
Differentiation of Zebrafish Melanophores Depends on Transcription Factors AP2 Alpha and AP2 Epsilon
A model of the gene-regulatory-network (GRN), governing growth, survival, and differentiation of melanocytes, has emerged from studies of mouse coat color mutants and melanoma cell lines. In this model, Transcription Factor Activator Protein 2 alpha (TFAP2A) contributes to melanocyte development by activating expression of the gene encoding the receptor tyrosine kinase Kit. Next, ligand-bound Kit stimulates a pathway activating transcription factor Microphthalmia (Mitf), which promotes differentiation and survival of melanocytes by activating expression of Tyrosinase family members, Bcl2, and other genes. The model predicts that in both Tfap2a and Kit null mutants there will be a phenotype of reduced melanocytes and that, because Tfap2a acts upstream of Kit, this phenotype will be more severe, or at least as severe as, in Tfap2a null mutants in comparison to Kit null mutants. Unexpectedly, this is not the case in zebrafish or mouse. Because many Tfap2 family members have identical DNA–binding specificity, we reasoned that another Tfap2 family member may work redundantly with Tfap2a in promoting Kit expression. We report that tfap2e is expressed in melanoblasts and melanophores in zebrafish embryos and that its orthologue, TFAP2E, is expressed in human melanocytes. We provide evidence that Tfap2e functions redundantly with Tfap2a to maintain kita expression in zebrafish embryonic melanophores. Further, we show that, in contrast to in kita mutants where embryonic melanophores appear to differentiate normally, in tfap2a/e doubly-deficient embryonic melanophores are small and under-melanized, although they retain expression of mitfa. Interestingly, forcing expression of mitfa in tfap2a/e doubly-deficient embryos partially restores melanophore differentiation. These findings reveal that Tfap2 activity, mediated redundantly by Tfap2a and Tfap2e, promotes melanophore differentiation in parallel with Mitf by an effector other than Kit. This work illustrates how analysis of single-gene mutants may fail to identify steps in a GRN that are affected by the redundant activity of related proteins.
Published in the journal:
. PLoS Genet 6(9): e32767. doi:10.1371/journal.pgen.1001122
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001122
Summary
A model of the gene-regulatory-network (GRN), governing growth, survival, and differentiation of melanocytes, has emerged from studies of mouse coat color mutants and melanoma cell lines. In this model, Transcription Factor Activator Protein 2 alpha (TFAP2A) contributes to melanocyte development by activating expression of the gene encoding the receptor tyrosine kinase Kit. Next, ligand-bound Kit stimulates a pathway activating transcription factor Microphthalmia (Mitf), which promotes differentiation and survival of melanocytes by activating expression of Tyrosinase family members, Bcl2, and other genes. The model predicts that in both Tfap2a and Kit null mutants there will be a phenotype of reduced melanocytes and that, because Tfap2a acts upstream of Kit, this phenotype will be more severe, or at least as severe as, in Tfap2a null mutants in comparison to Kit null mutants. Unexpectedly, this is not the case in zebrafish or mouse. Because many Tfap2 family members have identical DNA–binding specificity, we reasoned that another Tfap2 family member may work redundantly with Tfap2a in promoting Kit expression. We report that tfap2e is expressed in melanoblasts and melanophores in zebrafish embryos and that its orthologue, TFAP2E, is expressed in human melanocytes. We provide evidence that Tfap2e functions redundantly with Tfap2a to maintain kita expression in zebrafish embryonic melanophores. Further, we show that, in contrast to in kita mutants where embryonic melanophores appear to differentiate normally, in tfap2a/e doubly-deficient embryonic melanophores are small and under-melanized, although they retain expression of mitfa. Interestingly, forcing expression of mitfa in tfap2a/e doubly-deficient embryos partially restores melanophore differentiation. These findings reveal that Tfap2 activity, mediated redundantly by Tfap2a and Tfap2e, promotes melanophore differentiation in parallel with Mitf by an effector other than Kit. This work illustrates how analysis of single-gene mutants may fail to identify steps in a GRN that are affected by the redundant activity of related proteins.
Introduction
An important participant in the gene-regulatory-network (GRN) that governs the differentiation of melanocytes from neural crest precursors (i.e., the melanocyte GRN) is the class III receptor tyrosine kinase Kit. In mouse embryos, binding of this growth-factor receptor by its ligand, stem cell factor (SCF), promotes the growth, survival, migration, and possibly terminal differentiation of melanocytes [1]. Mouse embryos homozygous for hypomorphic alleles of Kit completely lack melanocytes (embryos homozygous for Kit null alleles die prior to pigmentation) [2]–[6]. While ligand-bound Kit stimulates many signal transduction pathways, its effects on melanocyte growth and differentiation appear to occur via the Ras/Raf/Map Kinase pathway. Activity of this pathway results in phosphorylation of Microphthalmia transcription factor (Mitf); phosphorylation of Mitf regulates its activity and stability [7], [8]. Within melanoblasts, Mitf promotes a) cell-cycle exit, by activating expression of the p21WAF1, a cyclin-dependent kinase inhibitor [9], b) cell survival, by upregulating the expression of BCL2 [10], and c) melanin synthesis, by activating expression of Tyrosinase (Tyr), Tyrosinase-related protein 1 (Tyrp1), and Tyrosinase-related protein 2 (Tyrp2, also known as Dopachrome tautomerase, Dct) [11]–[14]. Thus, Kit signaling is essential for normal melanocyte development, at least in part via its ability to stimulate Mitf activity. Of note, KIT levels are reported to be lower in metastatic melanoma cell lines than in benign nevi, and forced expression of KIT in these cells has been shown to induce apoptosis [15]. These findings highlight the importance of understanding the regulation of Kit expression within the melanocyte lineage.
While there is evidence that the KIT gene is dependent on direct stimulation by the Transcription Factor Activator Protein 2 alpha (TFAP2A) in melanoma, analyses of mutant model organisms indicate a more complex regulatory scenario within embryonic melanocytes. TFAP2A and other members of the TFAP2 family control cell fate specification, cell differentiation, cell survival and cell proliferation within neural crest, skin, breast epithelium, and other embryonic cell types and stem cells [16], [17]. Gel shift experiments showed that TFAP2A can bind an element 1.2 kb upstream of the KIT transcription start site, and expression driven by this enhancer in melanoma cells is lost when the TFAP2 binding sites are deleted [18]. Moreover, forced expression of the TFAP2A DNA binding domain, which presumably unseats endogenous TFAP2A and thus acts as a dominant negative AP2, prevents expression of KIT in these cells [18]. Mice lacking the Tfap2a gene do not live long enough to develop melanocytes, due to failure of body wall closure [19], [20]. However, in embryos with Wnt1-CRE-mediated deletion of Tfap2a specifically within the neural crest, melanocytes are absent from the belly [21]. Interestingly, this phenotype resembles that of heterozygous, not homozygous, Kit loss-of-function mutants, suggesting that loss of Tfap2a leads to a reduction rather than complete loss of Kit expression. Zebrafish have two orthologues of mammalian Kit, known as kita and kitb; only kita is expressed in the melanophore lineage [22]. In kita homozygous null mutants (i.e., kita mutants) relative to their wild-type counterparts, embryonic melanophores are reduced in number by about 40%, migrate less, and eventually undergo apoptosis [23]. In zebrafish tfap2a homozygous null mutants (i.e., tfap2a mutants), kita expression is reduced and embryonic melanophores exhibit reduced migration [24], [25]. However, in contrast to the melanophores in kita mutants, those in tfap2a mutants do not appear to die, at least as long these animals survive [23], [26]. The simplest explanation for this difference is that kita expression in melanophores is initially dependent on tfap2a but later becomes independent of it. How can the dominant negative AP2 block Kit expression while loss of Tfap2a only diminishes or delays it? Because many Tfap2 family members have the same DNA binding affinity, it is possible that another such family member cooperates with Tfap2a to activate Kit expression.
Here we show that Tfap2e, a homolog of Tfap2a with the equivalent DNA binding specificity, is expressed in zebrafish melanoblasts and in cultures of primary human melanocytes. With single and double knockdown studies, we show that while Tfap2e is not required for the development of embryonic melanophores, it functions redundantly with Tfap2a in maintaining kita expression in embryonic melanophores. Interestingly, in contrast to the situation in kita mutants, the melanophores in embryos doubly deficient for tfap2a/e fail to differentiate. These results imply that Tfap2 activity has targets other than kita that are important for melanophore development. We find that forced expression of mitfa partially restores melanophores in embryos lacking tfap2a and tfap2e, implying that the targets of Tfap2a/e function to stimulate Mitfa activity or act in parallel with it. These findings reveal unexpected roles for Tfap2 activity in the melanocyte GRN.
Results
tfap2e is expressed in zebrafish melanoblasts and cultured human melanocytes
To determine if a second Tfap2 family member is expressed in the melanoblast lineage, we identified orthologues of Tfap2b, Tfap2c, Tfap2d, and Tfap2e in a database of expressed sequence tags (www.ensembl.org), amplified partial clones of at least 1 kb from each to make a probe for in situ hybridization, and examined the expression of each in embryos that ranged in stage from 0.5 hours post fertilization (hpf), revealing maternal expression, to 48 hpf. Expression patterns of tfap2b and tfap2c have previously been reported [27], [28]. We did not detect expression of tfap2b, tfap2c, or tfap2d in melanoblasts or melanophores (Figure S1), so we did not pursue these orthologues in the context of melanophore development.
In 8-cell zebrafish embryos, maternal tfap2e transcripts were detected by both in situ hybridization and semi-quantitative RT-PCR (not shown). At 24 hpf, tfap2e expression was detected in several regions of the brain, including presumed olfactory bulb, as in mouse embryos [29], [30] (Figure 1A), and also within dispersed cells in the trunk that we assumed to be a subset of migrating neural crest cells (Figure 1B and 1D). At this stage, tfap2e expression was detectable in early-differentiating melanophores close to the ear (Figure 1C), suggesting that the dispersed, non-melanized cells expressing tfap2e were melanoblasts. To test this possibility, we probed homozygous mitfa null mutant embryos (i.e., mitfab692), which are devoid of melanoblasts [31], and found that tfap2e expression was absent from the dispersed cells in the trunk (Figure 1E-1G). This result was consistent with expression of tfap2e in melanoblasts. However, because mitfa is co-expressed with xdh and fms, two markers of xanthophore precursors [32], it was conceivable that tfap2e was expressed in the xanthophore lineage, in an Mitfa-dependent fashion. To test whether tfap2e is expressed in xanthophores, we processed embryos to simultaneously reveal expression of tfap2e mRNA and Pax7 protein, a marker of the xanthophore lineage [33]. We did not detect overlap of the two signals, which implies that tfap2e is not expressed in xanthophores (Figure 1H). In wild-type embryos at 36 hpf, tfap2e expression was present in the forebrain and presumed optic tectum, and expanded in the hindbrain relative to earlier stages (Figure 1I and 1J). However, at this stage expression was not detected in melanophores (Figure 1K). At 48 hpf, high-level tfap2e expression was also observed in the retina (Figure 1L).

To assess if melanocyte-specific expression of TFAP2E is conserved in humans, we performed quantitative RT-PCR on cDNA generated from various human cell lines. We detected higher levels of TFAP2E message in three independent isolates of primary melanocytes, consistent with microarray data indicating expression of TFAP2E in melanocytes and melanoma cell lines [34]. Expression in melanocytes was 2–10 fold higher than in a keratinocyte cell line, and approximately 50–100 fold higher than in a lymphocyte cell line (Figure 1M). In summary, tfap2e is expressed in zebrafish melanoblasts and in human melanocytes.
In tfap2a mutant embryos, kita expression is reduced in early melanophores but normal at later stages
As discussed in the Introduction, KITA has been reported to be a direct target of TFAP2A, and a dominant negative AP2 variant was found to block KIT expression in cultured cells [18]; however the status of kita expression in tfap2a mutants has not been fully investigated. In zebrafish tfap2a mutants or tfap2a MO-injected embryos at 28 hpf, kita expression in the melanophore lineage is reduced to undetectable levels as assessed by in situ hybridization [24], [25]. However because melanophores undergo cell death in kita mutants but do not do so in tfap2a mutants, it has been proposed that kita is expressed in the melanophore lineage of tfap2a mutants at a later stage [24]. To test this prediction, we crossed heterozygote tfap2a null mutants (i.e., lockjaw, tfap2ats213) and identified homozygous mutant offspring (hereafter, tfap2a mutants) at 28 hpf by virtue of their pigmentation phenotype. We fixed a fraction of these embryos at 28 hpf, and incubated the remainder in water containing phenylthiourea (PTU) to prevent melanin synthesis, until 36 hpf. We then processed all embryos to reveal kita expression. In tfap2a mutants at 28 hpf, kita expression in melanophores was undetectable by in situ hybridization (Figure 2C), as previously reported. However, at 36 hpf, kita expression was clearly visible in cells present in the dorsum of these embryos (Figure 2E). Thus normal kita expression in melanoblasts at 28 hpf is dependent on tfap2a, but later becomes independent of it. To explain these observations we hypothesized that Tfap2e compensates for the loss of Tfap2a and activates kita expression by 36 hpf.

To test whether Tfap2e maintains kita expression in tfap2a mutants, we first assessed tfap2e expression in tfap2a mutants, and found that it was expressed on schedule in migrating neural crest, as in wild-type embryos (Figure S2). Next we injected embryos with a morpholino (MO) targeting the tfap2e exon 3 splice donor site (i.e., tfap2e e3i3 MO) (Figure 2A). To confirm the efficacy of this MO towards its intended target, we harvested RNA from embryos injected with the tfap2e e3i3 MO, generated first-strand cDNA, and performed PCR using primers in exon 1 and exon 4. Sequencing of the major aberrant splice product revealed that the e3i3 MO causes deletion of exon 3 in its entirety, resulting in a frame shift and a severe truncation of the predicted protein that eliminates the DNA binding domain (Figure 2A). By semi-quantitative PCR, this MO appears to inhibit normal splicing of the majority of tfap2e transcripts at 36 hpf, but to act with greatly reduced efficiency at 3 days post fertilization (dpf) (Figure 2A). By 24 hpf, wild-type zebrafish embryos injected with tfap2e e3i3 MO showed evidence of cell death in the central nervous system (CNS), i.e., patches of opacity in the brain and spinal cord, but no other gross morphological defects; possibly this was due to non-specific toxicity of the MO to the embryo. Despite this cell death, the melanophores that developed in such embryos looked normal and were normally distributed (see below and Figure S3). tfap2e e3i3 MO-induced CNS cell death was reduced by co-injection of p53 MO, implying that Tfap2e has a role in cell survival in the CNS, or that the tfap2e e3i3 MO has non-specific toxicity towards the nervous system, which is true of many MOs (Figure S3) [35]. To preserve the morphology of embryos, in all experiments discussed hereafter we have included p53 MO with tfap2e e3i3 MO. Interestingly, in tfap2a mutants injected with the tfap2e e3i3 MO (hereafter, tfap2a/e doubly-deficient embryos), kita was absent from the dorsum at 36 hpf, although kita expression was readily detected in the cloaca and pharyngeal pouches (Figure 2G and not shown). These findings imply that in absence of Tfap2a, Tfap2e promotes kita expression in the melanophore lineage.
Simultaneous reduction of Tfap2a and Tfap2e inhibits melanophore development
Because of the sustained loss of kita expression in tfap2a/e doubly-deficient embryos, we expected that the phenotype in these embryos would be similar to that of kita homozygous null mutants, although perhaps not as severe because MO-mediated inhibition of gene expression is transient and partial; instead, however, we detected a much more severe phenotype. At 36 hpf, compared to the embryonic melanophores in their non-mutant siblings (Figure 3A), those in kita null mutants (i.e., kitab5) (Figure 3B) appeared normally melanized, but were reduced to about 60% of their normal numbers (because of a presumed defect in cell division) and did not migrate as extensively as their wild-type counterparts [23], [36]. In control MO-injected tfap2a mutants (Figure 3C), embryonic melanophores exhibited these same phenotypes. In tfap2e MO-injected sibling embryos (Figure 3D) there was no apparent melanophore phenotype. However, in tfap2a/e doubly-deficient embryos there were far fewer melanophores than present in control MO-injected tfap2a mutant embryos. Compared with control MO-injected tfap2a mutants, tfap2a/e doubly-deficient embryos had fewer pigmented melanophores in the dorsum and almost no visible melanophores on the lateral sides of the trunk or on the yolk sac (Figure 3E); this difference was still apparent at 84 hpf (not shown). In summary, whereas wild-type embryos injected with the tfap2e MO developed normally until at least 4 dpf, tfap2a/e doubly-deficient embryos displayed melanophore defects more severe than those of tfap2a or kita mutants. These findings suggest that Tfap2a and Tfap2e have partially redundant function in zebrafish melanophore development, and that this function exceeds the simple maintenance of kita expression.

To confirm the specificity of the tfap2e e3i3 MO-induced melanophore phenotypes, we co-injected mRNA encoding a glucocorticoid-fused version of Tfpa2a (tfap2aGR), whose nuclear transport is dexamethasone-inducible, or lacZ as a control, into embryos injected with MOs targeting tfap2a, tfap2e, and p53 (hereafter also termed tfap2a/e doubly-deficient embryos). Dexamethasone was added to both groups at 70% epiboly to avoid gastrulation defects caused by tfap2a over-expression [28]. Embryos were then scored for the rescue of under-melanized melanophores, seen in tfap2a/e doubly-deficient embryos, at 36 hpf. We found that tfap2aGR mRNA effectively rescued melanophores in tfap2a/e doubly-deficient embryos, whereas lacZ did not (Figure S4G and S4H). As an alternative approach for testing specificity, we purchased two additional independent tfap2e MOs—one targeting the exon 2 splice donor site (i.e., e2i2 MO) and the other the translation start site of the tfap2e gene (i.e., AUG MO) (Figure 2A). Injection of either the tfap2e e2i2 MO or the tfap2e AUG MO into wild-type embryos had no effect on melanophore development, although both induced some degree of nervous-system cell death. Upon injection of either the tfap2e e2i2 MO or tfap2e AUG MO into embryos derived from tfap2a mutant heterozygous parents, about one fourth of embryos exhibited the melanophore phenotype seen with the tfap2e e3i3 MO (Figure S4A-S4F); co-injection of p53 MO did not alter the melanophore phenotypes although it reduced nervous system cell death (not shown). These multiple tests of specificity strongly argue that the melanophore phenotypes we observe in tfap2e MO-injected embryos result from inhibition of tfap2e expression and not from off target effects.
Inhibition of tfap2e does not further reduce melanophore specification in tfap2a mutants
The reduced number of melanophores in tfap2a/e doubly-deficient embryos relative to tfap2a mutants could reflect a role for Tfap2a/e activity in the specification of melanoblasts or, alternatively, in either survival or differentiation of melanophores. To distinguish among these possibilities, we examined the expression of mitfa, an early marker of the melanoblast and xanthoblast lineages [31], [32]. At 29 hpf, mitfa-expressing cells are visible in the head and trunk of wild-type embryos injected with a control MO (Figure 4A). The number of mitfa-expressing cells is reduced by about half in tfap2a mutant embryos injected with a control MO (Figure 4B); this reduction results at least in part from the absence of kita in such mutants at this stage, because melanophores are reduced by this amount in kita mutants [23], as are mitfa-expressing cells (our unpublished observations). In tfap2e MO-injected, wild-type embryos, the number of mitfa cells is not grossly different from that in control MO-injected, wild-type embryos (Figure 4C). Interestingly, in tfap2a/e doubly-deficient embryos, the number of mitfa-expressing cells did not appear to be further decreased relative to that in control MO-injected tfap2a mutants (Figure 4D). To confirm these impressions, we counted mitfa-expressing cells over the hind yolk (see Materials and Methods) at 24 hpf, and compared the results for tfap2a mutants injected with control MO versus those injected with tfap2e MO; we found no significant difference (See Figure 4 legend for numbers). In addition, we used fluorescence-activated cell sorting (FACS) to count GFP-positive cells in dissociated mitfa:egfp transgenic embryos injected with MOs, and this analysis supported our findings from histology [37]. Thus, GFP-expressing cells were similarly reduced in tfap2a MO-injected and tfap2a/e doubly-deficient mitfa:egfp embryos (i.e., to about 40% of the number in controls), although the number of differentiated melanophores in tfap2a/e doubly-deficient embryos was clearly reduced relative to that in tfap2a MO injected embryos (Figure 4E, histogram). These findings imply that Tfap2 activity, provided by the redundant actions of Tfap2a and Tfap2e, is involved in a step of melanophore development that occurs subsequent to specification of the mitfa-positive lineage.

Tfap2a/e activity is required for melanophore differentiation
To determine which step in melanophore development depends on Tfap2 activity, we analyzed the expression of genes involved in melanophore differentiation: tyr, tyrp1b and dct [12]. In tfap2a mutant embryos at 29 hpf, the number of cells expressing each of these melanophore markers was reduced by about half relative to that in siblings, consistent with the previously described decrease in melanophores in tfap2a mutants (Figure 5A, 5E, 5I and 5C, 5G, 5K) [24], [25]. In tfap2e MO-injected embryos, the number of cells expressing each of these genes appeared to be normal (Figure 5B, 5F, and 5J), while in tfap2a/e doubly-deficient embryos their numbers were further reduced relative to that in tfap2a mutant embryos (Figure 5D, 5H, and 5L). To quantify this effect, we counted cells in embryos processed for in situ hybridization. We discovered that the reduction in gene expression was not equal in all cases. The number of cells expressing dct was most clearly and most consistently reduced in tfap2a/e doubly-deficient embryos, i.e., by approximately 47% relative to the number in tfap2a mutant embryos (Figure 5A-5D, and 5M). The reduction in tyrp1b and tyr expressing cells was more variable, with an average reduction of approximately 30% and 23%, respectively (Figure 5E-5L, and 5M).

The results described above indicate that when the expression of tfap2a and tfap2e is reduced, melanoblasts express mitfa but fail to progress to a stage at which they express normal levels of melanophore differentiation genes, such as dct, tyrp1b, and tyr. To test this model more quantitatively, we injected mitfa:egfp transgenic embryos [37] with either tfap2a MO or both tfap2a MO and tfap2e MO, dissociated them at 29 hpf, sorted and collected GFP-expressing cells, and measured the levels of various transcripts by quantitative RT-PCR (Figure 5N). Using this method, we saw a trend similar to that observed in the histology analysis: in GFP-positive cells sorted from tfap2a/e MO-injected embryos relative to those sorted from tfap2a MO-injected embryos, dct expression was reduced by approximately 45%, tyrp1b expression was reduced by 17%, and unexpectedly, tyr expression was not reduced. Taken together with the cell counts, these results reveal that Tfap2 activity, redundantly provided by Tfap2a and Tfap2e, promotes the differentiation of embryonic melanophores.
Loss of Tfap2a/e activity does not result in a cell-fate switch or early cell death
We tested the possibility that the loss of differentiated melanophores in tfap2a/e doubly-deficient embryos results from a fate switch of melanophores to xanthophores, because mitfa is co-expressed with c-fms, a marker of xanthophore precursors [32]. We injected embryos with a control MO, tfap2a MO, tfap2e MO, or tfap2a/e MOs, and at 36 hpf processed them to reveal expression of anti Pax7 IR, a marker of xanthophores [33] (Figure 6A-6C and not shown). While the numbers of xanthophores in these groups did not differ significantly (Figure 6D), melanophore differentiation was clearly affected in tfap2a/e doubly-deficient embryos. These findings suggest that loss of Tfap2 activity in the melanophore lineage does not result in a cell fate switch.
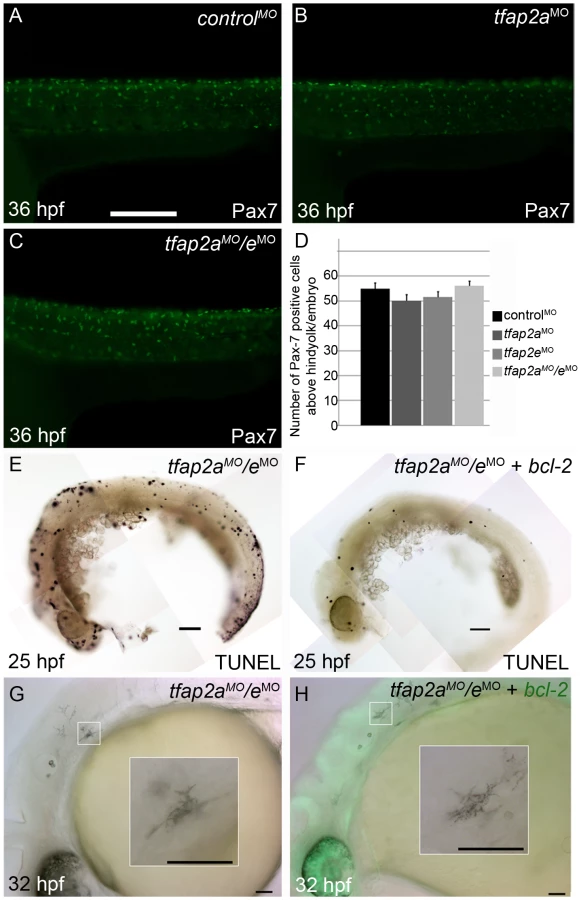
We also assessed whether melanophores in tfap2a/e doubly-deficient embryos undergo cell death, i.e. despite the presence of p53 MO. First, we co-injected embryos with MOs targeting tfap2a and tfap2e and with an mRNA encoding Bcl2, an inhibitor of apoptosis [38]. Injection of bcl2 mRNA reduced the number of cells expressing a marker of programmed cell death in control embryos at 25 hpf (Figure 6E and 6F), but had no effect on the melanophore phenotype in tfap2a/e doubly-deficient embryos (Figure 6G and 6H). Secondly, embryos were incubated in acridine orange (AO), which is taken up by dying cells, from 16 hpf to 30 hpf and assessed for the presence of AO-containing cells in the dorsal neural tube and migratory neural crest. Relative to control MO-injected wild-type embryos, control MO-injected tfap2a mutants had an elevated number of such cells, but these numbers were not detectably increased in tfap2e MO-injected tfap2a mutants (data not shown). These findings suggest that loss of Tfap2 activity in melanophores does not result in either a switch in cell fate specification or promotion of cell death, but more likely in inhibition of normal melanophore differentiation.
Tfap2a/e activity is cell-autonomously required for melanophore differentiation
In tfap2a mutants and MO-injected embryos, embryonic melanophores initially appear somewhat under-melanized [24], [25]. The tfap2a gene is expressed both in skin and neural crest, and we have reported evidence based on transplant studies that Tfap2a has both cell-autonomous and cell non-autonomous effects on melanophore differentiation [25]. Because tfap2e is expressed in melanoblasts but not skin, we assumed that the even poorer differentiation of melanophores in tfap2a/e doubly-deficient embryos is primarily a consequence of a cell autonomous role for Tfap2 activity. To confirm this prediction, we created genetic chimeras by carrying out transplantations at the blastula stage. Specifically, we transplanted cells from 4 hpf wild-type donors, which had been injected with a biotin-dextran as a lineage tracer, into 4 hpf hosts injected with tfap2a/e MO. We then reared the transplanted hosts to 48 hpf, and processed them for biotin staining to reveal the donor-derived cells. Melanophores lacking lineage tracer were indistinguishable from those seen in the untransplanted tfap2a/e MO-injected controls (Figure 7C-7F, arrows), whereas those positive for the lineage tracer were clearly darker, similar to wild-type controls (Figure 7A and 7B), indicating an increase in the level of melanin. In addition, they displayed a more normal morphology (Figure 7E and 7F, arrowheads). These findings indicate that normal melanophores can develop from wild-type cells that are flanked by tfap2a/e-deficient epidermis. This supports a cell-autonomous requirement for Tfap2a/e activity in melanophore differentiation.

Forced mitfa expression partially restores melanophores in tfap2a/e doubly-deficient embryos
Several signals are known to modulate Mitf transactivation activity [39], [40]. If Tfap2a/e is required for the expression of a component of such a signaling pathway, Mitfa activity might be reduced in tfap2a/e doubly-deficient embryos despite levels of mitfa mRNA being similar to those in tfap2a mutants. Alternatively, the Tfap2a/e effector required for melanocyte differentiation might be co-activated by Mitf. In either of these scenarios, forced mitfa expression might rescue melanophore differentiation in tfap2a/e doubly-deficient embryos. We injected tfap2a/e doubly-deficient embryos with a plasmid in which the sox10 promoter drives mitfa expression (sox10:mitfa) [41], and found sox10:mitfa-injection increased the number of tfap2a/e doubly-deficient embryos with differentiated melanophores (compare Figure 8B to Figure 8C, 8D). We observed an increase in the number of darkly-pigmented melanophores in tfap2a/e doubly-deficient embryos injected with sox10:mitfa compared to in tfap2a/e doubly-deficient embryos alone (Figure 8E). We also quantified the mean gray value of single melanophores in these embryos (as a measure of pigment density), within a defined region, using ImageJ software. We found that there was a significant reduction in the pigment density of tfap2a/e doubly-deficient embryo melanophores, compared to control MO-injected embryo melanophores, and that this density was restored in doubly-deficient embryos co-injected with sox10:mitfa (Figure 8F).

Since sox10 is expressed throughout the neural crest, we considered the possibility that sox10:mitfa might induce a conversion of neural crest to the melanoblast lineage, and that if this were to occur in neural crest that expressed another Tfap2 family member, normally differentiated melanophores might emerge in tfap2a/e doubly deficient embryos. However, arguing against this alternative model, we did not detect an increase in the number of melanophores in control-MO injected embryos co-injected with the sox10:mitfa plasmid (Figure 8E). Moreover, in this alternative model, tfap2b is the best candidate Tfap2 family member, as it is expressed in Rohon Beard sensory neurons [27], which are closely related to trunk neural crest [42], [43]. However, we found that even in embryos triply depleted of tfap2a/b/e using MOs, co-injection of sox10:mitfa plasmid elevated the number of normal-looking melanophores (our unpublished observation). Together these observations support the model that over-expression of mitfa can compensate for the role in melanophore differentiation normally played by Tfap2a/e, implying that the effector of Tfap2a/e-type activity necessary for melanophore differentiation acts upstream or in parallel with Mitfa.
Discussion
The phenotype of tfap2a/e double-knockdown embryos reflects multiple roles of Tfap2 activity in the melanophore lineage
Here we have presented two new findings relevant to the gene-regulatory-network (GRN) that governs the differentiation of zebrafish embryonic melanophores. First, kita expression in embryonic melanophores is positively regulated by Tfap2e, at least when Tfap2a levels have been reduced. Expression of tfap2a is present throughout the neural crest starting at the neurula stage, while the expression of tfap2e starts at approximately the time of neural crest delamination and appears to be restricted to melanoblasts [24], [25]. The relative timing of tfap2a and tfap2e expression explains why kita expression (in melanophores) in tfap2a mutants is reduced at 28 hpf, but present at later stages; Tfap2e compensates for the absence of Tfap2a but only after 28 hpf. The presence of TFAP2E expression in human melanocytes suggests that TFAP2A and TFAP2E have redundant or partially redundant function in mammalian melanocytes, as in fish melanophores. If so it would explain the observation, mentioned in the Introduction, that the coat color phenotype in mice with neural crest-specific deletion of Tfap2a is less severe than that of Kit homozygous null mutants [21].
The second unexpected finding is that Tfap2 activity (provided by Tfap2a and Tfap2e) promotes the differentiation of embryonic melanophores. This was revealed by reduced expression of the dct and tyrp1b mRNAs, as well as of melanin—changes that are evident in tfap2a mutants and more pronounced in tfap2a/e doubly-deficient embryos. Does Tfap2 activity also direct neural crest cells to join the melanophore sublineage? There is precedent for such a possibility, because Tfap2 activity provided by Tfap2a and Tfap2c appears to direct ectodermal precursors to join the neural crest lineage [28], [44]. In tfap2a single mutants, neural crest induction appears to occur normally, but mitfa-expressing cells, which are primarily melanoblasts, are reduced in number. This reduction may reflect a role for Tfap2 in melanophore specification or alternatively a reduction of Kita-mediated proliferation of melanoblasts. Whatever the explanation for reduced melanoblasts in tfap2a mutants, simultaneous reduction of tfap2a and tfap2e leads to a further reduction of melanophore numbers without a further reduction of mitfa-expressing cells, arguing Tfap2 promotes differentiation of melanoblasts to melanophores. While a reduction of melanophores without a reduction in mitfa-expressing cells might have been consistent with a cell fate change of melanophores to xanthophores (because markers of melanoblasts and xanthoblasts are briefly co-expressed [32]), xanthophore numbers are equivalent in tfap2a deficient and tfap2a/e doubly-deficient embryos, arguing against such a fate transformation. Does Tfap2 also promote survival of melanophores? We did not detect evidence of cell death of melanophores shortly after their differentiation in tfap2a/e doubly-deficient embryos. We predict that in embryos permanently deprived of both Tfap2a and Tfap2e melanophores would die as a consequence of the absence of Kita. However, because melanophores persist for several days in kita mutants, and this is longer than MOs are effective (see Figure 2A), it will be necessary to isolate a tfap2e mutant to test this prediction. Together these observations reveal that Tfap2 activity has multiple roles in melanophore development, including promoting melanophore differentiation.
Another result that will be important to revisit when a tfap2e mutant is available is the apparent heightened Tfap2-dependence of dct expression relative to tyr expression. Consistent with differential regulation of these related genes, in mice, Dct expression appears prior to Tyr expression, and this has also been suggested to be the case in zebrafish [45], [46]. However, because we knock-down tfap2e expression with an MO, the stronger effect on dct expression relative to on tyr expression may simply reflect loss of MO effectiveness over time. There may be a similar explanation for the inconsistent findings regarding tyr expression between the RNA in situ hybridization and the quantitative RT-PCR analyses. The cell dissociation protocol required for quantitative RT-PCR introduces a delay in the analysis of gene expression relative to that obtained using the RNA in situ hybridization protocol, giving further time for the MO to lose efficacy. Nevertheless, these results reveal that Tfap2 activity, redundantly provided by Tfap2a and Tfap2e, promotes the differentiation of embryonic melanophores.
Tfap2 and Mitfa may co-activate melanophore differentiation genes
How does Tfap2 activity, mediated by Tfap2a and Tfap2e, effect melanophore differentiation? In tfap2a/e doubly-deficient embryos, melanophore differentiation fails but can be rescued by forced expression of mitfa. One model to explain these findings is that Mitfa and Tfap2 normally co-activate genes important for melanophore differentiation, but in the absence of Tfap2, elevated levels of Mitfa can suffice to do so (Figure 9A). Thus, Tfap2 family members may directly activate genes involved in melanin synthesis, such as dct, tyrp1b, and possibly tyr, all of which are known to be Mitfa targets [47]–[49]. Consistent with this possibility, recent studies have identified conserved DNA elements adjacent to the dct and tyrp1b genes that have melanocyte enhancer activity [13], and some of these contain putative Tfap2 binding sites. Simultaneous inhibition of tyrp1a and tyrp1b blocks melanization of zebrafish melanophores, suggesting that tyrp1a/b may partially mediate Tfap2a/e activity within these cells [50]. A variation of this model is that, rather than Tfap2 itself functioning as a co-activator with Mitfa, the protein product of a gene stimulated by Tfap2 does so. For instance, Tfap2 activates expression of estrogen receptor alpha (ERα) [51], [52]. ERα, together with p300, interacts with Mitf to strongly activate the Dct promoter [53].

Tfap2 may indirectly promote Mitfa transactivation activity
It is also possible that the effector of Tfap2 activity is an enzyme that alters the activity, translation, or longevity of the Mitfa protein (Figure 9B). Thus, perhaps mitfa RNA levels are the same in tfap2a deficient vs. tfap2a/e deficient embryos, but Mitfa activity is reduced in the latter. For instance, the Tfap2-effector may be a receptor tyrosine kinase (RTK) whose activity results in posttranslational activation of Mitfa, i.e. similar to a proposed role of Kit [7], [8]. Supporting such a possibility, Kita itself is necessary for differentiation of embryonic melanophores in zebrafish in certain experimental conditions [54] [23]. A variety of RTKs are candidates for the Tfap2 effector in melanophore differentiation, including Erbb3 [55], [56], IGF1R [57], FGF receptor [58], c-Ret [59], and c-MET [60]. Two G-protein coupled receptors, which like RTKs can stimulate the MAP Kinase pathway, are also candidates. First, Endothelin receptor b (Ednrb signaling) promotes melanocyte differentiation in mammals, in part by activating MAP Kinase signaling and Mitfa [61]–[64]. While embryonic melanophores differentiate normally in zebrafish ednrb1 mutants [65], uncharacterized ednrb homologues are present in the zebrafish genome (e.g., on chromosome 9) and may function in embryonic melanophores. Second, Melanocortin 1 receptor (Mc1r) is necessary for normal levels of pigmentation in zebrafish [66] and in mammals [67], and MC1R expression may be directly regulated by TFAP2A, because it has been shown that TFAP2A binds DNA adjacent to the MC1R gene in HeLa cells (chromatin immunoprecipitation results) [68]. Finally, Tfap2 could normally repress expression of an Mitfa phosphatase, alter processing of the mitfa transcript, change Mitfa translation or change Mitfa protein stability. All these scenarios would result in similar mitfa mRNA levels in situ but weaker Mitfa activity when Tfap2 levels are reduced, and would potentially be by-passed by over-expression of mitfa mRNA. The direct targets of Tfap2 in melanocytes are currently under investigation.
Materials and Methods
Fish maintenance
Zebrafish embryos and adults were reared as described previously [69], in the University of Iowa Zebrafish Facility. Embryos were staged by hours or days post fertilization at 28.5°C (hpf or dpf) [70]. Homozygous mutant embryos were generated from heterozygous adults harboring a presumed null allele of tfap2a (lockjaw, tfap2ats213) [26], mitfa (mitfab692) [31], or kita (kitab5) [23], as indicated.
Generation of cDNAs and morpholinos
First-strand cDNA was synthesized from total RNA harvested from embryos at 4 hpf and 24 hpf as described [25]. A 1.4 kb full-length zebrafish tfap2e cDNA was amplified from the wild-type cDNA using the following primers: forward, 5′-GGA TTC ATG TTA GTC CAC TCC TAC TC-3′, reverse, 5′-TTA TTT GCG GTG CTT GAG CT-3′. This cDNA includes the entire open reading frame and was inserted into the pCR4-TOPO vector (Invitrogen, Carlsbad, CA). A 1.3 kb fragment of zebrafish tyrp1b cDNA was amplified from the wild-type 24 hpf cDNA using the following primers: forward, 5′-GAG AGC GGA TGA TAT AAG GAT GTG G-3′, reverse, 5′-GCC CAA TAG GAG CGT TTT CC-3′. This cDNA was inserted into pSC-A vector (Stratagene, La Jolla, CA).
In designing a tfap2e construct in which expression is disrupted, the exon 2 splice donor site and the exon 3 splice donor sites had to be inferred from comparison of the cDNA to the corresponding genomic sequence (http://uswest.ensembl.org/Danio_rerio/Info/Index). MOs complementary to these sites were ordered: tfap2e e2i2 MO, 5′-ATA CAA GAG TGA TTG AAC TCA CCT G-3′; tfap2e e3i3 MO, 5′-CAC ATG CAG ACT CTC ACC TTT CTT G-3′ (Gene Tools, Philomath, OR). In addition, a MO targeting the tfap2e translation start site (AUG MO) was designed, 5′-GCT GGA GTA GGA GTG GAC TAA CAT C-3′. MOs were reconstituted to 5 mg/ml in water and stored at room temperature (25°C). Immediately before use, they were diluted to 0.5 mg/ml in 0.2 M KCl. MOs (4–8 nl of diluted stock) were injected into the yolk underlying the blastomeres of embryos at the 1–4 cell stage. Upon injection of 3 ng or more of either MO, we saw evidence of non-specific toxicity, i.e., patches of opacity in the brain and spinal cord that did not develop when 5 ng of a p53 MO (5′-GCG CCA TTG CTT TGC AAG AAT TG-3′) was injected [71]. To assure strong penetrance while preventing non-specific toxicity, we used 3 ng/embryo of tfap2e e3i3 MO plus 5 ng/embryo of p53 MO to generate tfap2a−/eMO embryos. For double MO experiments (tfap2aMO/tfap2eMO), 3 ng of tfap2e e3i3 MO, 5 ng tfap2a e2i2 MO (5′-GAA ATT GCT TAC CTT TTT TGA TTA C-3′) and 5 ng of p53 MO were injected together. To test the efficacy of the tfap2e MOs, we used a pair of primers flanking a 305 bp fragment between exon 2 and exon 4 of tfap2e for RT-PCR (forward, 5′-CAC CAC GGC CTG GAT GAT ATT-3′; reverse, 5′-AGG ACT CCT CCA AGC AGC GA-3′). Additionally, where noted, a control MO (controlMO) was used for comparison (5′-CCT CTT ACC TCA GTT ACA ATT TAT A-3′).
Generating chimeric embryos
To create genetic chimeras, we injected donor embryos with 5 nl of 1% lysine-fixable biotinylated-dextran, 10,000 MW (Sigma, St. Louis, MO). At the sphere stage (4 hpf), about 100 cells were withdrawn from each donor embryo using a manual-drive syringe fitted with an oil-filled needle (Fine Science Tools, Vancouver, BC), and about 20 cells were inserted into each of several host embryos at the same stage. In placing these cells, we aimed for a position near the animal pole, to target clones to the ectoderm [72]. Host embryos were allowed to develop to 48 hpf, fixed, images taken, and then processed using an ABC kit (Vector Labs, Burlingame, CA) and DAB to reveal biotin as previously described [73], and subsequently photographed.
Analysis of gene expression
The following restriction fragments were used to generate DIG-labeled antisense RNA probes (Roche Diagnostics, Mannheim, Germany) for whole mount in situ hybridization: tfap2e, NotI/T3; tyrp1b, BamHI/T3; dct, EcoRI/T7 [74]; mitfa, EcoRI/T7 [31]. Standard procedures were followed as previously described [75]. For total cell counts, 10–20 embryos were analyzed per group (see figure legend).
For immunohistochemistry, a monoclonal anti-Pax7 antibody [33] was used at a 1∶25 dilution (supernatant obtained from the Developmental Studies Hybridoma Bank at the University of Iowa, USA). The primary antibody and an anti-DIG antibody were added during routine whole mount in situ hybridization. Following development of whole mount in situ hybridization with NBT/BCIP, the embryos were blocked and then incubated with an Alexa-488 conjugated goat-anti-rabbit secondary antibody, as previously described [76]. After several washes, the embryos were mounted in 50% glycerol/PBST, and photographed. Cell counts were performed on ten embryos per group, along the entire length of the hind yolk.
Dissociation of zebrafish embryos and FACS
Live embryos were reared to an appropriate stage, homogenized with a pestle, and dissociated with PBS containing trypsin and EDTA for 30 minutes at 33°C. After dissociation, cells were resuspended in PBS plus 3% fetal bovine serum (FBS). EGFP-positive cells were counted using a Becton Dickinson FACScan. For cell sorting, cells were dissociated as previously described, and subsequently sorted, on a Becton Dickinson FACS DiVa, directly into buffer RLT and β-mercaptoethanol for subsequent RNA isolation (RNeasy Plus Mini Kit, QIAGEN, Valencia, CA). FACScan cell counting, FACS DiVa cell sorting, and data analyses were conducted at the University of Iowa Flow Cytometry Facility.
Quantitative RT-PCR
The isolation and culture of normal melanocytes and keratinocytes was performed as described previously, Mel 1 and Ker [77], [78], Mel 2,3 [79] (see Figure 1M). Total messenger RNA was isolated using an RNeasy Plus Mini Kit (QIAGEN, Valencia, CA), along with on-column DNase digestion according to the manufacturer's instructions. Lymphocytes (Jurkat cells, clone E6-1) were obtained (ATCC, Manassas, VA), and total RNA was isolated using the PerfectPure RNA Kit (following manufacturer's instructions, 5 PRIME Inc., Gaithersburg, MD). RNA concentrations were determined using a NanoDrop spectrophotometer (Thermo Scientific) and diluted to equal concentrations. For complementary DNA (cDNA) reactions, approximately 200 ng of total RNA was added to 0.5 µg random hexamers, plus 2.5 µl of 10 mM dNTPs (Invitrogen; Carlsbad, CA), and brought to 30 µl with nuclease-free water. Reactions were heated to 65°C for 5 minutes, and cooled to 4.0°C for 5 minutes in a PTC-200 Peltier Thermo Cycler (MJ Research; Ramsey, MN). We then added 19 µl of a master mix containing 10 µl of 5x First-Strand buffer (Invitrogen), 5 µl of 0.1 M dithiothreitol, 20 units of RNasin (Promega, Madison, WI), and nuclease-free water to a volume of 19 µl. Reactions were incubated at 25°C for 10 minutes, and then at 37°C for 2 minutes. Then 1 µl of Moloney-murine leukemia virus Reverse Transcriptase (New England Biolabs, Ipswich, MA) or 1 µl nuclease-free water was added to each reaction. Reactions were carried out at 37°C for 2 hours, followed by incubation at 75°C for 15 minutes. PCR reactions (25 µl) were prepared with approximately 10 ng of cDNA, using the SYBR Green kit (Applied Biosystems, Foster City, CA) following the manufacturer's instructions. The following primers were used at a final concentration of 200 nM in separate PCR reactions: human TFAP2E (forward: 5′-AAT GTG ACG CTG CTG ACT TC-3′; reverse: 5′-GGT CCT GAG CCA TCA AGT CT-3′); or human GAPDH (forward: 5′-AGG TCG GAG TCA ACG GAT TTG-3′; reverse: 5′-GTG ATG GCA TGG ACT GTG GT-3′). Quantitative real-time PCR in Low 96-well plates (Bio-Rad, Hercules, CA) was conducted using a Bio-Rad thermal cycler (CFX96 Real-Time PCR Detection System) and following the default protocol. Primers were designed to flank large exon-intron boundaries to avoid the potential amplification of contaminating genomic DNA. Also, RNA samples not reverse-transcribed (-RT) were used as a negative control. The 2ΔΔCT method was used to determine relative levels of gene expression between samples (normalized to GAPDH) [80]. Experiments were performed in triplicate and mean and standard error were calculated. Following real-time PCR, melt-curve analysis was performed to determine reaction specificity. Similar methods were used for qRT-PCR of sorted cells, with the exception that approximately 20 ng of RNA was used for cDNA synthesis. The following primers were used at a final concentration of 200 nM in separate PCR reactions: tyr (forward: 5′-GGA TAC TTC ATG GTG CCC TT-3′; reverse: 5′-TCA GGA ACT CCT GCA CAA AC-3′); tyrp1b (forward: 5′-TAT GAG ACA CTG GGC ACC AT-3′; reverse: 5′-CAC CTG TGC CAT TGA GAA AC-3′); dct (forward: 5′-CCT CGA AGA ACT GGA CAA CA-3′; reverse: 5′ - CAA CAC CAA CAC GAT CAA CA-3′); and β-actin (forward: 5′-CGC GCA GGA GAT GGG AAC C-3′; reverse: 5′-CAA CGG AAA CGC TCA TTG C-3′). Again, the 2ΔΔCT method was used to determine relative levels of gene expression between samples, first normalizing both samples to β-actin, and then comparing relative gene expression levels in tfap2a/e doubly-deficient cells to those in tfap2a deficient cells.
TUNEL staining
Apoptotic cell death was revealed in whole embryos by terminal transferase dUTP nick-end labeling (TUNEL) as described [81]. The terminal transferase reaction was terminated by incubation at 70°C for 30 min, and embryos were processed with anti-FITC-alkaline phosphatase antibody and developed with NBT/BCIP, as for an RNA in situ hybridization.
Rescue experiments
For tfap2aGR mRNA rescue experiments, approximately 5 nL of 0.075 mg/mL tfap2aGR or lacZ encoding mRNA, transcribed in vitro (mMessage mMachine kit, Ambion, Austin, TX) was injected into one of four cells of embryos previously injected with tfap2a/e/p53 MOs (similar concentration as indicated before). Embryos were raised until they reached approximately 75% epiboly, at which point dexamethasone (dissolved in EtOH) was added to the fish water at a final concentration of 40 µM. For DNA rescue experiments, 5 nL of a 0.025 mg/ml plasmid encoding 4.9 Kb of the sox10 promoter driving full length mitfa [41] was injected at the one cell stage, followed by co-injection of various MO combinations (control MO and p53 MO or tfap2aMO,tfapeMO). Embryos were then raised until approximately 36 hpf and fixed in 4% paraformaldehyde overnight. Finally, embryos were rinsed in PBST, mounted in 3% methylcellulose, and photographed.
ImageJ analysis
To analyze the mean gray value of melanophores, embryos were first fixed at the appropriate stage in 4% paraformaldehyde overnight. Embryos were then rinsed in PBST and mounted in 3% methylcellulose, and images of single melanophores were taken near the otic vesicle at 40x. All lighting conditions remained constant throughout image capturing. 6–10 melanophores were imaged per embryo, and 10 embryos were analyzed per group (roughly 70–80 melanophores per group). Images were converted to a 32 bit gray image and then processed using the auto threshold function in ImageJ software (Version 1.40 g, National Institutes of Health, Bethesda, MD), creating an outline of the melanophore being analyzed. After application of the auto threshold function, a selection was created of the pixels highlighted, and a measurement reporting mean gray value for the given area was taken. An inverse of the selection was then created, highlighting the background (area not occupied by the melanophore), and a similar measurement was taken, reporting the mean gray value of the surrounding background. The difference was then calculated between the mean gray value of the melanophore and the surrounding background, resulting in the normalized mean gray value of the melanophore. Averages were then calculated for all melanophores measured per group, and standard deviation was calculated.
Cell counts
For mitfa-positive and TUNEL-positive cell counts, the entire region overlying the hind yolk was counted. For melanophore cell counts in sox10:mitfa rescue experiments, the total number of melanophores in the embryo body (excluding yolk and hind yolk) were counted. Embryos were fixed in 4% paraformaldehyde overnight, washed in PBST, and mounted in 3% methylcellulose for counts. Embryos were mounted and then counted blindly by an independent observer.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Wehrle-HallerB
2003 The role of Kit-ligand in melanocyte development and epidermal homeostasis. Pigment Cell Res 16 287 296
2. CableJ
JacksonIJ
SteelKP
1995 Mutations at the W locus affect survival of neural crest-derived melanocytes in the mouse. Mech Dev 50 139 150
3. GeisslerEN
McFarlandEC
RussellES
1981 Analysis of pleiotropism at the dominant white-spotting (W) locus of the house mouse: a description of ten new W alleles. Genetics 97 337 361
4. MackenzieMA
JordanSA
BuddPS
JacksonIJ
1997 Activation of the receptor tyrosine kinase Kit is required for the proliferation of melanoblasts in the mouse embryo. Dev Biol 192 99 107
5. ReithAD
RottapelR
GiddensE
BradyC
ForresterL
1990 W mutant mice with mild or severe developmental defects contain distinct point mutations in the kinase domain of the c-kit receptor. Genes Dev 4 390 400
6. Wehrle-HallerB
WestonJA
1995 Soluble and cell-bound forms of steel factor activity play distinct roles in melanocyte precursor dispersal and survival on the lateral neural crest migration pathway. Development 121 731 742
7. HemesathTJ
PriceER
TakemotoC
BadalianT
FisherDE
1998 MAP kinase links the transcription factor Microphthalmia to c-Kit signalling in melanocytes. Nature 391 298 301
8. PriceER
DingHF
BadalianT
BhattacharyaS
TakemotoC
1998 Lineage-specific signaling in melanocytes. C-kit stimulation recruits p300/CBP to microphthalmia. J Biol Chem 273 17983 17986
9. CarreiraS
GoodallJ
AksanI
La RoccaSA
GalibertMD
2005 Mitf cooperates with Rb1 and activates p21Cip1 expression to regulate cell cycle progression. Nature 433 764 769
10. McGillGG
HorstmannM
WidlundHR
DuJ
MotyckovaG
2002 Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell 109 707 718
11. GaggioliC
BuscaR
AbbeP
OrtonneJP
BallottiR
2003 Microphthalmia-associated transcription factor (MITF) is required but is not sufficient to induce the expression of melanogenic genes. Pigment Cell Res 16 374 382
12. LinJY
FisherDE
2007 Melanocyte biology and skin pigmentation. Nature 445 843 850
13. MurisierF
GuichardS
BeermannF
2006 A conserved transcriptional enhancer that specifies Tyrp1 expression to melanocytes. Dev Biol 298 644 655
14. ShibaharaS
YasumotoK
AmaeS
UdonoT
WatanabeK
2000 Regulation of pigment cell-specific gene expression by MITF. Pigment Cell Res 13 Suppl 8 98 102
15. HuangS
LucaM
GutmanM
McConkeyDJ
LangleyKE
1996 Enforced c-KIT expression renders highly metastatic human melanoma cells susceptible to stem cell factor-induced apoptosis and inhibits their tumorigenic and metastatic potential. Oncogene 13 2339 2347
16. EckertD
BuhlS
WeberS
JagerR
SchorleH
2005 The AP-2 family of transcription factors. Genome Biol 6 246
17. Hilger-EversheimK
MoserM
SchorleH
BuettnerR
2000 Regulatory roles of AP-2 transcription factors in vertebrate development, apoptosis and cell-cycle control. Gene 260 1 12
18. HuangS
JeanD
LucaM
TainskyMA
Bar-EliM
1998 Loss of AP-2 results in downregulation of c-KIT and enhancement of melanoma tumorigenicity and metastasis. Embo J 17 4358 4369
19. SchorleH
MeierP
BuchertM
JaenischR
MitchellPJ
1996 Transcription factor AP-2 essential for cranial closure and craniofacial development. Nature 381 235 238
20. ZhangJ
Hagopian-DonaldsonS
SerbedzijaG
ElsemoreJ
Plehn-DujowichD
1996 Neural tube, skeletal and body wall defects in mice lacking transcription factor AP-2. Nature 381 238 241
21. BrewerS
FengW
HuangJ
SullivanS
WilliamsT
2004 Wnt1-Cre-mediated deletion of AP-2alpha causes multiple neural crest-related defects. Dev Biol 267 135 152
22. MellgrenEM
JohnsonSL
2005 kitb, a second zebrafish ortholog of mouse Kit. Dev Genes Evol 215 470 477
23. ParichyDM
RawlsJF
PrattSJ
WhitfieldTT
JohnsonSL
1999 Zebrafish sparse corresponds to an orthologue of c-kit and is required for the morphogenesis of a subpopulation of melanocytes, but is not essential for hematopoiesis or primordial germ cell development. Development 126 3425 3436
24. KnightRD
JavidanY
NelsonS
ZhangT
SchillingT
2004 Skeletal and pigment cell defects in the lockjaw mutant reveal multiple roles for zebrafish tfap2a in neural crest development. Dev Dyn 229 87 98
25. O'BrienEK
d'AlenconC
BondeG
LiW
SchoenebeckJ
2004 Transcription factor Ap-2alpha is necessary for development of embryonic melanophores, autonomic neurons and pharyngeal skeleton in zebrafish. Dev Biol 265 246 261
26. KnightRD
NairS
NelsonSS
AfsharA
JavidanY
2003 lockjaw encodes a zebrafish tfap2a required for early neural crest development. Development 130 5755 5768
27. KnightRD
JavidanY
ZhangT
NelsonS
SchillingTF
2005 AP2-dependent signals from the ectoderm regulate craniofacial development in the zebrafish embryo. Development 132 3127 3138
28. LiW
CornellRA
2007 Redundant activities of Tfap2a and Tfap2c are required for neural crest induction and development of other non-neural ectoderm derivatives in zebrafish embryos. Dev Biol 304 338 354
29. FengW
WilliamsT
2003 Cloning and characterization of the mouse AP-2 epsilon gene: a novel family member expressed in the developing olfactory bulb. Mol Cell Neurosci 24 460 475
30. TummalaR
RomanoRA
FuchsE
SinhaS
2003 Molecular cloning and characterization of AP-2 epsilon, a fifth member of the AP-2 family. Gene 321 93 102
31. ListerJA
RobertsonCP
LepageT
JohnsonSL
RaibleDW
1999 nacre encodes a zebrafish microphthalmia-related protein that regulates neural-crest-derived pigment cell fate. Development 126 3757 3767
32. ParichyDM
RansomDG
PawB
ZonLI
JohnsonSL
2000 An orthologue of the kit-related gene fms is required for development of neural crest-derived xanthophores and a subpopulation of adult melanocytes in the zebrafish, Danio rerio. Development 127 3031 3044
33. MinchinJE
HughesSM
2008 Sequential actions of Pax3 and Pax7 drive xanthophore development in zebrafish neural crest. Dev Biol 317 508 522
34. SmithAP
HoekK
BeckerD
2005 Whole-genome expression profiling of the melanoma progression pathway reveals marked molecular differences between nevi/melanoma in situ and advanced-stage melanomas. Cancer Biol Ther 4 1018 1029
35. RobuME
LarsonJD
NaseviciusA
BeiraghiS
BrennerC
2007 p53 activation by knockdown technologies. PLoS Genet 3 e78 doi:10.1371/journal.pgen.0030078
36. EkkerSC
2008 Zinc finger-based knockout punches for zebrafish genes. Zebrafish 5 121 123
37. CurranK
RaibleDW
ListerJA
2009 Foxd3 controls melanophore specification in the zebrafish neural crest by regulation of Mitf. Dev Biol 332 408 417
38. HockenberyD
NunezG
MillimanC
SchreiberRD
KorsmeyerSJ
1990 Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 348 334 336
39. HouL
PavanWJ
2008 Transcriptional and signaling regulation in neural crest stem cell-derived melanocyte development: do all roads lead to Mitf? Cell Res 18 1163 1176
40. MitraD
FisherDE
2009 Transcriptional regulation in melanoma. Hematol Oncol Clin North Am 23 447 465, viii
41. ElworthyS
ListerJA
CarneyTJ
RaibleDW
KelshRN
2003 Transcriptional regulation of mitfa accounts for the sox10 requirement in zebrafish melanophore development. Development 130 2809 2818
42. ArtingerKB
ChitnisAB
MercolaM
DrieverW
1999 Zebrafish narrowminded suggests a genetic link between formation of neural crest and primary sensory neurons. Development 126 3969 3979
43. CornellRA
EisenJS
2002 Delta/Notch signaling promotes formation of zebrafish neural crest by repressing Neurogenin 1 function. Development 129 2639 2648
44. HoffmanTL
JavierAL
CampeauSA
KnightRD
SchillingTF
2007 Tfap2 transcription factors in zebrafish neural crest development and ectodermal evolution. J Exp Zool B Mol Dev Evol 308 679 691
45. HouL
ArnheiterH
PavanWJ
2006 Interspecies difference in the regulation of melanocyte development by SOX10 and MITF. Proc Natl Acad Sci U S A 103 9081 9085
46. SteelKP
DavidsonDR
JacksonIJ
1992 TRP-2/DT, a new early melanoblast marker, shows that steel growth factor (c-kit ligand) is a survival factor. Development 115 1111 1119
47. FangD
TsujiY
SetaluriV
2002 Selective down-regulation of tyrosinase family gene TYRP1 by inhibition of the activity of melanocyte transcription factor, MITF. Nucleic Acids Res 30 3096 3106
48. YasumotoK
TakedaK
SaitoH
WatanabeK
TakahashiK
2002 Microphthalmia-associated transcription factor interacts with LEF-1, a mediator of Wnt signaling. Embo J 21 2703 2714
49. YasumotoK
YokoyamaK
ShibataK
TomitaY
ShibaharaS
1994 Microphthalmia-associated transcription factor as a regulator for melanocyte-specific transcription of the human tyrosinase gene. Mol Cell Biol 14 8058 8070
50. BraaschI
LiedtkeD
VolffJN
SchartlM
2009 Pigmentary function and evolution of tyrp1 gene duplicates in fish. Pigment Cell Melanoma Res 22 839 850
51. BragancaJ
ElorantaJJ
BamforthSD
IbbittJC
HurstHC
2003 Physical and functional interactions among AP-2 transcription factors, p300/CREB-binding protein, and CITED2. J Biol Chem 278 16021 16029
52. McPhersonLA
BaichwalVR
WeigelRJ
1997 Identification of ERF-1 as a member of the AP2 transcription factor family. Proc Natl Acad Sci U S A 94 4342 4347
53. SchwahnDJ
TimchenkoNA
ShibaharaS
MedranoEE
2005 Dynamic regulation of the human dopachrome tautomerase promoter by MITF, ER-alpha and chromatin remodelers during proliferation and senescence of human melanocytes. Pigment Cell Res 18 203 213
54. MellgrenEM
JohnsonSL
2004 A requirement for kit in embryonic zebrafish melanocyte differentiation is revealed by melanoblast delay. Dev Genes Evol 214 493 502
55. BuacK
XuM
CroninJ
WeeraratnaAT
HewittSM
2009 NRG1/ERBB3 signaling in melanocyte development and melanoma: inhibition of differentiation and promotion of proliferation. Pigment Cell Melanoma Res 22 773 784
56. YangJW
LeeEY
KangKW
2006 ErbB2 overexpression in p53-inactivated mammary epithelial cells. FEBS Lett 580 6501 6508
57. YehAH
BohulaEA
MacaulayVM
2006 Human melanoma cells expressing V600E B-RAF are susceptible to IGF1R targeting by small interfering RNAs. Oncogene 25 6574 6581
58. BeckerD
LeePL
RodeckU
HerlynM
1992 Inhibition of the fibroblast growth factor receptor 1 (FGFR-1) gene in human melanocytes and malignant melanomas leads to inhibition of proliferation and signs indicative of differentiation. Oncogene 7 2303 2313
59. KatoM
TakedaK
KawamotoY
TsuzukiT
DaiY
2001 RET tyrosine kinase enhances hair growth in association with promotion of melanogenesis. Oncogene 20 7536 7541
60. HalabanR
RubinJS
FunasakaY
CobbM
BoultonT
1992 Met and hepatocyte growth factor/scatter factor signal transduction in normal melanocytes and melanoma cells. Oncogene 7 2195 2206
61. BaynashAG
HosodaK
GiaidA
RichardsonJA
EmotoN
1994 Interaction of endothelin-3 with endothelin-B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell 79 1277 1285
62. HosodaK
HammerRE
RichardsonJA
BaynashAG
CheungJC
1994 Targeted and natural (piebald-lethal) mutations of endothelin-B receptor gene produce megacolon associated with spotted coat color in mice. Cell 79 1267 1276
63. LevyC
KhaledM
FisherDE
2006 MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol Med 12 406 414
64. ReidK
TurnleyAM
MaxwellGD
KuriharaY
KuriharaH
1996 Multiple roles for endothelin in melanocyte development: regulation of progenitor number and stimulation of differentiation. Development 122 3911 3919
65. ParichyDM
MellgrenEM
RawlsJF
LopesSS
KelshRN
2000 Mutational analysis of endothelin receptor b1 (rose) during neural crest and pigment pattern development in the zebrafish Danio rerio. Dev Biol 227 294 306
66. GrossJB
BorowskyR
TabinCJ
2009 A novel role for Mc1r in the parallel evolution of depigmentation in independent populations of the cavefish Astyanax mexicanus. PLoS Genet 5 e1000326 doi:10.1371/journal.pgen.1000326
67. KadekaroAL
KantoH
KavanaghR
Abdel-MalekZA
2003 Significance of the melanocortin 1 receptor in regulating human melanocyte pigmentation, proliferation, and survival. Ann N Y Acad Sci 994 359 365
68. BirneyE
StamatoyannopoulosJA
DuttaA
GuigoR
GingerasTR
2007 Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447 799 816
69. WesterfieldM
1993 The Zebrafish Book. Eugene, OR University of Oregon Press
70. KimmelCB
BallardWW
KimmelSR
UllmannB
SchillingTF
1995 Stages of embryonic development of the zebrafish. Dev Dyn 203 253 310
71. McNeillMS
PaulsenJ
BondeG
BurnightE
HsuMY
2007 Cell death of melanophores in zebrafish trpm7 mutant embryos depends on melanin synthesis. J Invest Dermatol 127 2020 2030
72. KimmelCB
WargaRM
SchillingTF
1990 Origin and organization of the zebrafish fate map. Development 108 581 594
73. MoensCB
FritzA
1999 Techniques in neural development. Methods Cell Biol 59 253 272
74. KelshRN
SchmidB
EisenJS
2000 Genetic analysis of melanophore development in zebrafish embryos. Dev Biol 225 277 293
75. ThisseC
ThisseB
2008 High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc 3 59 69
76. JekelyG
ArendtD
2007 Cellular resolution expression profiling using confocal detection of NBT/BCIP precipitate by reflection microscopy. Biotechniques 42 751 755
77. HsuMY
ShihDT
MeierFE
Van BelleP
HsuJY
1998 Adenoviral gene transfer of beta3 integrin subunit induces conversion from radial to vertical growth phase in primary human melanoma. Am J Pathol 153 1435 1442
78. HsuMY
WheelockMJ
JohnsonKR
HerlynM
1996 Shifts in cadherin profiles between human normal melanocytes and melanomas. J Investig Dermatol Symp Proc 1 188 194
79. SongX
MosbyN
YangJ
XuA
Abdel-MalekZ
2009 alpha-MSH activates immediate defense responses to UV-induced oxidative stress in human melanocytes. Pigment Cell Melanoma Res 22 809 818
80. DussaultAA
PouliotM
2006 Rapid and simple comparison of messenger RNA levels using real-time PCR. Biol Proced Online 8 1 10
81. ReyesR
HaendelM
GrantD
MelanconE
EisenJS
2004 Slow degeneration of zebrafish Rohon-Beard neurons during programmed cell death. Dev Dyn 229 30 41
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 9
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Synthesizing and Salvaging NAD: Lessons Learned from
- Optimal Strategy for Competence Differentiation in Bacteria
- Long- and Short-Term Selective Forces on Malaria Parasite Genomes
- Identifying Signatures of Natural Selection in Tibetan and Andean Populations Using Dense Genome Scan Data
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy