
A Functional Phylogenomic View of the Seed Plants
A novel result of the current research is the development and implementation of a unique functional phylogenomic approach that explores the genomic origins of seed plant diversification. We first use 22,833 sets of orthologs from the nuclear genomes of 101 genera across land plants to reconstruct their phylogenetic relationships. One of the more salient results is the resolution of some enigmatic relationships in seed plant phylogeny, such as the placement of Gnetales as sister to the rest of the gymnosperms. In using this novel phylogenomic approach, we were also able to identify overrepresented functional gene ontology categories in genes that provide positive branch support for major nodes prompting new hypotheses for genes associated with the diversification of angiosperms. For example, RNA interference (RNAi) has played a significant role in the divergence of monocots from other angiosperms, which has experimental support in Arabidopsis and rice. This analysis also implied that the second largest subunit of RNA polymerase IV and V (NRPD2) played a prominent role in the divergence of gymnosperms. This hypothesis is supported by the lack of 24nt siRNA in conifers, the maternal control of small RNA in the seeds of flowering plants, and the emergence of double fertilization in angiosperms. Our approach takes advantage of genomic data to define orthologs, reconstruct relationships, and narrow down candidate genes involved in plant evolution within a phylogenomic view of species' diversification.
Published in the journal:
. PLoS Genet 7(12): e32767. doi:10.1371/journal.pgen.1002411
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002411
Summary
A novel result of the current research is the development and implementation of a unique functional phylogenomic approach that explores the genomic origins of seed plant diversification. We first use 22,833 sets of orthologs from the nuclear genomes of 101 genera across land plants to reconstruct their phylogenetic relationships. One of the more salient results is the resolution of some enigmatic relationships in seed plant phylogeny, such as the placement of Gnetales as sister to the rest of the gymnosperms. In using this novel phylogenomic approach, we were also able to identify overrepresented functional gene ontology categories in genes that provide positive branch support for major nodes prompting new hypotheses for genes associated with the diversification of angiosperms. For example, RNA interference (RNAi) has played a significant role in the divergence of monocots from other angiosperms, which has experimental support in Arabidopsis and rice. This analysis also implied that the second largest subunit of RNA polymerase IV and V (NRPD2) played a prominent role in the divergence of gymnosperms. This hypothesis is supported by the lack of 24nt siRNA in conifers, the maternal control of small RNA in the seeds of flowering plants, and the emergence of double fertilization in angiosperms. Our approach takes advantage of genomic data to define orthologs, reconstruct relationships, and narrow down candidate genes involved in plant evolution within a phylogenomic view of species' diversification.
Introduction
Attempts to clearly resolve the relationships among major seed plant groups using nuclear gene sequences have been hampered by the small number of completely sequenced genomes, the scarcity of ESTs for certain plant groups, and the lack of automated tools that can assemble and analyze large phylogenomic data sets. Existing phylogenetic hypotheses from molecular data, are often disputed due to the small sample of genes and/or taxa used in the analyses, regardless of the degree of support. Various conflicting topologies for the five basic seed plant groups have been obtained over time [1], [2]. Plant molecular phylogenetics has long relied on plastid genomes and only a few nuclear markers to infer relationships [1], [3]–[9]. Recently, progress has been made in generating plastid genome-based plant phylogenies [3], [10]–[16], but nuclear genome-scale analyses of plants have only recently started appearing in the literature [17]–[19]. The incorporation of nuclear phylogenomic information in plant phylogenetics would accomplish two important goals. First, the phylogenetic patterns discovered using nuclear genomic information could be used to corroborate the many well-supported plastid relationships, and to shed light on those relationships that are still at odds. Second, nuclear phylogenomic information can be used to derive new hypotheses for the function of plant genes that are relevant to major divergence events in plant evolution. In this study, we use phylogenetic information (emergent measures of phylogenetic support [20]) as the platform to identify candidate genes that may have played a role in plant adaptation. We first identify sets of orthologs from genomic sequences using a phylogenetic context [21]. We next use these orthologs to construct a total-evidence phylogeny and examine the distribution of their support metrics per node [20]. We then assess the statistical significance of Gene Ontology (GO) categories for gene lists that provide positive phylogenetic support to a node with functional processes of interest (e.g. seed development) [22]. The main premise of this approach is that genes (partitions) that are in agreement or in conflict with the overall evolutionary history of a particular node in a phylogeny, can be detected and used to derive hypotheses for the genes and biological processes potentially responsible for some of the more interesting organismal differences among the taxa in a phylogenetic analysis. We thus employ a phylogenomic approach to postulate hypotheses of gene function distributions and evolutionary mechanisms. These hypotheses can be validated experimentally in follow-up studies, focusing the effort of finding candidate genes and planning downstream experiments on those candidates based on a phylogenomic context. This functional phylogenomic approach is fundamentally different than classical phylogenetic analysis methods and also from current functional genomic methods that mine genomic information without incorporating a phylogenetic context in their search for both orthologs and candidate genes of functional importance. The present study is also a step toward generating an automated phylogenomic method for the entire nuclear component of plant genomes. Here, we use this approach to begin exploring and deriving hypotheses for the evolutionary mechanisms that underlie plant adaptation and diversification, as exemplified by the explosion of biodiversity within the seed plants, underlying Darwin's abominable mystery on the sudden appearance and rapid diversification of flowering seed plants, but also on the persistence of the gymnosperms over evolutionary time.
Results
Inferred seed plant phylogeny
We used OrthologID (OID) [21] – a program for automated, parsimony tree-based orthology determination, to identify 22,833 sets of orthologs from 150 plant species (see Materials and Methods for a description of the extended OID pipeline). These plant species, belonging to 101 different genera, represent a broad taxonomic range of angiosperms and extant gymnosperms. To reduce the size of the dataset for maximum likelihood (ML) analysis, and to remove partitions with the most missing data, we also constructed a matrix by only including genes with at least 30% representation across all genera. In this >30%-matrix, multiple taxa belonging to the same genus are collapsed into a single taxon. The average number of genera represented in each gene partition is 41 (40.6%) in this matrix. The cumulative distribution of gene partitions by taxon representation is shown in Figure S2. We performed maximum parsimony (MP) analysis on both matrices (MP-full and MP-30), as well as maximum likelihood analysis on the >30%-matrix (ML-30) (see Text S1). Figure 1 shows the phylogenetic tree generated from the ML-30 analysis.

The basic topologies for all three trees of the seed plants (MP-full, Figure S3; MP-30, Figure S4; and ML-30) are essentially identical. Node support based on bootstrap methods yielded a robust inferred phylogenetic tree overall. All three of our analyses (MP-full, MP-30, and ML-30) corroborate the same monophyletic groups of seed plants, as revealed in all previous morphological analyses and most molecular analyses, namely the seed plants, the cycads, the conifers, the gnetophytes, and the angiosperms. Moreover, all of our analyses support the gymnosperms as a monophyletic group (bootstrap = 100%). This is congruent with all comparable molecular data sets to date [1], [5], [11], [23], and in contrast to most morphological analyses, which retrieve gymnosperms as paraphyletic [7], [24], [25]. The differences between molecular-based topologies of the gymnosperms mainly involve the placement of the gnetophytes, that is with the gnetophytes as sister to the conifers [5], nested in conifers [1], [11], [13], [16], as sister to all other gymnosperms [23], or sister to all other seed plants [13], [26]. The position of the gnetophytes among seed plants, is indeed one of the most interesting unresolved issues in plant systematics, as reviewed in [2]. Our inferred genome-wide phylogeny (bootstrap = 100%) supports the gnetophytes as basal extant gymnosperms. This finding supports earlier hypotheses retrieved from individual gene trees such as rpoC1 and rbcL, as well as the non-coding regions of the inverted repeat representing the plastome [27]–[29], and from phytochrome genes [23], [30], AGAMOUS-like genes [31], [32], and FLORICAULA/LEAFY [33] representing the nuclear genome.
The topology of this phylogenomic view of the seed plants with a pectinate (i.e. maximally asymmetric, e.g. [34]) series of angiosperms, Gnetales, cycads + Ginkgo, and conifers has a impact on the interpretation of plant evolutionary changes, as characters are optimized in different ways. For example, in the previous view that cycads are sister to the rest of the gymnosperms, with ferns as sister to both flowering plants and gymnosperms, then the comparison is that angiosperms carpels are megasporophylls (seed-bearing sporophylls), and the angiosperm gynoecium is a simple strobilus (reproductive organ). By contrast, our phylogenomic view of the seed plants, Gnetales are sister to the rest of the gymnosperms with ferns as sister to all seed plants, and in this view each angiosperm carpel bearing ovules can be most parsimoniously interpreted as a simple strobilus. In this phylogenomic view, the angiosperm gynoecium would now be interpreted as a compound strobilus, with each carpel representing a bract enclosing an axillant ovule-bearing axis (Figure S6A). Another example of the impact of optimization is found with motile male gametes. Motile male gametes characterize all of the non-seed plant out-groups, and within seed plants are found only in cycads and Ginkgo. In our phylogenomic topology, motile male gametes would be independently and uniquely evolved (apomorphic) in cycads plus Ginkgo, and loss of motile male gametes in Gnetales and conifers would be ancestral in the gymnosperms (plesiomorphic). In contrast, if cycads were sister to the rest of the gymnosperms, the loss of motile male gametes would be convergent in conifers plus Gnetales and in the angiosperms, as an independently derived apomorphy. In this case, motile male gametes in cycads and Ginkgo would be a gymnosperm plesiomorphy (Figure S6B). Perhaps the most interesting aspect of these reversed character optimizations is that with the Gnetales sister to the rest of the gymnosperms as in our phylogenomic tree, the interpretation of character evolution is the same as in the Anthophyte hypothesis, where Gnetales is sister to angiosperms, and cycads are sister to all other seed plants. The optimization of motile male gametes of cycads and Ginkgo in our phylogenomic-based topology is thus equivalent to the optimization of characters required to explain the Gnetales as nested within the conifers. In this case, the reinsertion of the inverted repeat in the plastid genome of the Gnetales would be required after its loss in all of the conifers [11].
It is noteworthy that in this phylogenomic view of the seed plants, the basic topology of the angiosperm tree used by the Angiosperm Phylogeny Group (APG II, III) [35], [36] is supported with only minor changes (Figure S5). We retrieve the same topology of major groups on the pectinate backbone, starting with Amborella followed by Nuphar, magnoliids, ranunculids, caryophyllids, rosids, and asterids. The few discrepant nodes between our phylogenomic trees generated using different approaches (e.g. MP and ML), and between our tree and other major phylogenies, have low support, and are likely the consequence of ambiguous orthology statements due to missing data. For instance, Vaccinium is placed with low support (<65%) either sister to the caryophyllids in our ML-30 tree (Figure 1), or within the asterids (with caryophyllids as sister to both rosids and asterids) in the MP-30 tree (Figure S4), which is congruent with APG III (with caryophyllids as sister to asterids). Resolution of either hypothesis will probably depend on sampling the Cornales, of which core taxa have been placed within the asterids, and as sister to Ericales containing Vaccinium [35], [37]. Similarly, increased sampling of the alismatids and aroids will help confirm the position of Acorus within the angiosperms. Our findings of Acorus as sister to all other monocots, supports most analyses conducted with single and multiple gene trees [38]–[41], except for those that include atpA, which in some instances, place Acorus within the Alismatales aroids plus alismatids [42]–[44]. Within the rosids, the topology of MP-30 departs from that of APG III, by placing the Sapindales (Citrus and relatives) with the rosids I, versus rosids II in APG III and in our MP-full and ML-30 analyses. Our phylogenomic analysis identifies these controversial nodes and points to the taxa that need further sampling.
A controversial topology that is present and well supported (bootstrap = 100%) in all our of phylogenomic trees is the placement of the monocots between Nuphar and magnoliids. The monophyly and placement of the magnoliids has been enigmatic for some time, although recent work has suggested some resolution. The position of the monocots with respect to magnoliids has also been controversial. The three principal competing hypotheses for the position of the magnoliids are as sister to the eudicot lineage [45]–[47], as sister to the monocots plus the eudicots [38], [39], and as sister to the monocots [41]–[43]. By contrast, our phylogenomic tree firmly places the magnoliids as sister to the eudicots, and the monocots as sister to the magnoliids plus the eudicots, both with high support (100%). In fact, bootstrap support percentages along the backbone (the pectinate portion) of the angiosperms, also show that overall, our phylogenomic analysis provides a robust topology for this important group, based on a broad sampling of genomic and EST data.
Identifying candidate genes of functional significance within a phylogenomic framework
With the large number of genes in our phylogenomic matrix covering every Gene Ontology (GO) category for plants [48] (see Table S2), we are able to derive hypotheses for the functional evolution of genes in a phylogenomic context, by analyzing gene partitions that lend evidence of support at the major nodes on the tree [49]. Currently, one of the most widely used methods in testing for selection in phylogenomics, such as dN/dS [50], relies mostly on statistical methods of detection of natural selection. Partitioned phylogenetic support detects any positive or negative support that a gene or category of genes has on any given node on a phylogenetic tree. We propose that genes with positive partitioned support at nodes are a broader category of candidate genes for exploring function that would include genes evolving not only under positive Darwinian selection, but also under stochastic processes and even purifying selection. Once these candidate gene lists that provide positive branch support are derived, we can then examine them using positive selection scans to determine the functional categories of genes that are under selective pressure. Instead of requiring overwhelming statistical evidence of positive selection, our method identifies significant evolutionary trends by quantifying both phylogenetic congruence and incongruence, thus detecting potentially important genes that might be evolving neutrally or under negative selection. In this way, phylogenetic incongruence between a functional class of genes (e.g. RNA silencing genes) and the organismal phylogeny, would suggest that this given gene has experienced a unique evolutionary history relative to that of the organisms per se. Detection of such sequences is given by character information, meaning that no previous knowledge about the gene or gene function is required. Apart from being an unbiased approach, this method allows for the discovery of candidate genes with potential evolutionary and functional relevance, which can then be evaluated for evidence of selection using downstream validation and standard evolutionary tests [51], [52]. Here we provide a few examples of candidate genes for such validations.
For this analysis to be computationally feasible, and to exclude gene partitions with a large amount of missing taxa, while retaining as many partitions as possible for statistical analysis, we extracted a 9,787-gene matrix with >10% representation per partition and performed Partitioned Bremer Support (PBS) analysis [20]. This metric gives a relative measure of positive support on a gene-by-gene basis for each node. We identified 7,689 gene partitions with positive PBS values at one or more nodes in the simultaneous analysis tree. Of the 7,000+ genes, 4,803 of them have identifiable Arabidopsis orthologs, which were used to annotate the partitions with GO and MIPS [53] terms. To assign significance, we tested each node for overrepresented GO or MIPS categories within the list of gene partitions with positive PBS. As with any phylogenetic tree, missing genes as a result of incomplete EST coverage, and the proportion of matches to GO and MIPs categories, could shift PBS support. Importantly, the proportion of overrepresented genes with positive PBS is not just a numbers game, as it does not correlate with the number of genes per species in our matrix (e.g. Figure 1 shows Glycine and Phaseolus with a large EST set vs. Cucumis and Juglans regia, with a moderate EST set, but highly significant overrepresented genes with positive PBS). Furthermore, although the proportion of positive PBS varies within the phylogenomic matrix, it remains high even in nodes with lower numbers of genes and GO and MIPs terms (Figure S7).
This analysis of genes providing positive branch support at key nodes in the phylogenomic tree identified 29 overrepresented GO/MIPS term-node pairs (p<0.01), 87 such pairs (p<0.05), and 138 (p<0.10) (see Table S3 for the complete list and Figure 2 and Figure 3 for the distribution map of overrepresented GO/MIPS terms). The significant overrepresentation of genes in these GO/MIPS terms, points to potential candidate genes involved in metabolic and developmental traits associated with the evolution of taxa within these clades. Note that overrepresentation in this context is unrelated to levels of gene expression, referring instead to the overrepresentation of proteins in each functional category among those with amino acid sequences contributing positive PBS to gene partitions.
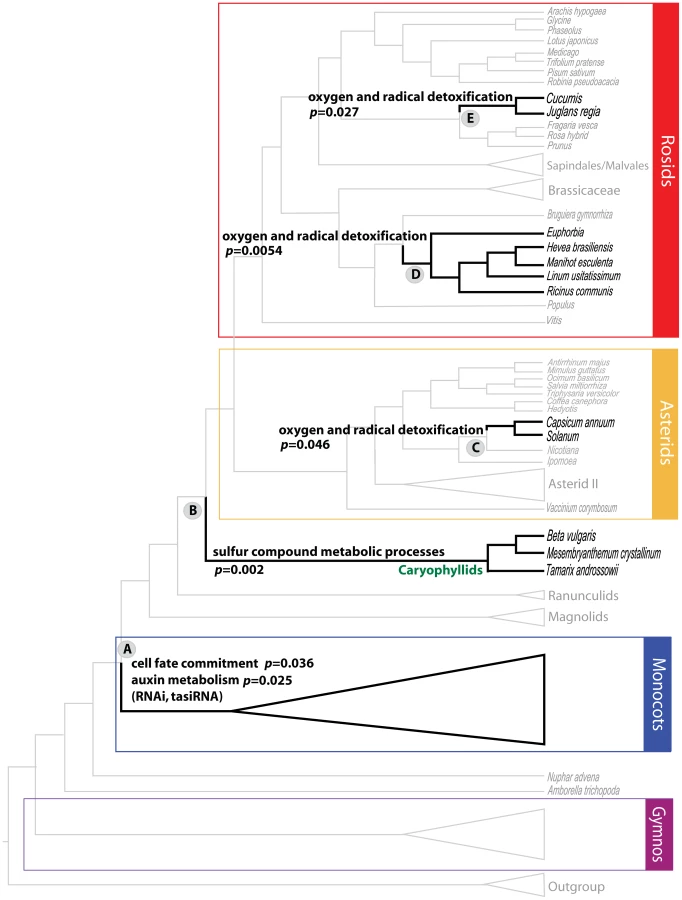

An example of a node with an overrepresentation of genes involved in a metabolic process is Node 15, which includes the caryophyllids, members of which include the salt/drought-tolerant plants Mesembryanthemum and Tamarix. This node shows significant overrepresentation of genes involved in “sulfur compound catabolic processes” (p = 0.002), for which experimental data relates both genes in this GO term (MGL1 and GGT3), to the trait of drought stress in Arabidopsis [54]–[56]. The examination of GO overrepresentation in a phylogenomic framework, confirms expected patterns of well-characterized genes in some taxa, and also allows us to identify similar gene functions in atypical candidates. Genes involved in “oxygen and radical detoxification” were overrepresented in Node 52 (p = 0.046), Node 19 (p = 0.00054), and Node 31 (p = 0.027). Species like tomato (Solanum lycopersicum) [57] and pepper (Capsicum annuum) [58] in Node 52, Euphorbia esula and E. tirucalli [58], [59] in Node 19, and walnut (Juglans regia) [60] in Node 31, are well known sources of detoxifying and antioxidant compounds. The predominance of glutathione-related genes involved in detoxification [61] in those clades is not surprising and demonstrates our approach in principle. The overrepresentation of glutathione peroxidase genes in other taxa such as melon (Cucumis melo) and cucumber (C. sativus) in Node 19, is thus worth examining further.
Another example of this functional phylogenomic approach identified genes belonging to “cell fate commitment” (p = 0.04) and “auxin metabolism” (p = 0.025) as overrepresented GO terms among genes with positive PBS at Node A (Figure 2), that defines all monocots except Acorus. These include three genes that encode proteins involved in cell fate decision, AGO1, KANADI and LACHESIS. LACHESIS controls the cell fate within female gametophytes in Arabidopsis, where mutants have supernumerary egg cells and are semi-sterile [62]. LACHESIS is a homolog of yeast PRP4, a kinase that influences mRNA splicing. AGO1 is the key effector endonuclease for multiple aspects of RNAi, including cleavage and translational inhibition of target messages via microRNA and trans-acting short interfering RNA (tasiRNA). Weak alleles of AGO1 in Arabidopsis have drastic effects on leaf polarity [63]. KANADI is also a master regulator of leaf polarity, along with the auxin response factor ARF3 (a target of tasiRNA). The mRNA export factor homolog SDE5 also contributes positive PBS to the monocot clade, and is required for trans-acting siRNA accumulation [64], along with the RNA-dependent RNA polymerase RDR6, which was found to contribute positive PBS to the monocot clade in a smaller study of 17 taxa [65]. Mutants in the tasiRNA pathway disrupt the eponymous monocotyledonous embryo of rice, which displays radial asymmetry, far more severely than the symmetric dicotyledonous embryo of Arabidopsis [66], perhaps because tasiRNA act non-autonomously in Arabidopsis. Thus, it is hypothesized from this phylogenomic analysis that RNAi had a significant influence in the divergence of monocots and magnoliids plus eudicots from the ancestral angiosperm. This is an exciting hypothesis derived from the functional phylogenomic analysis of the seed plants, as we are increasingly aware of the importance of siRNAs and transcriptional gene silencing pathways of RNAi for plant evolution [67].
In perhaps the most important hypothesis derived from the phylogenomic approach we describe, the plant-specific RNA polymerase subunit NRPD2 contributes PBS to several nodes among lower plants, including conifers and Marchantia/moss, but especially to gymnosperms as a group, for which there are 21 steps of support, which is among the highest 2% of all genes with positive support for the gymnosperm clade. In Arabidopsis, NRPD2 is a subunit of both RNA polymerase IV and RNA polymerase V, both of which are required for 24nt siRNA biogenesis and for RNA-directed DNA methylation. Remarkably, 24nt small RNAs, which correspond to transposons and heterochromatic repeats in angiosperms, are absent from Pinus contorta, the only gymnosperm in which they have been examined [68]. They are nonetheless found in non-seed plants such as Physcomitrella and Selaginella [69]. Our phylogenomic analysis implicates NRPD2 in the loss of 24nt siRNA from gymnosperms that have very large unmethylated genomes [70], consistent with a loss of transposon control via siRNA.
In support of this derived hypothesis, high levels of maternal 24nt siRNA are found in the endosperm of developing Arabidopsis seeds in which NRPD2 is highly expressed [71]. In angiosperms, fertilization of both the egg and the central cell nucleus (double fertilization) lead to embryo and endosperm development, respectively, while in gymnosperms, the megagametophyte develops maternally without fertilization [71]. Interestingly, maternal mutants in Arabidopsis that disrupt this small RNA pathway are defective in transposon defense and develop unreduced gametophytes [72], the first step in maternal endosperm formation. We propose therefore that transposon-defense, mediated by small RNA, is responsible, in part, for the emergence of novel reproductive strategies within the flowering plants.
Along with NRPD2, a total of 297 genes in the phylogenomic tree provide PBS for the gymnosperm clade, while 407 genes provide PBS for the angiosperm clade, but the vast majority of the remaining ∼7,000 genes in this seed plant matrix provide support for individual nodes in the tree. These candidate genes lay the ground for new testable hypotheses concerning the evolutionary changes in function that may be of relevance in the astounding radiation of flowering plants, potentially underscoring Darwin's ‘abominable mystery’ of seed plant radiation [73].
In our effort to examine patterns of natural selection across the seed plants, we used established measures of synonymous (dS) and nonsynonymous (dN) nucleotide substitution rates – not in a gross fashion across the whole gene sequence, but rather on a codon-by-codon basis. The rate ratio dN/dS is a commonly used measure of selective pressure that has been expanded to incorporate sequence and codon evolution models, as well as branch rate variation [50]. dS is vulnerable to substitution saturation, and is not reliably estimated for very divergent taxa, even below the genus level [74]. In this study we are dealing with extant spermatophyte taxa that diverged near the Devonian–Carboniferous boundary at ca. 350 Ma, with their daughter groups diversifying after the Carboniferous (gymnosperms and conifers), and in the Jurassic in the last 200 million years (Myr) (angiosperms) [75], therefore substitution saturation is expected at synonymous substitutions. In an attempt to circumvent dS estimation issues, and thus undefined codon-specific dN/dS values, we allowed for the non-synonymous evolutionary rate to vary along the phylogeny, and more specifically, in the two subtrees united by the node where markedly high positive PBS scores where detected. We selected genus Euphorbia (angiosperms, eudicots, rosids, Malpighiales) as a case study. This is one of the most taxonomically rich plant genera likely encompassing ∼3000 species (http://www.plantsystematics.org) [76], that occupies habitats distributed worldwide and displays a marked degree of morphological and anatomical variation. The pantropical and very speciose family Euphorbiaceae (∼6300 species [76], 245 genera [77]) may have diverged from other Malpighiales in the Lower Cretaceous Aptian age around 119.4–101.1 Myr before present), but diversification within the family is much more recent, e.g. within Acalyphoideae within the last ∼70 Myr [78]. Note that Acalyphoideae is sister to the rest of the Euphorbiaceae including Euphorbia (see Figures 3 and 4 in [79]) that is itself nested inside Euphorbiaceae with an origin estimated at ∼38 Myr ago with most of its diversification having occurred in the interval 30–10 Myr before present (see Figure S1G in [80]). Euphorbia exhibited high positive PBS scores for 13 proteins involved in oxygen and radical detoxification (MIPS functional category 32.07.07). Codon-wise estimates of synonymous rates showed a highly positively skewed distribution within each gene, with discrepant median and mean values (dS across genes: median range = 2.1–3.8, mean range = 89–685, skewness = 3.31–11.05), thus reinforcing our hesitation to use dS and subsequently dN/dS (Table S6). Gene-wide estimates of dN/dS [81] were well below 1.0 showing no evidence of the action of positive selection with rate ratio values ranging from 0.138 to 0.41 (Table S6). Contrasting dN between the subtree leading to Euphorbia and the “background” subtree, we found statistical evidence (p<0.05) of non-synonymous rate variation in 3 to 25 codons per gene. More specifically, we detected a general trend of non-synonymous rate acceleration in the subtree containing Euphorbia. A more complex picture became apparent in the case of 2 of those 13 genes (At1g76080 and At1g76080), where around half of the codons that showed significant evidence of dN rate change decelerated in the Euphorbia-containing clade. Detailed results are provided in Table S6.
Discussion
Using a phylogenetic matrix with broad taxonomic sampling and gene representation, we are able to provide support for some of the more controversial topologies within plants, and in particular within the various hypotheses of gymnosperm evolution. From a phylogenomic perspective, we suggest hypotheses on genes and their evolutionary processes might be related to patterns in plant diversification. By focusing on the clade-specific variation of phylogenetic characters in a multi-gene matrix, we can determine the effect of individual genes or groups of genes within a particular gene category, on support metrics and their statistical correlation with functional processes of interest (such as seed development and gene silencing). Specifically, we can pinpoint the amino acid sequences that support individual branches in the tree. This enables us to investigate the genetic mechanisms that underlie the rapid radiation of the angiosperms and the persistence of the gymnosperms on a subset of candidate genes in follow-up studies. Our tree-based method, combined with maximum likelihood methods of non-synonymous and synonymous evolutionary rate variation, isolated 14 genes involved in oxygen and radical detoxification from one of the most speciose plant genera, exhibiting evidence for changes in selective pressure through non-synonymous rate heterogeneity. Most importantly, our functional phylogenomic method has shed light on the evolution of very large gymnosperm genomes, and on maternal endosperm development via the role of small interfering RNA in transposon defense and in asexual development.
In all, we demonstrate how a functional phylogenomic approach can be used to postulate hypotheses of gene function distributions and evolutionary mechanisms. Our framework sets the groundwork for future molecular biology, ecological genomics, and evolutionary development research, which will refine the hypothesized role of the genes we have identified herein. Furthermore, with the increasing amount of genomic sequence data available, we expect to see increased resolution of the seed plant tree, and more genes of importance to the evolution of major clades to be discovered using the phylogenomic methodology described herein.
Materials and Methods
Sequences and orthologs
Sequences were collected from the gene sets of 5 completely sequenced plant genomes and ESTs of 145 other plant species with at least 2,000 unigenes. The complete genomes include Arabidopsis thaliana (TAIR), Oryza sativa (JCVI), Populus trichocarpa (JGI), Vitis vinifera (Genoscope), and Physcomitrella patens (JGI). Unigenes were obtained from the TIGR Plant Transcript Assemblies (http://plantta.jcvi.org).
Orthology was determined using an extended OrthologID pipeline (Figure S1). The original OrthologID pipeline [21] only utilizes complete genomes in the generation of “guide trees” which are used to classify ESTs and determine their orthologs. However, the limited number of completely sequenced plant genomes to date would hamper the accuracy of EST placement when large numbers of ESTs from diverse plant species are classified into gene family trees with limited taxonomic representation. To alleviate this problem, we also included 17 extra species (ingroup and outgroup) with high number of ESTs in the generation of “guide trees”. These extra species represent a full spectrum of plant lineages and include: Adiantum capillus-veneris, Aquilegia formosa, Amborella trichopoda, Ceratopteris richardii, Cichorium intybus, Coffea canephora, Gossypium hirsutum, Liriodendron tulipifera, Marchantia polymorpha, Medicago truncatula, Nuphar advena, Pinus taeda, Saruma henryi, Solanum tuberosum, Welwitschia mirabilis, Zamia fischeri, and Zingiber officinale. As in the original OrthologID pipeline, gene families were clustered using a 1e-20 BLAST E-value cutoff, and were aligned with MAFFT [82] using three different sets of parameters with ambiguous regions culled. OrthologID generated guide trees from these gene families using parsimony. ESTs from all other species were then classified into gene families using the following stepwise shortest-tree method extrapolated from OrthologID: Each EST was identified with a gene family using BLAST. The complete set of ESTs that belong to a gene family were sorted in decreasing order of similarity according to their highest BLAST e-values against gene family members in the guide tree. Each EST was then inserted into the fixed guide tree in the aforementioned order. At every iteration the tree with the shortest length (most parsimonious) with respect to the guide tree is chosen. Finally, we determined sets of orthologs from the gene family tree by extracting the largest non-overlapping subsets of genes that are orthologous according to the topology. In cases where there is a 1-to-many or many-to-many ortholog relationship, each of the multiple orthologs is treated as equal and only a randomly selected one from each species is included in the matrix. In the >30%-matrix, multiple species of the same genus are represented by a single taxon. For each gene partition in this matrix, the gene sequence from the most ancestral of the species belonging to the same genus, as identified by the MP-full tree, is chosen to be the representative for that genus (taxon).
Phylogenetic analysis
We assembled an alignment phylogenetic matrix of amino acid residues using the ortholog sets determined above and partitioned by gene. Only genes with at least four taxa present were included, resulting in a matrix with 22,833 partitions and 10,768,363 characters. Parsimony analysis was performed on the full alignment matrix and the >30%-representation genus alignment matrix using PAUP* v4b10 [83] and TNT [84]. The most efficient parsimony tree search strategy used the tree fusion method [85]: 100 jackknife resamplings (proportion = 0.3679) were searched with subtree pruning-regrafting holding two trees per resampling. The collected jackknife trees were then submitted to 100 rounds of tree fusion. Parsimony ratchet [86] also resulted in the same shortest trees (Figures S3 and S4). Node support was evaluated with 2,000 bootstrap pseudoreplicates and summarized on a 50% majority-rule consensus tree. Partitioned Bremer support (PBS) analysis was done on a submatrix that included only gene partitions with at least 10% taxon-representation. We use PBS to assess the direction of support (positive, negative, or neutral) of a particular gene to the various branches or nodes in a phylogeny. PBS is defined as follows: for a particular combined data set, a particular node (branch), and a particular data partition, PBS is the minimum number of character steps for that partition on the shortest topologies for the combined data set that do not contain that node, minus the minimum number of character steps for that partition on the shortest topologies for the combined data set that do contain that node [20]. Values for these metrics can be positive, zero or negative, and indicate the direction of support for the overall concatenated hypothesis: a positive PBS value indicates that the partition provides support for the node. Negative PBS means that the length of partition is shorter on an alternative tree (i.e. that partition provides contradictory evidence). The sum of PBS values for each data partition always equals Bremer Support for combined data [87]. We used TreeRot v3 [88] to generate PBS values for each partition-node pair.
We performed maximum likelihood (ML) inference of phylogeny on the >30% representation genus matrix using the fine-grained parallel Pthreads (POSIX Threads Library) [89] and MPI (Message Passing Interface) [90], [91] implementations of the 2009 development version of RAxML [92]. Our analysis represents the largest ML-based phylogenetic inference with respect to main memory requirements (89.2 GB) conducted to date. We employed both the JTT substitution matrix [93] with empirical amino acid residue frequencies (F), and the general time-reversible (GTR) substitution matrix [94] estimated directly from the genus-only alignment (Table S5). Both amino acid substitution models yielded the same overall topology. The JTT+F model was selected as the best-fit model based on its likelihood score among 22 models overall (11 with fixed residue frequencies and 11 with empirical residue frequencies calculated from the data in hand). We investigated the effect of the starting topology on the ML tree search and determined the best-scoring ML tree by employing ten random and ten randomized stepwise-addition MP trees in order to assess convergence to the final best ML topology, as well as the best single MP tree produced in our parsimony analysis. The way among-site rate heterogeneity was modeled had an impact on the final likelihood score; the CAT approximation model [95] with 25 per-site rate categories produced a better likelihood score than the Γ-distributed rate heterogeneity model with four discrete rates [96] in all cases where MP starting trees were used, while the GAMMA model performed better in 60% of the inferences when random starting trees were used (Table S4). Using the MP tree as a starting tree produced better likelihood scores for both rate heterogeneity models in conjunction with the GTR substitution model (logLikCAT = –35,802,562; logLikGAMMA = –35,802,416) (Table S4). Node support was quantified by means of 223 rapid non-parametric bootstrap pseudoreplicates (RBS) [97]. In order to determine if we conducted a sufficient number of RBS replicates we applied the novel bootstrap convergence test [98] implemented in RAxML a posteriori to our collection of 223 RBS trees. The Weighted and Frequency Criteria (WC and FC, respectively) [98] suggested that more than 74 and 122 replicates, respectively, would not induce significant changes on node support. Unlike majority-rule consensus trees, these support values indicate the percentage of pseudoreplicates in which the nodes of the best ML tree are present. Additional information on the ML analysis can be found in Text S1.
Gene Ontology analysis
In order to functionally characterize the genes that are providing positive support to the phylogenetic tree, we identified the Arabidopsis orthologs of gene partitions with positive PBS at each node and determined GO [99] and MIPS [100] terms that are statistically overrepresented at each node. The analysis was performed using the BioMaps tool [101] available on VirtualPlant (http://www.virtualplant.org) with the hypergeometric statistics using the union of genes with positive PBS at all nodes as the background population, with a correction for multiple hypothesis testing. Terms with a p-value less than 0.05 were considered statistically significant. The Gene Ontology version of 31 May 2008 and the MIPS Functional Catalogue (FunCat) database version of 27 May 2008 were used. Given that we only include gene partitions with positive PBS in our functional GO term analysis, but do not use the actual PBS value in our statistical analysis, the use of a randomly selected ortholog in a gene partition in the case of multiple co-orthologs will likely have no effect on our results, except for the cases where the PBS values are very close to zero. In those scenarios, using a different co-ortholog may cause the PBS value to change from positive to non-positive, or vice versa, and therefore be excluded or included in the GO analysis. However, in a very large dataset, we expect the randomness to have a cancelling effect when we look at high-level GO categories that include many genes. Similarly, nodes with low support in our guide trees may affect orthology determination and subsequent GO term analysis. However, we expect the effect to be minimal due to our large sample size.
Selection analysis
The extent of the pressure of natural selection was measured by estimating the ratio of the rate of nonsynonymous substitutions (dN) to the synonymous substitution rate (dS). A dN/dS rate value near one indicates neutral evolution, while deviations exceeding one are suggestive of positive selection, and positive values below one are considered to be evidence of purifying or negative selection as a result of strong structural or functional constraints at the protein level [102]. Functional protein-encoding genes tend to be subject to negative selection across their codons [103], therefore when only a few codons are positively selected [104] the measure of natural selection is averaged across all codons as a gene-wide dN/dS rate ratio. Our inference of episodic instances of positive selection becomes more powerful if, instead of averaging, we allow for dN/dS rate ratios to vary along the sequence alignment on a per-codon basis and across the phylogenetic tree [105], [106]. For this reason we estimated dN, dS, and dN/dS in a maximum likelihood framework as implemented in the latest development build of HyPhy v2.0 [107] (http://www.hyphy.org). Coding sequence evolution was modeled using the generalized Muse–Gaut (MG94) [108] model crossed with the Hasegawa–Kishino–Yano (HKY85) [109] nucleotide substitution model. The selective pressure at each codon site was quantified using the fixed effects likelihood (FEL) method [110] that estimates separately dN and dS rates for each codon and subsequently contrasts them through a likelihood ratio test (LRT). So as to avoid unpredictably biased dN/dS results because of saturation of the synonymous substitution rate dS across such a deep evolutionary timescale examined here, we chose to compare the variation of nonsynonymous rate dN along the phylogeny alongside gene-wide dN/dS estimates [81]. We selected a set of genes whose corresponding amino acid sequences exhibited strong, positive PBS (PBS>10) and belonged to the same MIPS term, such as 13 genes involved in oxygen and radical detoxification supporting consistently the node leading to the genus Euphorbia (Arabidopsis gene symbols: At1g65820, At1g76080, At2g47730, At1g64500, At3g54960, At3g15360, At4g33040, At1g20620, At4g31870, At1g19570, At3g27820, At5g23310, At2g31570). By allowing dN to vary in the two subsequent subtrees (Euphorbia-containing clade vs. the rest of the tree), we discounted possible dS biases and examined whether there is statistical evidence through a LRT for a change in nonsynonymous rates.
Computing
We performed maximum parsimony (MP) and Bremer support analysis on a 64-node Linux cluster at NYU. MP bootstrap analysis was performed on the 2,000-node BlueHelix HPCC facility at CSHL. In order to analyze this challenging dataset under maximum likelihood (ML) we used several clusters, multi-core nodes, and supercomputers: the Woodcrest Cluster at the Regionales Rechenzentrum Erlangen in Germany (868 Intel Woodcrest cores, Infiniband interconnect), the Infiniband Cluster at the Technical University of Munich (128 AMD Opteron cores, Infiniband interconnect), the AMD Barcelona multi-core nodes at the Swiss Federal Institute of Technology in Lausanne (2 16-core AMD Barcelona nodes), and the SGI ALTIX 4700 supercomputer at the Leibniz Rechenzentrum in Munich (8192 Intel Itanium cores, custom interconnect). The selection ML analysis was carried out on an Apple Mac Pro 12-core with Intel Xeon 2.66 GHz processors and 8 GB of RAM (1333 MHz DDR3) running 20 processes for each inference.
The complete phylogenomic matrix and trees are available at the BIGPLANT website (http://nypg.bio.nyu.edu).
Supporting Information
Zdroje
1. BurleighJGMathewsS 2004 Phylogenetic signal in nucleotide data from seed plants: implications for resolving the seed plant tree of life. Am J Bot 91 1599 1613
2. MathewsS 2009 Phylogenetic relationships among seed plants: persistent questions and the limits of molecular data. Am J Bot 96 228 236
3. BarkmanTJMcNealJRLimSHCoatGCroomHB 2007 Mitochondrial DNA suggests at least 11 origins of parasitism in angiosperms and reveals genomic chimerism in parasitic plants. BMC Evol Biol 7 248
4. Bouchenak-KhelladiYSalaminNSavolainenVForestFBankM 2008 Large multi-gene phylogenetic trees of the grasses (Poaceae): progress towards complete tribal and generic level sampling. Mol Phylogenet Evol 47 488 505
5. BoweLMCoatGdePamphilisCW 2000 Phylogeny of seed plants based on all three genomic compartments: extant gymnosperms are monophyletic and Gnetales' closest relatives are conifers. Proc Natl Acad Sci U S A 97 4092 4097
6. BurleighJGHiluKWSoltisDE 2009 Inferring phylogenies with incomplete data sets: a 5-gene, 567-taxon analysis of angiosperms. BMC Evol Biol 9 61
7. ChaseMWSoltisDEOlmsteadRGMorganDLesDH 1993 Phylogenetics of seed plants: an analysis of nucleotide sequences from the plastid gene rbcL. Ann Missouri Bot Gard 80 528 580
8. SmithSADonoghueMJ 2008 Rates of molecular evolution are linked to life history in flowering plants. Science 322 86 89
9. ZhuXYChaseMWQiuYLKongHZDilcherDL 2007 Mitochondrial matR sequences help to resolve deep phylogenetic relationships in rosids. BMC Evol Biol 7 217
10. Leebens-MackJRaubesonLACuiLKuehlJVFourcadeMH 2005 Identifying the basal angiosperm node in chloroplast genome phylogenies: sampling one's way out of the Felsenstein zone. Mol Biol Evol 22 1948 1963
11. BraukmannTWKuzminaMStefanovicS 2009 Loss of all plastid ndh genes in Gnetales and conifers: extent and evolutionary significance for the seed plant phylogeny. Curr Genet 55 323 337
12. JansenRKCaiZRaubesonLADaniellHDepamphilisCW 2007 Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc Natl Acad Sci U S A 104 19369 19374
13. McCoySRKuehlJVBooreJLRaubesonLA 2008 The complete plastid genome sequence of Welwitschia mirabilis: an unusually compact plastome with accelerated divergence rates. BMC Evol Biol 8 130
14. MooreMJBellCDSoltisPSSoltisDE 2007 Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc Natl Acad Sci U S A 104 19363 19368
15. QiuYLLiLWangBChenZKnoopV 2006 The deepest divergences in land plants inferred from phylogenomic evidence. Proc Natl Acad Sci U S A 103 15511 15516
16. ZhongBYonezawaTZhongYHasegawaM 2010 The position of Gnetales among seed plants: overcoming pitfalls of chloroplast phylogenomics. Mol Biol Evol 27 2855 2863
17. BurleighJGBansalMSEulensteinOHartmannSWeheA 2011 Genome-scale phylogenetics: inferring the plant tree of life from 18,896 gene trees. Syst Biol 60 117 125
18. FinetCTimmeREDelwicheCFMarlétazF 2010 Multigene phylogeny of the green lineage reveals the origin and diversification of land plants. Curr Biol 20 2217 2222
19. SandersonMMcMahonM 2007 Inferring angiosperm phylogeny from EST data with widespread gene duplication. BMC Evol Biol 7 S3
20. BakerRHDeSalleR 1997 Multiple sources of character information and the phylogeny of Hawaiian drosophilids. Syst Biol 46 654 673
21. ChiuJCLeeEKEganMGSarkarINCoruzziGM 2006 OrthologID: automation of genome-scale ortholog identification within a parsimony framework. Bioinformatics 22 699 707
22. KatariMSNowickiSDAceitunoFFNeroDKelferJ 2010 VirtualPlant: a software platform to support systems biology research. Plant Physiol 152 500 515
23. SchmidtMSchneider-PoetschHA 2002 The evolution of gymnosperms redrawn by phytochrome genes: the Gnetatae appear at the base of the gymnosperms. J Mol Evol 54 715 724
24. NixonKCCrepetWLStevensonDFriisEM 1994 A reevaluation of seed plant phylogeny. Ann Missouri Bot Gard 81 484 533
25. RothwellGWSerbetR 1994 Lignophyte phylogeny and the evolution of spermatophytes: a numerical cladistic analysis. Syst Bot 19 443 482
26. AlbertVABacklundABremerKChaseMWManhartJR 1994 Functional constraints and rbcL evidence for land plant phylogeny. Ann Missouri Bot Gard 81 534 567
27. GoremykinVBobrovaVPahnkeJTroitskyAAntonovA 1996 Noncoding sequences from the slowly evolving chloroplast inverted repeat in addition to rbcL data do not support gnetalean affinities of angiosperms. Mol Biol Evol 13 383 396
28. HasebeMKofujiRItoMKatoMIwatsukiK 1992 Phylogeny of gymnosperms inferred from rbcL gene sequences. J Plant Res 105 673 679
29. SamigullinTKMartinWFTroitskyAVAntonovAS 1999 Molecular data from the chloroplast rpoC1 gene suggest a deep and distinct dichotomy of contemporary spermatophytes into two monophyla: gymnosperms (including Gnetales) and angiosperms. J Mol Evol 49 310 315
30. MathewsSDonoghueMJ. Analyses of phytochrome data from seed plants: exploration of conflicting results from parsimony and Bayesian approaches; 2002 Aug 2-7; Madison, WI
31. BeckerATheissenG 2003 The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol Phylogenet Evol 29 464 489
32. WinterKUBeckerAMunsterTKimJTSaedlerH 1999 MADS-box genes reveal that gnetophytes are more closely related to conifers than to flowering plants. Proc Natl Acad Sci U S A 96 7342 7347
33. FrohlichMWParkerDS 2000 The mostly male theory of flower evolutionary origins: from genes to fossils. Syst Bot 25 155 170
34. PearsonPN 1999 Apomorphy distribution is an important aspect of cladogram symmetry. Syst Biol 48 399 406
35. The Angiosperm Phylogeny Group 2003 An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG II. Bot J Linn Soc 141 399 436
36. The Angiosperm Phylogeny Group 2009 An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot J Linn Soc 161 105 121
37. WikstromNSavolainenVChaseMW 2001 Evolution of the angiosperms: calibrating the family tree. Proc R Soc B Biol Sci 268 2211 2220
38. ChaseMWFayMFDeveyDSRønstedNDaviesJ 2006 Multi-gene analyses of monocot relationships: a summary. Aliso 22 63 76
39. ChaseMWSoltisDESoltisPSRudallPJFayMF 2000 Higher-level systematics of the monocotyledons: an assessment of current knowledge and a new classification. WilsonKLMorrisonDA Monocots: Systematics and Evolution Melbourne CSIRO 1 16
40. ChaseMWStevensonDWWilkinPRudallPJ 1995 Monocot systematics: a combined analysis. RudallPJCribbPJCutlerDFHumphriesCJ Monocotyledons: Systematics and Evolution: Royal Botanical Gardens, Kew 685 730
41. DuvallMRLearnGHJrEguiarteLECleggMT 1993 Phylogenetic analysis of rbcL sequences identifies Acorus calamus as the primal extant monocotyledon. Proc Natl Acad Sci U S A 90 4641 4644
42. DavisJIPetersenGSebergOStevensonDWHardyCR 2006 Are mitochondrial genes useful for the analysis of monocot relationships? Taxon 55 857 870
43. DavisJIStevensonDWPetersenGSebergOCampbellLM 2004 A phylogeny of the monocots, as inferred from rbcL and atpA sequence variation, and a comparison of methods for calculating jackknife and bootstrap values. Syst Bot 29 467 510
44. StevensonDDavisJFreudensteinJVHardyCRSimmonsMP 2000 A phylogenetic analysis of the monocotyledons based on morphological and molecular character sets, with comments on the placement of Acorus and Hydatellaceae. WilsonKLMorrisonDA Monocots: Systematics and Evolution Melbourne CSIRO 17 24
45. SoltisDESoltisPSChaseMWMortMEAlbachDC 2000 Angiosperm phylogeny inferred from 18S rDNA, rbcL, and atpB sequences. Bot J Linn Soc 133 381 461
46. SoltisPSSoltisDEZanisMJKimS 2000 Basal lineages of angiosperms: relationships and implications for floral evolution. Intl J Plant Sci 161 S97 S107
47. ZanisMJSoltisDESoltisPSMathewsSDonoghueMJ 2002 The root of the angiosperms revisited. Proc Natl Acad Sci U S A 99 6848 6853
48. AshburnerMBallCABlakeJABotsteinDButlerH 2000 Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25 25 29
49. RosenfeldJADeSalleRLeeEKO'GradyP 2008 Using whole genome presence/absence data to untangle function in 12 Drosophila genomes. Fly 2 291 299
50. NielsenR 2005 Statistical Methods in Molecular Evolution. New York Springer 504
51. BiswasSAkeyJM 2006 Genomic insights into positive selection. Trends Genet 22 437 446
52. YangZBielawskiJP 2000 Statistical methods for detecting molecular adaptation. Trends Ecol Evol 15 496 503
53. MewesHWDietmannSFrishmanDGregoryRMannhauptG 2008 MIPS: analysis and annotation of genome information in 2007. Nucleic Acids Res 36 D196 201
54. Ohkama-OhtsuNZhaoPXiangCOliverDJ 2007 Glutathione conjugates in the vacuole are degraded by γ-glutamyl transpeptidase GGT3 in Arabidopsis. Plant J 49 878 888
55. RébeilléFJabrinSBlignyRLoizeauKGambonnetB 2006 Methionine catabolism in Arabidopsis cells is initiated by a γ-cleavage process and leads to S-methylcysteine and isoleucine syntheses. Proc Natl Acad Sci U S A 103 15687 15692
56. RizhskyLLiangHShumanJShulaevVDavletovaS 2004 When defense pathways collide. The response of Arabidopsis to a combination of drought and heat stress. Plant Physiol 134 1683 1696
57. CervillaLMBlascoBRiosJJRomeroLRuizJM 2007 Oxidative stress and antioxidants in tomato (Solanum lycopersicum) plants subjected to boron toxicity. Ann Bot 100 747 756
58. MateosRMLeonAMSandalioLMGomezMdel RioLA 2003 Peroxisomes from pepper fruits (Capsicum annuum L.): purification, characterisation and antioxidant activity. J Plant Physiol 160 1507 1516
59. AndersonJVDavisDG 2004 Abiotic stress alters transcript profiles and activity of glutathione S-transferase, glutathione peroxidase, and glutathione reductase in Euphorbia esula. Physiol Plantarum 120 421 433
60. BhatiaKRahmanSAliMRaisuddinS 2006 In vitro antioxidant activity of Juglans regia L. bark extract and its protective effect on cyclophosphamide-induced urotoxicity in mice. Redox Rep 11 273 279
61. Rodriguez MillaMAMaurerARodriguez HueteAGustafsonJP 2003 Glutathione peroxidase genes in Arabidopsis are ubiquitous and regulated by abiotic stresses through diverse signaling pathways. Plant J 36 602 615
62. Gross-HardtRKagiCBaumannNMooreJMBaskarR 2007 LACHESIS restricts gametic cell fate in the female gametophyte of Arabidopsis. PLoS Biol 5 e47 doi:10.1371/journal.pbio.0050047
63. KidnerCAMartienssenRA 2004 Spatially restricted microRNA directs leaf polarity through ARGONAUTE1. Nature 428 81 84
64. Hernandez-PinzonIYelinaNESchwachFStudholmeDJBaulcombeD 2007 SDE5, the putative homologue of a human mRNA export factor, is required for transgene silencing and accumulation of trans-acting endogenous siRNA. Plant J 50 140 148
65. Cibrián-JaramilloADe la Torre-BarcenaJELeeEKKatariMSLittleDP 2010 Using phylogenomic patterns and gene ontology to identify proteins of importance in plant evolution. Genome Biol Evol 2 225 239
66. NagasakiHItohJHayashiKHibaraKSatoh-NagasawaN 2007 The small interfering RNA production pathway is required for shoot meristem initiation in rice. Proc Natl Acad Sci U S A 104 14867 14871
67. MartienssenR 2010 Molecular biology. Small RNA makes its move. Science 328 834 835
68. MorinRDAksayGDolgosheinaEEbhardtHAMagriniV 2008 Comparative analysis of the small RNA transcriptomes of Pinus contorta and Oryza sativa. Genome Res 18 571 584
69. AxtellMJSnyderJABartelDP 2007 Common functions for diverse small RNAs of land plants. Plant Cell 19 1750 1769
70. RabinowiczPDCitekRBudimanMANunbergABedellJA 2005 Differential methylation of genes and repeats in land plants. Genome Res 15 1431 1440
71. MosherRAMelnykCWKellyKADunnRMStudholmeDJ 2009 Uniparental expression of PolIV-dependent siRNAs in developing endosperm of Arabidopsis. Nature 460 283 286
72. Olmedo-MonfilVDuran-FigueroaNArteaga-VazquezMDemesa-ArevaloEAutranD 2010 Control of female gamete formation by a small RNA pathway in Arabidopsis. Nature 464 628 632
73. FriedmanWE 2009 The meaning of Darwin's ‘abominable mystery’. Am J Bot 96 5 21
74. WeedallGDPolleySDConwayDJ 2008 Gene-specific signatures of elevated non-synonymous substitution rates correlate poorly across the Plasmodium genus. PLoS ONE 3 e2281 doi:10.1371/journal.pone.0002281
75. MagallónSASandersonMJSoltisP 2005 Angiosperm divergence times: the effect of genes, codon positions, and time constraints. Evolution 59 1653 1670
76. GovaertsRFrodinDGRadcliffe-SmithA 2000 World checklist and bibliography of Euphorbiaceae (with Pandaceae). 4 Volumes. London Royal Botanic Gardens, Kew
77. Radcliffe-SmithA 2001 Genera Euphorbiacearum. London Royal Botanic Gardens, Kew 464
78. DavisCCWebbCOWurdackKJJaramilloCADonoghueMJ 2005 Explosive radiation of Malpighiales supports a mid-Cretaceous origin of modern tropical rain forests. Am Nat 165 E36 E65
79. WurdackKJHoffmannPChaseMW 2005 Molecular phylogenetic analysis of uniovulate Euphorbiaceae (Euphorbiaceae sensu stricto) using plastid rbcL and trnL-F DNA sequences. Am J Bot 92 1397 1420
80. ChristinP-AOsborneCPSageRFArakakiMEdwardsEJ 2011 C4 eudicots are not younger than C4 monocots. J Exp Bot 62 3171 3181
81. GoldmanNYangZ 1994 A codon-based model of nucleotide substitution for protein-coding DNA sequence. Mol Biol Evol 11 725 736
82. KatohKTohH 2008 Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform 9 286 298
83. SwoffordDL 2003 PAUP*: Phylogenetic Analysis Using Parsimony (and other methods). Sunderland, , MA Sinauer Associates
84. GoloboffPAFarrisJSNixonKC 2008 TNT, a free program for phylogenetic analysis. Cladistics 24 774 786
85. GoloboffPA 1999 Analyzing large data sets in reasonable times: solutions for composite optima. Cladistics 15 415 428
86. NixonKC 1999 The parsimony ratchet, a new method for rapid parsimony analysis. Cladistics 15 407 414
87. GatesyJO'GradyPBakerRH 1999 Corroboration among data sets in simultaneous analysis: hidden support for phylogenetic relationships among higher level artiodactyl taxa. Cladistics 15 271 313
88. SorensonMDFranzosaEA 2007 TreeRot. 3 ed. Boston Boston University
89. StamatakisAOttM 2008 Efficient computation of the phylogenetic likelihood function on multi-gene alignments and multi-core architectures. Phil Trans R Soc B Biol Sci 363 3977 3984
90. OttMZolaJStamatakisAAluruS 2007 Large-scale maximum likelihood-based phylogenetic analysis on the IBM BlueGene/L. Proceedings of the 2007 ACM/IEEE Conference on Supercomputing Reno, , NV ACM
91. StamatakisAOttM 2008 Exploiting fine-grained parallelism in the phylogenetic likelihood function with MPI, Pthreads, and OpenMP: a performance study. Pattern Recognition in Bioinformatics Berlin Springer 424 435
92. StamatakisA 2006 RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22 2688 2690
93. JonesDTTaylorWRThorntonJM 1992 The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8 275 282
94. LanaveCPreparataGSacconeCSerioG 1984 A new method for calculating evolutionary substitution rates. J Mol Evol 20 86 93
95. StamatakisA 2006 Phylogenetic models of rate heterogeneity: a high performance computing perspective. IEEE International Parallel and Distributed Processing Symposium. Rhodes, Greece
96. YangZ 1994 Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J Mol Evol 39 306 314
97. StamatakisAHooverPRougemontJ 2008 A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol 57 758 771
98. PattengaleNDAlipourMBininda-EmondsORPMoretBMEStamatakisA 2010 How many bootstrap replicates are necessary? J Comput Biol 17 337 354
99. AshburnerMBallCABlakeJABotsteinDButlerH 2000 Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25 25 29
100. MewesHWDietmannSFrishmanDGregoryRMannhauptG 2008 MIPS: analysis and annotation of genome information in 2007. Nucl Acids Res 36 D196 201
101. WangRTischnerRGutierrezRAHoffmanMXingX 2004 Genomic analysis of the nitrate response using a nitrate reductase-null mutant of Arabidopsis. Plant Physiol 136 2512 2522
102. YangZ 2006 Computational Molecular Evolution. Oxford Oxford University Press 357
103. SharpPM 1997 In search of molecular darwinism. Nature 385 111 112
104. GoldingGBDeanAM 1998 The structural basis of molecular adaptation. Mol Biol Evol 15 355 369
105. YangZ 1998 Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol Biol Evol 15 568 573
106. YangZNielsenRGoldmanNPedersenAM 2000 Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155 431 449
107. Kosakovsky PondSLFrostSDWMuseSV 2005 HyPhy: hypothesis testing using phylogenies. Bioinformatics 21 676 679
108. MuseSVGautBS 1994 A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol Biol Evol 11 715 724
109. HasegawaMKishinoHYanoT 1985 Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22 160 174
110. Kosakovsky PondSLFrostSDW 2005 Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22 1208 1222
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 12
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Targeted Proteolysis of Plectin Isoform 1a Accounts for Hemidesmosome Dysfunction in Mice Mimicking the Dominant Skin Blistering Disease EBS-Ogna
- The RNA Silencing Enzyme RNA Polymerase V Is Required for Plant Immunity
- The FGFR4-G388R Polymorphism Promotes Mitochondrial STAT3 Serine Phosphorylation to Facilitate Pituitary Growth Hormone Cell Tumorigenesis
- Target Site Recognition by a Diversity-Generating Retroelement
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy