
Widespread Cotranslational Formation of Protein Complexes
Most cellular processes are conducted by multi-protein complexes. However, little is known about how these complexes are assembled. In particular, it is not known if they are formed while one or more members of the complexes are being translated (cotranslational assembly). We took a genomic approach to address this question, by systematically identifying mRNAs associated with specific proteins. In a sample of 31 proteins from Schizosaccharomyces pombe that did not contain RNA–binding domains, we found that ∼38% copurify with mRNAs that encode interacting proteins. For example, the cyclin-dependent kinase Cdc2p associates with the rum1 and cdc18 mRNAs, which encode, respectively, an inhibitor of Cdc2p kinase activity and an essential regulator of DNA replication. Both proteins interact with Cdc2p and are key cell cycle regulators. We obtained analogous results with proteins with different structures and cellular functions (kinesins, protein kinases, transcription factors, proteasome components, etc.). We showed that copurification of a bait protein and of specific mRNAs was dependent on the presence of the proteins encoded by the interacting mRNAs and on polysomal integrity. These results indicate that these observed associations reflect the cotranslational interaction between the bait and the nascent proteins encoded by the interacting mRNAs. Therefore, we show that the cotranslational formation of protein–protein interactions is a widespread phenomenon.
Published in the journal:
. PLoS Genet 7(12): e32767. doi:10.1371/journal.pgen.1002398
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002398
Summary
Most cellular processes are conducted by multi-protein complexes. However, little is known about how these complexes are assembled. In particular, it is not known if they are formed while one or more members of the complexes are being translated (cotranslational assembly). We took a genomic approach to address this question, by systematically identifying mRNAs associated with specific proteins. In a sample of 31 proteins from Schizosaccharomyces pombe that did not contain RNA–binding domains, we found that ∼38% copurify with mRNAs that encode interacting proteins. For example, the cyclin-dependent kinase Cdc2p associates with the rum1 and cdc18 mRNAs, which encode, respectively, an inhibitor of Cdc2p kinase activity and an essential regulator of DNA replication. Both proteins interact with Cdc2p and are key cell cycle regulators. We obtained analogous results with proteins with different structures and cellular functions (kinesins, protein kinases, transcription factors, proteasome components, etc.). We showed that copurification of a bait protein and of specific mRNAs was dependent on the presence of the proteins encoded by the interacting mRNAs and on polysomal integrity. These results indicate that these observed associations reflect the cotranslational interaction between the bait and the nascent proteins encoded by the interacting mRNAs. Therefore, we show that the cotranslational formation of protein–protein interactions is a widespread phenomenon.
Introduction
The majority of cellular proteins function as subunits in larger protein complexes. However, very little is known about how protein complexes form in vivo. One possibility is that proteins are fully translated and released into the cytoplasm before finding their interacting partners (posttranslational assembly). Alternatively, protein-protein interactions could form as one or several of the interacting proteins are being translated (cotranslational assembly).
There are indications that some cytoskeletal proteins, including vimentin, myosin and titin, assemble cotranslationally into insoluble filaments [1]. The formation of some multimeric membrane channels also appears to take place cotranslationally [2], [3]. There are also a few examples of cotranslational assembly of soluble proteins: the p53 and NF-κB transcription factors form homodimers, which are thought to be generated by cotranslational interactions within a single polysome [4], [5]. Importantly, the majority of these examples involve the assembly of a single protein into higher order structures. A number of recent studies have shown that the use of immunoprecipitation coupled with microarray analysis (RIp-chip, for Ribonucleoprotein Immunoprecipitation analysed with DNA chips) can be used to study cotranslational pathways involved in protein biosynthesis [6], [7], [8]. In this approach, a protein is purified together with associated RNAs, and the mRNAs are identified using DNA microarrays. When this method is applied to proteins associated with polysomes, it allows the identification of mRNAs cotranslationally associated with the bait protein. Using this technique we recently showed that the Rng3p myosin-specific chaperone associates cotranslationally with all five myosin heavy chains in the fission yeast Schizosaccharomyces pombe [6]. Another study in the budding yeast Saccharomyces cerevisiae found that the SET1 mRNA is part of a complex containing four components of the SET1C histone methyltransferase complex. The protein-RNA interactions were dependent on active translation, suggesting that the complex between these proteins was formed cotranslationally [7]. Apart from these few examples, very little is known about the prevalence of cotranslational assembly in the formation of protein complexes. Importantly, systematic approaches to identify and characterise this phenomenon (such as RIp-chip) have not been applied to large numbers of proteins.
To address these questions we carried out RIp-chip experiments with 31 proteins with different functions and structures. We found that more than 12 of the proteins interacted specifically with small numbers of mRNAs (between 1 and 3), most of which encoded proteins that are known or predicted to interact with the bait proteins. We examined the protein-RNA interactions of three proteins in detail: in all cases we found that the interactions required the presence of the protein encoded by the associated mRNA as well as active translation. These data demonstrate that these protein mRNA interactions reflect the cotranslational formation of protein-protein interactions, and suggest that this is a widespread phenomenon.
Results
The Tea2p Kinesin and the Tip1 CLIP-170 protein interact cotranslationally
As part of a project to identify RNAs associated with molecular motors in S. pombe, we performed RIp-chip experiments with the Tea2p kinesin. Surprisingly, Tea2p copurified specifically with only two mRNAs: tea2 and tip1 (Figure 1A). tip1 encodes a protein of the CLIP-170 family that interacts physically with Tea2p and is transported by it along cytoplasmic microtubules [9]. We considered three models that could explain the association between Tea2p and the tip1 mRNA: in models 1 and 2 (Figure 1B), Tea2p could interact with specific sequences on the tip1 mRNA, either directly (model 1) or through a sequence-specific RNA-binding protein (model 2). In model 3, Tea2p interacts with the Tip1p nascent peptide, and thus pull downs the tip1 mRNA as part of the whole polysome. In this case, the complex between Tea2p and Tip1p forms cotranslationally. The different models can be distinguished from each other experimentally by their dependence on the Tip1 protein for the interaction: in models 1 and 2 (recognition of RNA sequences), the interaction between Tea2p and tip1 mRNA should be independent of the presence of Tip1p. In model 3 (cotranslational assembly), the association should only occur if Tip1p is present.
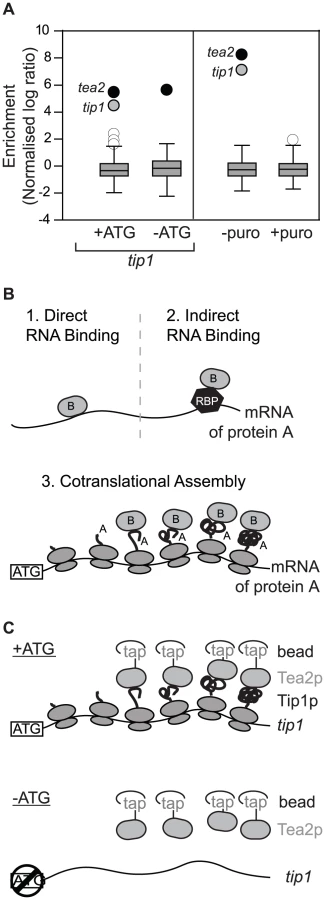
To discriminate between these possibilities we used a strain expressing tip1 RNA but not Tip1 protein (Figure 1C, Figure S1, Figure S2). The strain was generated by mutating a single nucleotide in the initiation codon of tip1 (−ATG-tip1), and expressing the resulting construct in cells in which the endogenous tip1 gene had been deleted (see Materials and Methods). As a control, a similar strain was constructed expressing wild type tip1 (+ATG-tip1). While Tea2p and tip1 mRNA copurified in +ATG-tip1 cells, the interaction was completely lost in −ATG-tip1 cells (Figure 1A and Table S1). By contrast, tea2 mRNA was precipitated to a similar extent in +ATG-tip1 and −ATG-tip1 cells (Figure 1A and Table S1). These data demonstrate that association of Tea2p with the tip1 mRNA is dependent on expression of Tip1p, and strongly suggests that Tea2p and Tip1p bind to each other cotranslationally. This model makes a further prediction, namely that the interaction between Tea2p and tip1 should be dependent on the integrity of polysomes. To test this idea we performed RIp-chip experiments after disrupting polysomes in vivo and in vitro. Treatment of S. pombe cells with puromycin leads to polysome disassembly and the release of nascent peptides [10], [11]. In cells incubated with puromycin, the interactions between Tea2p and both tea2 and tip1 mRNAs were entirely lost (Figure 1A and Table S1). Treatment of extracts with EDTA, which chelates magnesium and causes polysome disassembly, also disrupted the association between Tea2p and tip1 and tea2 mRNAs (Table S1). All together, these experiments strongly suggest that the complex between the Tea2 and Tip1 proteins forms cotranslationally. To test if this phenomenon is bidirectional (i.e. if Tip1p interacts with Tea2p as tea2 mRNA is being translated), we carried out RIp-chip experiments with Tip1p. In this case, Tip1p coprecipitated with its own mRNA, but not with that of tea2 (Figure S3 and Table S2).
Many proteins associate with mRNAs encoding interacting proteins
We wondered whether cotranslational assembly is a common mechanism for the formation of protein complexes. To address this question we analyzed 31 proteins by RIp-chip (Figure S3 and Table S2). We tested a variety of proteins, none of which contained canonical RNA-binding domains. Our baits included protein kinases, transcription factors, components of the proteasome, kinesins and several members of the actin related protein (Arp) family. Of the 31 bait proteins probed, 10 showed no significant association with any RNA, 9 proteins coprecipitated with only their own mRNAs, and 12 proteins reproducibly pulled down other mRNAs (Figure S3 and Table S2). Notably, the majority of associated mRNAs encoded known or suspected protein interactors of the corresponding bait proteins (Figure 2 and Table 1).


A striking example is provided by Cdc2p, the ortholog of CDK1 in higher eukaryotes. Cdc2p is the only cyclin-dependent kinase in fission yeast and is an essential regulator of cell cycle progression. Cdc2p interacted with two mRNAs: rum1, which encodes a CDK (Cyclin-Dependent Kinase) inhibitor that associates with Cdc2p and inhibits its kinase activity [12], and cdc18, which encodes an essential DNA replication factor (a homologue of budding yeast CDC6) [13]. Both Cdc18p and Rum1p are also direct targets of Cdc2p.
Another protein kinase, Sty1p, which is a MAP kinase that mediates most stress responses in fission yeast, interacted with three mRNAs, pyp2, cip2 and its own transcript. Pyp2p is a protein tyrosine phosphatase that directly binds and dephosphorylates Sty1p [14]. Cip2p is an RNA-binding protein thought to be regulated by Sty1p (however, no direct protein-protein interaction has been demonstrated) [15]. A predicted component of the 19S proteasome regulatory subunit, Rpn12p/Mts3p, associated with mRNAs encoding other subunits of the 19S proteasome (rpn1301, rpn1302) and a protein required for the assembly of the proteasome core and regulatory subunits (ecm29). A second component of the 19S proteasome, Rpt2p/Mts2p, interacted with the ubp6 and rhp23 mRNAs. Ubp6p is a proteasome-associated ubiquitin C-terminal hydrolase, while Rhp23p contains a ubiquitin-like N-terminus. The budding yeast orthologs of these two proteins (Ubp6 and Rad23, respectively) copurify with components of the proteasome. We also looked at two transcription factor of the b-ZIP family, Atf1p and Pcr1p, which can form heterodimers with each other [16]. Atf1p interacted with pcr1 and its own mRNA, while Pcr1p only pulled down its cognate mRNA. Finally, Mnh1p, the S. pombe ortholog of the Mago nashi protein (SPBC3B9.08c, a component of the splicing-dependent exon–exon junction complex) associated with the mRNA of mni1 (SPBC19C7.01), which encodes a protein that contains a Mago nashi-binding domain.
These data suggested that the mRNAs associated to a protein can provide insight into their cellular function and the protein complexes they belong to. To test this idea we examined all 10 members of the Actin-related protein family, which are structurally similar to actin but involved in functions as varied as chromatin remodelling, actin polymerisation or microtubular transport (as part of dynactin). These functions are carried out as part of different protein complexes that have been well characterised [17], [18], providing an excellent model to test this hypothesis. Half of the probed proteins pulled down specific mRNAs other than their own. The dynactin Arps (Arp1p and Arp10p) were the only members of this family that did not to associate with any mRNA. S. pombe nuclear Arps are components of several chromatin-remodelling complexes (SWI/SNF, INO80, NuA4 and Swr1C). Arp6p, a component of the Swr1C chromatin-remodelling complex, copurified with the alp5 mRNA (Alp5p is also part of this complex, but also of Ino80 and NuA4) [18]. Arp9p and Arp42p are members of the SWI/SNF and RSC complexes [17], and both proteins associated with mRNAs encoding two SNF helicases (snf21 and snf22). The Ino80 complex contains three Arps (Arp8p, Arp5p and Alp5p) [18]. Of these, Arp8p associated with the ino80 mRNA, while Arp5p and Alp5p pulled down only their own mRNAs. Unexpectedly, Arp2p, a member of the family that regulates actin polymerisation, copurified with the mRNAs of two unrelated Arps (arp8 and arp9). These proteins have very different functions and are not expected to interact directly with Arp2p. Therefore, the nature of the protein-RNA interactions of the Arps could allow the assignment of one protein (Arp6p) to one of several related complexes, and the unambiguous allocation of three proteins to a specific complex (Arp8p, Arp9p and Arp42p).
Cotranslational formation of protein complexes is widespread
These results demonstrate that a large fraction of proteins copurify with mRNAs encoding interacting proteins. To confirm that, as in the case of Tea2p/tip1, these interactions reflect cotranslational formation of the corresponding protein complexes, we characterised in more detail the interactions between Sty1p/cip2 and Cdc2p/rum1. We followed the same strategy described above for Tea2p/tip1. First, we made non-translatable versions of the rum1 and cip2 mRNAs (Figure S1 and Figure S2). Second, we performed RIp-chip experiments after in vivo treatment of the cells with puromycin, or after in vitro incubation of the extracts with EDTA. Sty1p did not associate with −ATG-cip2, while the association with another mRNA (pyp2) was unaffected by the mutation. By contrast, neither cip2 nor pyp2 copurified with Sty1p after puromycin or EDTA treatments (Figure 3A and Table S1). Similarly, the association between Cdc2p and rum1 was lost in −ATG-rum1, but the interaction with cdc18 was not. As expected, both associations were disrupted by puromycin and EDTA incubations (Figure 3B and Table S1).

Discussion
Our data show that around 38% of a randomly selected set of proteins that do not contain canonical RNA-binding domains specifically copurify with small number of mRNAs (between 1 and 3). Remarkably, the majority of these mRNAs encode proteins that are closely related to the proteins used as bait, either as known direct interactors or as members of the same multiprotein complex. In some cases (rum1, pyp2), the proteins are key regulators of the bait protein. This phenomenon is not limited to a specific type of protein (we have observed it with protein kinases, transcription factors, actin-related proteins, kinesins, etc), nor is it restricted to specific cellular processes (the proteins we tested function in microtubular transport, cell cycle control, proteolysis, stress responses, chromatin remodelling and splicing). In fact, the functional association between bait proteins and their associated mRNAs could conceivably be used to gain insight into the function of the bait protein (if the proteins encoded by the interacting mRNAs have known functions) or to identify interacting partners. Although previous work had hinted at the importance of cotranslationally assembly (especially for homomultimers), this is the first demonstration that cotranslational assembly is a widespread and ubiquitous process.
We notice that the protein-mRNA interactions we report are extremely specific. For example, Cdc2p interacts with and is regulated by many proteins, including at least four cyclins, protein phosphatases (Cdc25p), protein kinases (Wee1p) and kinase inhibitors (Rum1p). In addition, Cdc2p is thought to have dozen of targets. However, Cdc2p associates specifically with two mRNAs. This suggests that only a fraction of all protein-protein interactions are formed cotranslationally. Interestingly, both proteins encoded by the Cdc2p-bound mRNAs are phosphorylated by Cdc2p and degraded through the same mechanism [19], possibly pointing at a role of cotranslational assembly in the control of protein stability. The preference for certain nascent peptides to undergo cotranslational assembly could simply be a reflection of their levels in the cell (which, in turn, would depend on the abundance of their cognate mRNAs and their translation rates). However, Cdc2p associates with rum1 but not with cdc13 (which encodes a B type cyclin that binds to Cdc2p), despite the fact that rum1 mRNA levels are almost 6-fold lower that those of cdc13 and both mRNAs are associated with similar number of ribosomes (3.7 for rum1 and 4.2 for cdc13) [20]. Therefore, the specificity of cotranslational assembly seems to be conferred by factors other than the abundance of the interacting nascent chains.
It has recently been shown that many proteins that lack RNA-binding domains can interact directly with specific mRNA sets [21], [22], [23]. We do not believe that our observations reflect direct binding: first, most of the proteins identified in those studies interacted with much larger numbers of mRNAs; second, in the few cases in which those interactions were analysed more thoroughly, they were shown to be resistant to EDTA, arguing against cotranslational assembly [23]. By contrast, the three examples we analysed in detail using a variety of approaches were consistent with cotranslational assembly. It is formally possible that our results reflect direct binding to RNA that is dependent on active translation through an unidentified mechanism, but we consider this possibility highly unlikely.
The cotranslational formation of protein complexes could be required for multiple reasons in the cell. It is possible that some interactions can only form before a given member of the complex has folded completely. Indeed, it is relatively frequent that recombinant proteins need to be coexpressed in order for them to form a complex. Second, some proteins are unstable in the absence of their partner. In this case, cotranslational assembly would stabilise the protein by reducing the time during which a protein is susceptible to degradation. This explanation has been proposed for the SET1C histone methyltransferase complex [7]. Consistently, Pcr1p is unstable in the absence Atf1p [24]. Finally, some proteins could be toxic when not part of a complex. Again, early formation of a complex would reduce this potential toxicity.
Unexpectedly, we have found that a large fraction of protein-protein interactions are likely to be formed cotranslationally, and we demonstrate that the RIp-chip strategy can provide a genome-wide view of this phenomenon. The protein-RNA networks we present here add another layer of complexity to the formation and regulation of protein complexes in eukaryotic cells.
Materials and Methods
Yeast methods and experimental design
Standard methods were used for fission yeast growth and manipulation [25]. Proteins were TAP-tagged using a one-step PCR method in haploid cells except for alp5 and arp10, where ∼400 nucleotides of ORF and 3′UTR sequences were cloned into the pFA6a-2xTap-Kan vector [26], [27]. All construct were transformed into haploid cells with the exceptions of arp1 (integrated in pat1 diploids) and alp5 and arp10 (transformed into wild type diploids). Successful tagging was verified by western blot as described below. All epitope-tagged strains grew normally and displayed normal cell shape, showing that the tagged proteins were functional. A complete list of the strains used in this work is presented in Table S3. Integration of constructs at the leu1 locus was performed as described [28].
Construction of −ATG mutants
All constructs were tagged integrated into the leu1 locus of a strain in which the endogenous copy of the mutated gene had been deleted (Table S3). The length of the flanking sequences required to include endogenous 5′ and 3′ UTR was estimated from high throughput sequencing data [29]. For every gene two constructs were made, one containing a mutated ATG and a control carrying the normal initiation codon. The presence of the mutations was confirmed by sequencing. In all three cases, the control construct complemented the phenotype of the corresponding deletion strain (Figure S2).
The cip2 ORF and 989 bp downstream were amplified by PCR from genomic DNA using primers that added a SalI and an EcoRI site to the 5′ and 3′ end, respectively, and cloned into pBluescript II. Two constructs were made: in the wild type control the 5′ primer contained the endogenous ATG, while in the −ATG one the ATG was mutated to AGG. The cip2 promoter (560 bp) region was amplified by PCR as a KpnI/SalI fragment and cloned upstream of the ORF. The leu1 gene was amplified by PCR as a XmaI/NotI and cloned into the vectors above. The plasmids were linearised with NruI before transformation into leu1-32 cip2Δ cells.
The tip1 ORF and 154 bp downstream were amplified by PCR from genomic DNA as PacI/AscI fragment and cloned into pFA6a. Two constructs were made: in the wild type control the 5′ primer contained the endogenous ATG, while in the −ATG one the ATG was mutated to TTT. The tip1 promoter region (865 bp) was amplified by PCR as a BamHI/PacI fragment and cloned into the vector above. The whole construct containing the tip1 ORF and flanking regions was cloned into pJK148 (containing a leu1 marker) as a KpnI/BamHI fragment. The construct was linearised and transformed as described above into leu1-32 tip1Δ cells.
For rum1 we used site-directed mutagenesis to mutate the first five ATGs of the ORF to CTGs. However, we found that this construct was still able to complement the sterility phenotype of a rum1Δ strain. This suggested that translation was taking place from a cryptic start site (different from ATG). Therefore, we made a construct in which all seven in-frame ATGs in the rum1 ORF were mutated to stop codons. For this construct a DNA fragment was synthesised (GENEART) that contained the whole ORF and flanking sequences (from the NheI to the SphI restriction sites), and in which all 7 in-frame ATGs had been replaced with the stop codon TGA. The digested DNA fragment was used to replace the corresponding section in pJET2.1 containing a leu1 marker and 2.6 kb of rum1 ORF and UTR sequences. −ATG and +ATG rum1 plasmids were linearised with NruI and transformed into leu1-32 rum1Δ cells.
In contrast to the situation with rum1, mutation of the first ATG of cip2 and tip1 was sufficient to create a loss-of-function phenotype indistinguishable to that of the deletion mutant. As both genes contain several ATGs downstream of the annotated initiation codon, we cannot completely rule out the possibility that in the –ATG mutants there is initiation from downstream ATGs (or from other cryptic sites different from ATG), leading to the formation of N-terminally truncated peptides. However, any such peptides would not be functional (Table S3).
Protein detection
Expression of TAP-tagged proteins was verified by western blot using peroxidase-anti-peroxidase soluble complexes (Sigma) to detect the protein A-binding domains of TAP. Myc tags were detected using the 9E11 monoclonal (Abcam).
RIp-chip
We followed a previously published protocol [6]. Immunoprecipitation of TAP-tagged proteins was carried out using monoclonal antibodies against protein A (Sigma), and myc-tagged proteins were purified using the 9E11 monoclonal antibody (Abcam). All experiments were performed with vegetative cells except those using Arp1-TAP, Alp5-TAP and Arp10-TAP, which were carried out under meiotic conditions. Puromycin experiments were conducted by adding a final concentration of 1 mM puromycin to cell cultures, followed by incubation at 32°C for 15 minutes. In addition, immunoprecipitation buffers contained 1 mM puromycin and 2 mM GTP. In control experiments the buffers contained only 2 mM GTP. EDTA treatment was performed as previously described [6].
Labelling and microarray experiments
Total RNA purified from the cell extract was used as a reference in all experiments. 20 µg of total RNA and all the RNA from the IP were labelled using the SuperScript Plus Direct cDNA Labelling System (Invitrogen). Labelled cDNAs were hybridised to PCR S. pombe DNA microarrays or to custom-designed oligonucleotide microarrays manufactured by Agilent as described [30], [31]. Microarrays were scanned with a GenePix 4000A microarray scanner and analysed with GenePix Pro 5.0 (Molecular Devices).
Data analysis
Only probes corresponding to coding sequences were considered. Spots with unreliable signals were removed as follows: for the PCR microarrays, spots that did not show a minimum of 55% of pixels above the median background signal plus two standard deviations in the immunoprecipitate and 90% of pixels in the total RNA (or at least 90% in the channel of the IP) were removed; for the Agilent microarrays the corresponding thresholds were 70%, 98% and 98%, respectively. Median log10 ratios were used for the analysis. We compiled a list of common mRNA unspecific contaminants, which were removed from the analysis. Selection of enriched RNAs in the RIp-chip experiments was carried out as described [6], by choosing genes whose enrichment ratios were at least two standard deviations above the median enrichment of all genes. Only mRNAs that passed this threshold in every independent biological experiment were considered enriched (Table 1). Assuming a normal distribution of the enrichments, the expected fraction of false positives using this threshold with a single experiment would be ∼0.05. However, as we only selected RNAs enriched in each of 2–4 repeats, this number is reduced to ∼2.5×10−3 to ∼6.25×10−6. All RIp-chip experiments in which mRNAs different from the one encoding the bait were detected were carried out at least twice. Other RIp-chips (negatives or containing only the cognate mRNA of the bait) were performed once or twice. All repeats were independent biological experiments and dyes were swapped for at least one experiment with each protein.
Data deposition
All raw and normalised microarray data have been deposited in ArrayExpress (accession number E-TABM-1158). Dataset S1 contains normalised data and experimental details for all RIp-chip experiments reported in this manuscript.
Supporting Information
Zdroje
1. FultonABL'EcuyerT 1993 Cotranslational assembly of some cytoskeletal proteins: implications and prospects. J Cell Sci 105 867 871
2. LuJRobinsonJMEdwardsDDeutschC 2001 T1-T1 interactions occur in ER membranes while nascent Kv peptides are still attached to ribosomes. Biochemistry 40 10934 10946
3. PhartiyalPJonesEMRobertsonGA 2007 Heteromeric assembly of human ether-a-go-go-related gene (hERG) 1a/1b channels occurs cotranslationally via N-terminal interactions. J Biol Chem 282 9874 9882
4. LinLDeMartinoGNGreeneWC 2000 Cotranslational dimerization of the Rel homology domain of NF-kappaB1 generates p50-p105 heterodimers and is required for effective p50 production. Embo J 19 4712 4722
5. NichollsCDMcLureKGShieldsMALeePW 2002 Biogenesis of p53 involves cotranslational dimerization of monomers and posttranslational dimerization of dimers. Implications on the dominant negative effect. J Biol Chem 277 12937 12945
6. AmorimMJMataJ 2009 Rng3, a member of the UCS family of myosin co-chaperones, associates with myosin heavy chains cotranslationally. EMBO Rep 10 186 191
7. HalbachAZhangHWengiAJablonskaZGruberIM 2009 Cotranslational assembly of the yeast SET1C histone methyltransferase complex. Embo J 28 2959 2970
8. MataJ 2010 Genome-wide mapping of myosin protein-RNA networks suggests the existence of specialized protein production sites. Faseb J 24 479 484
9. BuschKEHaylesJNursePBrunnerD 2004 Tea2p kinesin is involved in spatial microtubule organization by transporting tip1p on microtubules. Dev Cell 6 831 843
10. KellerCWoolcockKHessDBuhlerM 2010 Proteomic and functional analysis of the noncanonical poly(A) polymerase Cid14. Rna 16 1124 1129
11. LemieuxCBachandF 2009 Cotranscriptional recruitment of the nuclear poly(A)-binding protein Pab2 to nascent transcripts and association with translating mRNPs. Nucleic Acids Res 37 3418 3430
12. Correa-BordesJNurseP 1995 p25rum1 orders S phase and mitosis by acting as an inhibitor of the p34cdc2 mitotic kinase. Cell 83 1001 1009
13. KellyTJMartinGSForsburgSLStephenRJRussoA 1993 The fission yeast cdc18+ gene product couples S phase to START and mitosis. Cell 74 371 382
14. MillarJBBuckVWilkinsonMG 1995 Pyp1 and Pyp2 PTPases dephosphorylate an osmosensing MAP kinase controlling cell size at division in fission yeast. Genes Dev 9 2117 2130
15. MartinVRodriguez-GabrielMAMcDonaldWHWattSYatesJR3rd 2006 Cip1 and Cip2 are novel RNA-recognition-motif proteins that counteract Csx1 function during oxidative stress. Mol Biol Cell 17 1176 1183
16. KonNKrawchukMDWarrenBGSmithGRWahlsWP 1997 Transcription factor Mts1/Mts2 (Atf1/Pcr1, Gad7/Pcr1) activates the M26 meiotic recombination hotspot in Schizosaccharomyces pombe. Proc Natl Acad Sci U S A 94 13765 13770
17. MonahanBJVillenJMargueratSBahlerJGygiSP 2008 Fission yeast SWI/SNF and RSC complexes show compositional and functional differences from budding yeast. Nat Struct Mol Biol 15 873 880
18. ShevchenkoARoguevASchaftDBuchananLHabermannB 2008 Chromatin Central: towards the comparative proteome by accurate mapping of the yeast proteomic environment. Genome Biol 9 R167
19. KominamiKTodaT 1997 Fission yeast WD-repeat protein pop1 regulates genome ploidy through ubiquitin-proteasome-mediated degradation of the CDK inhibitor Rum1 and the S-phase initiator Cdc18. Genes Dev 11 1548 1560
20. LacknerDHBeilharzTHMargueratSMataJWattS 2007 A network of multiple regulatory layers shapes gene expression in fission yeast. Mol Cell 26 145 155
21. MittalNScherrerTGerberAPJangaSC 2010 Interplay between Posttranscriptional and Posttranslational Interactions of RNA-Binding Proteins. J Mol Biol
22. ScherrerTMittalNJangaSCGerberAP 2010 A screen for RNA-binding proteins in yeast indicates dual functions for many enzymes. PLoS ONE 5 e15499 doi:10.1371/journal.pone.0015499
23. TsvetanovaNGKlassDMSalzmanJBrownPO 2010 Proteome-wide search reveals unexpected RNA-binding proteins in Saccharomyces cerevisiae. PLoS ONE 5 e12671 doi:10.1371/journal.pone.0012671
24. LawrenceCLMaekawaHWorthingtonJLReiterWWilkinsonCR 2007 Regulation of Schizosaccharomyces pombe Atf1 protein levels by Sty1-mediated phosphorylation and heterodimerization with Pcr1. J Biol Chem 282 5160 5170
25. ForsburgSLRhindN 2006 Basic methods for fission yeast. Yeast 23 173 183
26. BählerJWuJQLongtineMSShahNGMcKenzieA3rd 1998 Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14 943 951
27. TastoJJCarnahanRHMcDonaldWHGouldKL 2001 Vectors and gene targeting modules for tandem affinity purification in Schizosaccharomyces pombe. Yeast 18 657 662
28. KeeneyJBBoekeJD 1994 Efficient targeted integration at leu1-32 and ura4-294 in Schizosaccharomyces pombe. Genetics 136 849 856
29. WilhelmBTMargueratSWattSSchubertFWoodV 2008 Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature 453 1239 1243
30. AmorimMJCotobalCDuncanCMataJ 2010 Global coordination of transcriptional control and mRNA decay during cellular differentiation. Mol Syst Biol 6 380
31. LyneRBurnsGMataJPenkettCJRusticiG 2003 Whole-genome microarrays of fission yeast: characteristics, accuracy, reproducibility, and processing of array data. BMC Genomics 4 27
32. BrowningHHaylesJMataJAvelineLNurseP 2000 Tea2p is a kinesin-like protein required to generate polarized growth in fission yeast. J Cell Biol 151 15 28
33. SimanisVNurseP 1986 The cell cycle control gene cdc2+ of fission yeast encodes a protein kinase potentially regulated by phosphorylation. Cell 45 261 268
34. ShiozakiKRussellP 1995 Cell-cycle control linked to extracellular environment by MAP kinase pathway in fission yeast. Nature 378 739 743
35. AslettMWoodV 2006 Gene Ontology annotation status of the fission yeast genome: preliminary coverage approaches 100%. Yeast 23 913 919
36. TakedaTTodaTKominamiKKohnosuAYanagidaM 1995 Schizosaccharomyces pombe atf1+ encodes a transcription factor required for sexual development and entry into stationary phase. EMBO J 14 6193 6208
37. MorrellJLMorphewMGouldKL 1999 A mutant of Arp2p causes partial disassembly of the Arp2/3 complex and loss of cortical actin function in fission yeast. Mol Biol Cell 10 4201 4215
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 12
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Targeted Proteolysis of Plectin Isoform 1a Accounts for Hemidesmosome Dysfunction in Mice Mimicking the Dominant Skin Blistering Disease EBS-Ogna
- The RNA Silencing Enzyme RNA Polymerase V Is Required for Plant Immunity
- The FGFR4-G388R Polymorphism Promotes Mitochondrial STAT3 Serine Phosphorylation to Facilitate Pituitary Growth Hormone Cell Tumorigenesis
- Target Site Recognition by a Diversity-Generating Retroelement
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy