extensive in yku70Δ rad9Δ than in yku70Δ G1-arrested cells (Figure 3), the decrease of the 12 kb HO-cut band signal was much more apparent in yku70Δ rad9Δ than in yku70Δ G1 cells, whereas the 2.5 kb HO-cut band signal decreased with similar kinetics in both G1 cell cultures (Figure 4B and 4D). Cdk1 kinase activity, which was present in all exponentially growing cells, was not required for accumulation of the repair products in yku70Δ rad9Δ cells, as it was undetectable in all G1-arrested cell cultures throughout the experiment (Figure 4E).

SSA requires the strand-annealing activity of the Rad52 protein, but it occurs independently of Rad51 [16]. Consistent with the SSA repair mode, formation of the repair products in G1-arrested yku70Δ rad9Δ cells was abolished by RAD52 deletion (Figure 4F), whereas it was unaffected by RAD51 deletion (Figure 4G). As a DSB flanked by direct repeats could be repaired, at least in principle, also by Rad51-dependent BIR [15], the finding that yku70Δ rad9Δ and yku70Δ rad9Δ rad51Δ G1 cells accumulated the 8 kb repair product with similar kinetics (Figure 4G) indicates that SSA is responsible for this repair event. Thus, we conclude that the lack of Rad9 increases the ability of yku70Δ cells to carry out DSB repair by SSA in G1, likely by enhancing the efficiency of DSB resection.
If competence for SSA-mediated DSB repair relies solely on 3′-ended ssDNA generation, then this repair process should take place with similar efficiency in G1 - and G2-arrested yku70Δ rad9Δ cells. As this expectation is based on the assumption that G1 - and G2-arrested yku70Δ rad9Δ cells resect DSB ends with similar efficiencies, we compared resection (Figure 5B and 5C) and SSA (Figure 5B and 5D) in yku70Δ rad9Δ cells arrested either in G1 or in G2 (Figure 5A) during break induction. Disappearance of the 2.5 kb and 12 kb HO-cut bands occurred with similar kinetics in G1 - and G2-arrested yku70Δ rad9Δ cells (Figure 5B and 5C), which also accumulated similar amounts of the 8 kb SSA repair product (Figure 5B and 5D). As expected, Cdk1 kinase activity was undetectable in yku70Δ rad9Δ cells during the α-factor arrest, whereas it was high in nocodazole-arrested G2 cells (Figure 5E). Thus, DSB resection is the limiting step in DSB repair by SSA.

If SSA is generally restricted to G2 only because high Cdk1 activity allows DSB resection, then inactivation of Cdk1 in G2 should prevent SSA in wild type but not in yku70Δ rad9Δ cells, where DSB resection occurs independently of Cdk1. Thus, we compared DSB repair by SSA in G2-arrested wild type and yku70Δ rad9Δ cells expressing high levels of a stable version of the mitotic Clb-Cdk1 inhibitor Sic1 (Sic1ntΔ) [35]. Consistent with the hypothesis that Cdk1 promotes SSA by regulating the resection step, Sic1 overproduction inhibited SSA repair in G2-arrested wild type cells but not in yku70Δ rad9Δ cells. In fact, the 8 kb SSA repair product accumulated in yku70Δ rad9Δ GAL-SIC1ntΔ cells (Figure 5F and 5G), which showed a decrease of both the 2.5 kb and 12 kb HO-cut band signals (Figure 5F and 5H). By contrast, the same repair product was only barely detectable in G2-arrested GAL-SIC1ntΔ cells, where the HO-cut band signals remained high throughout the experiment (Figure 5F–5H).
The lack of Yku70 allows noncrossover recombination in G1
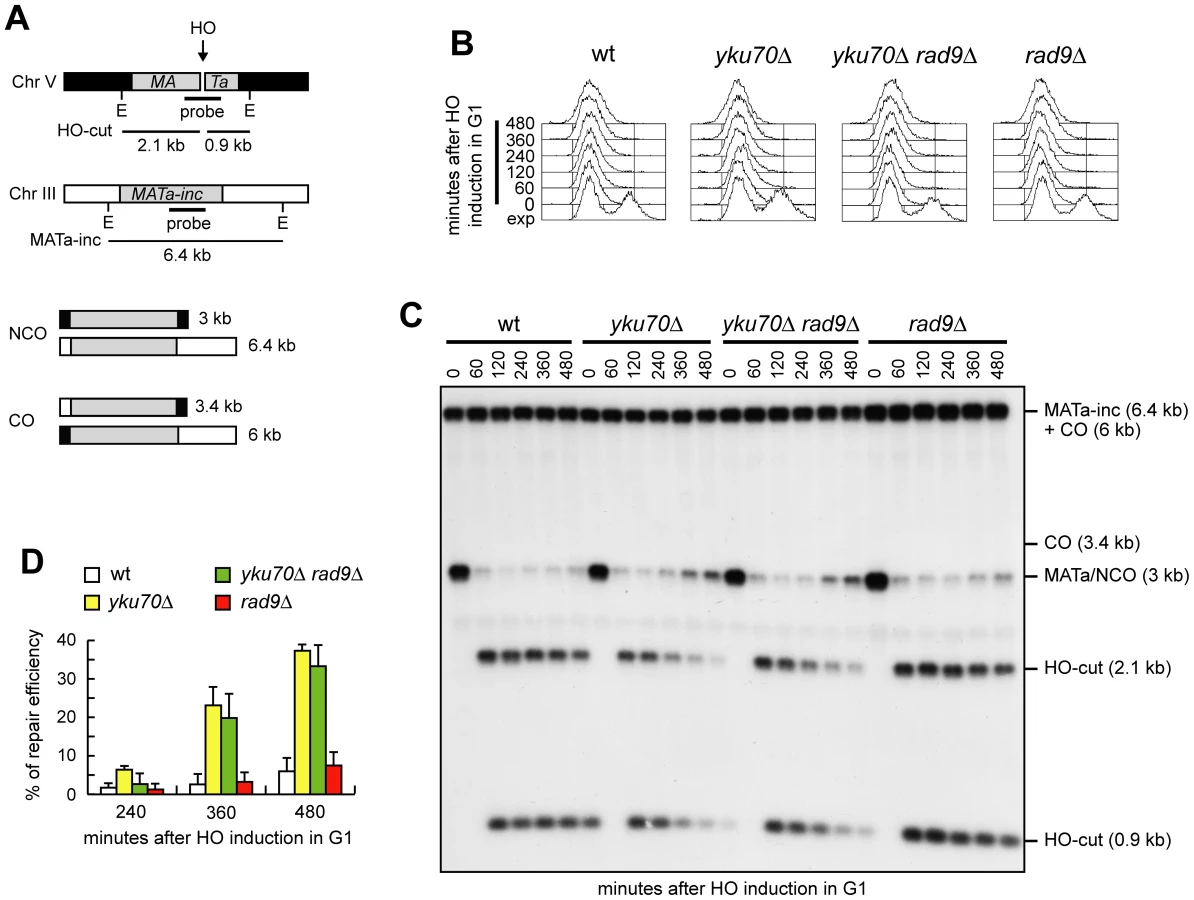
When both ends of a DSB share homology with an intact DNA sequence, repair by Rad51-dependent recombination pathways leads to the formation of noncrossover or crossover products. We investigated whether generation of 3′-ended ssDNA can bypass Cdk1 requirement also in this process. To detect crossovers and noncrossovers at the molecular level, we used a haploid strain that bears two copies of the MATa sequence (Figure 6A) [28], [36]. One copy is located ectopically on chromosome V and carries the recognition site for the HO endonuclease, while the endogenous copy on chromosome III carries a single base pair mutation that prevents HO recognition (MATa-inc). Upon galactose addition, the HO-induced DSB can be repaired by Rad51-dependent HR using the uncleavable MATa-inc sequence as a donor. This repair event can occur either with or without an accompanying crossover (Figure 6A) with the proportion of crossovers being 5–6% among the overall repair events [28], [36]. We induced HO expression in α-factor-arrested cells that were kept arrested in G1 in the presence of galactose (Figure 6B). Galactose was maintained in the medium to cleave the HO sites that were eventually reconstituted by NHEJ-mediated DSB repair. The 3 kb MATa band resulting from recombination events that are not associated to crossovers re-accumulated in both yku70Δ and yku70Δ rad9Δ G1 cells, but not in wild type and rad9Δ G1 cells (Figure 6C and 6D). The repair efficiency in both yku70Δ and yku70Δ rad9Δ G1 cells was around 40% after 8 hours (Figure 6C and 6D), reaching 80–90% after 24 hours (data not shown). This finding indicates that the absence of Yku is sufficient for noncrossover HR events to take place despite of the low Cdk1 activity.
Cdk1 requirement for crossover recombination
Interestingly, the 3.4 kb chromosomal band expected in the experiment above in case of crossover products was not detectable in any G1 cell culture (Figure 6C), suggesting a role for Cdk1 in promoting crossover outcomes that is different from its function in DSB resection. We then compared the products of interchromosomal recombination in G1 - and G2-arrested wild type and yku70Δ rad9Δ cells (Figure 7A). As expected, Cdk1 kinase activity remained undetectable in all α-factor arrested cell cultures, whereas it was high in G2-arrested cells (Figure 7B). The overall DSB repair efficiency of G1-arrested yku70Δ rad9Δ cells was similar to that of G2-arrested wild type and yku70Δ rad9Δ cells (Figure 7C and 7D). However, while no crossover events were detectable in yku70Δ rad9Δ G1 cells, ∼4–5% of repair events were associated to crossovers in both wild type and yku70Δ rad9Δ G2 cells, as indicated by the appearance of the 3.4 kb crossover band (Figure 7C and 7E). Thus, yku70Δ rad9Δ G1 cells appear to be specifically defective in generating crossover products. This inability was not due to the absence of Yku and/or Rad9, because similar amounts of crossover products were detectable in wild type and yku70Δ rad9Δ G2-arrested cells (high Cdk1 activity) (Figure 7C and 7E). These results suggest that Cdk1 has a function in promoting crossover recombination that is independent of its role in DSB resection.

If the inability to perform crossover recombination in G1 were due to the lack of Cdk1 activation, then ectopic expression of active Cdk1 should allow crossover recombination in G1, whereas Cdk1 inhibition should prevent crossover formation in G2. We then constructed wild type and yku70Δ rad9Δ strains carrying the system in Figure 6A and expressing a stable version of the mitotic cyclin CLB2 under the control of the GAL promoter (GAL-CLB2dbΔ). This Clb2 variant forms active Clb2-Cdk1 complexes also during G1, because it lacks the destruction box, and therefore it is not subjected to B-type cyclin-specific proteolysis [37]. Strikingly, when both DSB formation and Clb2dbΔ overproduction were induced in G1-arrested cell cultures by galactose addition (Figure 8A), crossover products became detectable in both GAL-CLB2dbΔ and yku70Δ rad9Δ GAL-CLB2dbΔ cells, whereas they were not present in wild type and yku70Δ rad9Δ cells under the same conditions (Figure 8B and 8C).

To assess whether Cdk1 inhibition prevented crossover formation in G2, we compared the products of interchromosomal recombination in G2-arrested yku70Δ rad9Δ and yku70Δ rad9Δ GAL-SIC1ntΔ cells (Figure 8D), the latter expressing high levels of a stable version of the Cdk1 inhibitor Sic1 (Sic1ntΔ) [35]. When both DSB formation and Sic1ntΔ overproduction were induced in G2-arrested cell cultures by galactose addition, crossover products accumulated, as expected, in yku70Δ rad9Δ cells, but they were undetectable in yku70Δ rad9Δ GAL-SIC1ntΔ cells (Figure 8E and 8F). Thus, Sic1-mediated Cdk1 inhibition prevents generation of crossover products in G2, whereas ectopic Cdk1 activation leads to crossover recombination in G1, supporting the hypothesis that Cdk1 activity is required to promote crossover HR events even when DSB resection is allowed by the absence of Yku and Rad9.
Discussion
HR is highly coordinated with the cell cycle: it takes place predominantly during the S and G2 phases, when the presence of a sister chromatid provides a donor template and high Cdk1 activity promotes DSB end resection to expose ssDNA that is necessary to initiate HR [17], [18], [21], [30]. To study whether Cdk1 plays additional role(s) in HR, we asked whether generation of ssDNA at the DSB ends is sufficient to bypass Cdk1 requirement for HR. Because the lack of either Yku or Rad9 allows Cdk1-independent generation of 3′-ended ssDNA at DSB ends [24], [26], we investigated whether cells lacking Yku and/or Rad9 could repair a DSB by HR when Cdk1 activity is low. We found that DSB repair by SSA can take place in G1-arrested yku70Δ cells. The ability of these cells to carry out SSA in G1 depends on Cdk1-mediated generation of 3′-ended ssDNA at the DSB ends. In fact, the lack of Rad9 increases efficiency of both resection and SSA in yku70Δ G1 cells. Furthermore, Cdk1 inhibition prevents SSA in G2 wild type cells, but not in yku70Δ rad9Δ G2 cells, where DSB resection occurs independently of Cdk1. We also found that G1-arrested yku70Δ and yku70Δ rad9Δ cells can undergo interchromosomal recombination events that are not accompanied by crossovers. Thus, Cdk1 requirement for carrying out SSA and noncrossover recombination is bypassed by DSB resection, indicating that Cdk1 promotes these HR events essentially by regulating the resection step.
Rad52 is essential for both SSA and noncrossover recombination events, while only the latter require the assembly of Rad51 nucleoprotein filaments, which promote homologous pairing and strand exchange (reviewed in [1]–[3]). As the function of Cdk1 in DSB repair by SSA and noncrossover recombination is primarily the regulation of the resection step, neither Rad51 nor Rad52 appear to require Cdk1 activity to exert their biochemical activities.
Interestingly, although RAD9 deletion was shown to allow MRX-dependent DSB resection in G2 cells that overproduced the Cdk1 inhibitor Sic1 [26], the lack of Rad9 did not increase DSB resection or HR-mediated DSB repair in G1 compared to wild type cells. Thus, although Rad9 provides a barrier to resection in yku70Δ G1 cells, its lack is not sufficient, by itself, to escape the inhibitory effect of Yku on DSB resection in G1. This finding is consistent with previous data showing that the resection block imposed by Yku is relieved in G2 [33], [34]. It also indicates that Rad9 prevents DSB resection in all cell cycle phases, but its inhibitory effect in G1 becomes apparent only in the absence of Yku.
Surprisingly, we found that G1-arrested yku70Δ rad9Δ cells are specifically impaired in the formation of crossovers by interchromosomal recombination. Expression of an activated form of Cdk1 allows crossover recombination in both wild type and yku70Δ rad9Δ G1 cells, whereas inhibition of Cdk1 activity in G2-arrested yku70Δ rad9Δ cells prevents crossover formation without affecting noncrossover outcomes. These findings are consistent with a role of Cdk1 in promoting crossover recombination that is independent of its function in DSB resection.
How does Cdk1 promote crossover outcomes? The choice between crossover and noncrossover is tightly regulated [38]. Meiotic recombination results frequently in crossovers [39], while DSB repair in mitotic cells is mostly not associated with crossovers [6]. An explanation of these differences could be that specific mechanisms limit crossovers during mitotic homologous recombination. Indeed, dissociation of the D-loop intermediates gives rise to noncrossover products, and this process is promoted by the helicases Srs2 and Mph1 [7], [28], [36], [40]. Furthermore, noncrossover outcomes can arise also from the dissolution of dHJ intermediates that requires the combined activity of the BLM/Sgs1 helicase, which drives migration of the constrained dHJs, and the Top3-Rmi1 complex, which decatenates the interlinked strands between the two HJs [7]–[9]. One possibility is that Cdk1 promotes crossover recombination by inhibiting proteins specifically involved in limiting crossover generation (i.e. Sgs1, Top3-Rmi1, Srs2 and Mph1). A similar mechanism seems to act during meiotic recombination, where proteins required for homologous chromosome synapsis have been proposed to antagonize the anti-crossover activity of Sgs1 [41]. However, none of the above anti-crossover proteins have been reported to undergo Cdk1-dependent inhibitory phosphorylation. On the other hand, crossovers arise from dHJ intermediate cleavage, which involves the resolvases Mus81-Mms4, Slx1-Slx4, Yen1 and Rad1-Rad10 (reviewed in [42]), suggesting that Cdk1 might promote crossover recombination by stimulating dHJ resolution. Consistent with this hypothesis, the Yen1 and Mms4 resolvases appear to be phosphorylated by Cdk1 [43], raising the possibility that they might represent Cdk1 targets in dHJ resolution. Further studies will be required to assess whether Cdk1-dependent phosphorylation of these proteins has a role in regulating crossover formation.
In conclusion, Cdk1 controls primarily DSB resection to allow SSA and noncrossover recombination, while crossover outcomes appear to require additional Cdk1-promoted events. As mitotic crossovers have the potential for deleterious genome rearrangements, their Cdk1-dependent regulation can provide an additional safety mechanism, ensuring that the rare mitotic recombination events accompanied by crossing over at least occur in S/G2, when a sister chromatid is available as appropriate donor.
Materials and Methods
Yeast strains
Strain genotypes are listed in Table S1. Strains JKM139, YMV86 and YMV45 were kindly provided by J. Haber (Brandeis University, Waltham, USA). Strains YMV86 and YMV45 are isogenic to YFP17 (matΔ::hisG hmlΔ::ADE1 hmrΔ::ADE1 ade1 lys5 ura3-52 trp1 ho ade3::GAL-HO leu2::cs) except for the presence of a LEU2 fragment inserted, respectively, 0.7 kb or 4.6 kb centromere-distal to leu2::cs [32]. Strain tGI354 was kindly provided by G. Liberi (IFOM, Milano, Italy) and J. Haber [28]. To induce a persistent G1 arrest with α-factor, all strains used in this study carried the deletion of the BAR1 gene, which encodes a protease that degrades the α-factor. Deletions of the YKU70, RAD9, RAD51, RAD52 and BAR1 genes were generated by one-step PCR disruption method. YMV86, YMV45 and tGI354 derivatives strains carrying a fully functional CDC28-HA allele at the CDC28 chromosomal locus were generated by one-step PCR tagging method. A plasmid carrying the GAL-CLB2dbΔ allele was kindly provided by R. Visintin (IEO, Milan, Italy) and was used to integrate the GAL-CLB2dbΔ fusion at the URA3 locus in the tGI354 derivative strains. Strain YLL3019, carrying the GAL-SIC1ntΔ allele integrated at the URA3 locus, was obtained by transforming strain tGI354 rad9Δ yku70Δ with ApaI-digested plasmid pLD1, kindly provided by J. Diffley (Clare Hall Laboratories, South Mimms, United Kingdom). The GAL-SIC1ntΔ fusion was cloned into a TRP1-based integrative plasmid that was used to integrate the fusion at the TRP1 locus in the YMV45 derivative strains. Integration accuracy was verified by Southern blot analysis. Cells were grown in YEP medium (1% yeast extract, 2% bactopeptone) supplemented with 2% raffinose (YEP+raf) or 2% raffinose and 3% galactose (YEP+raf+gal).
Kinase assay
For Cdk1 kinase assays, protein extracts were prepared as described previously [44]. HA-tagged Cdk1 was immunoprecipitated with anti-HA antibody from 150 µg of protein extracts and the kinase activity in the immunoprecipitates was measured on histone H1 [45].
DSB resection and repair
DSB formation and repair in YMV86 and YMV45 strains were detected by Southern blot analysis using an Asp718-SalI fragment containing part of the LEU2 gene as a probe. DSB end resection at the MAT locus in JKM139 derivative strains was analyzed on alkaline agarose gels as described in [24], by using a single-stranded probe complementary to the unresected DSB strand. This probe was obtained by in vitro transcription using Promega Riboprobe System-T7 and plasmid pML514 as a template. Plasmid pML514 was constructed by inserting in the pGEM7Zf EcoRI site a 900-bp fragment containing part of the MATα locus (coordinates 200870 to 201587 on chromosome III). Quantitative analysis of DSB resection was performed by calculating the ratio of band intensities for ssDNA and total amount of DSB products. DSB repair in tGI354 strain was detected as described in [28]. To determine the amount of noncrossover and crossover products, the normalized intensity of the corresponding bands at different time points after DSB formation was divided by the normalized intensity of the uncut MATa band at time zero before HO induction (100%). The repair efficiency (NCO+CO) was normalized with respect to the efficiency of DSB formation by subtracting the value calculated 2 hours after HO induction (maximum efficiency of DSB formation) from the values calculated at the subsequent time points after galactose addition.
Supporting Information
Zdroje
1. PâquesFHaberJE 1999 Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 63 349 404
2. KroghBOSymingtonLS 2004 Recombination proteins in yeast. Annu Rev Genet 38 233 271
3. San FilippoJSungPKleinH 2008 Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 77 229 257
4. LiXStithCMBurgersPMHeyerWD 2009 PCNA is required for initiation of recombination-associated DNA synthesis by DNA polymerase delta. Mol Cell 36 704 713
5. SzostakJWOrr-WeaverTLRothsteinRJStahlFW 1983 The double-strand-break repair model for recombination. Cell 33 25 35
6. BzymekMThayerNHOhSDKlecknerNHunterN 2010 Double Holliday junctions are intermediates of DNA break repair. Nature 464 937 941
7. IraGMalkovaALiberiGFoianiMHaberJE 2003 Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell 115 401 411
8. WuLHicksonID 2003 The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426 870 874
9. LoYCPaffettKSAmitOClikemanJASterkR 2006 Sgs1 regulates gene conversion tract lengths and crossovers independently of its helicase activity. Mol Cell Biol 26 4086 4094
10. StrathernJNKlarAJHicksJBAbrahamJAIvyJM 1982 Homothallic switching of yeast mating type cassettes is initiated by a double-stranded cut in the MAT locus. Cell 31 183 192
11. NassifNPenneyJPalSEngelsWRGloorGB 1994 Efficient copying of nonhomologous sequences from ectopic sites via P-element-induced gap repair. Mol Cell Biol 14 1613 1625
12. FergusonDOHollomanWK 1996 Recombinational repair of gaps in DNA is asymmetric in Ustilago maydis and can be explained by a migrating D-loop model. Proc Natl Acad Sci USA 93 5419 5424
13. AllersTLichtenM 2001 Differential timing and control of noncrossover and crossover recombination during meiosis. Cell 106 47 57
14. HunterNKlecknerN 2001 The single-end invasion: an asymmetric intermediate at the double-strand break to double-Holliday junction transition of meiotic recombination. Cell 106 59 70
15. JainSSugawaraNLydeardJVazeMTanguy Le GacN 2009 A recombination execution checkpoint regulates the choice of homologous recombination pathway during DNA double-strand break repair. Genes Dev 23 291 303
16. IvanovELSugawaraNFishman-LobellJHaberJE 1996 Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics 142 693 704
17. AylonYLiefshitzBKupiecM 2004 The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J 23 4868 4875
18. IraGPellicioliABalijjaAWangXFioraniS 2004 DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 431 1011 1017
19. LongheseMPBonettiDManfriniNClericiM 2010 Mechanisms and regulation of DNA end resection. EMBO J 29 2864 2874
20. MimitouEPSymingtonLS 2011 DNA end resection-unraveling the tail. DNA Repair 10 344 348
21. HuertasPCortés-LedesmaFSartoriAAAguileraAJacksonSP 2008 CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 455 689 692
22. ManfriniNGueriniICitterioALucchiniGLongheseMP 2010 Processing of meiotic DNA double strand breaks requires cyclin-dependent kinase and multiple nucleases. J Biol Chem 285 11628 11637
23. LeeSEMooreJKHolmesAUmezuKKolodnerRD 1998 Saccharomyces Ku70, Mre11/Rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 94 399 409
24. ClericiMMantieroDGueriniILucchiniGLongheseMP 2008 The Yku70–Yku80 complex contributes to regulate double-strand break processing and checkpoint activation during the cell cycle. EMBO Rep 9 810 818
25. LydallDWeinertT 1995 Yeast checkpoint genes in DNA damage processing: implications for repair and arrest. Science 270 1488 1491
26. LazzaroFSapountziVGranataMPellicioliAVazeM 2008 Histone methyltransferase Dot1 and Rad9 inhibit single-stranded DNA accumulation at DSBs and uncapped telomeres. EMBO J 27 1502 1512
27. BarlowJHRothsteinR 2009 Rad52 recruitment is DNA replication independent and regulated by Cdc28 and the Mec1 kinase. EMBO J 28 1121 1130
28. SaponaroMCallahanDZhengXKrejciLHaberJE 2010 Cdk1 targets Srs2 to complete synthesis-dependent strand annealing and to promote recombinational repair. PLoS Genet 6 e1000858 doi:10.1371/journal.pgen.1000858
29. EsashiFChristNGannonJLiuYHuntT 2005 CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 434 598 604
30. CaspariTMurrayJMCarrAM 2002 Cdc2-cyclin B kinase activity links Crb2 and Rqh1-topoisomerase III. Genes Dev 16 1195 1208
31. Fishman-LobellJRudinNHaberJE 1992 Two alternative pathways of double-strand break repair that are kinetically separable and independently modulated. Mol Cell Biol 12 1292 1303
32. VazeMBPellicioliALeeSEIraGLiberiG 2002 Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol Cell 10 373 385
33. BonettiDClericiMManfriniNLucchiniGLongheseMP 2010 The MRX complex plays multiple functions in resection of Yku - and Rif2-protected DNA ends. PLoS ONE 5 e14142 doi:10.1371/journal.pone.0014142
34. ShimEYChungWHNicoletteMLZhangYDavisM 2010 Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J 29 3370 3380
35. DesdouetsCSantocanaleCDruryLSPerkinsGFoianiM 1998 Evidence for a Cdc6-independent mitotic resetting event involving DNA polymerase α. EMBO J 17 4139 4146
36. PrakashRSatoryDDrayEPapushaASchellerJ 2009 Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev 23 67 79
37. AmonAIrnigerSNasmythK 1994 Closing the cell cycle circle in yeast: G2 cyclin proteolysis initiated at mitosis persists until the activation of G1 cyclins in the next cycle. Cell 77 1037 1050
38. MartiniEDiazRLHunterNKeeneyS 2006 Crossover homeostasis in yeast meiosis. Cell 126 285 295
39. YoudsJLBoultonSJ 2011 The choice in meiosis - defining the factors that influence crossover or non-crossover formation. J Cell Sci 124 501 513
40. RobertTDervinsDFabreFGangloffS 2006 Mrc1 and Srs2 are major actors in the regulation of spontaneous crossover. EMBO J 25 2837 2846
41. JessopLRockmillBRoederGSLichtenM 2006 Meiotic chromosome synapsis-promoting proteins antagonize the anti-crossover activity of Sgs1. PLoS Genet 2 e155 doi:10.1371/journal.pgen.0020155
42. SvendsenJMHarperJW 2010 GEN1/Yen1 and the SLX4 complex: Solutions to the problem of Holliday junction resolution. Genes Dev 24 521 536
43. UbersaxJAWoodburyELQuangPNParazMBlethrowJD 2003 Targets of the cyclin-dependent kinase Cdk1. Nature 425 859 864
44. SchwobEBöhmTMendenhallMDNasmythK 1994 The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell 79 233 244
45. SuranaUAmonADowzerCMcGrewJByersB 1993 Destruction of the CDC28/CLB mitotic kinase is not required for the metaphase to anaphase transition in budding yeast. EMBO J 12 1969 1978
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 8
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- An EMT–Driven Alternative Splicing Program Occurs in Human Breast Cancer and Modulates Cellular Phenotype
- Chromosome Painting Reveals Asynaptic Full Alignment of Homologs and HIM-8–Dependent Remodeling of Chromosome Territories during Meiosis
- A Pre-mRNA–Associating Factor Links Endogenous siRNAs to Chromatin Regulation
- Discovery of Sexual Dimorphisms in Metabolic and Genetic Biomarkers
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy