
Age-Related Neuronal Degeneration: Complementary Roles of Nucleotide Excision Repair and Transcription-Coupled Repair in Preventing Neuropathology
Neuronal degeneration is a hallmark of many DNA repair syndromes. Yet, how DNA damage causes neuronal degeneration and whether defects in different repair systems affect the brain differently is largely unknown. Here, we performed a systematic detailed analysis of neurodegenerative changes in mouse models deficient in nucleotide excision repair (NER) and transcription-coupled repair (TCR), two partially overlapping DNA repair systems that remove helix-distorting and transcription-blocking lesions, respectively, and that are associated with the UV-sensitive syndromes xeroderma pigmentosum (XP) and Cockayne syndrome (CS). TCR–deficient Csa−/− and Csb−/− CS mice showed activated microglia cells surrounding oligodendrocytes in regions with myelinated axons throughout the nervous system. This white matter microglia activation was not observed in NER–deficient Xpa−/− and Xpc−/− XP mice, but also occurred in XpdXPCS mice carrying a point mutation (G602D) in the Xpd gene that is associated with a combined XPCS disorder and causes a partial NER and TCR defect. The white matter abnormalities in TCR–deficient mice are compatible with focal dysmyelination in CS patients. Both TCR–deficient and NER–deficient mice showed no evidence for neuronal degeneration apart from p53 activation in sporadic (Csa−/−, Csb−/−) or highly sporadic (Xpa−/−, Xpc−/−) neurons and astrocytes. To examine to what extent overlap occurs between both repair systems, we generated TCR–deficient mice with selective inactivation of NER in postnatal neurons. These mice develop dramatic age-related cumulative neuronal loss indicating DNA damage substrate overlap and synergism between TCR and NER pathways in neurons, and they uncover the occurrence of spontaneous DNA injury that may trigger neuronal degeneration. We propose that, while Csa−/− and Csb−/− TCR–deficient mice represent powerful animal models to study the mechanisms underlying myelin abnormalities in CS, neuron-specific inactivation of NER in TCR–deficient mice represents a valuable model for the role of NER in neuronal maintenance and survival.
Published in the journal:
. PLoS Genet 7(12): e32767. doi:10.1371/journal.pgen.1002405
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002405
Summary
Neuronal degeneration is a hallmark of many DNA repair syndromes. Yet, how DNA damage causes neuronal degeneration and whether defects in different repair systems affect the brain differently is largely unknown. Here, we performed a systematic detailed analysis of neurodegenerative changes in mouse models deficient in nucleotide excision repair (NER) and transcription-coupled repair (TCR), two partially overlapping DNA repair systems that remove helix-distorting and transcription-blocking lesions, respectively, and that are associated with the UV-sensitive syndromes xeroderma pigmentosum (XP) and Cockayne syndrome (CS). TCR–deficient Csa−/− and Csb−/− CS mice showed activated microglia cells surrounding oligodendrocytes in regions with myelinated axons throughout the nervous system. This white matter microglia activation was not observed in NER–deficient Xpa−/− and Xpc−/− XP mice, but also occurred in XpdXPCS mice carrying a point mutation (G602D) in the Xpd gene that is associated with a combined XPCS disorder and causes a partial NER and TCR defect. The white matter abnormalities in TCR–deficient mice are compatible with focal dysmyelination in CS patients. Both TCR–deficient and NER–deficient mice showed no evidence for neuronal degeneration apart from p53 activation in sporadic (Csa−/−, Csb−/−) or highly sporadic (Xpa−/−, Xpc−/−) neurons and astrocytes. To examine to what extent overlap occurs between both repair systems, we generated TCR–deficient mice with selective inactivation of NER in postnatal neurons. These mice develop dramatic age-related cumulative neuronal loss indicating DNA damage substrate overlap and synergism between TCR and NER pathways in neurons, and they uncover the occurrence of spontaneous DNA injury that may trigger neuronal degeneration. We propose that, while Csa−/− and Csb−/− TCR–deficient mice represent powerful animal models to study the mechanisms underlying myelin abnormalities in CS, neuron-specific inactivation of NER in TCR–deficient mice represents a valuable model for the role of NER in neuronal maintenance and survival.
Introduction
DNA is continuously damaged by spontaneous hydrolytic decay, endogenous metabolites (e.g. reactive oxygen species, malondialdehyde), and environmental genotoxins. DNA lesions can give rise to irreversible mutations and chromosomal aberrations that may trigger carcinogenesis. Alternatively, DNA damage can cause replicative senescence and cell death, which promotes the process of aging [1]. Cumulative DNA damage has also been implicated in the functional deterioration and degeneration of long-living post-mitotic cells such as neurons [2]. To counteract the harmful effects of DNA injuries, cells have a variety of DNA surveillance and repair systems. The importance of these genome maintenance pathways for human health is well illustrated by a heterogeneous set of inherited syndromes that are associated with defects in specific DNA repair pathways resulting in cancer predisposition, developmental abnormalities, accelerated aging and neurodevelopmental or neurodegenerative abnormalities [1], [3]–[5].
Nucleotide excision repair (NER) is a key DNA repair pathway for removal of UV-induced DNA damage and a wide range of other helix-distorting lesions, including bulky chemical adducts and specific types of oxidative damage [1]. In NER the DNA lesion is removed as a part of a 25–30 nucleotide single-strand fragment excised via a multi-step reaction followed by resynthesis of the excised strand [4], [6]–[8]. NER can be divided into two subpathways that differ in the damage recognition step: While global genome NER (GG-NER) removes distorting DNA damage throughout the genome, transcription-coupled NER (TC-NER) specifically targets transcription-blocking lesions in the template strand of active genes to allow recovery of transcription after damage induction [1], [4], [7], [8]. Several NER proteins have functions beyond NER, which is particularly evident for the transcription/repair factor TFIIH, which is required for the local opening of the damaged DNA in NER, but in addition plays an essential role in transcription. Furthermore, several lines of evidence indicate that TC-NER components are involved in repair of transcription-blocking lesions independent of the NER core complex, putatively via recruitment of other repair mechanisms. The term transcription-coupled repair (TCR) has been used to designate this broader, still poorly defined repair process [7], [9], [10].
NER gene defects are associated with a heterogeneous set of rare clinical syndromes, whose characteristics can be explained by the type of NER pathway that is affected or by defects in additional functions of these NER components in other DNA repair pathways or transcription. Selective defects in GG-NER, resulting from mutations in the XPC and XPE (also termed UV-DDB2) genes encoding GG-NER-specific damage recognition proteins, cause xeroderma pigmentosum (XP), a photosensitivity syndrome characterized by UV-hypersensitivity, pigmentation abnormalities and UV-induced skin cancer predisposition [11], [12]. Cancer-predisposition in XP-C patients is explained by bulky lesions that accumulate over the entire genome causing mutations after replication [1]. Selective defects in TC-NER result from mutations in the genes encoding the proteins CSA or CSB, both of which are selectively recruited to stalled RNA polymerase II [13]. Mutations in CSB and CSA are associated with Cockayne syndrome (CS), a progeroid disorder characterized by cachectic dwarfism and progressive neurological abnormalities, in addition to skin photosensitivity [14]–[16]. CS patients do not show cancer predisposition, which is explained by the normal function of GG-NER, and indicates that TC-NER is not required for preventing cancer. On the other hand, most CS pathological features cannot be explained by the sole loss of TC-NER function as they do not occur in XP-A patients, which show a combined GG-NER/TC-NER deficiency, resulting from mutations in the gene encoding for the core NER protein XPA. Thus XP-A patients present with UV-hypersensitivity and skin cancer predisposition, like XP-C patients, usually in combination with progressive neurological abnormalities (see below) [17]–[19], but they do not develop cachectic dwarfism and other progeroid features of CS patients. This has led to the notion that the CS phenotype is largely the consequence of an overall TCR defect, i.e., the inability to rescue transcription arrested by NER - and non-NER-types of DNA damage [7], [20], [21]. In addition it has been suggested that CS is associated with transcriptional abnormalities independent of DNA lesions [7], [22], [23].
The complementarity of NER and TCR DNA repair pathways and disorders resulting from deficiencies in these processes is also illustrated by XPCS patients, which display both XP and CS symptoms. XPCS is caused by mutations in the XPB or XPD genes, both encoding helicases of the transcription/repair factor TFIIH, or in the XPG gene, encoding the endonuclease that mediates the 3′ incision of the excision step [24]–[27]. Mutations in the XPB and XPD genes may also cause pure XP or trichothiodystrophy (TTD), a disorder that is characterized by sulphur-deficient hair, in association with a variable spectrum of abnormalities that usually include neurodevelopmental deficits. Mutations that cause XP preferentially afflict the NER activity of TFIIH, while TTD mutations destabilize the TFIIH complex causing exhaustion of TFIIH in specific cell types [27]. The occurrence of CS symptoms in association with specific XPB and XPD mutations point to functions beyond NER and basal transcription presumably linked to non-NER TCR activities akin to CSA and CSB [22], [27]–[31]. XPG mutations associated with XPCS have been proposed to destabilize the interaction between XPG and TFIIH, while mutations causing XP disrupt its endonuclease activity, further pointing to a non-NER activity underlying CS symptoms [25], [26], [32].
The presence of progressive juvenile or adult onset neurological abnormalities in XP-A patients has provided a strong hint that the NER pathway is important for neuronal function and maintenance [14], [19], [33]–[36]. The neurological symptoms are characterized by progressive sensory and motor deficits, as well as cognitive deterioration and emotional abnormalities, and are associated with widespread neuronal degeneration in multiple brain areas and the spinal cord [17]–[19]. XP-C patients (who are only deficient in GG-NER) do not develop overt neurological symptoms, indicating that the neurodegenerative changes follow from TC-NER or a combined GG-NER and TC-NER dysfunction. A dominant role of the TC-NER pathway in the nervous system is also suggested by the occurrence of XP-A-like progressive neurological abnormalities in CS patients. However, in CS patients neuropathological changes are primarily characterized by myelin abnormalities, while neurons and their axons seem relatively unaffected [14], [35], [37], [38]. An additional complicating factor is formed by patients carrying CSA or CSB mutations that develop UV-sensitive syndrome, a disorder that is characterized by the skin abnormalities of CS in the absence of other CS features. The lack of typical CS features in these patients has been linked to residual TCR activities required for repair of oxidative DNA lesions, while TC-NER of UV-induced DNA lesions was deficient [39]. In sum, the data from XP and CS patients indicate that combined deficiency of GG-NER and TC-NER as in XP-A patients predominantly afflicts neurons, while deficiencies of TCR predominantly cause myelin problems. However, the precise mechanisms underlying the differential cellular vulnerabilities in XP and CS nervous system are still poorly defined, in particular in CS where distinct degenerative mechanisms may operate in oligodendrocytes and neurons [7], [22].
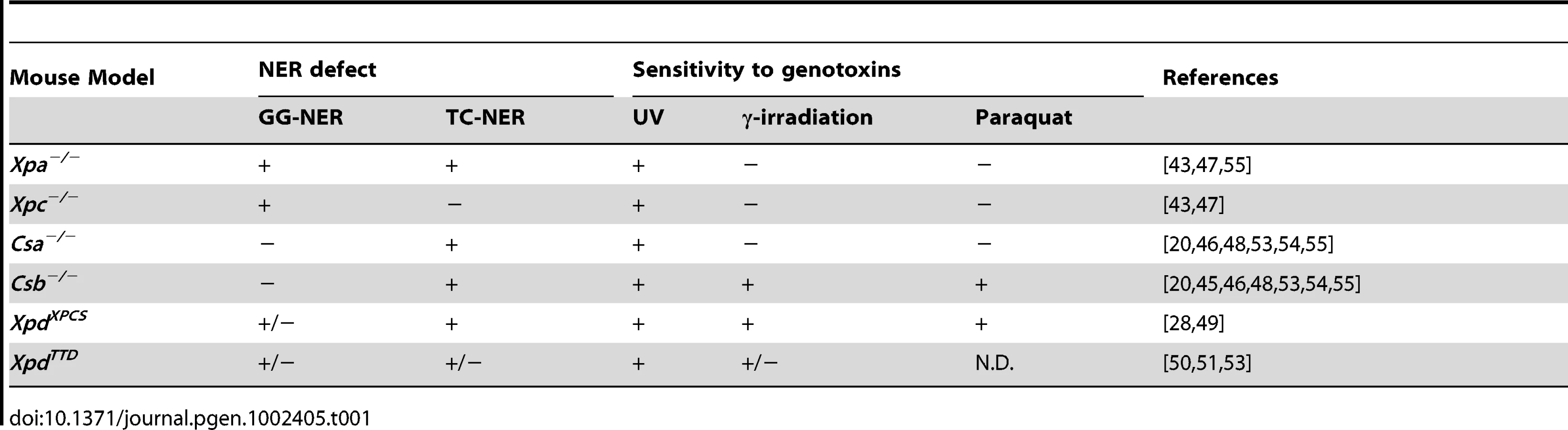
Although previous studies have shown that mouse models for XP, CS, XP-CS and TTD reliably recapitulate the repair defect (i.e. GG-NER and/or TC-NER/TCR), UV-sensitivity and skin cancer predisposition associated with the corresponding NER syndromes (Table 1 and Table 2), this does not apply to the neurological features [40]. In particular, Xpa−/− mice fail to exhibit obvious neurological symptoms and neuropathological changes observed in human XP-A [41]–[44]. Likewise, Csa−/− and Csb−/− mouse models for CS, except for photoreceptor-loss, do not show overt neurological abnormalities [45], [46]. However, a detailed systematic neuropathological analysis is still lacking. In the present study we reexamined various NER and TCR mutant mouse models for neurodegenerative abnormalities to assess and dissect the contribution of the different repair systems in preventing neurodegeneration. To permit analysis of the direct contribution of DNA repair defects to neurological functioning, in the absence of pathology elsewhere in the body due to systemic DNA repair deficiency, we have generated a Cre-lox-based conditional Xpa mouse model that enables inactivation of the Xpa gene selectively in neurons. In particular, we used this novel mouse model to examine the effect of combined NER and TCR deficiency on neuronal survival.

Results
Sporadic p53 activation in astrocytes and neurons throughout the central nervous system of Cockayne syndrome mice
As a first step to study the role of NER and TCR in maintaining neuronal integrity, we have re-examined six previously reported mutant mouse models for the presence of neuropathological abnormalities. These mouse lines consisted of Xpc−/− mice, in which only GG-NER is completely inactive [43], [47]; Xpa−/− mice, in which both GG-NER and TC-NER are fully deficient [41], [43]; Csa−/− and Csb−/− mice, in which TC-NER and presumably the entire TCR pathway is abrogated, but which have proficient GG-NER [45], [46], [48]; XpdXPCS mice (homozygous for the G602D XPCS mutation in the Xpd gene), which carry a partial GG-NER and a partial TCR defect [49]; and XpdTTD mice carrying Xpd alleles with the R722W TTD mutation, that also have a partial GG-NER and TCR defect, in addition to TFIIH instability causing transcriptional insufficiency in terminally differentiated cells with consequent brittle hair and nails (Table 1 and Table 2) [50], [51]. Consistent with their respective NER-deficiencies the mutant mice manifest various degrees of increased susceptibility to UV - and 7,12-dimethylbenz[a]anthracene (DMBA)-induced skin carcinogenesis (Table 1 and Table 2). TCR-deficient mice to varying extent exhibit other symptoms like reduced growth, osteoporosis, photoreceptor loss, liver and kidney aging, and reduced lifespan (Table 2) [20], [28], [44]–[56].
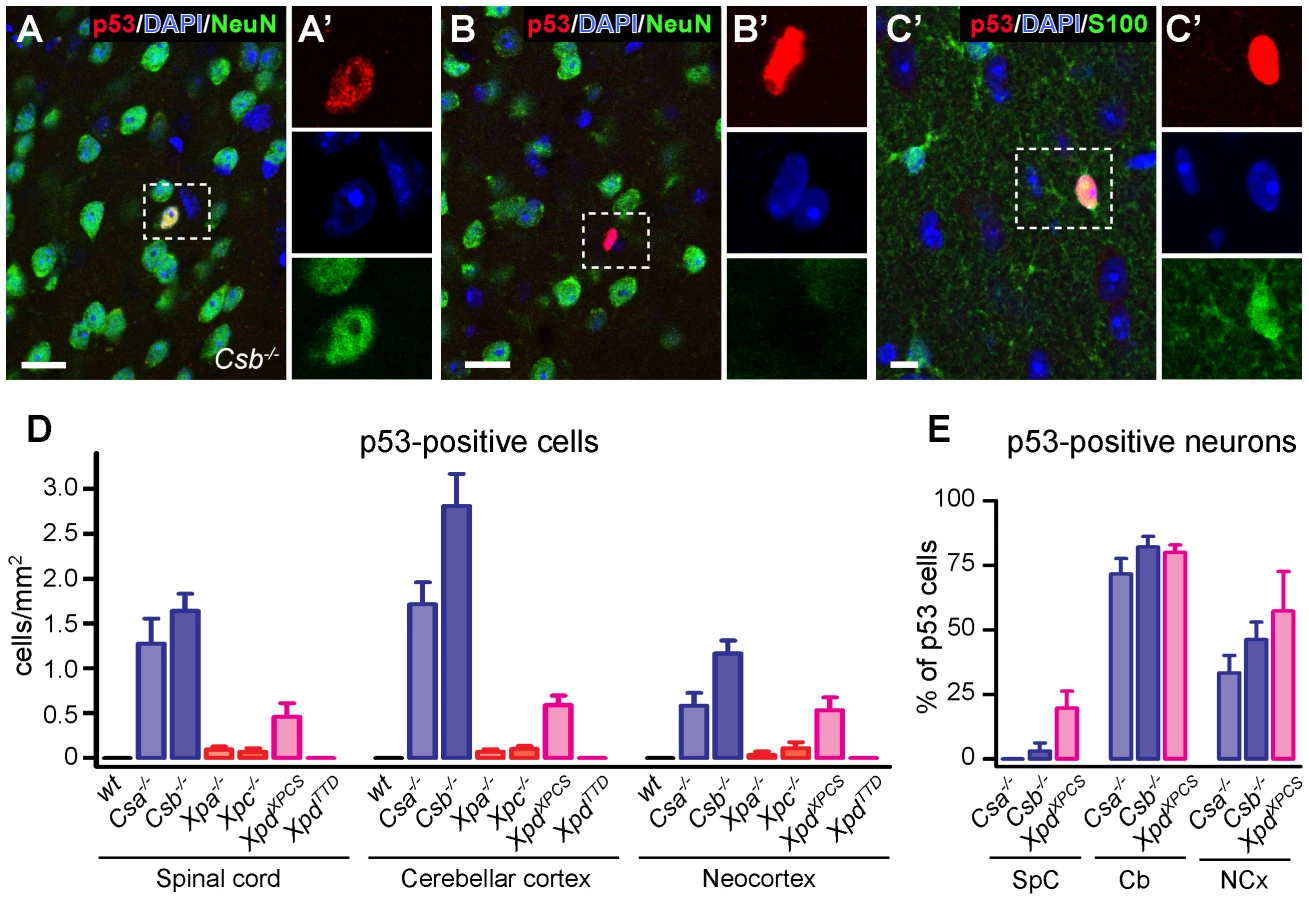
Consistent with previous reports [43]–[50] analysis of thionin - and hematoxylin/eosin-stained brain sections revealed that the gross anatomy and histological organization of all central nervous system regions of aforementioned mutant mice were indistinguishable from wild type animals at 6 months of age, precluding overt neurodevelopmental deficits or neuronal degeneration. Also the central nervous system of 70–100 week old Csb−/− and Xpa−/− mice appeared normal. Therefore, to examine the possible occurrence of subtle abnormalities, we employed immunohistological approaches. First, to determine whether central nervous system cells of the NER-deficient mice experience genotoxic stress, we studied the expression of the transcription factor p53, which is activated by multiple types of DNA damage [57]. p53-immunoreactive cells were not detected in the central nervous system of wild-type and XpdTTD mice. Instead, occasional cells with p53-immunoreactive nuclei were observed throughout the nervous system of Csa−/−, Csb−/− and XpdXPCS mice, while even more sporadic p53 induction was visible in Xpa−/− and Xpc−/− animals (Figure 1). p53-staining was associated with neurons (NeuN-positive cells; Figure 1A), astrocytes (GFAP - or S100-positive cells; Figure 1C and Figure S1), and sometimes oligodendrocytes (APC-positive cells; Figure S1). The relative amount of glial versus neuronal p53-staining varied per brain region, and to some extent per mouse model: in cerebellar cortex the large majority of p53-positive cells consisted of neurons, in the neocortex p53-positive cells were neurons or non-neuronal cells in equal amounts, while in spinal cord and the brainstem reticular formation p53-immunoreactivity was predominantly or almost exclusively associated with glia cells (Figure 1E and Figure S1). p53-immunoreactive neurons generally showed normal nuclear morphologies, in contrast to most p53-immunoreactive glial cells. For instance, a subset of p53-positive astrocytes in spinal cord and the brainstem reticular formation showed nuclei with a DAPI-negative centre that was intensely p53-positive (Figure S1). Another nuclear abnormality of p53-positive glia consisted of a larger nuclear size (Figure S1). Also in Xpa−/− and Xpc−/− nervous systems p53-positive cells consisted of both neurons and astrocytes, but their frequency was too low to allow systematic analysis of the relative proportion of neuronal versus non-neuronal cells in different brain areas. Taken together the data indicate that mice with a complete (Csa−/−, Csb−/−) or severe partial (XpdXPCS) TCR defect show nuclear p53 expression in sporadic neurons and glia throughout the nervous system, pointing to the occurrence of genotoxic stress.
Microglia activation and astrocytosis in white matter regions of Cockayne syndrome Csa−/−, Csb−/−, and XpdXPCS mice
To further investigate the presence of degenerative changes in the nervous system of NER-deficient mice we examined microglia cells which proliferate and acquire activated morphologies in conditions of neuronal and glial damage [58], [59]. Immunostaining for Iba-1, a marker of all microglia cells and Mac2 (also known as galectin-3), a protein selectively expressed by activated phagocytosing microglia [58], did not, or only sporadically, reveal activated microglia cells in the nervous system of wild-type, Xpa−/−, Xpc−/− and XpdTTD mice (Figure 2 and Figure S2). In contrast, prominent levels of activated microglia were present throughout the nervous system of Csa−/−, Csb−/− mice and, to a somewhat lesser extent, XpdXPCS mice (Figure 2, Figures S2 and S3). Typically, Mac2-positive microglia cells occurred in small clusters in areas with myelinated fibers. Thus, in the forebrain Mac2-positive microglial cells were concentrated in the corpus callosum, the anterior commissure, the capsula interna and the fornix, while in the caudal brain high levels of activated microglia occurred in the cerebellar white matter, throughout the reticular formation and in fiber tracts in the brainstem (Figure 2, Figure S2 and Figure S3). We performed a more in-dept analysis of the time of onset and course of these features in Csb−/− mice at different ages. Prominent levels of Mac2-positive cells were already present before 10 weeks of age (not shown), and the density of Mac2-positive cells did not show a distinct increase with age (Figure 2G).

To examine whether microglia activation is paralleled by changes in astrocytes, we also stained for glial fibrillary acidic protein (GFAP), an astrocytic protein up-regulated under conditions of neuronal injury. Increased GFAP staining was observed in the nervous system of mouse mutants that also showed microglial cell activation, i.e. Csa−/−, Csb−/− and XpdXPCS mice (Figure 2I–2O). Increased GFAP staining was most prominent in the brainstem reticular formation and spinal cord (Figure S4). No obvious changes in GFAP staining were noted in some white matter areas such as the corpus callosum and the capsula interna of Csa−/−, Csb−/− and XpdXPCS mice, which may be explained by relatively higher baseline GFAP-immunoreactivity in these areas in wild-type mice. To further examine astrocytic changes in white matter areas, we examined the expression of Hsp25 (also known as Hsp27 or Hspb1), a small heat shock protein that is expressed at high levels in a subset of astrocytes in conditions of injury [60]. Indeed Csa−/−, Csb−/− and XpdXPCS, but not Xpa−/−, Xpc−/− and XpdTTD nervous systems showed the appearance of intensely Hsp25-immunoreactive astrocytes in multiple regions including the brainstem reticular formation, spinal cord, the white matter of cerebellum and the corpus callosum forebrain (Figure S4).
The above data indicate that Csa−/−, Csb−/− and XpdXPCS mice show microglia and astrocyte activation in multiple central nervous system areas, indicative of the occurrence of cellular degeneration or another detrimental process. As these mutant mice also displayed p53-immunoreactive cells, we performed double labeling of p53 with Mac2 to determine whether p53-immunoreactive cells are contacted by phagocytosing microglia cells. However, Mac2-positive microglia cells were never found in the vicinity of p53 cells (Figure 3A). Instead, Mac2-positive microglia were frequently in close proximity of oligodendrocytes (Olig2 and APC-positive; Figure 3B, 3C), that otherwise showed a healthy appearance with normal DAPI-stained nuclei. These data indicate that glial abnormalities may be associated with subtle alterations in oligodendrocytes. To examine whether the presence of activated microglia was associated with myelin abnormalities, we performed double labeling of neurofilament-H and myelin basic protein to outline axons and their myelin sheets. No differences in myelin basic protein staining were observed in spinal white matter and corpus callosum of wild-type and Csb−/− mice (not shown).

Finally, to determine whether Csa−/−, Csb−/− and XpdXPCS mice show increased levels of cell death of oligodendrocytes or other cells, we stained for active caspase 3, which is a final executioner caspase associated with multiple cell death pathways [61]. Very sporadically caspase 3 immunoreactive cells were observed in the nervous system of all mutant mouse models; all positive cells showing morphologies compatible with glial cells (Figure 3D, 3E). Quantitative analysis in spinal cord indicated that the number of active caspase 3-positive cells, although still very low, was higher in Csa−/−, Csb−/− and XpdXPCS mice as compared to the other genotypes (Figure 3F). It was not possible to determine whether the active caspase 3 cells represented oligodendrocytes or astrocytes because they did not stain for cellular markers such as GFAP, S100, APC ad NeuN. Consistent with active caspase 3 staining, a silver degeneration procedure, that outlines degenerating neurons and their processes, indicated that none of the NER mice showed detectable levels of neuronal degeneration at 26 weeks of age. Taken together the data show that Csa−/−, Csb−/− and XpdXPCS mice develop prominent microglia cell activation as well as astrocytic changes that may be mostly triggered by subtle oligodendrocyte abnormalities.
Neuron-specific inactivation of Xpa causes age-dependent neuronal loss in Csb−/− mice
The NER mouse models investigated above exhibited either no detectable neuronal abnormalities or evidence for only subtle neuronal dysfunction and degeneration, indicating that inactivation of NER or TCR pathways by itself is not sufficient to significantly affect long-term survival of neurons in mice. Previous studies disclosed synergistic deleterious effects of intercrossing XP (Xpa−/− or Xpc−/−) with CS (Csa−/−, Csb−/−, XpdXPCS) mice, resulting in double mutants with very short life span and dramatic progeroid features [49], [62]–[64]. This raises the possibility that neuronal degeneration may be achieved by inactivation of multiple NER components afflicting both NER and TCR pathways. However, the very short lifespan as well as the serious systemic abnormalities of the double mutant mice precludes systematic analysis of neuronal degeneration, which could also be an indirect consequence of impaired function of other organs and systems. To address this issue, we generated a Cre-lox-based conditional Xpa mouse model that enables selective inactivation of the Xpa gene in postnatal neurons of CS mouse lines and hence to study the effect of combined TCR and NER inactivation in neurons of adult mice that do not suffer from other severe deficits.
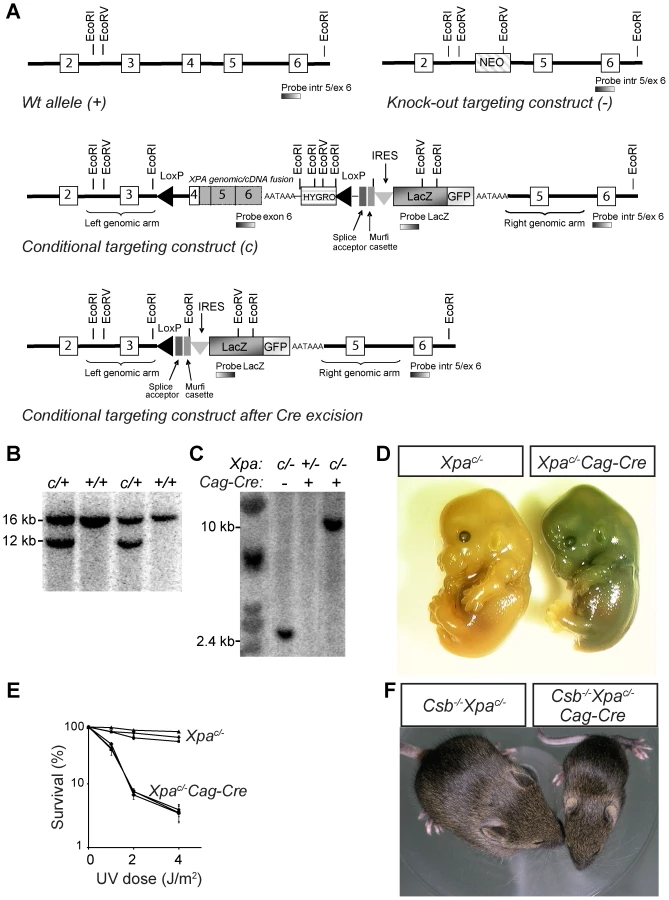
To establish a conditional Xpa knockout mouse model, we generated a targeting construct in which exon 4 of the Xpa gene is fused in frame to the mouse Xpa cDNA containing the remaining coding sequence and including a synthetic polyA sequence, followed by a PGK promoter-driven hygromycin selectable marker gene, and a LacZ-GFP fusion gene (Figure 4A). The splice acceptor-Murfi cassette ensures proper splicing and translational stops in all frames respectively when the Xpa gene is knocked out (Figure 4A). The functionality of this conditional genomic-cDNA fusion allele was tested in UV-hypersensitive Xpa−/− ES cells (Figure S5). These experiments showed that the Xpac conditional allele fully averted the UV-hypersensitivity of Xpa−/− ES cells (Figure S5). Next Xpac/+ ES cells, obtained by transfection of IB10 ES cells (Figure 4B), were used for blastocyst injections and subsequent generation of Xpac/+ mice. To determine whether Cre recombinase was capable of excising the floxed Xpa sequence in vivo, we generated Xpac/−/Cag-Cre mice by crossing Xpac/+ mice with Xpa+/− mice carrying a Cag-promotor driven Cre transgene (Cag-Cre), which drives Cre recombinase expression immediately after conception [65]. Southern blot analysis showed Cre-recombinase excision of the floxed sequence in Xpac/−/Cag-Cre embryos at ∼100% efficiency (Figure 4C). Consistent with ubiquitous recombination, Xpac/−/Cag-Cre embryos stained blue upon X-gal staining due to LacZ expression, while Xpac/− embryos remained unstained (Figure 4D). Xpac/−/Cag-Cre mouse embryonic fibroblasts (MEFs) like Xpa−/− MEFs [41] showed severe UV-hypersensitivity, while Xpac/− MEFs show wild-type UV-resistance, consistent with Cre-dependent inactivation of the conditional allele (Figure 4E). Next, we crossed Csb−/−/Xpac/+ and Csb−/−/Xpa+/−/Cag-Cre mice to obtain Csb−/−/Xpac/− and Csb−/−/Xpac/−/Cag-Cre mice. In line with the phenotype of Csb−/−/Xpa−/− mice [63], Csb−/−/Xpac/−Cag-Cre pups displayed severe postnatal growth deficits, cachexia, disturbed gait, and death before weaning, while Csb−/−/Xpac/− littermates did not develop overt pathology (Figure 4F). Taken together, these data demonstrate that we have generated a valid conditional Xpa mouse model that enables us to study the Csb−/−/Xpa−/− phenotype in a cell or tissue-specific manner.
To study the effect of Xpa inactivation in the absence of Csb, specifically in postnatal neurons, we crossed Csb−/−/Xpac/− mice with a calcium/calmodulin-dependent protein kinase IIα (CamKIIα) Cre transgenic line that expresses Cre-recombinase selectively in postnatal neurons throughout the forebrain [66], [67]. Forebrain-specific recombination was confirmed by PCR and analysis of LacZ expression. Csb−/−/Xpac/−/CamKIIα-Cre mice grew into young adulthood without any noticeable phenotype, showed a normal body weight and appearance at the age of 6 months, but from the age of 9–12 months became smaller and exhibited reduced weight as compared to littermates with other genotypes, i.e. CamKIIα-Cre, Csb−/−CamKIIα-Cre, Xpac/−CamKIIα-Cre and Csb−/−/Xpac/− littermates (Figure 5A, Figure S6). In addition, from 9–12 months of age, Csb−/−/Xpac/−/CamKII-Cre mice started to display seizure behavior, characterized by episodes of immobility. Subsequently, Csb−/−/Xpac/−/CamKII-Cre mice became moribund, all animals dying prematurely between the age of 12 and 22 months (Figure 5B), while animals from littermates with other genotypes survived up to 24 months (the oldest age examined). Analysis of locomotor behavior using the accelerating rotarod assay demonstrated that Csb−/−/Xpac/−/CamKIIα-Cre mice performed within the normal range at the age of 6 months, but showed reduced performance at 12 months (Figure S6). For further analysis of behavioral abnormalities, we used an open-field exploratory test. This test revealed that Csb−/−/Xpac/−/CamKIIα-Cre mice avoided exploration of the central part of the open field, which is considered a measure of anxiety-related behavior [68]. Total movement time and distance were the same as for the other groups ruling out impaired mobility as explanation for the difference in the test. The ratio of the ambulatory activity in the center and the total walking distance was already reduced at 3 months of age, and further declined at 6 and 12 months of age (Figure 5C).

Macroscopic examination of the brain of Csb−/−/Xpac/−/CamKIIα-Cre mice revealed no obvious changes at 3 months, mild atrophy of the cortex at 6 months, and severe atrophy of the cortex at older age (Figure S6). The sizes of olfactory bulbs, cerebellum and spinal cord were the same as in other groups. Analysis of coronal sections of 12–16 month-old Csb−/−/Xpac/−/CamKIIα-Cre brains showed a large reduction in cortical thickness, atrophy of other telencephalic areas (i.e. hippocampus, caudatus-putamen and septum), and dramatically enlarged lateral ventricles (Figure 5D, 5E). No abnormalities were observed in non-telencephalic areas, consistent with specific inactivation of the conditional Xpa allele in forebrain neurons. Atrophy of telencephalic areas was paralleled by a marked increase in GFAP immunoreactivity, while GFAP staining in other brain areas was the same as in Csb−/−/Xpac/− and Csb−/− mice (Figure 5F). Atrophied brain areas also showed loss of the neuronal somato-dendritic marker microtubule-associated protein 2 (MAP2), in particular in the hippocampal CA1 region, indicative of neuronal degeneration (Figure S7).
Staining for p53 revealed a prominent increase in the number of p53 immunoreactive neurons in the forebrain of Csb−/−/Xpac/−/CamKIIα-Cre mice as compared to Csb−/− mice and other genotypes that showed essentially no p53 immunoreactive cells (Figure 6A, 6D). In addition, Csb−/−/Xpac/−/CamKIIα-Cre forebrain exhibited a strong increase in neurons expressing ATF3 (Figure 6B, 6D), a stress-inducible transcription factor that is induced following genotoxic stress via p53-dependent and -independent pathways [69], [70]. Finally, direct evidence for neuronal degeneration was obtained by staining for active caspase 3 and by using a silver staining procedure: active caspase 3 staining revealed intensely stained neuronal profiles (Figure 6C, 6D). Similarly, the silver degeneration staining method outlined infrequent argyrophylic neuronal profiles, reflecting neurons that are in the process of dying. In addition, the silver staining uncovered high levels of argyrophilic axonal degeneration in the corpus callosum, the fimbria-fornix, the anterior commissure, and the cortifugal fiber bundles coursing in the capsula interna, the cerebral peduncle and the pyramidal tract (Figure 6E), which is consistent with the selective occurrence of neuronal degeneration in forebrain neurons. Together these data indicate that Csb−/−/Xpac/−/CamKII-Cre mice display chronic neuronal degeneration that in the long term has resulted in severe neuronal loss and atrophy of the forebrain regions.

The distribution of degenerative changes in Csb−/−/Xpac/−/CamKII-Cre mice is consistent with the specific inactivation of Xpa in forebrain neurons induced by CamKII-promotor driven Cre-recombinase expression [66], [67], and highlights the vulnerability of Csb-deficient forebrain neurons to loss of Xpa function. To determine the effect of Xpa inactivation in other neuronal populations of the Csb−/− brain, we crossed Csb−/−Xpac/− mice with a postnatal Purkinje cell specific Cre (L7-Cre) transgenic line [71] to obtain Csb−/−/Xpac/−/L7-Cre mice. Analysis of motor behavior with accelerating rotarod revealed no or very mild motor abnormalities in Csb−/−/Xpac/−/L7-Cre mice at the age of 3 and 6 months (the oldest age examined). However, neuropathological analysis disclosed multiple signs of selective Purkinje cell degeneration resembling pathological changes in forebrain neurons of Csb−/−/Xpac/−/CamKII-Cre mice (Figure 6). Abnormalities included the presence of argyrophilic axonal degeneration, specifically in the cerebellar white matter and cerebellar nuclei, i.e. the areas that contain Purkinje cell axons (Figure 6F), and sporadic argyrophilic debris in the molecular and Purkinje cell layer, while no argyrophilic changes occurred in other brain areas. In addition, the Purkinje and molecular layers also showed a strong increase in GFAP-immunoreactivity (Figure 6G), while staining for calbindin, a protein that in the cerebellum is selectively expressed in Purkinje cells, revealed calbindin-negative regions in the molecular layer, indicative of loss of Purkinje cells (Figure 6H). Furthermore, a subset of Purkinje cells (with morphologies varying from relatively normal to severely atrophic cells) displayed strong nuclear ATF3 staining (Figure 6I), which was distinct from the non-specific cytoplasmic staining of Purkinje cells produced by the ATF3 antibody. Nuclear ATF3 staining was not observed in Purkinje cells (nor other cerebellar neurons) of wild-type, Csb−/−, Csb−/−/Xpac/+/L7Cre, Csb−/−/Xpac/− and Xpac/−/L7-Cre, as well as forebrain-specific Csb−/−/Xpac/−/CamKII-Cre mice. Quantification of ATF3-immunoreactive Purkinje cells in mid-sagittal sections of 6 month-old Csb−/−/Xpac/−/L7-Cre mice (n = 3) indicated that 1.3±0.6% (Mean ± SE) of Purkinje cells were ATF3-positive. Finally, staining for active caspase 3 revealed infrequent (<1 in 5000) positive Purkinje cells (Figure 6J). In conclusion, the data obtained with Csb−/−/Xpac/−/L7-Cre mice further demonstrate that the addition of an Xpa defect to Csb-deficient neurons results in pronounced neuronal degeneration.
Discussion
To determine the importance of the NER and TCR DNA repair pathways for neuronal integrity and survival, we first conducted a comparative analysis of neuropathological abnormalities in six mouse models for the human syndromes XP, CS, XPCS and TTD. The major finding of this analysis is that CS-like/TCR-deficient Csa−/−, Csb−/− mice and, to a lesser extent, XpdXPCS mice, develop a characteristic set of mild degenerative changes that was predominantly characterized by microglia activation in regions with myelinated axons, in the absence of obvious signs of axonal degeneration. This microglia activation did not occur in the XP-like GG-NER-defective Xpc−/− and total NER-defective Xpa−/− mutant animals, nor in the TTD-like XpdTTD mouse. In view of very limited signs of neuronal degeneration in NER - and TCR-deficient mice, respectively, we also investigated the effect of combined NER - and TCR-deficiency on neuronal survival. For this purpose we generated a Cre-lox based conditional Xpa knockout mouse that was crossed with neuron-specific Cre lines and TCR-deficient Csb−/− mice. These experiments showed that combining NER and TCR-defects in neurons causes progressive neuronal degeneration and disclose a functional overlap as well as functional complementarity of the NER and TCR repair pathways.
Identification of a CS–like neuropathological phenotype in TCR–deficient mouse mutants
The abnormalities identified in the CS (Csa−/− and Csb−/−), and XPCS (XpdXPCS) mice consisted of 1) the presence of activated phagocytosing microglia cells in regions containing myelinated axons such as the corpus callosum, the brainstem reticular formation and the spinal cord; and 2) sporadic cells with intense p53-immunoreactive nuclei. Microglia activation was frequently accompanied by signs of astrocytosis indicative of a central nervous tissue injury response. We did not find an association between microglia activation and p53-positive cells, and neither was microglia activation associated with detectable axonal degeneration. However, activated microglia cells were often in close contact with oligodendrocytes. These data indicate that microglia activation follows from oligodendrocyte or myelin abnormalities. Previous electron microscopic analysis did not reveal abnormalities in the morphology and thickness of myelin sheets in Csb−/− mice [45], and in the current study, apart from evidence suggesting a minor increase in apoptosis of oligodendrocytes, we did not identify other overt oligodendrocytic abnormalities. Hence, the precise cellular abnormality that triggers microglia activation in myelinated regions of Csa−/−, Csb−/−, and XpdXPCS mice remains to be determined. Importantly, however, the presence of activated microglia is consistent with the notion that irregular patchy myelination with minimal axonal degeneration is a dominant neuropathological hallmark of CS [14], [22], [35], [37], [72]. Hence, our findings together with human neuropathological data strongly indicate that oligodendrocyte abnormalities are a prime defect in CS. We also show that XpdTTD mice, unlike XpdXPCS mice, do not develop microglia activation in myelinated areas. This is in line with the notion that myelin abnormalities in TTD patients and XpdTTD mice result from developmental deficits and arise via different mechanisms than in CS patients [14], [22]. Our data further illustrate that specific point mutations in the Xpd gene result in different cellular deficits and associated pathologies in the mouse, mimicking the different pathologies in patients [27], [31], [49].
The second abnormality that we identified in the CS (Csa−/− and Csb−/−) and the XPCS (XpdXPCS) mouse nervous systems consisted of sporadically distributed p53-immunoreactive neurons and astrocytes, and, albeit very infrequent, oligodendrocytes. p53-immunoreactive cells occurred in all brain areas, but the proportion of neuronal versus glial p53-immunoreactive cells varied among brain areas. Thus, in cortex and cerebellum a large proportion of p53-positive cells are neurons while in the brain stem and spinal cord the far majority, if not all, p53-positive cells are glial cells. The expression of p53, which is known to be activated by multiple types of DNA damage and which mediates neuronal degeneration [57], provides indirect evidence for the occurrence of genotoxic stress, which can be explained by cumulative DNA damage resulting from compromised DNA repair. Interestingly, a subset of p53-positive astrocytes showed abnormal nuclear morphologies, which is compatible with reports of astrocytic nuclear abnormalities in CS patients [38], [72], [73], and further indicate that astrocytes are vulnerable to loss of TCR function.
We did not observe abnormal nuclear morphology in p53-immunoreactive neurons, nor did we obtain direct evidence for ongoing neuronal death using two neuropathological markers for dying neurons, i.e. active caspase 3 immunoreactivity, and silver degeneration staining. However, the process of death and removal of individual neurons may occur within a few hours, making the in vivo detection of asynchronous sporadically distributed cell death challenging [74], [75]. Hence, our methods do not exclude the possibility of a low frequency of ongoing neuronal degeneration. The lack of an obvious neurodegenerative phenotype in the CS mouse models is compatible with the neuropathology of CS patients indicating relatively modest neuronal degeneration in most brain areas [35], [38], [72], [73]. Interestingly, cerebellar granule cells, which are among the most severely affected populations of neurons in CS patients [76], most frequently showed p53 immunoreactivity in the CS mice, indicative of a differential vulnerability of cerebellar granule cells to loss of TCR function in both CS patients and mouse models. Furthermore, p53-immunoreactive granule cells have been demonstrated in autopsy cases of CS [76].
Together our data indicate that Csa−/−, Csb−/−, and XpdXPCS mice reproduce the major aspects of CS neuropathology, albeit in a mild form, which may explain the absence of macroscopic neuropathological and obvious neurological deficits associated with CS. In this context it would be interesting to know whether patients with UV-sensitivity syndrome (UVSS), also carrying mutations in CSA and CSB genes, develop the same mild abnormalities. The presence of activated microglia in UVSS patients would support the notion of a continuum of CS phenotypes ranging from CS type II to UVSS [16] as also suggested by a CS patient with a CSB null mutation displaying adult-onset neurological symptoms [37]. As the pathologies of Csa−/−, Csb−/−, and XpdXPCS mice are relatively similar, our data also indicate that the CS neurodegenerative features can not be explained by molecular mechanisms that do not include all three proteins. Furthermore, the data indicate that the CS neurodegenerative changes result from deficits in a shared non-NER activity of these proteins as Xpa−/− mice with complete loss of GG-NER and TC-NER function did not reproduce the neuropathological features that we observed in Csa−/−, Csb−/−, and XpdXPCS mice (see below). This is consistent with a broader TCR process, which encompasses transcription-coupled repair of non-NER/non-distorting transcription-blocking lesions involving CS and TFIIH proteins.
A marginal nervous system phenotype in GG-NER – and entirely NER–deficient mice
The GG-NER-defective Xpc−/− and total NER-defective Xpa−/− mutant mice at 26 weeks of age showed very low levels of p53-immunoreactive neurons and astrocytes, which nevertheless was higher than in XpdTTD and wild-type mice of the same age, in which we did not detect any cells with nuclear p53 immunoreactivity throughout the nervous system. These data suggest that Xpa−/− and Xpc−/− mice have a central nervous system phenotype, albeit marginal. In case of Xpc−/− mice the phenotype is compatible with that of XP-C patients that, although neurologically and cognitively asymptomatic, may develop mild neurodegenerative changes [19]. However, in Xpa−/− mice the phenotype is very different from the severe progressive neurodegenerative changes of many XP-A patients, which develop juvenile or adult progressive neuronal degeneration throughout the central and peripheral nervous system depending on the severity of NER dysfunction [17], [19], [33], [34], [36]. Neurons from Xpa−/− mice display considerably increased sensitivity to UV radiation [77] and the cross-linking agent cisplatin [78], consistent with loss of NER function and excluding redundancy of NER activity by other proteins at least for the lesions induced by these agents. The discrepancies between human and rodents may follow from differences in the rate of production and type of DNA lesions caused by endogenous metabolites, and from the shorter lifespan of mice.
Synergistic effects of NER and TCR deficiencies in neuronal degeneration
To investigate the effect of combined NER and TCR-deficiency on neuronal survival, we generated a Cre-lox-based conditional Xpa mouse model to inactivate Xpa selectively in postnatal neurons in Csb−/− mice. The use of a conditional Xpa mouse model was required in view of our previous findings that global Csb−/−/Xpa−/− double mutant animals show degenerative changes in multiple organs as well as a very short life span [62], [63], precluding prolonged analysis of neurodegeneration and separation from direct and indirect consequences. Our data show that Csb−/− mice with neuron-specific inactivation of Xpa develop progressive neuronal degeneration, indicating that the XPA protein (and the NER pathway as a whole) is essential for the survival of mouse neurons in the absence of the CSB protein. The time course and distribution of neurodegenerative changes indicate that the affected neurons degenerate asynchronously over a prolonged time window. When Xpa is inactivated in forebrain neurons of Csb-deficient animals, mild behavioral abnormalities were observed at 3 months of age, while death, associated with severe atrophy of forebrain areas, occurred between 12–21 months of age. Analysis of the distribution of dying neurons, as identified by active caspase 3 or ATF3 staining, showed that the level of ongoing neuronal degeneration at a given time point was low. Similarly, in Csb-deficient mice with selective inactivation of Xpa in Purkinje cells which were analyzed at a single time point, a subset of Purkinje cells had disappeared (identified as loss of calbindin staining), a very small subset was in the process degenerating or dying (ATF3 and caspase 3 staining), while a subset showed a normal appearance consistent with asynchronous degeneration. Such an asynchronous neuronal degeneration is consistent with cell death resulting from the accumulation of stochastic DNA damage [79], [80], and strongly resembles the pattern of neuronal degeneration in Ercc1Δ/− mice that are impaired in several DNA repair systems, i.e. nucleotide excision repair, interstrand crosslink repair and double strand break repair [81].
Together, the data with conditional Xpa/Csb-deficient mice indicate that adult neurons in rodents are vulnerable to endogenous DNA lesions when deficient in both NER and TCR, but are able to cope with these lesions when either the TCR or NER pathway are defective. While the NER and TCR pathway share the TC-NER activity, they have non-overlapping activities consisting of GG-NER and the still poorly defined non-NER TCR activities. In neurons, factors of the GG-NER machinery, in particular XPC, have been shown to operate in a specialized type of transcription-associated repair, termed domain-associated repair (DAR). DAR operates on both strands in active genes, including regions of a gene that RNA polymerase II does not reach and has been proposed to complement TCR [80], and it may possibly mask or compensate for the loss of TCR [80]. This is supported by the demonstration that XPC-deficient mice that are selectively deficient in GG-NER when crossed with CSB-deficient mice have a similar phenotype as Csb−/−Xpa−/− mice [64]. Our data indicate that Xpc−/− and Xpa−/− animals develop similar marginal central nervous system phenotypes consisting of highly sporadic p53-positive cells. Together these data suggest that in mice Xpc is equally important as Xpa for the central nervous system. Also in man, XPC-deficiency may result in subtle neurodegenerative changes [19], although XPA-deficiencies results in much more severe neurodegenerative phenotypes [14], [17], [19].
Non-NER TCR has been proposed to operate in conditions of specific transcription-blocking oxidative DNA lesions, putatively via recruitment of alternative DNA repair pathways [7], [9], [10]. This explains why cells from CS mice and CS patients, unlike XPA-deficient cells, show increased vulnerability to some types of oxidative stress, and may more readily accumulate oxidative DNA lesions [9], [20], [39], [82]. In addition, increased levels of oxidative DNA lesions have been reported in brain tissue of Csb−/− mice [10]. Thus, the inability to cope with oxidative lesions may explain the pathological phenotype of CS mice, as well as the severe degenerative phenotype in the conditional Xpa-deficient Csb−/− mice. However, the precise identity of DNA lesions and the question whether the CS phenotype truly results from a repair deficiency remains to be further explored.
In summary, our data indicate that the GG-NER, TC-NER, and non-NER TCR mechanisms operate together in maintaining the integrity of neurons, and that the absence of one pathway aggravates the risk for deficiencies in other pathways, explaining the severe neurodegenerative phenotype in double mutants. The extent to which a combined deficiency of NER and TCR is detrimental to non-neuronal nervous systems cells remains to be determined in future studies by selectively inactivating Xpa in these cells in TCR deficient mice. We propose that neuron-specific inactivation of Xpa - in Csb-deficient mice represents a powerful model for studying XP neurological disease and the role of NER in neurons. As neurologic symptoms seen in XP are hallmark features of age-related neurodegenerative diseases these mice may also reproduce aspects of accelerated aging.
Materials and Methods
Mutant mice
Experiments were performed in accordance with the “Principles of laboratory animal care” (NIH publication no. 86-23) and the guidelines approved by the Erasmus University animal care committee. Animals used were Xpc−/−, knock-out for the Xpc gene [47], Xpa−/−, knock-out for the Xpa gene [41], Csa−/−, knock-out for the Csa gene [46], Csb−/−, in which the CS1AN patient mutation is mimicked resulting in a null mouse [45], XpdXPCS, homozygous for the G602D XPCS point mutation in the Xpd gene [49] and XpdTTD, carrying Xpd alleles with the R722W TTD mutation [50] bred in a pure C57BL/6J background.
To obtain a conditional Xpa knockout mouse model, we generated a targeting construct in which exon 4 was fused in frame to the mouse Xpa cDNA (containing the remaining coding sequence and including a synthetic polyA sequence), followed by a PGK promoter-driven hygromycin selectable marker gene (Figure 4A). A genomic clone containing 10 kb of the 129ola mouse Xpa locus (pMMXP3-6#13; [41]), was used to re-clone an approximately 10 kb size BamHI fragment, containing exon 3 to 6, into the psp72 vector. Following XbaI digestion, part of exon 4 and intron 4 was replaced by a cassette containing the mouse Xpa cDNA including the natural 3′ UTR and polyadenylation signal followed by a PGK promoter-driven hygromycin selectable marker and a LoxP site respectively. Next, the SmaI site downstream of the LoxP site was used to introduce a cassette containing a splice acceptor sequence (SA), an ochre stopcodon multiple reading frame insertion (Murfi) linker, a ribosomal entry site (IRES), and a LacZ/GFP fusion reporter gene (as a blunted SalI fragment). The SmaI site in intron 3 was used to insert a blunted XhoI-SalI loxP fragment from pGEM30 (kindly provided by Dr. W. Gu, University of Cologne). This targeting construct, which was designated pIP-Xpa-con, contains homologous arms of 4 kb at the 5′ end and 5 kb at the 3′ end.
The 129Ola-derived ES cell line IB10 was electroporated with NotI linearized pIP-Xpa-con DNA and cultured in gelatin-coated dishes as described before [45]. Hygromycin (Roche, 843555) was added 24 hr after electroporation to a final concentration of 100 µg/ml. Cells were maintained under selection for 7–8 days, after which clones were isolated and expanded in 24-well plates. Genomic DNA from individual hygromycin-resistant clones was digested with EcoRI and analyzed by Southern blotting using a 500 bp DraI fragment (“intron 5/exon 6” probe; obtained from a 7.5 kb PCR fragment spanning exon 5 and 6). EcoRI digested DNA from targeted ES clones was subsequently screened with the hygromycin (cDNA) probe to confirm proper homologous recombination at the 5′ end of the targeting construct.
For the generation of Xpac/− ES cells, we followed the same procedure as described above, except that Xpa−/− ES cells [54] were used. To test the functionality of the loxP sites, Xpac/+ ES cells were electroporated with a purCre plasmid (kindly provided by Dr. M. Jaegle, Erasmus MC) and cultured on gelatin dishes as described. Puromycin (Sigma, P7255) was added 24 hr after electroporation to a final concentration of 100 µg/ml. Cells were maintained under selection for 3 days. Genomic DNA from individual puromycin-resistant clones was digested with EcoRV and analyzed by Southern blotting using a 500 bp PCR fragment of the LacZ gene. Properly targeted IB10 ES clones were karyotyped and cells from two independent clones (selected for the presence of 40 chromosomes) were injected into 3.5-day-old C57BL/6J blastocysts. Male chimeric mice were mated with C57BL/6J females to obtain heterozygote offspring.
Heterozygous males and females were bred to Xpa+/− as well as Csb−/+ animals to ultimately obtain Xpac/−, Xpac/+ and Csb−/−/Xpac/− animals. Genotyping was initially performed by Southern blot analysis of genomic DNA obtained from tail biopsies of 8–10-day-old born pups. A description of PCR-based genotyping methods is given below. Xpac/− and Csb−/−/Xpac/− animals were also interbred with Cag-Cre [65], CamKII-Cre (line L7ag#13) [66], [67], and L7-Cre (line L7Cre-2, [71]) Cre-recombinase transgenic mice, which were kindly provided by A. de Wit (ErasmusMC), S. Zeitlin (Columbia University), and J.J. Barski (Max-Planck-Institute of Neurobiology, Martinsried, Germany), respectively.
Primary mouse embryonic fibroblasts from the various single and double mutant mouse models (three independent lines per genotype) were isolated from day 13.5 embryos and cultured as described before [83].
Mice and cells were genotyped by PCR for the wild-type and (conditional) mutant Xpa or Csb alleles using a primer mix that (per genotype) amplifies both the wild-type and targeted alleles in a single reaction [41]. The presence or absence of the conditional Xpa allele was detected by PCR using primers XpaFex3 (5′-TTT GAT CTG CCA ACG TGT G-3′) and XpaRex4 (5′-GCT TCG CTT CTG TCT TGG T-3′). The presence or absence of the Cre transgene was detected by PCR using primers 5′-GCA CGT TCA CCG GCA TCA AC-3′ and 5′-CGA TGC AAC GAG TGA TGA GGT TC-3′. Both products were amplified with the same PCR program: 5 min. 93°C, 1 min. 93°C, 1 min. 58°C, 2.5 min. 72°C (35 cycles of the latter three steps), 5 min. 72°C.
LacZ staining of embryos and cells
Cells or embryos were fixed for 30 minutes at 4°C in a buffer containing 1% paraformaldehyde, and subsequently washed 3×15 minutes with PBS/0.01% NP40. Cells or embryos were stained overnight at 37°C in dark in a staining solution containing 3.1 mM K3Fe(CN)6, 3.1 mM K4Fe(CN)6, 0.15 M NaCl, 1 mM MgCl2 and 1 mg/ml X-gal (Roche Applied Sciences, USA, IN). For tissues, the same procedure was used, except that the fixation time was extended to 3 hours.
DNA repair assays
Seeded cultures at a density of 1000 spontaneously immortalized MEFs on a 6 cm dish were exposed to different doses of UV-C (254 nm, Philips TUV lamp). The cells were allowed to grow for another 7 days after which the resulting clones were fixed, stained and counted. For each independent cell line, the amount of surviving clones at each dose of UV, 3 dishes per dose, was calculated as the percentage of clones on the plate without UV.
Behavioral assays
For the open field test, animals were placed for 30 min in a square (26×26×26 cm) open field box, equipped with photobeam sensors (TruScan E63 10–12, Coulbourn Instruments), and attached to a computer to record the following ambulatory parameters: total distance, center distance, total move time, center time and corner time. Each test session lasted 30 minutes, and data were collected in 5 minute intervals. The anxiety ratio was calculated by dividing center distance by total distance. Rotarod analyses were performed as described previously [84].
Immunohistochemical and histopathological procedures
Mice were anesthetized with pentobarbital and perfused transcardially with 4% paraformaldehyde, and brains were dissected out, weighed, and postfixed overnight in 4% paraformaldehyde at 4°C. For standard histological analyses brains were paraffin-embedded, sectioned at 4 µm and stained with haematoxylin/eosin solution. For other staining procedures brain specimen were embedded in gelatin blocks [81] and sectioned at 40 µm with a freezing microtome. Sections were processed, free floating, using immunofluorescence or a standard avidin-biotin–immunoperoxidase complex method (ABC; Vector Laboratories) with diaminobenzidine (0.05%) as the chromogen. In addition, a selected number of frozen sections were processed for a silver staining procedure that selectively labels dying neurons and their processes [81].
Immunoperoxidase-stained sections were analyzed and photographed using a Leica (Nussloch, Germany) DM-RB microscope and a Leica DC300 digital camera. Sections stained for immunofluorescence were analyzed with a Zeiss (Oberkochen, Germany) LSM 510 confocal laser scanning microscope using 40x/1.3 and 63x/1.4 oil-immersion objectives.
Primary antibodies reported in this study are as follows: mouse anti-APC (Calbiochem, clone CC-1, 1∶2000); rabbit anti-activating transcription factor 3 (ATF3; Santa Cruz Biotechnology, Santa Cruz, 1∶1000); rabbit anti-cleaved caspase 3 (Asp175; Cell Signaling Technology, 1∶200); mouse anti-calbindin (Sigma, clone CB-955, 1∶10000); rabbit anti-GFAP (DAKO, 1∶5000); mouse anti-GFAP (Sigma, clone G-A-5, 1∶20000); rabbit anti-HSP25 (Stressgen, 1∶7000); rabbit anti-Iba1 (WAKO Chemicals, 1∶ 2000); rat anti-Mac2 (Cedarlane, 1∶2000); mouse anti-MAP2 (Millipore, clone AP20, 1∶200); rat anti-myelin basic protein (Millipore, MAB386, 1∶500); mouse anti-NeuN (Millipore MAB377, 1∶2000); rabbit anti-neurofilament-H (Millipore, 1∶2000); rabbit anti-olig2 (IBL, 1∶2000); rabbit anti-p53 (Leica, 1∶2000); mouse anti-S100B (Sigma, clone 1B2, 1∶2000); and guinea pig anti-VGLUT1 (Millipore, 1∶2000). For avidin-biotin–peroxidase immunocytochemistry biotinylated secondary antibodies from Vector Laboratories (Burlingame, CA) diluted 1∶200 were used. FITC-, cyanine 3 (Cy3)-, and Cy5-conjugated secondary antibodies raised in donkey (Jackson ImmunoResearch, West Grove, PA) diluted at 1∶200 were used for immunofluorescence.
Immunoperoxidase-stained sections were analyzed and photographed using a Leica DM-RB microscope and a Leica DC300 digital camera. To determine the relative staining intensity of GFAP staining, sections were photographed using a 5× objective, and optical densities were determined from TIFF files using MetaMorph 4.6 image analysis software. Optical densities determined in rectangular areas of 200×250 µm. To minimize variability resulting from the staining procedure this analysis was performed with sections stained in a single immunostaining session.
Statistical analyses
Statistical analyses were done with GraphPad Prism software (San Diego, USA). Means from different age groups, and different transgenic mouse lines were compared using one-way-ANOVA with Tukey's post tests.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HoeijmakersJH 2009 DNA damage, aging, and cancer. N Engl J Med 361 1475 1485
2. NouspikelT 2007 DNA repair in differentiated cells: some new answers to old questions. Neuroscience 145 1213 1221
3. RassUAhelIWestSC 2007 Defective DNA repair and neurodegenerative disease. Cell 130 991 1004
4. FriedbergECAguileraAGellertMHanawaltPCHaysJB 2006 DNA repair: from molecular mechanism to human disease. DNA Repair (Amst) 5 986 996
5. McKinnonPJ 2009 DNA repair deficiency and neurological disease. Nat Rev Neurosci 10 100 112
6. HoeijmakersJH 2001 Genome maintenance mechanisms for preventing cancer. Nature 411 366 374
7. HanawaltPCSpivakG 2008 Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol 9 958 970
8. CleaverJELamETRevetI 2009 Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat Rev Genet 10 756 768
9. StevnsnerTMuftuogluMAamannMDBohrVA 2008 The role of Cockayne Syndrome group B (CSB) protein in base excision repair and aging. Mech Ageing Dev 129 441 448
10. MuftuogluMde Souza-PintoNCDoganAAamannMStevnsnerT 2009 Cockayne Syndrome Group B Protein Stimulates Repair of Formamidopyrimidines by NEIL1 DNA Glycosylase. J Biol Chem 284 9270 9279
11. SoufirNGedCBourillonAAusterlitzFCheminC 2010 A prevalent mutation with founder effect in xeroderma pigmentosum group C from north Africa. J Invest Dermatol 130 1537 1542
12. KhanSGOhKSEmmertSImotoKTamuraD 2009 XPC initiation codon mutation in xeroderma pigmentosum patients with and without neurological symptoms. DNA Repair (Amst) 8 114 125
13. FousteriMVermeulenWvan ZeelandAAMullendersLH 2006 Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol Cell 23 471 482
14. KraemerKHPatronasNJSchiffmannRBrooksBPTamuraD 2007 Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship. Neuroscience 145 1388 1396
15. NanceMABerrySA 1992 Cockayne syndrome: review of 140 cases. Am J Med Genet 42 68 84
16. LaugelVDallozCDurandMSauvanaudFKristensenU 2010 Mutation update for the CSB/ERCC6 and CSA/ERCC8 genes involved in Cockayne syndrome. Hum Mutat 31 113 126
17. MimakiTItohNAbeJTagawaTSatoK 1986 Neurological manifestations in xeroderma pigmentosum. Ann Neurol 20 70 75
18. KraemerKHLeeMMScottoJ 1987 Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol 123 241 250
19. AnttinenAKouluLNikoskelainenEPortinRKurkiT 2008 Neurological symptoms and natural course of xeroderma pigmentosum. Brain 131 1979 1989
20. de WaardHde WitJAndressooJOvan OostromCTRiisB 2004 Different effects of CSA and CSB deficiency on sensitivity to oxidative DNA damage. Mol Cell Biol 24 7941 7948
21. NardoTOnedaRSpivakGVazBMortierL 2009 A UV-sensitive syndrome patient with a specific CSA mutation reveals separable roles for CSA in response to UV and oxidative DNA damage. Proc Natl Acad Sci U S A
22. BrooksPJChengTFCooperL 2008 Do all of the neurologic diseases in patients with DNA repair gene mutations result from the accumulation of DNA damage? DNA Repair (Amst) 7 834 848
23. LakeRJGeykoAHemashettarGZhaoYFanHY 2010 UV-induced association of the CSB remodeling protein with chromatin requires ATP-dependent relief of N-terminal autorepression. Mol Cell 37 235 246
24. ScharerOD 2008 Hot topics in DNA repair: the molecular basis for different disease states caused by mutations in TFIIH and XPG. DNA Repair (Amst) 7 339 344
25. ItoSKuraokaIChymkowitchPCompeETakedachiA 2007 XPG stabilizes TFIIH, allowing transactivation of nuclear receptors: implications for Cockayne syndrome in XP-G/CS patients. Mol Cell 26 231 243
26. ShiomiNMoriMKitoSHaradaYNTanakaK 2005 Severe growth retardation and short life span of double-mutant mice lacking Xpa and exon 15 of Xpg. DNA Repair (Amst) 4 351 357
27. LehmannAR 2008 XPD structure reveals its secrets. DNA Repair (Amst) 7 1912 1915
28. AndressooJOJansJde WitJCoinFHoogstratenD 2006 Rescue of progeria in trichothiodystrophy by homozygous lethal Xpd alleles. PLoS Biol 4 e322 doi:10.1371/journal.pbio.0040322
29. AndressooJOWeedaGde WitJMitchellJRBeemsRB 2009 An Xpb mouse model for combined xeroderma pigmentosum and cockayne syndrome reveals progeroid features upon further attenuation of DNA repair. Mol Cell Biol 29 1276 1290
30. CoinFOksenychVEglyJM 2007 Distinct roles for the XPB/p52 and XPD/p44 subcomplexes of TFIIH in damaged DNA opening during nucleotide excision repair. Mol Cell 26 245 256
31. StefaniniMBottaELanzafameMOrioliD 2010 Trichothiodystrophy: from basic mechanisms to clinical implications. DNA Repair (Amst) 9 2 10
32. ScharerOD 2008 XPG: its products and biological roles. Adv Exp Med Biol 637 83 92
33. AndrewsADBarrettSFRobbinsJH 1976 Relation of D.N.A. repair processes to pathological ageing of the nervous system in xeroderma pigmentosum. Lancet 1 1318 1320
34. MaedaTSatoKMinamiHTaguchiHYoshikawaK 1995 Chronological difference in walking impairment among Japanese group A xeroderma pigmentosum (XP-A) patients with various combinations of mutation sites. Clin Genet 48 225 231
35. ItohMHayashiMShiodaKMinagawaMIsaF 1999 Neurodegeneration in hereditary nucleotide repair disorders. Brain Dev 21 326 333
36. MimakiTNittaMSaijoMTachiNMinamiR 1996 Truncated XPA protein detected in atypical group A xeroderma pigmentosum. Acta Paediatr 85 511 513
37. HashimotoSSugaTKudoEIhnHUchinoM 2008 Adult-onset neurological degeneration in a patient with Cockayne syndrome and a null mutation in the CSB gene. J Invest Dermatol 128 1597 1599
38. RapinIWeidenheimKLindenbaumYRosenbaumPMerchantSN 2006 Cockayne syndrome in adults: review with clinical and pathologic study of a new case. J Child Neurol 21 991 1006
39. SpivakGHanawaltPC 2006 Host cell reactivation of plasmids containing oxidative DNA lesions is defective in Cockayne syndrome but normal in UV-sensitive syndrome fibroblasts. DNA Repair (Amst) 5 13 22
40. NiedernhoferLJ 2008 Nucleotide excision repair deficient mouse models and neurological disease. DNA Repair (Amst) 7 1180 1189
41. de VriesAvan OostromCTHofhuisFMDortantPMBergRJ 1995 Increased susceptibility to ultraviolet-B and carcinogens of mice lacking the DNA excision repair gene XPA. Nature 377 169 173
42. NakaneHTakeuchiSYubaSSaijoMNakatsuY 1995 High incidence of ultraviolet-B-or chemical-carcinogen-induced skin tumours in mice lacking the xeroderma pigmentosum group A gene. Nature 377 165 168
43. MelisJPWijnhovenSWBeemsRBRoodbergenMvan den BergJ 2008 Mouse models for xeroderma pigmentosum group A and group C show divergent cancer phenotypes. Cancer Res 68 1347 1353
44. NakaneHHirotaSBrooksPJNakabeppuYNakatsuY 2008 Impaired spermatogenesis and elevated spontaneous tumorigenesis in xeroderma pigmentosum group A gene (Xpa)-deficient mice. DNA Repair (Amst) 7 1938 1950
45. van der HorstGTvan SteegHBergRJvan GoolAJde WitJ 1997 Defective transcription-coupled repair in Cockayne syndrome B mice is associated with skin cancer predisposition. Cell 89 425 435
46. van der HorstGTMeiraLGorgelsTGde WitJVelasco-MiguelS 2002 UVB radiation-induced cancer predisposition in Cockayne syndrome group A (Csa) mutant mice. DNA Repair (Amst) 1 143 157
47. CheoDLRuvenHJMeiraLBHammerREBurnsDK 1997 Characterization of defective nucleotide excision repair in XPC mutant mice. Mutat Res 374 1 9
48. GorgelsTGvan der PluijmIBrandtRMGarinisGAvan SteegH 2007 Retinal degeneration and ionizing radiation hypersensitivity in a mouse model for Cockayne syndrome. Mol Cell Biol 27 1433 1441
49. AndressooJOMitchellJRde WitJHoogstratenDVolkerM 2006 An Xpd mouse model for the combined xeroderma pigmentosum/Cockayne syndrome exhibiting both cancer predisposition and segmental progeria. Cancer Cell 10 121 132
50. de BoerJde WitJvan SteegHBergRJMorreauH 1998 A mouse model for the basal transcription/DNA repair syndrome trichothiodystrophy. Mol Cell 1 981 990
51. de BoerJvan SteegHBergRJGarssenJde WitJ 1999 Mouse model for the DNA repair/basal transcription disorder trichothiodystrophy reveals cancer predisposition. Cancer Res 59 3489 3494
52. WijnhovenSWBeemsRBRoodbergenMvan den BergJLohmanPH 2005 Accelerated aging pathology in ad libitum fed Xpd(TTD) mice is accompanied by features suggestive of caloric restriction. DNA Repair (Amst) 4 1314 1324
53. DolleMEBusuttilRAGarciaAMWijnhovenSvan DrunenE 2006 Increased genomic instability is not a prerequisite for shortened lifespan in DNA repair deficient mice. Mutat Res 596 22 35
54. de WaardHde WitJGorgelsTGvan den AardwegGAndressooJO 2003 Cell type-specific hypersensitivity to oxidative damage in CSB and XPA mice. DNA Repair (Amst) 2 13 25
55. de WaardHSonneveldEde WitJEsveldt-van LangeRHoeijmakersJH 2008 Cell-type-specific consequences of nucleotide excision repair deficiencies: Embryonic stem cells versus fibroblasts. DNA Repair (Amst) 7 1659 1669
56. WijnhovenSWHoogervorstEMde WaardHvan der HorstGTvan SteegH 2007 Tissue specific mutagenic and carcinogenic responses in NER defective mouse models. Mutat Res 614 77 94
57. LevineAJHuWFengZ 2006 The P53 pathway: what questions remain to be explored? Cell Death Differ 13 1027 1036
58. RotshenkerSReichertFGitikMHaklaiRElad-SfadiaG 2008 Galectin-3/MAC-2, Ras and PI3K activate complement receptor-3 and scavenger receptor-AI/II mediated myelin phagocytosis in microglia. Glia 56 1607 1613
59. StreitWJWalterSAPennellNA 1999 Reactive microgliosis. Prog Neurobiol 57 563 581
60. IwakiTIwakiATateishiJSakakiYGoldmanJE 1993 Alpha B-crystallin and 27-kd heat shock protein are regulated by stress conditions in the central nervous system and accumulate in Rosenthal fibers. Am J Pathol 143 487 495
61. LiHYuanJ 1999 Deciphering the pathways of life and death. Curr Opin Cell Biol 11 261 266
62. MuraiMEnokidoYInamuraNYoshinoMNakatsuY 2001 Early postnatal ataxia and abnormal cerebellar development in mice lacking Xeroderma pigmentosum Group A and Cockayne syndrome Group B DNA repair genes. Proc Natl Acad Sci U S A 98 13379 13384
63. van der PluijmIGarinisGABrandtRMGorgelsTGWijnhovenSW 2007 Impaired genome maintenance suppresses the growth hormone–insulin-like growth factor 1 axis in mice with Cockayne syndrome. PLoS Biol 5 e2 doi:10.1371/journal.pbio.0050002
64. LaposaRRHuangEJCleaverJE 2007 Increased apoptosis, p53 up-regulation, and cerebellar neuronal degeneration in repair-deficient Cockayne syndrome mice. Proc Natl Acad Sci U S A 104 1389 1394
65. SakaiKMiyazakiJ 1997 A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochem Biophys Res Commun 237 318 324
66. DragatsisIZeitlinS 2000 CaMKIIalpha-Cre transgene expression and recombination patterns in the mouse brain. Genesis 26 133 135
67. FukuiHDiazFGarciaSMoraesCT 2007 Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 104 14163 14168
68. HolmesAYangRJCrawleyJN 2002 Evaluation of an anxiety-related phenotype in galanin overexpressing transgenic mice. J Mol Neurosci 18 151 165
69. FanFJinSAmundsonSATongTFanW 2002 ATF3 induction following DNA damage is regulated by distinct signaling pathways and over-expression of ATF3 protein suppresses cells growth. Oncogene 21 7488 7496
70. TurchiLFarehMAberdamEKitajimaSSimpsonF 2009 ATF3 and p15PAF are novel gatekeepers of genomic integrity upon UV stress. Cell Death Differ 16 728 737
71. BarskiJJDethleffsenKMeyerM 2000 Cre recombinase expression in cerebellar Purkinje cells. Genesis 28 93 98
72. SofferDGrotskyHWRapinISuzukiK 1979 Cockayne syndrome: unusual neuropathological findings and review of the literature. Ann Neurol 6 340 348
73. LeechRWBrumbackRAMillerRHOtsukaFTaroneRE 1985 Cockayne syndrome: clinicopathologic and tissue culture studies of affected siblings. J Neuropathol Exp Neurol 44 507 519
74. RothKA 2001 Caspases, apoptosis, and Alzheimer disease: causation, correlation, and confusion. J Neuropathol Exp Neurol 60 829 838
75. GueganCPrzedborskiS 2003 Programmed cell death in amyotrophic lateral sclerosis. J Clin Invest 111 153 161
76. KohjiTHayashiMShiodaKMinagawaMMorimatsuY 1998 Cerebellar neurodegeneration in human hereditary DNA repair disorders. Neurosci Lett 243 133 136
77. EnokidoYInamuraNArakiTSatohTNakaneH 1997 Loss of the xeroderma pigmentosum group A gene (XPA) enhances apoptosis of cultured cerebellar neurons induced by UV but not by low-K+ medium. J Neurochem 69 246 251
78. DzagnidzeAKatsaravaZMakhalovaJLiedertBYoonMS 2007 Repair capacity for platinum-DNA adducts determines the severity of cisplatin-induced peripheral neuropathy. J Neurosci 27 9451 9457
79. BrooksPJ 2008 The 8,5′-cyclopurine-2′-deoxynucleosides: candidate neurodegenerative DNA lesions in xeroderma pigmentosum, and unique probes of transcription and nucleotide excision repair. DNA Repair (Amst) 7 1168 1179
80. NouspikelT 2008 Nucleotide excision repair and neurological diseases. DNA Repair (Amst) 7 1155 1167
81. de WaardMCvan der PluijmIZuiderveen BorgesiusNComleyLHHaasdijkED 2010 Age-related motor neuron degeneration in DNA repair-deficient Ercc1 mice. Acta Neuropathol 120 461 475
82. Pastoriza-GallegoMArmierJSarasinA 2007 Transcription through 8-oxoguanine in DNA repair-proficient and Csb(−)/Ogg1(−) DNA repair-deficient mouse embryonic fibroblasts is dependent upon promoter strength and sequence context. Mutagenesis 22 343 351
83. NgJMVrielingHSugasawaKOomsMPGrootegoedJA 2002 Developmental defects and male sterility in mice lacking the ubiquitin-like DNA repair gene mHR23B. Mol Cell Biol 22 1233 1245
84. KadotaniHHiranoTMasugiMNakamuraKNakaoK 1996 Motor discoordination results from combined gene disruption of the NMDA receptor NR2A and NR2C subunits, but not from single disruption of the NR2A or NR2C subunit. J Neurosci 16 7859 7867
85. PaxinosGFranklinKBJ 2001 The mouse brain in stereotaxic coordinates London Academic Press
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 12
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Targeted Proteolysis of Plectin Isoform 1a Accounts for Hemidesmosome Dysfunction in Mice Mimicking the Dominant Skin Blistering Disease EBS-Ogna
- The RNA Silencing Enzyme RNA Polymerase V Is Required for Plant Immunity
- The FGFR4-G388R Polymorphism Promotes Mitochondrial STAT3 Serine Phosphorylation to Facilitate Pituitary Growth Hormone Cell Tumorigenesis
- Target Site Recognition by a Diversity-Generating Retroelement
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy