
Acute Multiple Organ Failure in Adult Mice Deleted for the Developmental Regulator Wt1
There is much interest in the mechanisms that regulate adult tissue homeostasis and their relationship to processes governing foetal development. Mice deleted for the Wilms' tumour gene, Wt1, lack kidneys, gonads, and spleen and die at mid-gestation due to defective coronary vasculature. Wt1 is vital for maintaining the mesenchymal–epithelial balance in these tissues and is required for the epithelial-to-mesenchyme transition (EMT) that generates coronary vascular progenitors. Although Wt1 is only expressed in rare cell populations in adults including glomerular podocytes, 1% of bone marrow cells, and mesothelium, we hypothesised that this might be important for homeostasis of adult tissues; hence, we deleted the gene ubiquitously in young and adult mice. Within just a few days, the mice suffered glomerulosclerosis, atrophy of the exocrine pancreas and spleen, severe reduction in bone and fat, and failure of erythropoiesis. FACS and culture experiments showed that Wt1 has an intrinsic role in both haematopoietic and mesenchymal stem cell lineages and suggest that defects within these contribute to the phenotypes we observe. We propose that glomerulosclerosis arises in part through down regulation of nephrin, a known Wt1 target gene. Protein profiling in mutant serum showed that there was no systemic inflammatory or nutritional response in the mutant mice. However, there was a dramatic reduction in circulating IGF-1 levels, which is likely to contribute to the bone and fat phenotypes. The reduction of IGF-1 did not result from a decrease in circulating GH, and there is no apparent pathology of the pituitary and adrenal glands. These findings 1) suggest that Wt1 is a major regulator of the homeostasis of some adult tissues, through both local and systemic actions; 2) highlight the differences between foetal and adult tissue regulation; 3) point to the importance of adult mesenchyme in tissue turnover.
Published in the journal:
. PLoS Genet 7(12): e32767. doi:10.1371/journal.pgen.1002404
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002404
Summary
There is much interest in the mechanisms that regulate adult tissue homeostasis and their relationship to processes governing foetal development. Mice deleted for the Wilms' tumour gene, Wt1, lack kidneys, gonads, and spleen and die at mid-gestation due to defective coronary vasculature. Wt1 is vital for maintaining the mesenchymal–epithelial balance in these tissues and is required for the epithelial-to-mesenchyme transition (EMT) that generates coronary vascular progenitors. Although Wt1 is only expressed in rare cell populations in adults including glomerular podocytes, 1% of bone marrow cells, and mesothelium, we hypothesised that this might be important for homeostasis of adult tissues; hence, we deleted the gene ubiquitously in young and adult mice. Within just a few days, the mice suffered glomerulosclerosis, atrophy of the exocrine pancreas and spleen, severe reduction in bone and fat, and failure of erythropoiesis. FACS and culture experiments showed that Wt1 has an intrinsic role in both haematopoietic and mesenchymal stem cell lineages and suggest that defects within these contribute to the phenotypes we observe. We propose that glomerulosclerosis arises in part through down regulation of nephrin, a known Wt1 target gene. Protein profiling in mutant serum showed that there was no systemic inflammatory or nutritional response in the mutant mice. However, there was a dramatic reduction in circulating IGF-1 levels, which is likely to contribute to the bone and fat phenotypes. The reduction of IGF-1 did not result from a decrease in circulating GH, and there is no apparent pathology of the pituitary and adrenal glands. These findings 1) suggest that Wt1 is a major regulator of the homeostasis of some adult tissues, through both local and systemic actions; 2) highlight the differences between foetal and adult tissue regulation; 3) point to the importance of adult mesenchyme in tissue turnover.
Introduction
Although much is known about the mechanisms that govern cellular differentiation during development, we know less about the processes that regulate cell turnover and homeostasis in the adult. Perhaps the exceptions to this rule are rapidly turning over tissues such as intestine, skin and haematopoietic tissue. Recently it has been shown that genes required for regulating differentiation during foetal development may not be used in regulating turnover of the same tissues in the adult [1], [2].
Mutation of the Wilms tumour gene, WT1, in humans may lead to the eponymous paediatric kidney cancer, glomerulosclerosis of the kidney and gonadal dysgenesis, which can manifest as male to female sex reversal [3]. During foetal development, Wt1 is expressed in the kidney, gonads, spleen, the mesothelium which surrounds most organs as well as ill-defined body mesenchyme. Knockout mice lack kidneys, gonads, and spleen and the animals die at mid-gestation through the lack of coronary vasculature formation [4]. There are no apparent defects of the skeletal, haematopoietic, digestive, or metabolic systems.
Recently we have shown that Wt1 is a key regulator of the balance between the epithelial and mesenchymal states in a number of developing organs. Whereas it is required for the mesenchymal to epithelial transition (MET) underlying the formation of kidney nephrons, in the heart it is essential for the reverse process, the epithelial to mesenchyme transition (EMT) required for the production of proliferating cardiovascular progenitors from the epicardium (a mesothelium) [5]. In a similar vein Wt1 expressing mesothelial cells in the intestine and lung produce mesenchymal progenitors for vascular smooth muscle [6], [7]. Furthermore, very recent evidence proves that, in the developing liver, Wt1 expressing mesothelial cells provide the precursors for stellate cells [8], [9], [10]. Stellate cells in the liver and the pancreas have aroused much interest through their ability to regulate tissue fibrosis, via the production of cytokines [11], [12]. They are also important for the progression of pancreatic cancer [13].
In the adult, Wt1 is expressed in very few tissues in a small percent of cells. These include the mesothelium surrounding a number of visceral organs [14], the glomerular podocyte cells of the kidney, Sertoli/granulosa cells in the testes/ovaries [15], [16], [17] and 1% of bone marrow (BM) cells (with properties of restricted haematopoietic progenitors) [18]. Nothern Blot analysis has shown that Wt1 is also expressed in a variety of epithelial cells including spleen, lung and heart. Our own data, including those provided in this paper suggest that this mainly reflects expression in the mesothelial lining of these tissues. We speculated that the expression of Wt1 in these rare sites in the adult could have functional significance, for the following reasons. Firstly, given the importance of the mesothelium as a source of progenitor cells, requiring Wt1 function during development, we hypothesised that mesothelia might perform a similar function in the adult and this might require Wt1.
Secondly, Wt1 is essential for the formation and maturation of podocytes [19]. We hypothesised that continued expression of Wt1 in the adult would reflect a role in kidney maintenance.
Thirdly, WT1 is mutated or overexpressed in acute myeloid leukaemia (AML) [20]. However, Wt1 is not required for foetal haematopoiesis [21]. Given Wt1 expression in adult bone marrow and association with leukaemia, we surmised that Wt1 might play a role in adult haematopoiesis.
Finally, WT1 is expressed at high levels in most adult cancers studied [22], though expression has not been detected in the normal tissue counterparts. It has been proposed that WT1 might be an oncogene in adult cancer in contrast to its function as a tumour suppressor in paediatric kidney cancer [3]. As a prelude to testing this, it was necessary first to determine whether the gene is essential for normal development or maintenance of the epithelia from which these tumours arise.
To address these propositions, we deleted the Wt1 gene ubiquitously in adult mice. While our findings inform on these issues, the results far exceeded our expectations. The range, severity, and rapidity of the phenotypes observed were dramatic and unexpected and raise major questions about adult tissue homeostasis.
Results
Tamoxifen-mediated deletion of Wt1
To enable inducible deletion of Wt1 in the adult, we generated tamoxifen inducible Wt1 KOs by crossing CAGG promoter driven Cre-ER™ mice with our homozygous Wt1 conditional mice, where the first exon of Wt1 is flanked by loxP sites [5]. Successful Wt1 deletion was demonstrated by recombination PCR and the depletion of Wt1 expression in mesothelia (Figure S1 and Figure S2). Deletion of Wt1 in the mesothelium did not affect the integrity of the tissue (Figure S3). The health status of the mutant animals deteriorated quickly and all the mice had to be culled by 10 days post-induction (p.i.). Prior to death, the mutant mice presented dramatic phenotypes; they were less active and oedemic. Upon dissection, fluid was sometimes found in the abdominal cavity and in the subcutaneous tissues. Detailed gravimetric analysis showed that there was a reduction in the spleen to body weight ratio as well as in the heart to body weight ratio (Table 1). Subsequent histological analysis of internal organs revealed pale kidneys, severe spleen and pancreas atrophy, and deficiency of fat tissues. For most tissues, mice treated at 3, 10, or 13 weeks of age developed the same phenotypes. The only exception to this involved fat, as we discuss in more detail later. Before considering each phenotype, it is important to emphasise that not all tissues showed overt signs of damage. For example, we observed no obvious macroscopic changes to the lung, liver or intestine - three tissues often involved in systemic inflammatory responses. Furthermore, although there was a 30% reduction in the heart/body weight ratio there was no obvious cardiovascular pathology (Table 1).

Deletion of Wt1 leads to acute glomerulosclerosis
Wt1 is crucial for kidney development as the conventional Wt1-null embryos suffer from renal agenesis [4]. Upon induction of Wt1 deletion in our model, expression of Wt1 in the podocytes was completely depleted (Figure 1B) and the mutant mice were shown to have severe proteinuria (Table 2). H&E staining showed that the tubules were filled with protein casts (Figure 1A, arrow). The mutant kidneys had well developed glomerulopathy with cytopathic changes in podocytes and parietal epithelium. There was almost complete loss of synaptopodin and nephrin expression in the podocytes in the mutant kidneys (Figure 1C and 1D). EM studies showed that the foot processes of the podocytes were completely lost in the mutant kidneys (Figure 1E, day 10 post-injection). The development of the kidney phenotype in our model was extremely rapid. Five days post-tamoxifen injection, H&E stained kidney sections showed normal histology while podocyte effacement started to appear (Figure 1F). At day 7 post-injection, protein casts in the tubules were already present and the glomeruli started showing signs of degeneration (Figure S4a). Finally, plasma levels of urea and creatinine were normal at day 5 p.i., started to rise at day 7 p.i., and were significantly elevated at day 10 p.i (Table 2). In our model, mice that were heterozygous for the Wt1 conditional allele (CAGG-CreER™; Wt1loxP/+) did not exhibit any kidney abnormalities after tamoxifen-mediated deletion of Wt1. In addition, tamoxifen treated mice that were only positive for the CAGG-CreER™ allele and wild type for the Wt1 loxP sites (i.e. CAGG-CreER™ positive; Wt1+/+) were also included as controls and did not demonstrate any phenotypes. The kidney phenotype in our model is similar to other nephrotic syndrome mice where podocytes are damaged [23], [24], [25], [26]. However, none of these other mouse models presented any of the other phenotypes we describe below apart from the kidney defects. Most importantly, Wt1 has been deleted specifically in adult podocytes. These animals develop glomeruloscelosis similar to that described here but did not develop the other phenotypes we report below. Furthermore, the mice survived well beyond the timeframe reported here [27].


Wt1 is expressed at E9 in the urogenital ridge and subsequently in the sex cords of the genital ridge in mice and it is a crucial factor for gonad development and sex determination [28]. In adult mice, Wt1 is expressed in Sertoli cells in the testes and granulosa cells in the ovaries [15]. We observed a reduction in the size of the testes and ovaries; however the difference was not statistically significant (Table 1). None of the testis markers studied showed any difference in expression patterns (Figure S5).
Deletion of Wt1 leads to an aberrant haematopoietic system
Asplenia in the conventional Wt1-null mice correlates with enhanced apoptosis in the primordial spleen cells [29]. In the adult Wt1 KO model, the mutant spleen was much paler and smaller in size compared with the control spleen (Figure 2A, arrow). There was a reduction in the number of proliferating cells in the mutant spleen; however the number of cells expressing an apoptotic marker (active caspase 3) remained unchanged (Figure S8A–S8D). The spleen to body weight ratio was reduced by 60% in the mutants of both the young (Figure 2D, 3 week old, p-value = 0.003; 8 controls and 5 mutants were analysed) and mature groups (Figure 2D, p-value = 0.000, 9 controls and 12 mutants were analysed).

The mutant mice had diminished extramedullary haematopoiesis within the red pulp compartment while white pulp remained largely unaffected (Figure 2B, 2C). FACS analysis showed an almost complete absence of erythrocytes (Ter-119 positive) in the mutant spleens (Figure 2E, 0.69±0.17% in the mutant c.f. 55.7±3.9% in the control spleen, p-value = 0.024; five controls and three mutants were analysed) and in Wt1-mutant bone marrow (Figure 2E, 7.3±3.1% in the mutant c.f. 30.3±4.0% in the control bone marrow, p-value = 0.025; five controls and three mutants were analysed).
An intrinsic defect in the mutant haematopoietic system
Maturation of red blood cells requires erythropoietin (EPO) [30], which is synthesised mainly in the kidney. Furthermore, Wt1 has been shown to transcriptionally activate the EPO gene [31]. To determine whether the defect in erythropoiesis is intrinsic to the haematopoietic system, we cultured the mutant bone marrow cells in a methylcellulose-based system where a complete set of factors for supporting haematopoietic differentiation is provided in the medium. After two weeks in culture, despite the presence of all the required growth factors, the Wt1-mutant bone marrow cells failed to differentiate into the erythrocyte lineage, while the control bone marrow cells, as expected, did form red blood cells (Figure 2F, 5.0%±1.87% in the mutant compared with 31.3%±9.6% in the control; five controls and three mutants were analysed, p-value = 0.05).
To address whether this defect in erythropoiesis reflects a cell autonomous role for Wt1 in haematopoiesis, we set out to characterise the 1% of bone marrow cells that express Wt1. Using the Wt1-GFP knockin mouse (Wt1GFP/+), we FACS sorted GFP positive cells from the bone marrow of Wt1GFP/+ mice and cultured them in a methylcellulose-based system. It has been shown previously that some Wt1-expressing cells in the bone marrow express markers characteristic of short-term haematopoietic stem cells (Ter119−CD45+Mac-1loc-kit+Sca-1+) [18] but the differentiation potential of these cells was not investigated. Hence we investigated the potential of these Wt1-GFP cells to differentiate to different haematopoietic lineages in culture. First we stained the GFP positive BM cells with a set of haematopoietic stem cell markers (CD150, CD48, and CD244) [32] and showed that approximately 50% of GFP positive BM cells were in the population of oligolineage-restricted progenitors (CD150−CD48+CD244−). Before culturing, no GFP-positive cells were positive for Ter-119 or Cd11b and only a few percent of the cells expressed CD45. After two weeks in culture, the GFP-positive cells were able to form Ter119 (red blood cells), CD45 (white blood cells), and CD11b (granulocytes) positive cells (Figure 2G). From this we can conclude that the Wt1-expressing cells are oligolineage-restricted progenitors.
We then set out to test if the reduction of erythrocytes reflected a decrease in the number of erythrocyte progenitors (Pre CFU-E) using the high resolution myeloerythroid progenitor cell staging method described by Pronk et al [33]. Representative flow cytometric profiles are shown in Figure 3. We saw a significant reduction in the % of Pre CFU-E in the mutant spleen (Figure 3, 0.27±0.06 in the controls and 0.03±0.008 in the mutants, p-value = 0.001; 7 control and 8 mutant mice were analysed). Erythrocyte progenitor cells branch from megakaryocyte-erythrocyte progenitors (PreMegE). Another progenitor that branches from PreMegE is the Megakaryocyte progenitor (MkP) which produces platelets. Both MkP and Pre MegE were reduced significantly in the mutant spleen (Figure 3) However, the number of platelets in the circulation was not affected (control platelet number is 880.5±89.9 K/µL and mutant platelet number is 817.5±164 K/µL). Mutant mice did not show any obvious bleeding tendencies. The half life of platelets is about 35 hours [34]. Platelet deficiency may have developed if the mice had survived longer.

Deletion of Wt1 leads to rapid bone loss
We observed abnormalities of the growth plate in both the tibias and femurs of Wt1-mutant mice. The vascular invasion zones were irregular and anaemic (Figure 4A, indicated by arrow). The proliferative zone chondrocytes of the mutant mice were irregular with less surrounding territorial matrix than control mice (Figure 4A). The inner (marrow) surface of the long bone from the mutant mice was ragged compared with control mice (Figure 4B, arrows), suggesting increased bone resorption. We then analysed the bone architecture of femurs, tibias, and spine 9 days after induction of Wt1 deletion using μCT (Figure 4C). The 3D movie of the trabecular bone loss is shown in Videos S1 and S2. Trabecular bone volume was reduced by 30% in the mutants (Figure 4D), mostly due to a reduction in trabecular number and a small reduction in trabecular thickness. Furthermore, trabecular connectivity was also reduced. Taken together, these changes in bone architecture would be expected to lead to a substantial reduction in bone strength (Figure 4D). The bone loss observed could be due to either reduced bone growth or increased bone absorption. However, bone formation is a relatively slow process, and in view of the rapidity of the phenotype observed here it seemed that increased bone resorption was the more likely cause. We therefore stained sections of the long bones for the osteoclast marker TRAcP and observed dramatically increased numbers of osteoclasts on the bone surface of the Wt1-mutant bones (Figure 4E). To test if these bone phenotypes might reflect an intrinsic role for Wt1 in the osteoclast and osteoblast lineages, we harvested fresh bone marrow cells from the mutant mice, induced Wt1 deletion by treating the bone marrow cells with 4-OH tamoxifen for three days and cultured the cells in media supplemented with M-CSF and RANKL to induce osteoclast differentiation. Surprisingly and in contrast to the in vivo study, mutant bone marrow cells in which Wt1 had been deleted by tamoxifen treatment were less capable of forming osteoclasts in vitro (Figure 4F, p-value = 0.05 and 0.029 at 10 and 30 µg/ml of RANKL respectively; three separate experiments were performed each using bone marrow pooled from 2–3 control or mutant mice). When we used a similar in vitro approach using culture medium inducing osteoblastic differentiation, we observed that Wt1-mutant osteoblasts had reduced bone differentiation ability as levels of the osteoblast marker enzyme alkaline phosphatase were reduced (Figure 4G, p-value = 0.037; three separate experiments were performed). These results suggest that Wt1 plays an intrinsic role in both osteoclast and osteoblast differentiation, and that the loss of Wt1 is likely to disturb bone homeostasis.

Fat reduction following Wt1 deletion
The Wt1-mutant mice also displayed reduction in the size of fat pads. In addition to the abdominal fat pads which mainly comprise white adipocytes, interscapular brown adipocytes were also atrophic and had fewer lipid cytoplasmic vacuoles than controls (Figure 5A–5J). Although the trend of fat loss was consistent in mutant mice, the reduction of fat pad size seemed to be more variable in the older group of animals (13 weeks, Figure 5K, arrows). In some mutant animals, the reduction in the size of fat pads was observed in both the interscapular and abdominal fat pads, while in other mutants the lipid vacuole size reduction was seen in the abdominal fat pads but not in the interscapular fat pads. The weight of fat pads in the mutant mice did not reflect their actual size because of the oedema (data not shown), and we therefore analysed fat pad volume using whole body μCT scans. Mice were scanned at the start (before tamoxifen injection) and the end of the experiment (9 days after induction). Results from the μCT scan confirmed the substantial fat loss in the mutants (Figure S7, arrows). There was no difference in the number of apoptotic and proliferating cells in the fat pads between mutant and control mice. Histological analysis of the adipose tissues showed that the reduction in the size of fat pads reflected a decrease in the vacuole size of the adipose tissues, as seen in the abdominal fat pads (Figure 5L–5M, p<0.05; three controls and three mutant mice were analysed). Consistent with this loss of fat, there was a significant reduction in the level of AP2 expression in mutant abdominal fat pads (Figure 5N, p-value = 0.05; three controls and three mutants were analysed).

Wt1 expression in fat has not been reported previously. However, here we show that Wt1 is expressed in the mesentery, epididymal, and retroperitoneal fat pads, but not at detectable levels in the abdominal fat pad nor in the interscapular brown adipose tissue (Figure 5O, 5P). Given the fact that adipocytes and osteoblasts have a common origin in the bone marrow, we examined whether there was any alteration in the number of adipocytes in the bone marrow. Labelling adipocytes using AdipoRed, we found a reduction in the number of adipocytes in the mutant bone marrow (Figure 5Q, p-value = 0.021; four controls and four mutants were analysed).
As adipocytes and osteoblasts arise from the stromal mesenchymal population in the bone marrow, we speculated that Wt1 loss might lead to a disturbance in this population which can be quantified using an antibody to Stro-1. We did in fact find a significant (five fold) increase in this population of cells following Wt1 loss (Figure 4H, p-value = 0.02; four controls and four mutants were analysed).
Deletion of Wt1 leads to atrophy in the exocrine pancreas
Figure 6G–6J (arrows) shows the successful depletion of Wt1 expression in the pancreatic mesothelium. The pancreas from the mutant mice was severely atrophied. H&E staining demonstrated that there was a substantial amount of cell loss in the exocrine tissues while the endocrine pancreas remained largely unaffected (Figure 6A, 6B). Acini in the mutant pancreas were loosely packed and acinar cells appeared atrophied and presented less eosinophilic cytoplasmic staining, suggesting a reduced zymogen content. Residual acinar epithelial cells were rounded and less cohesive with neighbouring cells. Similar aberrant histology started to appear at day 7 after Cre activation (Figure S4B). We saw an increase in the number of apoptotic cells in the mutant pancreas (Figure 6C, 6D) while the number of proliferating cells remained unchanged (Figure 6E, 6F). Although the pathology of our model shares many similarities to pancreatitis mouse models, there was no elevation of serum amylase level in the Wt1-mutant mice (Table 2). Given the severity of the pancreas phenotype, it is surprising to see the lack of any elevation of serum amylase. However, this probably reflects the short space of time between the onset of the phenotype and death of the mice. Pancreatitis involves inflammation of the pancreatic tissues and in Wt1-mutant mice we observed a low-grade inflammation in much of the pancreas and scattered foci of more severe active inflammation. In the Wt1-mutant pancreas, the presence of infiltrating macrophages was confirmed by staining with macrophage marker F4/80 (Figure S6E, S6F); however, staining of CD11b, Gr1, and CD3 were absent (data not shown). Both insulin and amylase expression were normal in the mutant pancreas sections (Figure S6A–S6D).
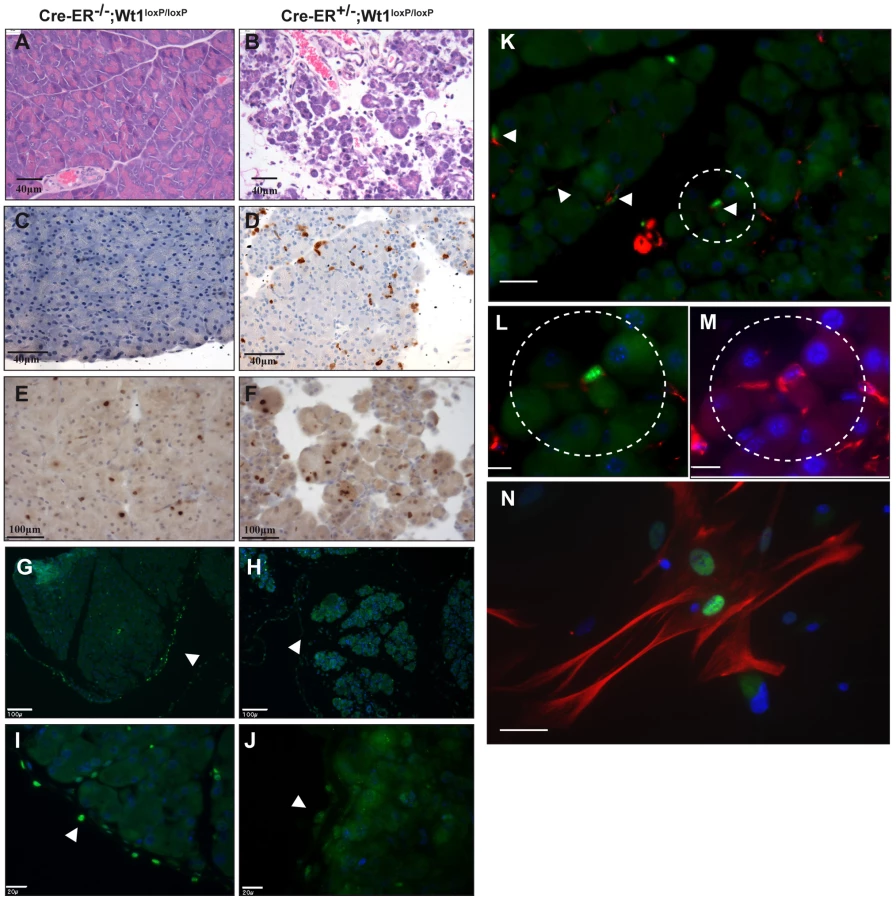
To try to gain more insight into the origin of the pancreatic phenotype, we examined more closely the cell types that express Wt1 in the exocrine pancreas. Pancreatic stellate cells (PSCs) have been implicated in pancreatitis and pancreatic cancer. We show Wt1 is expressed in the mesothelial lining of the pancreas as well as in PSCs. Desmin is a marker for PSCs [35]. The interstitial cells that express Wt1 also express desmin, and this was demonstrated in sectioned pancreata (Figure 6K–6M) and in cultured PSCs (Figure 6N).
Serum protein profiling reveals no systemic inflammatory or nutritional response but dramatic reduction in IGF-1 levels
One possible explanation for the dramatic and acute nature of the phenotypes observed in these mice is a systemic inflammatory response, even though analysis of the diseased pancreas did not suggest this. Furthermore, even though the animals appeared to show no signs of distress and their stomachs were full at 9–10 days, it is possible that the bone and fat defects were due to nutritional deprivation. To assess these possibilities, we carried out quantitative analysis of 40 cytokines and 38 adipokines in mutant versus wildtype serum using antibody arrays. Perhaps surprisingly, given the severity of the phenotypes there was no statistically significant change in the levels of any inflammatory cytokines (Figure 7A; three controls and three mutants were analysed), arguing that the phenotypes were not due to a systemic inflammatory response. As a positive control to test that the arrays were working, we treated the mice with LPS and then assayed cytokine levels. There was a 23 fold induction in MIP-2, an 11 fold induction of JE, a 6 fold induction of KC, and a 3 fold induction of TNFα (Figure 7B). These findings demonstrate that the assays work and are able to measure an acute systemic inflammatory response. Similarly, there was no indication of nutritional deprivation. Following calorific restriction, there is reported to be a 60–80% reduction in serum leptin levels [36], [37], a 65% reduction in TNFα [38], a 100% increase in AgRP/FIAF [39], and a 75% increase in the levels of adiponectin [40]. We saw no significant changes in any of these molecules (Table 3 and Figure 7C), supporting the idea that the mice were not suffering nutritional deprivation and, in turn, this was not causing any of the phenotypes. However, we did observe a dramatic 85% reduction in the levels of IGF-1 and 3.5 fold increase in the levels of FGF21 (Figure 7C). This could in part account for the bone and fat phenotypes respectively. To investigate if the reduction of IGF-1 levels could due to global growth hormone deficiency, we measured circulating growth hormone (GH) using ELISA. We observed a slight elevation of GH levels in the mutant serum (Figure 7J; three controls and five mutants were analysed, p-value = 0.025). Histology analysis showed absence of any pathological abnormalities in the pituitary and adrenal glands (Figure 7D–7I).


Discussion
The multiple organ disturbance observed in adult mice deleted for Wt1 is striking, and, we believe, unprecedented in terms of severity and rapidity of onset. There is perhaps no need to point out that most of these phenotypes have relevance for diseases common in adults, even though our starting point was a gene more or less defined for its role in the development of several organs. Our study shows that Wt1 plays a key role in regulating the production or turnover of red blood cells, bone and fat in the adult. Despite intensive analysis of Wt1-null foetuses, including those surviving to 18 days gestation, no developmental defects in these tissues were found previously [4], [29]. Thus our study contributes to the growing body of evidence that adult tissues may employ different or additional players compared to foetal development. Wt1 is among a list of genes whose methylation increases with age in a genome-wide CpG island methylation profiling study [41]. Therefore Wt1 expression levels may well decrease with age. It will be important to determine whether Wt1 levels in these key cell populations reduce during aging or under different environmental influences. If so, this could contribute to disease-related phenotypes described here.
Although there is much future work needed to elucidate the mechanisms underlying these phenotypes, there are several conclusions we can draw at present. Perhaps, surprisingly, we could detect no significant changes in serum cytokine levels, arguing that the phenotypes we observe are unlikely to be due to a systemic inflammatory response, even though this is often associated with damage to the tissues that are affected in the Wt1 mutant mice. As we argue below, the phenotypes involving the kidney and erythrocytes reflect an intrinsic function of Wt1 in these tissues or their progenitors. On the other hand, we believe loss of fat and bone is likely to be a combination of systemic and local factors.
The phenotypes involving the haematopoietic system and bone, have their origins wholly or partly within the bone marrow itself. Wt1 is expressed in a restricted haematopoietic progenitor population and its loss leads to disturbance in red blood cell and osteoclast production. This is consistent with the previous finding that Wt1 expression is upregulated during early myeloid differentiation (particularly in the common myeloid progenitors and megakaryocyte-erythroid progenitors) [18]. In keeping with this, we found the levels of PreMegE, MkP, and Pre CFU-E were significantly decreased in mutant spleen, Given the association of Wt1 with AML, we might have expected an imbalance in the myeloid compartment. Preliminary analysis has not demonstrated a reduction in the absolute number of monocytes and granulocytes in the circulation of Wt1 mutant mice. However, this may have only become evident if the mice had survived longer.
The bone loss in most part is likely to result from the increase in osteoclasts that we observed in the bone marrow. Paradoxically, mutant mice showed a reduction in osteoclast formation ability in vitro. The bone marrow compartment in which we saw an increase in the number of osteoclasts consists of a mixed population of cells. The mesenchymal stromal cells and haemaatopoietic stem cells are in close proximity in the bone marrow and there is known to be crosstalk between these cell types [42], [43]. Our in vitro osteoclast formation cell culture system started with a restricted population of cells (bone marrow stromal cells). The in vivo and in vitro difference could be due to factor(s) that are present in the bone marrow but absent in the in vitro culturing system.
However, we also found that Wt1 is required for osteoblast synthesis in bone marrow culture pointing to a role in the mesenchymal lineage. Consistent with this, our preliminary experiments have shown that non-haematopoietic Wt1-GFP positive cells from the bone marrow stroma are able to differentiate to bone and fat (unpublished observations). Furthermore, we show here that Wt1 loss also leads to an increase in Stro1 positive stromal mesenchymal stem cells, which may explain partly the disturbance in adipocyte and osteoblast production in the bone marrow. Our serum protein analysis showed a dramatic reduction of IGF-1 levels and this might be expected to contribute to the bone loss phenotype. Interestingly, deletion of IGF-1 specifically in the liver, the major source of synthesis, only leads to a 75% reduction in circulating IGF-1 levels and there is no apparent phenotype [44]. However, mice that are double homozygous mutant for IGF-1 and the binding protein acid labile subunit (ALS) [45] show an 85% reduction in IGF1 - levels and a similar degree of bone thinning to that seen in our Wt1 adult knockout mice. Hence, it seems reasonable to conclude that the 85% reduction of IGF-1 levels in our mutant mice is a major factor behind the bone phenotype. In the Wt1 mutant mice, the IGF-1 levels are much lower than those observed when IGF-1 is deleted specifically in the liver, so either Wt1 is required for IGF-1 expression in non-hepatocytes, or for factors that stabilise IGF-1 in the serum. Growth hormone, produced by the pituitary gland, is a major regulator of IGF-1 levels. One possibility was that the reduction in IGF-1 level was due to defects in the pituitary axis and downregulation of GH. However, we detected no pathological abnormalities in the pituitary and adrenal glands, and if anything GH levels were increased.
Obesity is a major health problem and there is considerable topical interest in the factors that regulate fat levels. Loss of Wt1 not only leads to reduced adipocyte production in the bone marrow but also to rapid systemic loss of fat, with dramatically reduced vacuole size. There are several reasons why we believe this fat loss is not due to under-nourishment. Fat vacuole reduction was already apparent 7 days after tamoxifen injection, at which time the health status of the animals was normal. Nine days post-injection, the mutant mice still actively sought food and their stomachs were full on autopsy. Importantly, there was no change in the levels of circulating leptin, adiponectin, TNFα, and AgRP/FIAF, all of which would be expected to change dramatically after one or two days of calorific restriction. There was a reduction in the level of lipocalin 2 in mutant serum (Figure 7C). Lipocalin 2 is abundantly produced from adipocytes [46], [47]. The reduction of lipocalin 2 could be caused by the reduced volume of adipose tissues in mutant mice. Taken together our findings provide evidence that Wt1 may influence both the formation and maintenance of adipocytes. The fat loss is extremely rapid and given that Wt1 only appears to be expressed in a proportion of fat pads affected, it seems likely that systemic factors might be involved. We found that the levels of circulating FGF21 increased by 3.5 fold in the mutant animals and this would be expected to induce some fat loss [48].
The RT-PCR result showed that Wt1 expression was detected in fat pads (Figure 5O). In preliminary experiments to address whether this reflects expression in mature adipocytes or the stromal vascular compartments, we digested and fractioned fat tissues from the Wt1-GFP knockin mice into the floating mature adipocyte layer and the stromal vascular fraction. The majority of the GFP signal was seen in the stromal vascular fraction (unpublished data). This supports the idea that systemic or local paracrine factors dependent on Wt1 are regulating adipocyte homeostasis.
The effect of Wt1 loss on bone and fat turnover is interesting in the context of Wilms' tumours. We and others have shown that the 15–20% subset of Wilms' tumours arising through WT1 loss are more likely to be stromal (mesenchymal) predominant and often contain ectopic tissues, including bone, fat, cartilage and muscle [49], [50], [51]. Taken together this and our new findings underline the key role of the mesenchyme and Wt1 in tissue turnover and maintenance.
With regard to the pancreatic atrophy, this does not appear to be typical pancreatitis as there was no increase in serum α-amylase. However, as discussed above, amylase level may have increased if the mice had lived longer. Serum cytokine profiles showed that there was no systemic inflammatory response in the mutant mice. In line with this there was no observable pathology in liver, lung and intestine, all tissues susceptible to inflammation. It remains to be seen whether the severe pancreatic atrophy is due to loss of Wt1 function within the tissue itself. We can exclude an effect through loss of Wt1 function in the islet or acinar cells as deletion of the gene specifically in these cell types using PDX1-Cre did not lead to overt pathology in the pancreas or elsewhere (P. Hohenstein, V. Brunton, M. Frame, O. Samson and N. Hastie unpublished observations). One possibility is that the pancreatic atrophy arises through activation of the sub-population of stellate cells that express Wt1 although further study is required to investigate this hypothesis. Activated stellate cells produce cytokines [52] and we speculate that these may be responsible for destroying the acinar cells. Given the published data on foetal liver [8], the parallel between pancreatic and hepatic stellate cells, and the role of Wt1 in generating vascular progenitors from the epicardium by EMT [5], we hypothesise that a proportion of pancreatic stellate cells arise from the mesothelium, via an EMT, once more pointing to the role of this tissue as a source of mesenchymal progenitor cells.
Despite the accumulating knowledge about the importance of Wt1 at multiple stages of kidney development, the function of Wt1 in the podocytes of mature glomeruli has remained the subject of some speculation. Even though children and adult mice with Wt1 mutations characteristic of Denys-Drash and Frasier syndrome develop glomerulosclerosis, it was always possible that the damage had its origin in utero, rather than reflecting a continued function for Wt1 in the maintenance of the adult kidney. Our results provide the first evidence that Wt1 is crucial for maintaining the integrity of mature podocytes. Our model allowed us to test whether the glomerulosclerosis we observed arises through abnormalities of cell proliferation or the differentiation state of the mature podocytes. We did not see major changes in proliferation or apoptosis in the mutant glomeruli deleted for Wt1 (using proliferation marker anti-phosph-histone H3 and apoptosis marker active caspase 3, Figure S8E–S8H). However, we showed that loss of Wt1 expression resulted in damage to the foot processes of the podocytes therefore causing a morphological alteration. Nephrin is necessary for the renal filtration barrier and is also a known downstream target of Wt1 during kidney development [53]. Consistent with this we found that nephrin expression levels reduce dramatically after Wt1 deletion, indicating that the transcriptional regulation of nephrin by Wt1 continues into adult life. Here we show that Wt1, known to be a key regulator of nephrogenesis, is also vital for the maintenance of adult glomerular structure and function, something that has been the subject of speculation but not proven until now.
Clearly these findings should be followed up using tissue specific Cre lines. However, at present suitable Cre lines are not available for several of the crucial lineages we wished to investigate, including the mesothelium and mesenchymal stem cells. In the mean time, we have been able to use cultures to show that several of the phenotypes we observed are intrinsic to the bone marrow.
The results presented in this study open new avenues of research into mesenchymal cell function in adult tissues. The cell types that express Wt1 in adult tissues e.g. the hepatic and pancreatic stellate cells and bone marrow progenitors are mesenchymal. The other major cell types expressing Wt1, namely the podocytes and mesothelia are considered epithelial, but are unusual in expressing high levels of mesenchymal markers, such as vimentin. Given our findings, it is interesting to speculate on the possible relationships between the cell types expressing and requiring Wt1 in these different tissues. Different reports have shown that stellate cells may arise from the mesothelium and bone marrow [10], [54]. Our studies suggest that Wt1 may have a function in both stellate cells and bone marrow mesenchymal stem cells. Stellate cells, like the epicardially-derived cells requiring Wt1, synthesise retinoic acid. One of the striking features of stellate cells is the presence of vitamin A (retinoid) droplets and this becomes lost upon stellate cell activation. In the epicardium we have shown that RALDH2 levels and RA are reduced when Wt1 is deleted and that RALDH2 is a direct transcriptional target of Wt1 [9]. We have shown that Wt1 is required for the EMT that generates RA-synthesising coronary vascular progenitors from the epicardium and it is interesting that an EMT is required for activation of stellate cells. It is also notable that stellate cells synthesise high levels of fat and it will be interesting to see if the Wt1 expressing cells in fat have similarities to stellate cells and mesenchymal bone marrow cells.
Finally our findings may also have implications for cancer therapy. There is a growing number of studies developing anti-WT1 immune therapy for common cancers predicated on the belief that WT1 is expressed at high levels in cancers [20], [55], but very low levels in the normal adult. Our findings raise questions about this approach as damage to these normal Wt1-expressing tissues might have adverse effects.
Materials and Methods
Generation of Wt1-conditional knockout mice
Mice were housed and bred in animal facilities at the MRC HGU and the University of Edinburgh. Animals were kept in compliance with Home Office regulations. The Wt1-conditional line was made in our group [5]. To obtain [CAGG-CreER™ positive, Wt1loxP/loxP] and [CAGG-CreER™ negative, Wt1loxP/loxP] transgenic mice, [CAGG-CreER™ positive] males were mated with Wt1loxP/loxP females, and the resulting offspring intercrossed. Wt1-GFP knockin mice used in this study were kindly provided by Professor H Sugiyama [18].
Tamoxifen-induced Wt1 deletion in [CAGG-CreER™, Wt1loxP/loxP] mice
Cre recombinase was induced by intraperitoneal administration of tamoxifen (4 mg/40 g body weight for 5 days; Sigma). All animal work was carried out under the permission of license. To delete Wt1 in vitro, cells were treated with 4-OH tamoxifen (1 µM, Sigma) for three days.
Full methods are described in Text S1. Antibodies and primers are listed in Tables S1 and S2.
Supporting Information
Zdroje
1. LepperCConwaySJFanCM 2009 Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements.
Nature 460
627 631
2. KimISaundersTLMorrisonSJ 2007 Sox17 dependence distinguishes the transcriptional regulation of fetal from adult hematopoietic stem cells. Cell
130 470
483
3. HohensteinPHastieND 2006 The many facets of the Wilms' tumour gene, WT1.
Hum Mol Genet 15 Spec No 2
R196 201
4. KreidbergJASariolaHLoringJMMaedaMPelletierJ 1993 WT-1 is required for early kidney development. Cell
74 679
691
5. Martinez-EstradaOMLetticeLAEssafiAGuadixJASlightJ 2010 Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of Snail and E-cadherin. Nat Genet
42 89 93
6. WilmBIpenbergAHastieNDBurchJBBaderDM 2005 The serosal mesothelium is a major source of smooth muscle cells of the gut vasculature.
Development 132
5317 5328
7. QueJWilmBHasegawaHWangFBaderD 2008 Mesothelium contributes to vascular smooth muscle and mesenchyme during lung development.
Proc Natl Acad Sci U S A 105
16626 16630
8. IjpenbergAPerez-PomaresJMGuadixJACarmonaRPortillo-SanchezV 2007 Wt1 and retinoic acid signaling are essential for stellate cell development and liver morphogenesis. Dev Biol
312 157
170
9. GuadixJARuiz-VillalbaALetticeLVelecelaVMunoz-ChapuliR 2011 Wt1 controls retinoic acid signalling in embryonic epicardium through transcriptional activation of Raldh2. Development
138 1093
1097
10. AsahinaKZhouBPuWTTsukamotoH 2011 Septum transversum-derived mesothelium gives rise to hepatic stellate cells and perivascular mesenchymal cells in developing mouse liver. Hepatology
53 983
995
11. FriedmanSL 2008 Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver.
Physiol Rev 88
125 172
12. MasamuneAShimosegawaT 2009 Signal transduction in pancreatic stellate cells. J Gastroenterol 44 249
260
13. VonlaufenAJoshiSQuCPhillipsPAXuZ 2008 Pancreatic stellate cells: partners in crime with pancreatic cancer cells.
Cancer Res 68
2085 2093
14. WalkerCRuttenFYuanXPassHMewDM 1994 Wilms' tumor suppressor gene expression in rat and human mesothelioma.
Cancer Res 54
3101 3106
15. ArmstrongJFPritchard-JonesKBickmoreWAHastieNDBardJB 1993 The expression of the Wilms' tumour gene, WT1, in the developing mammalian embryo. Mech Dev 40
85 97
16. RaoMKPhamJImamJSMacLeanJAMuraliD 2006 Tissue-specific RNAi reveals that WT1 expression in nurse cells controls germ cell survival and spermatogenesis. Genes Dev
20 147
152
17. PelletierJSchallingMBucklerAJRogersAHaberDA 1991 Expression of the Wilms' tumor gene WT1 in the murine urogenital system.
Genes Dev 5
1345 1356
18. HosenNShirakataTNishidaSYanagiharaMTsuboiA 2007 The Wilms' tumor gene WT1-GFP knock-in mouse reveals the dynamic regulation of WT1 expression in normal and leukemic hematopoiesis.
Leukemia 21
1783 1791
19. HammesAGuoJKLutschGLehesteJRLandrockD 2001 Two splice variants of the Wilms' tumor 1 gene have distinct functions during sex determination and nephron formation. Cell
106 319
329
20. RosenfeldCCheeverMAGaigerA 2003 WT1 in acute leukemia, chronic myelogenous leukemia and myelodysplastic syndrome: therapeutic potential of WT1 targeted therapies.
Leukemia 17 1301
1312
21. King-UnderwoodLLittleSBakerMClutterbuckRDelassusS 2005 Wt1 is not essential for hematopoiesis in the mouse.
Leuk Res 29 803
812
22. NakatsukaSOjiYHoriuchiTKandaTKitagawaM 2006 Immunohistochemical detection of WT1 protein in a variety of cancer cells.
Mod Pathol 19
804 814
23. MolletGRateladeJBoyerOMudaAOMorissetL 2009 Podocin inactivation in mature kidneys causes focal segmental glomerulosclerosis and nephrotic syndrome. J Am Soc Nephrol
20 2181
2189
24. GoldbergSAdair-KirkTLSeniorRMMinerJH 2010 Maintenance of Glomerular Filtration Barrier Integrity Requires Laminin {alpha}5.
J Am Soc Nephrol
25. DaiCStolzDBBastackySISt-ArnaudRWuC 2006 Essential role of integrin-linked kinase in podocyte biology: Bridging the integrin and slit diaphragm signaling. J Am Soc Nephrol 17 2164
2175
26. PhilippeAWeberSEsquivelELHoubronCHamardG 2008 A missense mutation in podocin leads to early and severe renal disease in mice.
Kidney Int 73
1038 1047
27. SierigRKruspeDKastnerJLckCWitzgallR The Wilms tumour protein is required for kidney function in adult mice 2009;
Edinburgh, Scotland, UK S152
28. ShimamuraRFraizerGCTrapmanJLau YfCSaundersGF 1997 The Wilms' tumor gene WT1 can regulate genes involved in sex determination and differentiation: SRY, Mullerian-inhibiting substance, and the androgen receptor. Clin Cancer Res
3 2571 2580
29. HerzerUCrocollABartonDHowellsNEnglertC 1999 The Wilms tumor suppressor gene wt1 is required for development of the spleen. Curr Biol 9
837 840
30. KouryMJ 2005 Erythropoietin: the story of hypoxia and a finely regulated hematopoietic hormone.
Exp Hematol 33
1263 1270
31. DameCKirschnerKMBartzKVWallachTHusselsCS 2006 Wilms tumor suppressor, Wt1, is a transcriptional activator of the erythropoietin gene. Blood 107
4282 4290
32. WagersAJ 2005 Stem cell grand SLAM. Cell
121 967 970
33. PronkCJRossiDJManssonRAttemaJLNorddahlGL 2007 Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy. Cell Stem Cell
1 428 442
34. FuentesRWangYHirschJWangCRauovaL Infusion of mature megakaryocytes into mice yields functional platelets. J Clin Invest 120 3917
3922
35. ApteMVHaberPSApplegateTLNortonIDMcCaughanGW 1998 Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture.
Gut 43
128 133
36. AhimaRSPrabakaranDMantzorosCQuDLowellB 1996 Role of leptin in the neuroendocrine response to fasting.
Nature 382
250 252
37. FaggioniRMoserAFeingoldKRGrunfeldC 2000 Reduced leptin levels in starvation increase susceptibility to endotoxic shock.
Am J Pathol 156
1781 1787
38. GaetkeLMOzHSFrederichRCMcClainCJ 2003 Anti-TNF-alpha antibody normalizes serum leptin in IL-2 deficient mice.
J Am Coll Nutr 22
415 420
39. KerstenSMandardSTanNSEscherPMetzgerD 2000 Characterization of the fasting-induced adipose factor FIAF, a novel peroxisome proliferator-activated receptor target gene. J Biol Chem
275 28488
28493
40. ShinmuraKTamakiKSaitoKNakanoYTobeT 2007 Cardioprotective effects of short-term caloric restriction are mediated by adiponectin via activation of AMP-activated protein kinase. Circulation
116 2809
2817
41. MaegawaSHinkalGKimHSShenLZhangL Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res 20 332
340
42. CalviLMAdamsGBWeibrechtKWWeberJMOlsonDP 2003 Osteoblastic cells regulate the haematopoietic stem cell niche.
Nature 425 841
846
43. SacchettiBFunariAMichienziSDi CesareSPiersantiS 2007 Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment.
Cell 131
324 336
44. SjogrenKLiuJLBladKSkrticSVidalO 1999 Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice.
Proc Natl Acad Sci U S A 96
7088 7092
45. YakarSRosenCJBeamerWGAckert-BicknellCLWuY 2002 Circulating levels of IGF-1 directly regulate bone growth and density.
J Clin Invest 110
771 781
46. JessenBAStevensGJ 2002 Expression profiling during adipocyte differentiation of 3T3-L1 fibroblasts.
Gene 299
95 100
47. KratchmarovaIKalumeDEBlagoevBSchererPEPodtelejnikovAV 2002 A proteomic approach for identification of secreted proteins during the differentiation of 3T3-L1 preadipocytes to adipocytes. Mol Cell Proteomics 1 213
222
48. InagakiTDutchakPZhaoGDingXGautronL 2007 Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab
5 415 425
49. MiyagawaKKentJMooreACharlieuJPLittleMH 1998 Loss of WT1 function leads to ectopic myogenesis in Wilms' tumour.
Nat Genet 18
15 17
50. SchumacherVSchneiderSFiggeAWildhardtGHarmsD 1997 Correlation of germ-line mutations and two-hit inactivation of the WT1 gene with Wilms tumors of stromal-predominant histology. Proc Natl Acad Sci U S A 94 3972
3977
51. SchumacherVSchuhenSSonnerSWeirichALeuschnerI 2003 Two molecular subgroups of Wilms' tumors with or without WT1 mutations.
Clin Cancer Res 9
2005 2014
52. OmaryMBLugeaALoweAWPandolSJ 2007 The pancreatic stellate cell: a star on the rise in pancreatic diseases.
J Clin Invest 117
50 59
53. WagnerNWagnerKDXingYScholzHSchedlA 2004 The major podocyte protein nephrin is transcriptionally activated by the Wilms' tumor suppressor WT1. J Am Soc Nephrol
15 3044
3051
54. MiyataEMasuyaMYoshidaSNakamuraSKatoK 2008 Hematopoietic origin of hepatic stellate cells in the adult liver.
Blood 111
2427 2435
55. OkaYTsuboiAElisseevaOANakajimaHFujikiF 2007 WT1 peptide cancer vaccine for patients with hematopoietic malignancies and solid cancers. ScientificWorldJournal
7 649 665
56. CohenDESupinskiAMBonkowskiMSDonmezGGuarenteLP 2009 Neuronal SIRT1 regulates endocrine and behavioral responses to calorie restriction.
Genes Dev 23
2812 2817
57. HeinrichsCColliMYanovskiJALaueLGerstlNA 1997 Effects of fasting on the growth plate: systemic and local mechanisms.
Endocrinology 138
5359 5365
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 12
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Targeted Proteolysis of Plectin Isoform 1a Accounts for Hemidesmosome Dysfunction in Mice Mimicking the Dominant Skin Blistering Disease EBS-Ogna
- The RNA Silencing Enzyme RNA Polymerase V Is Required for Plant Immunity
- The FGFR4-G388R Polymorphism Promotes Mitochondrial STAT3 Serine Phosphorylation to Facilitate Pituitary Growth Hormone Cell Tumorigenesis
- Target Site Recognition by a Diversity-Generating Retroelement
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy