
An Insect Herbivore Microbiome with High Plant Biomass-Degrading Capacity
Herbivores can gain indirect access to recalcitrant carbon present in plant cell walls through symbiotic associations with lignocellulolytic microbes. A paradigmatic example is the leaf-cutter ant (Tribe: Attini), which uses fresh leaves to cultivate a fungus for food in specialized gardens. Using a combination of sugar composition analyses, metagenomics, and whole-genome sequencing, we reveal that the fungus garden microbiome of leaf-cutter ants is composed of a diverse community of bacteria with high plant biomass-degrading capacity. Comparison of this microbiome's predicted carbohydrate-degrading enzyme profile with other metagenomes shows closest similarity to the bovine rumen, indicating evolutionary convergence of plant biomass degrading potential between two important herbivorous animals. Genomic and physiological characterization of two dominant bacteria in the fungus garden microbiome provides evidence of their capacity to degrade cellulose. Given the recent interest in cellulosic biofuels, understanding how large-scale and rapid plant biomass degradation occurs in a highly evolved insect herbivore is of particular relevance for bioenergy.
Published in the journal:
. PLoS Genet 6(9): e32767. doi:10.1371/journal.pgen.1001129
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001129
Summary
Herbivores can gain indirect access to recalcitrant carbon present in plant cell walls through symbiotic associations with lignocellulolytic microbes. A paradigmatic example is the leaf-cutter ant (Tribe: Attini), which uses fresh leaves to cultivate a fungus for food in specialized gardens. Using a combination of sugar composition analyses, metagenomics, and whole-genome sequencing, we reveal that the fungus garden microbiome of leaf-cutter ants is composed of a diverse community of bacteria with high plant biomass-degrading capacity. Comparison of this microbiome's predicted carbohydrate-degrading enzyme profile with other metagenomes shows closest similarity to the bovine rumen, indicating evolutionary convergence of plant biomass degrading potential between two important herbivorous animals. Genomic and physiological characterization of two dominant bacteria in the fungus garden microbiome provides evidence of their capacity to degrade cellulose. Given the recent interest in cellulosic biofuels, understanding how large-scale and rapid plant biomass degradation occurs in a highly evolved insect herbivore is of particular relevance for bioenergy.
Introduction
Plant cell walls contain the largest reservoirs of organic carbon on Earth [1]. This carbon is largely inaccessible to most organisms, occurring in the form of cellulose, hemicelluloses, and lignin. Certain bacteria and fungi are capable of deconstructing these recalcitrant plant polymers, and thus play a critical role in nutrient cycling in the biosphere. Lignocellulolytic microbes form symbiotic relationships with animals that feed on plant biomass, providing their hosts with access to nutrients in return for a constant supply of plant polymers. Recent microbiome studies have revealed how these communities mediate plant biomass deconstruction in animals, including detritivores [2], ruminants [3], and omnivores [4]–[6]. Here, we characterize the microbiome of an important Neotropical herbivore, the leaf-cutter ant Atta colombica.
Leaf-cutter ants in the genus Atta are one of the most conspicuous and widespread insects in the New World tropics, forming massive colonies composed of millions of workers. Mature colonies forage hundreds of kilograms in leaves each year (Figure 1A), substantially altering forest ecosystems and contributing to nutrient cycling [7]. Leaf-cutter ants do not feed directly on harvested leaves; rather, they use leaf fragments as substrate to cultivate a mutualistic fungus in specialized subterranean gardens (Figure 1B and 1C). The fungus serves as the primary food source for the colony and in return is provided with substrate, protection from competitors, and dispersal through colony founding [7]–[9]. Despite the impact of these ants on tropical ecosystems, and the critical role leaves play in Atta colonies reaching immense sizes, our current understanding of plant biomass deconstruction within fungus gardens is limited.

Results/Discussion
The primary function of leaf-cutter ant fungus gardens is to convert plant biomass into nutrients for the ants: it serves as the ants' external digestive system [10]. Fungus gardens have a clear distinction between the top layer, which retains the green, harvested state of plant leaves; and the bottom layer, which contains mature fungus and partially-degraded plant material. This difference is due to the temporal process of plant biomass transformation by the ants; freshly-harvested leaves are integrated into the garden top, while material at the bottom is removed by the ants and placed into specialized refuse dumps. Plant biomass degradation in the garden is thought to be mediated exclusively by the ants' mutualistic fungus (order: Agaricales), but its recently reported inability to degrade cellulose [11] poses the question as to what plant polymers are degraded in the fungus garden matrix. We sampled the top and bottom layers of fungus gardens from five colonies of Atta colombica leaf-cutter ants in Gamboa, Panama and performed sugar composition analyses. Our quantification of plant biomass polymer content from these layers revealed that crystalline cellulose and sugars representing various plant polysaccharides, such as hemicelluloses, decreased in content from garden top to bottom (Figure 1D and 1E), whereas lignin did not (Figure 1F). Cellulose in particular, had one of the highest percent decreases, dropping by an average content of 30% from the top to the bottom of the garden.
Our finding that certain plant cell wall polymers are consumed in the fungus garden, including cellulose, which is not known to be degraded by the fungal cultivar, suggests that other microbes may be partially responsible for this deconstruction; a prediction consistent with previous reports of cellulase activity of unknown origin within the fungus garden [12], [13]. We explored this possibility by characterizing the fungus garden microbial communities of three A. colombica leaf-cutter ant colonies using near-full length 16S rDNA clone sequencing, short-read SSU rDNA pyrotag sequencing, and whole community metagenome sequencing. A total of 703 and 2,794 near full-length bacterial 16S rDNA sequences were generated for fungus garden top and bottom layers, respectively (Table S1), and short-read pyrotag sequencing of the same samples yielded 8,968 and 11,362 sequences, respectively. PCR using full-length Archaea-specific primers failed to amplify Archaeal 16S rDNA. Community metagenome sequencing of whole fungus gardens using pyrosequencing [14] generated over 401 Mb of sequence (Table S2), and assembly resulted in 155,000 contigs and 200,000 singletons, totaling 130 Mb.
These DNA sequences indicate the presence of a diverse community of bacteria in leaf-cutter ant fungus gardens (Figure 2, Figure S1, Figure S2). Full-length 16S rDNA libraries contained 132 phylotypes (97% sequence identity) from 9 phyla in garden tops (Figure 2A, Table S3), and 197 phylotypes from 8 phyla in garden bottoms (Figure 2B, Table S3). Comparison of the phylogenetic diversity between top and bottom layer samples using UniFrac [15] indicates that the top layer diversity is different from bottom layer diversity (Figure S3). Both top and bottom layers were dominated by phylotypes in the α-proteobacteria, β-proteobacteria, γ-proteobacteria, Actinobacteria, and Bacteroidetes (Figure 2 and Figures S4, S5, S6, S7, and S8), which collectively contributed 80% (117 of 148 phylotypes) and 85% (185 of 217 phylotypes) of the bacterial diversity detected from top and bottom samples, respectively. A comparison of total generated sequences from these phyla further confirms that these phylotypes are abundant, with 92% (645 of 703 clones) and 91% (2540 of 2794 clones) of all sequenced clones belonging to these 5 lineages for top and bottom samples, respectively. Data from 16S rDNA short-read sequences also confirmed these findings, and further revealed rare phylotypes not found in the full-length analysis, including members of the candidate phyla NC10 [16], OP10 [17], and TM6 [18] (Table S4). Bacterial diversity comparisons among colonies and vertical layers revealed a number of consistent phylotypes, the majority of which are γ-proteobacteria (Figure S9, Figure S10, Table S5). Interestingly, the full-length 16S rDNA libraries revealed phylotypes in the Gemmatimonadetes and candidate phylum SPAM [19] (2 phylotypes each; Figure 2A) exclusive in garden tops, whereas phylotypes in the Chloroflexi and candidate phylum TM7 [20] (1 phylotype each; Figure 2B) were only detected in the garden bottoms. The short-read 16S rDNA sequences confirmed these findings (Table S4), suggesting that specific phyla may play specialized roles within vertical layers of the garden.

Our phylotype diversity analyses were further confirmed through community metagenomics, which does not suffer from the PCR bias inherent to 16S rDNA sequencing [21]. Phylogenetic binning of our community metagenome (Table 1 and Table S6) using a number of different approaches including the program PhymmBL [22], indicates that the fungus garden is dominated by γ-proteobacteria (30% of total bacterial sequences), α-proteobacteria (16%), Actinobacteria (9%), δ-proteobacteria (7%), and β-proteobacteria (7%) (Figure S11, Table S7, Text S1). In particular, the most highly represented sequences are from γ-proteobacterial genera in the family Enterobacteriaceae. Our phylogenetic binning analysis also revealed DNA sequences predicted to be derived from insects, fungi, and plants (Figure S12, Table S2, Table S8, Text S1). It is likely that these sequences originate from the ants, their fungal symbiont, and their primary plant feedstuffs, although genome sequences are currently not available for comparison.

To identify how the fungus garden microbial community associated with leaf-cutter ants mediates plant polymer degradation, we performed a carbohydrate-active enzyme (CAZy) [23] characterization of the garden community metagenome. This analysis identified 69 gene modules across 28 families of glycosyl hydrolases, carbohydrate esterases, and polysaccharide lyases (Table 2). In total, 58% of the sequences predicted to code for enzymes putatively involved in plant polymer degradation, including cellulose and hemicellulose, were of bacterial origin. These enzymes include β-mannosidases (GH1), α-galactosidases (GH1, GH4, GH57), and cellulases (β-1,4-glucanase; GH8), suggesting that bacteria are important contributors to plant polymer degradation within leaf-cutter ant fungus gardens.
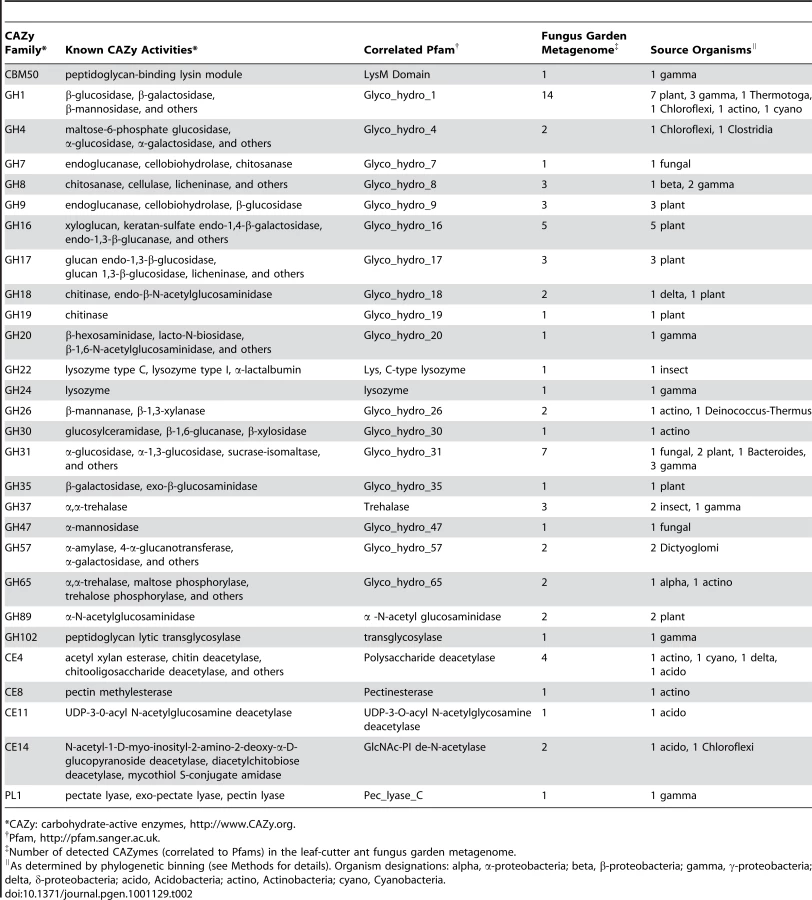
We further explored the underlying mechanisms for plant biomass deconstruction in leaf-cutter ants by comparing the predicted bacterial CAZy profile of the fungus garden metagenome with those of 13 other metagenomes from similar environments that exhibit biomass degradation including animal guts and soil. Clustering analysis of these profiles showed that the fungus garden metagenome groups closest to bovine rumen [3] (Figure 3A). Comparison of shared CAZymes between these two metagenomes revealed enzymes involved in amylose (GH57), galactan (GH4), mannan (GH1), maltose (GH65), pectin (CE8), and xylan (CE4, GH26, GH31) deconstruction (Table S9). Many of these oligosaccharide polymers are components of hemicelluloses and other carbohydrates known to be degraded in both bovine rumen [24] and leaf-cutter ant fungus gardens (Figure 1D). Our CAZy profile clustering reveals the importance and similarity of carbohydrate degradation in these two microbiomes, as these metagenomes did not group together in a similar clustering analysis involving entire gene content (Figure 3B, Figure S13, Table S10, Table S11, Text S2).

Despite leaf-cutter ant fungus gardens and bovine rumen utilizing similar plant biomass, leaves and grass, the microbial communities in these systems are markedly different. In the bovine rumen, the majority of resident bacteria are in the genera Prevotella (phylum Bacteroidetes), Fibrobacter (phylum Fibrobacteres), and Ruminococcus (phylum Firmicutes) [25], whereas leaf-cutter ant fungus gardens primarily contain bacteria from the Proteobacteria (Figure 2, Table S7). The similarity in carbohydrate-degrading potential between these two microbiomes is surprising, and the difference in their bacterial communities suggests that there is evolutionary convergence of enzymatic approaches for the deconstruction of at least some plant polymers. Given that there currently are a limited number of plant biomass degrading metagenomes available for comparison, and that the microbiomes used in our analysis were generated using different sequencing technologies and DNA extraction methods, which we are unable to account for (a difficulty that has been previously noted [26]), it is likely that future work may reveal other microbiomes exhibiting CAZyme profiles more similar to leaf-cutter ant fungus gardens than the bovine rumen. Nevertheless, this analysis provides insights into how two microbial communities that utilize similar plant biomass deconstruct polysaccharides.
To further examine the role of cellulolytic bacteria in leaf-cutter ant fungus gardens we characterized representative isolates of Klebsiella and Pantoea, the two most abundant bacterial genera identified in our community metagenome (Table 1, Table S6). We sequenced the genomes and analyzed the predicted proteomes of Klebsiella variicola At-22 and Pantoea sp. At-9b (Table S12); two isolates obtained from the fungus gardens of Atta cephalotes leaf-cutter ants. Both genomes contained a number of sequences predicted to code for enzymes known to be involved in plant polymer degradation, including cellulases (β-1,4-glucanase; GH8), β-galactosidases (GH2), chitinases (GH18), α-xylosidases (GH31), α-mannosidases (GH47), α-rhamnosidases (GH78), and pectinesterases (CE8) (Table S13, Table S14). Bioassays on pure cultures of these bacteria further revealed their capacity to degrade cellulose (Table S15), suggesting that Klebsiella and Pantoea may play a role as cellulose-degrading symbionts in the gardens of leaf-cutter ants. The symbiosis between these bacteria and leaf-cutter ants is further supported by previous work, which showed they can be consistently isolated from fungus gardens across the diversity and geography of leaf-cutter ants [10]. Indeed, these bacteria appear to be responsible for a significant amount of the nitrogen that is fixed in leaf-cutter fungus gardens; nitrogen that has been shown to be integrated into the ants [10]. Our finding that Klebsiella and Pantoea are the most abundant bacteria present in the gardens of A. colombica; genomic and physiological support for their capacity to degrade cellulose; and previous reports of their contributions to fixed nitrogen in leaf-cutter ant fungus gardens, provides evidence that these bacteria are important symbionts of leaf-cutter ants.
Because our fungus garden metagenome and Klebsiella and Pantoea genomes originate from different Atta species, we examined the potential strain diversity of these symbionts by performing a recruitment analysis [27]. This was done by comparing the community metagenome reads against the microbial genome collection and our Klebsiella and Pantoea genomes (Figure 4A and 4B). Of all 887 genomes analyzed, the genus Pantoea had the highest number of recruited reads (2,064), while Klebsiella had the third highest (1,226) (Table S16). Mapping of the recruited reads specific to Klebsiella variicola At-22 and Pantoea sp. At-9b onto their respective genomes showed markedly different results. For Klebsiella, 90% of the reads recruited to Klebsiella variicola At-22 had sequence identities >98%, indicating that both Atta species possess Klebsiella symbionts with highly-similar genomes (Figure 4A, Figure S14, Table S16). In contrast, only 4% of the Pantoea recruited reads had sequence identities >98% (Figure 4B, Figure S14, Table S16). This supports previous findings that multiple Pantoea species exist in leaf-cutter ant fungus gardens [10]. Further comparison of the two γ-proteobacteria GH8 cellulases identified in the community metagenome (Table 2) against the genomes of Klebsiella variicola At-22 and Pantoea sp. At-9b showed that they matched sequences in these genomes with identities of 99% and 87%, respectively. These data indicate that these two symbionts are present in the fungus gardens of both Atta species where they may play a role as cellulose-degrading symbionts.

Conclusions
Our study presents the first functional metagenomic characterization of the microbiome of an insect herbivore. We reveal that the microbial community within the fungus gardens of leaf-cutter ants contains not only the fungal cultivar, but a diverse assembly of bacteria dominated by γ-proteobacteria in the family Enterobacteriaceae. We further show that these bacteria likely participate in the symbiotic degradation of plant biomass in the fungus garden, indicating that the fungal cultivar is not solely responsible for this process, as has been previously assumed. This suggests a model of plant biomass degradation in the fungus garden that includes both bacteria and the fungal cultivar, and we speculate that persistent cellulose-degrading bacterial symbionts like Klebsiella and Pantoea could work in concert with the fungal cultivar to deconstruct plant polymers.
As an external digestive system, the fungus garden of leaf-cutter ants parallels the role of the gut in other plant biomass degrading systems like bovines and termites. The presence of a bacterial community dominated by Proteobacteria in leaf-cutter ant fungus gardens is similar to the gut microbiota reported for other insect herbivores, suggesting that bacteria in this phylum may be widespread in their association with herbivorous insects [28]–[30]. However, in contrast to other insect herbivores, the external nature of the leaf-cutter ant digestive system removes the restrictions imposed by the physical limitations of internal guts. This feature is likely responsible for these ants achieving massive colony sizes that harvest vast quantities of plant biomass to support their extensive agricultural operations. As a result, these herbivores have a considerable impact on their surrounding ecosystem by contributing significantly to the cycling of carbon and nutrients in the Neotropics. This study of the leaf-cutter ant fungus garden microbiome illustrates how a natural and highly-evolved microbial community deconstructs plant biomass, and may promote the technological goal of converting cellulosic plant biomass into renewable biofuels.
Materials and Methods
Sample Collection
A total of 25 fungus gardens from 5 healthy colonies (5 gardens each) of the leaf-cutter ant Atta colombica were collected at the end of May and beginning of June, 2008. These colonies are located along Pipeline Road in Soberanía National Park, Panama (latitude 9° 7′ 0″ N, longitude 79° 42′ 0″ W) and designated N9, N11, N12, N13, and N14, respectively. Each fungus garden was vertically cross-sectioned into thirds with the top third designated as the “top” of the garden and the bottom third designated as the “bottom” of the garden. All material was frozen and transported back to the University of Wisconsin-Madison where it was stored at −20°C prior to processing.
Sugar Composition and Lignin Analysis
From all 5 colonies (3 gardens per colony), 5 independent samples from fungus garden tops and bottoms of each garden were collected for sugar composition analysis. Thus, a total of 75 fungus garden samples each from the top and bottom were used for this part of our study. This material was tested for crystalline cellulose and hemicellulose (matrix polysaccharide) content as follows.
Cellulose content of fungus garden plant biomass was determined by first washing each sample using Updegraff reagent [31], which removes matrix polysaccharides such as hemicelluloses, pectins and amorphous glucan. The remaining residue, containing only crystalline cellulose, was hydrolyzed using Saeman hydrolysis [32]. The resulting glucose monosaccharides were then quantified with an anthrone colourmetric assay as previously described [32].
For the composition of the matrix polysaccharide content, the following components were tested: arabinose, fucose, galactose, glucose, rhamnose, mannose and xylose. Quantification of these sugars were performed by treating finely ground materials with solvents to remove pigments, proteins, lipids, and DNA from the material as previous described [33]. The residue was de-starched with an amylase treatment, resulting in only cell wall material. This material was then treated with 2M trifluoroacetic acid solubilizing the matrix polysaccharides in form of their monosaccharides, and subsequently derivatized into their corresponding alditol-acetates, which were separated and quantified by GC-MS as previously described [34].
The same set of samples used for sugar composition analysis was also used for lignin content analysis, as previously described [35]. Briefly, all samples were dried to 60°C and ground using a 1-mm cyclone mill and analyzed for total non-lignin organic matter, lignin, and ash (organic and inorganic) content. Total carbohydrate content was assessed through a two-step acid hydrolysis with neutral sugars quantified using GC and uronic acids quantified using colorimetry. Klason lignin was quantified from the ash-free residue from the two-step acid hydrolysis. Ash content was quantified by combustion at 450°C for 18 h and the average µg/mg of material was calculated.
DNA Extraction
Total DNA was extracted in preparation for either 16S rDNA sequencing or community metagenomic sequencing. For 16S rDNA sequencing, a total of 5 gardens each from 3 Atta colombica colonies (N9, N11, and N12) were used. A total of 1 g (wet weight) of fungus garden material was sampled from the top layer of each garden corresponding to each colony, for a combined final weight of 5 g of fungus garden material. Total DNA from this sample was then extracted using a MoBio Power Soil DNA Extraction Kit (MOBIO Laboratories, Carlsbad, CA, USA). The same procedures were performed for all fungus garden bottom layer samples for all 3 colonies.
For community metagenomic sequencing, total community DNA was extracted from 5 whole fungus gardens each from all 5 Atta colombica colonies used in this study. A total of 1 g of fungus garden material was sampled from top, middle, and bottom layers from all fungus gardens and combined to produce a final sample weight of 75 g. This material was then enriched for bacteria using a modification of a previously-described protocol [36]. Briefly, total fungus garden material was buffered in 1X PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4) containing 0.1% Tween and then centrifuged for 5 minutes at 40×g. This resulted in a 3-layer mixture containing leaf-material at the top, fungal mass in the middle, and bacteria at the bottom. The top and middle layers were carefully removed, buffered with 1X PBS containing 0.1% Tween, and washed using the same centrifugation method an additional 3 times. The final mixture was then centrifuged for 30 minutes at 2800×g, re-suspended in 1X PBS containing 0.1% Tween and filtered through a 100 um filter. Total DNA from this resulting sample was then extracted using a Qiagen DNeasy Plant Maxi Kit (Qiagen Sciences, Germantown, MD, USA).
16S rDNA Full-Length and Pyrotag Sequencing
Extracted DNA from fungus gardens was PCR amplified (20 cycles) using full-length universal bacterial (27F [5′-AGA GTT TGA TCC TGG CTC AG-3′] and 1391R [5′ - GAC GGG CRG TGW GTR CA-3′]) and archaeal (4aF [5′ - TCC GGT TGA TCC TGC CRG-3′] and 1391R [5′ - GAC GGG CRG TGW GTR CA-3′]) primers and cloned into the pCR4-TOPO vector (Invitrogen) (See http://my.jgi.doe.gov/general/protocols/SOP_16S18S_rRNA_PCR_Library_Creation.pdf). This was then sequenced using standard Sanger-based capillary sequencing and assembled as previously described [2] (http://www.jgi.doe.gov/sequencing/protocols/prots_production.html). These same samples were then pyrosequenced by first PCR amplifying all samples with prokaryote-specific primers corresponding to the V6-V8 region (1492R [5′ - TAC GCY TAC CTT GTT ACG ACT T - 3′] and 926F [5′ - AAA CTY AAA KGA ATT GAC GG - 3′] fused to 5-base barcodes (reverse primer only) and 454-Titanium adapter sequences) and then sequenced on a Roche 454 FLX GS Titanium pyrosequencer [14]. All 16S rDNA sequences generated in this study are deposited in GenBank with accessions HM545912–HM556124 and HM556125–HM559218 for near full-length 16S rDNA sequences and pyrotagged 16S rDNA sequences, respectively.
Phylogenetic Analysis of 16S rDNA Sequences
Assembled full-length 16S contigs were first compared against the National Center for Biotechnological Information's (NCBI) non-redundant nucleotide (nt) and environmental nucleotide (env_nt) databases (accessed: 05/01/2009) using BLAST [37] to verify that all sequences were bacterial. A small number of eukaryotic 18S sequences belonging to the fungus the ants cultivate, Leucoagaricus gongylophorus, which were likely amplified due to the cross-reactivity of the 16S primers, were removed. No sequences identified as archaeal were detected from our library generated using archaeal-specific primers, and only bacterial sequences were amplified. Sequences were prepared for alignment by orienting each sequence in the same direction using the computer program Orientation Checker [38], putative chimeras were removed using Bellerophon [39], and each set was de-replicated to remove exact duplicates.
Finalized sets for each sample were then analyzed using the ARB [40] software environment as follows. All full-length 16S rDNA sequences were imported and then aligned using the ARB fast-aligner tool [40] against a user-constructed PT-Server (constructed from the SILVA [41] 16S SSU rDNA preconfigured ARB reference database with 7,682 columns and 134,095 bacterial sequences; accessed: 01/15/2009). The full alignment was manually curated using the ARBprimary editor (ARB_EDIT4) in preparation for phylogenetic and community analysis. Once an acceptable alignment was obtained we created a PHYLIP [42] distance matrix in ARB using the filter-by-base-frequency method (column filter; minimal similarity = 50%; gaps ignored if occurred in >50% of the samples; 1,320 valid columns). The PHYLIP distance matrix was exported to the MOTHUR software package v.1.5.0 [43] for community analysis and OTU designation. Briefly, the distance matrix was read into MOTHUR and clustered using the furthest neighbor algorithm. From here, we performed rarefaction, rank-abundance, species abundance, and shared analyses. Representative sequences from each OTU at 97% were re-imported into ARB for phylogenetic analysis (Figure S4, S5, S6, S7, and S8). We used a Maximum Likelihood (RAxML [44]) method for all phylogenetic analyses (normal hill-climbing search algorithm) and the above-mentioned method for positional filtering. Closest taxonomic assignment of clones was performed using the Ribosomal Database Project (RDP) [45] by comparing sequences against the type strain database (Table S5).
For pyrotagged short-read 16S rDNA sequences, all sequences were compared against the National Center for Biotechnological Information's (NCBI) non-redundant nucleotide (nt) and environmental nucleotide (env_nt) databases (accessed: 05/01/2009) using BLASTN. Sequences were then classified as either bacterial, archaeal, or eukaryotic, and only those bacterial sequences (20,330) were retained for further analysis.
These sequences were then processed through Orientation Checker, chimeras removed using the program Mallard [38], and subsequently analyzed using MOTHUR in the following fashion. First the entire dataset was de-replicated to eliminate duplicate sequences. The remaining sequences were aligned in MOTHUR against the Greengenes [46] reference alignment (core_set_aligned.imputed.fasta; 7,682 columns, accessed: 09/11/2009) using the Needleman alignment method with the following parameters: k-tuple size = 9; match = +1; mismatch penalty = −3; gap extension penalty = −1; gap opening penalty = −5. Sequences were then screened to eliminate those shorter than 400 bp (gaps included). Filtration eliminated 7,062 columns resulting in a total alignment size of 620 bp (gaps included). The remaining dataset was again de-replicated to eliminate duplicate sequences and we constructed a furthest-neighbor distance matrix in MOTHUR using the twice de-replicated, filtered, alignment. All subsequent analyses (rarefaction, rank-abundance, species abundance, and shared analyses) were performed in MOTHUR using this distance matrix.
UniFrac Analysis
A UniFrac [15] analysis was performed on all full-length 16S rDNA samples generated in this study, including 3 from the top and 3 from the bottom of fungus gardens. MOTHUR was used to generate phylip distance matrices and the computer program Clearcut [47] was then employed to construct neighbor-joining trees. UniFrac was then used to compare these samples as shown in Figure S3.
Community Metagenome Sequencing and Assembly
Whole community DNA was used to create a shotgun library which was then sequenced using a single pyrosequencing plate on a Roche 454 FLX GS Titanium sequencer. Assembly of the data was performed using the 454 de novo assembler software with default parameters. Total amounts of data generated and statistical coverage is presented in Table S2. Raw sequence reads generated for this microbiome are deposited in NCBI's Short Read Archive under Study Accession SRP001011.1, and assembled contigs and singletons have been deposited into DDBJ/EMBL/GenBank under the accession ADWX00000000.
Community Metagenome Phylogenetic Binning
The complete set of assembled contigs and singletons representing the fungus garden community metagenome was phylogenetically binned using the following approach. First, the metagenome was compared against NCBI's non-redundant nucleotide (nt) and environmental nucleotide (env_nt) databases (accessed: 05/01/2009) using BLASTN (e-value: 1e-05) and the top hit was retained. The designated phylogenetic classification of the top hit for each contig and singleton was then assessed and binned into one of the following 4 sets: Bacterial, Eukaryotic, Viral, or Unknown. We then performed in-depth phylogenetic binning of the bacterial portion of the fungus garden community metagenome using the current microbial genome collection (http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi, accessed: 05/15/2009). We reasoned that using the current microbial genome collection is a likely a more accurate metric for classifying the bacterial set at the genus level because each genome in this collection is correctly annotated and the current iteration of this collection contains both phylogenetic breadth and depth for many represented genera. As a result, we performed two different phylogenetic bins using the current microbial genome collection.
First, GeneMark [48] was used to predict open reading frames and their corresponding translated proteins of the bacterial portion of the fungus garden community metagenome using a generic bacterial gene model. This predicted proteome was then compared against a local database containing all proteomes in the current microbial genome collection (http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi, accessed: 05/15/2009) supplemented with the predicted proteomes of two bacterial strains (Klebsiella variicola At-22 and Pantoea sp. At-9b, see below) isolated from the fungus gardens of a related leaf-cutting ant species, Atta cephalotes. Comparison of the fungus garden proteome against our microbial reference database was done using BLASTP (e-value: 1e-05) and the phylogenetic identity of the top hit was recorded. The total number of proteins was then tabulated to the genus level. Total nucleotide coding content for each predicted protein was then calculated to determine the total amount of nucleotide represented in each bin.
Second, we performed phylogenetic binning on the bacterial portion of the fungus garden metagenome using the entire nucleotide content of the current microbial genome collection (http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi, accessed: 05/15/2009), and again supplemented with the nucleotide content from the draft genome sequences of our two bacterial isolates from Atta cephalotes leaf-cutter ant fungus gardens. Using complete nucleotide content of the current microbial genome collection is advantageous because it includes both coding and intergenic regions, and provides a more robust measure of phylogenetic identity. We compared the entire bacterial portion of the fungus garden metagenome against this database using BLASTN (e-value: 1e-05) and the phylogenetic identity of the top hit was recorded. The total number of contigs and singletons was then tabulated to the genus level and the corresponding nucleotide amounts were also calculated. Furthermore, we performed this same analysis using all high-quality reads from our fungus garden community metagenome. Finally, we employed the phylogenetic binning program PhymmBL [22], which resulted in similar phylogenetic binning results as our comparison against the sequenced genome collection.
GC Content Analysis
We performed GC content analysis on the Bacterial, Eukaryotic, and Unclassified phylogenetic bins of the leaf-cutter ant fungus garden community metagenome. For the bacterial set, we divided the sequences according to the NCBI Taxonomic Groups Acidobacteria, Actinobacteria, α-proteobacteria, Bacteroidetes, β-proteobacteria, and γ-proteobacteria. We then calculated their GC content, and tabulated the total number of sequences within each group corresponding to each percentage as shown in Figure S11. For Eukaryotic sequences, these were divided into fungal, metazoan, and plant classifications and GC content analysis was also performed as shown in Figure S12. Furthermore, this same analysis was performed for the unclassified portion of the community metagenome and plotted alongside our Eukaryotic GC content analysis.
Carbohydrate-Active Enzyme Annotation Analysis
The predicted proteome from the bacterial portion of the fungus garden community metagenome was annotated using the carbohydrate active enzyme (CAZy) database [23] as follows. A local database of all proteins corresponding to each CAZy family from the CAZy online database (http://www.cazy.org/, accessed: 06/01/2009) was constructed, and this was used to align the predicted proteome of the bacterial portion of the fungus garden community metagenome using BLASTP (e-value of 1e-05). This proteome was then annotated against the protein family (Pfam [49]) database (ftp://ftp.ncbi.nih.gov/pub/mmdb/cdd/, accessed: 05/01/2009) using RPSBLAST [50] (e-value: 1e-05). A CAZy to Pfam correlation list was then compiled based on the secondary annotations provided through the CAZy online database. Finally, only those proteins that had significant BLAST hits to a protein from our local CAZy database and its corresponding Pfam were retained and designated as a carbohydrate-associated enzyme.
A similar process was performed using the eukaryotic portion of the fungus garden metagenome. However, because of the difficulty in accurately predicting proteins from this subset, due to the lack of good gene models, we compared the contigs and singletons in this subset to our local CAZy and Pfam databases using BLASTX (e-value: 1e-05). Only those hits with significant matches to a protein from our local CAZy database, and its corresponding Pfam were retained and designated as a carbohydrate-associated enzyme in this set.
Comparative COG and CAZy Cluster Analysis
To determine the similarity of the fungus garden community metagenome with respect to other sequenced metagenomes, we performed a comparative analysis using protein domain and carbohydrate enzyme content as a comparative metric, as previously described [51]. In general, the predicted proteome from the bacterial portion of the fungus garden metagenome was annotated according to clusters of orthologous groups (COGs [52]) database (ftp://ftp.ncbi.nih.gov/pub/mmdb/cdd/, accessed: 05/01/2009) using RPSBLAST (e-value: 1e-05). The predicted proteomes from the following 13 metagenomes were also annotated in the same manner: bovine rumen [3], chicken cecum [53], fish gut and slime [54], gutless worm [55], human gut (Gill) [6], human gut (Kurokawa) [56], Minnesota soil [51], lean mouse [5], obese mouse [5], termite hindgut [2], wastewater sludge USA [57], sastewater sludge OZ [57], and whale fall [51]. The COG profiles from all of these metagenomes were divided according to their COG gene category designations and plotted as a proportion as shown in Figure S13. Cluster analysis of COG profiles for these metagenomes were performed as follows. A matrix was generated with each row corresponding to a metagenome and each column corresponding to a COG ID. The proportion of each COG with respect to the total number of annotated COGs in that metagenome was calculated and populated in the appropriate cell of the matrix. Spearman's rank correlation was then applied to this matrix to generate a similarity matrix correlating each metagenome to each other based on the similarity of each metagenome's COG profile. A distance matrix was then calculated using the neighbor program from the computer suite Phylip [42] (using the UPGMA method), and the resulting unrooted tree was visualized using the phylodendron tree drawing program (http://iubio.bio.indiana.edu/treeapp/, accessed 07/25/2009). This same analysis was also performed using protein domains (Pfam) and no discernable difference in metagenome groupings was detected (data not shown).
A similar approach was used for clustering these metagenomes according to CAZy content. Each metagenome's predicted proteome was annotated using CAZy and correlated to its Pfam annotation as described above. Because each protein potentially encodes for domain that belong to multiple CAZy families (i.e. a protein may contain both a GH and a CBM), we assigned multiple CAZy annotations to a particular protein. A carbohydrate enzyme matrix was then constructed with each row corresponding to a metagenome sample and each column corresponding to a CAZy family. Each cell in this matrix was then populated with the proportion of each CAZy family with respect to the total number of annotated CAZy families in each respective metagenome. Generation of an unrooted tree using this matrix was then constructed using the same procedure outlined for clustering based on the protein domain content metric.
Draft Genome Sequencing, Assembly, and Annotation
Pure isolates of Klebsiella variicola At-22 and Pantoea sp. At-9b were cultured from the fungus gardens of the leaf-cutter ant Atta cephalotes, as previously described [10]. Genomic DNA from these isolates were extracted, as previously described [10]. Draft genomes of Klebsiella variicola At-22 and Pantoea sp. At-9b were sequenced at the U.S. Department of Energy Joint Genome Institute (JGI) using a random shotgun approach through a combination of 454 standard and paired-end pyrosequencing (454 Life Sciences, a Roche Company) and 36 bp read Illumina sequencing (Illumina, Inc.). Sequencing using 454 was performed to an average depth of coverage of 30X for both Klebsiella and Pantoea. All general aspects of library construction and sequencing performed at the JGI can be found at http://www.jgi.doe.gov. A draft assembly for Klebsiella variicola At-22 was compiled based on 459,192 reads; for Pantoea sp. At9b, a draft assembly was constructed using 557,748 reads. The Phred/Phrap/Consed software package (http://www.phrap.com) was used for sequence assembly and quality assessment of both drafts [58]–[60]. After the shotgun stage, reads were assembled with parallel Phrap (High Performance Software LLC). Automated annotation of these draft genomes were performed by the Computational Biology and Bioinformatics Group of the Biosciences Division of the U.S. Department of Energy Oak Ridge National Laboratory as described at http://genome.ornl.gov/. The draft genome sequence and annotation for Klebsiella variicola At-22 and Pantoea sp. At-9b were deposited in GenBank under accession numbers CP001891 and ACYJ00000000, respectively.
Recruitment Analysis
The full set of reads used for the assembly of the fungus garden community metagenome was used to generate a recruitment plot against the draft genomes of Klebsiella variicola At-22 and Pantoea sp. At-9b, two isolates we cultured from the fungus garden of the leaf-cutter ant Atta cephalotes [10], as previously described [27] . Briefly, the contigs from each draft genome were concatenated together in ascending size to produce a “pseudogenome”, and the reads from the fungus garden community metagenome were aligned against a database containing both pseudogenomes, and all genomes from the current microbial genome collection (http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi, accessed: 05/15/2009) using BLASTN. The top hit for each read was retained, and categorized to each genome. We then mapped reads corresponding to Klebsiella variicola At-22 and Pantoea sp. At-9b onto each organism's respective psuedogenome and further binned them according to their sequence identities as follows: 95%–100%, 90%–95%, 85%–90%, 80%–85%, and 70%–80%. Visualization of the mapped reads onto each respective draft genome was performed using the DNAPlotter software package [61].
CAZy Analysis of Draft Genomes
A CAZy analysis was performed on the predicted proteomes of Klebsiella variicola At-22 and Pantoea sp. At-9b using the same approach as described for CAZy analysis of the leaf-cutter ant fungus garden community metagenome. Furthermore, both GH8 cellulases from each of these genomes were compared against the CAZyme of the fungus garden community metagenome at the nucleotide level using BLASTN (e-value: 1e-05).
Cellulose Degradation Bioassays
Bioassays were performed on pure cultures of Klebsiella variicola At-22 and Pantoea sp. At-9b to determine their capacity to degrade cellulose. These include carboxymethyl cellulose (CMC) assays and growth on microcrystalline cellulose. CMC assays were performed as previously described [62]. Briefly, pure cultures of both bacteria were inoculated onto yeast malt extract agar (YMEA, 4 g yeast extract, 10 g Bacto Peptone, 4 g Dextrose, 15 g agar) and grown at 25°C for 2 days. Single colonies were then spotted onto carboxymethyl cellulose plates (15 g agar, 5 g carboxymethyl cellulose [Calbiochem, La Jolla, CA]). Detection of cellulose degradation on CMC was performed using congo red, and the ability of each isolate's capacity for cellulose degradation was measured based on the zone of clearing present on the plate. Growth on microcrystalline cellulose was performed by inoculating 10 µl of pure culture into 150 µl of microcrystalline cellulose broth (1 L water and 5 g cellulose powder microcrystalline cellulose [MP Biomedicals, Solon, OH]) and growth was measured using a DTX 880 Multimode Detector Plate Reader (Beckman Coulter Inc., Fullerton, CA) at an absorbance of 595 for 2 days. Positive growth on microcrystalline cellulose was correlated to an increase in the measured absorbance over this period.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. SticklenMB
2008 Plant genetic engineering for biofuel production: towards affordable cellulosic ethanol. Nat Rev Genet 9 433 443
2. WarneckeF
LuginbuhlP
IvanovaN
GhassemianM
RichardsonTH
2007 Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450 560 565
3. BrulcJM
AntonopoulosDA
MillerME
WilsonMK
YannarellAC
2009 Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc Natl Acad Sci U S A 106 1948 1953
4. LeyRE
HamadyM
LozuponeC
TurnbaughPJ
RameyRR
2008 Evolution of mammals and their gut microbes. Science 320 1647 1651
5. TurnbaughPJ
LeyRE
MahowaldMA
MagriniV
MardisER
2006 An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444 1027 1031
6. GillSR
PopM
DeboyRT
EckburgPB
TurnbaughPJ
2006 Metagenomic analysis of the human distal gut microbiome. Science 312 1355 1359
7. WirthR
HerzH
RyelRJ
BeyschlagW
HolldoblerB
2003 Herbivory of leaf-cutting ants. A case study on Atta colombica in the tropical rain forest of Panama. Springer xvi Berlin, Heidelberg 230
8. WeberNA
1966 Fungus-growing ants. Science 153 587 604
9. CurrieCR
StuartAE
2001 Weeding and grooming of pathogens in agriculture by ants. Proc R Soc London Ser B Biol Sci 268 1033 1039
10. Pinto-TomásAA
AndersenMA
SuenG
StevensonDM
ChuFST
2009 Symbiotic Nitrogen Fixation in the Fungus Gardens of Leaf-cutter Ants. Science 326 1120 1123
11. AbrilAB
BucherEH
2002 Evidence that the fungus cultured by leaf-cutting ants does not metabolize cellulose. Ecology Letters 5 325 328
12. SchiottM
De Fine LichtHH
LangeL
BoomsmaJJ
2008 Towards a molecular understanding of symbiont function: identification of a fungal gene for the degradation of xylan in the fungus gardens of leaf-cutting ants. BMC Microbiol 8 40
13. ErthalMJr
SilvaCP
CooperRM
SamuelsRI
2009 Hydrolytic enzymes of leaf-cutting ant fungi. Comp Biochem Physiol B Biochem Mol Biol 152 54 59
14. MarguliesM
EgholmM
AltmanWE
AttiyaS
BaderJS
2005 Genome sequencing in microfabricated high-density picolitre reactors. Nature 437 376 380
15. LozuponeC
HamadyM
KnightR
2006 UniFrac - An online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 7 371
16. HolmesAJ
TujulaNA
HolleyM
ContosA
JamesJM
2001 Phylogenetic structure of unusual aquatic microbial formations in Nullarbor caves, Australia. Environ Microbiol 3 256 264
17. HugenholtzP
PitulleC
HershbergerKL
PaceNR
1998 Novel division level bacterial diversity in a Yellowstone hot spring. J Bacteriol 180 366 376
18. RheimsH
RaineyFA
StackebrandtE
1996 A molecular approach to search for diversity among bacteria in the environment. Journal of Industrial Microbiology and Biotechnology 17 159 169
19. LipsonDA
SchmidtSK
2004 Seasonal changes in an alpine soil bacterial community in the colorado rocky mountains. Appl Environ Microbiol 70 2867 2879
20. HugenholtzP
GoebelBM
PaceNR
1998 Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180 4765 4774
21. von MeringC
HugenholtzP
RaesJ
TringeSG
DoerksT
2007 Quantitative phylogenetic assessment of microbial communities in diverse environments. Science 315 1126 1130
22. BradyA
SalzbergSL
2009 Phymm and PhymmBL: metagenomic phylogenetic classification with interpolated Markov models. Nat Methods 6 673 676
23. CantarelBL
CoutinhoPM
RancurelC
BernardT
LombardV
2009 The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res 37 D233 238
24. WeimerPJ
RussellJB
MuckRE
2009 Lessons from the cow: what the ruminant animal can teach us about consolidated bioprocessing of cellulosic biomass. Bioresour Technol 100 5323 5331
25. WeimerPJ
StevensonDM
MertensDR
ThomasEE
2008 Effect of monensin feeding and withdrawal on populations of individual bacterial species in the rumen of lactating dairy cows fed high-starch rations. Appl Microbiol Biotechnol 80 135 145
26. PfisterCA
MeyerF
AntonopoulosDA
2010 Metagenomic Profiling of a Microbial Assemblage Associated with the California Mussel: A Node in Networks of Carbon and Nitrogen Cycling. PLoS ONE 5 e10518doi:10.1371/journal.pone.0010518
27. RuschDB
HalpernAL
SuttonG
HeidelbergKB
WilliamsonS
2007 The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol 5 e77 doi:10.1371/journal.pbio.0050077
28. DillonRJ
DillonVM
2004 The gut bacteria of insects: nonpathogenic interactions. Annu Rev Entomol 49 71 92
29. BroderickNA
RaffaKF
GoodmanRM
HandelsmanJ
2004 Census of the bacterial community of the gypsy moth larval midgut by using culturing and culture-independent methods. Appl Environ Microbiol 70 293 300
30. RussellJA
MoreauCS
Goldman-HuertasB
FujiwaraM
LohmanDJ
2009 Bacterial gut symbionts are tightly linked with the evolution of herbivory in ants. Proc Natl Acad Sci U S A
31. UpdegraffDM
1969 Semimicro determination of cellulose in biological materials. Analytical Biochemistry 32 420 424
32. SelvendraRR
O'NeillMA
1987 Isolation and analysis of cell walls from plant material.
DavidG
Methods of Biochemical Analysis: John Wiley & Sons 25 153
33. YorkWS
DarvillAG
McNeilT
StevensonTT
AlbersheimP
1985 Isolation and characterization of plant cell walls and cell wall components. Methods in Enzymology 118 3 40
34. AlbersheimP
NevinsDJ
EnglishPD
KarrA
1967 A method for the analysis of sugars in plant cell wall polysaccharides by gas-liquid chromatography. Carbohydrate Research 5 340 345
35. JungHJ
VarelVH
WeimerPJ
RalphJ
1999 Accuracy of Klason lignin and acid detergent lignin methods as assessed by bomb calorimetry. J Agric Food Chem 47 2005 2008
36. ApajalahtiJHA
SarkilahtiLK
MakiBRE
HeikkinenJP
NurminenPH
1998 Effective Recovery of Bacterial DNA and Percent-Guanine-Plus-Cytosine-Based Analysis of Community Structure in the Gastrointestinal Tract of Broiler Chickens. Applied and Environmental Microbiology 64 4084
37. AltschulSF
MaddenTL
SchafferAA
ZhangJ
ZhangZ
1997 Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25 3389 3402
38. AshelfordKE
ChuzhanovaNA
FryJC
JonesAJ
WeightmanAJ
2006 New screening software shows that most recent large 16S rRNA gene clone libraries contain chimeras. Appl Environ Microbiol 72 5734 5741
39. HuberT
FaulknerG
HugenholtzP
2004 Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 20 2317 2319
40. LudwigW
StrunkO
WestramR
RichterL
MeierH
2004 ARB: a software environment for sequence data. Nucleic Acids Res 32 1363 1371
41. PruesseE
QuastC
KnittelK
FuchsBM
LudwigW
2007 SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35 7188 7196
42. FelensteinJ
1989 PHYLIP - Phylogeny Inference Package (Version 3.2). Cladistics 5
43. SchlossPD
WestcottSL
RyabinT
HallJR
HartmannM
2009 Introducing mothur: Open Source, Platform-independent, Community-supported Software for Describing and Comparing Microbial Communities. Appl Environ Microbiol
44. StamatakisA
LudwigT
MeierH
2005 RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21 456 463
45. ColeJR
WangQ
CardenasE
FishJ
ChaiB
2009 The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Research 37 D141
46. DeSantisTZ
HugenholtzP
LarsenN
RojasM
BrodieEL
2006 Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72 5069 5072
47. ShenemanL
EvansJ
FosterJA
2006 Clearcut: a fast implementation of relaxed neighbor joining. Bioinformatics 22 2823
48. BesemerJ
BorodovskyM
2005 GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res 33 W451 454
49. FinnRD
TateJ
MistryJ
CoggillPC
SammutSJ
2008 The Pfam protein families database. Nucleic Acids Res 36 D281 288
50. Marchler-BauerA
AndersonJB
DerbyshireMK
DeWeese-ScottC
GonzalesNR
2007 CDD: a conserved domain database for interactive domain family analysis. Nucleic Acids Res 35 D237 240
51. TringeSG
von MeringC
KobayashiA
SalamovAA
ChenK
2005 Comparative metagenomics of microbial communities. Science 308 554 557
52. TatusovRL
GalperinMY
NataleDA
KooninEV
2000 The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28 33 36
53. QuA
BrulcJM
WilsonMK
LawBF
TheoretJR
2008 Comparative metagenomics reveals host specific metavirulomes and horizontal gene transfer elements in the chicken cecum microbiome. PLoS One 3 e2945 doi:10.1371/journal.pone.0002945
54. DinsdaleEA
EdwardsRA
HallD
AnglyF
BreitbartM
2008 Functional metagenomic profiling of nine biomes. Nature 452 629 632
55. WoykeT
TeelingH
IvanovaNN
HuntemannM
RichterM
2006 Symbiosis insights through metagenomic analysis of a microbial consortium. Nature 443 950 955
56. KurokawaK
ItohT
KuwaharaT
OshimaK
TohH
2007 Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res 14 169 181
57. Garcia MartinH
IvanovaN
KuninV
WarneckeF
BarryKW
2006 Metagenomic analysis of two enhanced biological phosphorus removal (EBPR) sludge communities. Nat Biotechnol 24 1263 1269
58. GordonD
AbajianC
GreenP
1998 Consed: a graphical tool for sequence finishing. Genome Res 8 195 202
59. EwingB
GreenP
1998 Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 8 186 194
60. EwingB
HillierL
WendlMC
GreenP
1998 Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 8 175 185
61. CarverT
ThomsonN
BleasbyA
BerrimanM
ParkhillJ
2009 DNAPlotter: circular and linear interactive genome visualization. Bioinformatics 25 119 120
62. UlrichA
KlimkeG
WirthS
2008 Diversity and activity of cellulose-decomposing bacteria, isolated from a sandy and a loamy soil after long-term manure application. Microb Ecol 55 512 522
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 9
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Synthesizing and Salvaging NAD: Lessons Learned from
- Optimal Strategy for Competence Differentiation in Bacteria
- Long- and Short-Term Selective Forces on Malaria Parasite Genomes
- Identifying Signatures of Natural Selection in Tibetan and Andean Populations Using Dense Genome Scan Data
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy