
Genome-Wide Double-Stranded RNA Sequencing Reveals the Functional Significance of Base-Paired RNAs in
The functional structure of all biologically active molecules is dependent on intra - and inter-molecular interactions. This is especially evident for RNA molecules whose functionality, maturation, and regulation require formation of correct secondary structure through encoded base-pairing interactions. Unfortunately, intra - and inter-molecular base-pairing information is lacking for most RNAs. Here, we marry classical nuclease-based structure mapping techniques with high-throughput sequencing technology to interrogate all base-paired RNA in Arabidopsis thaliana and identify ∼200 new small (sm)RNA–producing substrates of RNA–DEPENDENT RNA POLYMERASE6. Our comprehensive analysis of paired RNAs reveals conserved functionality within introns and both 5′ and 3′ untranslated regions (UTRs) of mRNAs, as well as a novel population of functional RNAs, many of which are the precursors of smRNAs. Finally, we identify intra-molecular base-pairing interactions to produce a genome-wide collection of RNA secondary structure models. Although our methodology reveals the pairing status of RNA molecules in the absence of cellular proteins, previous studies have demonstrated that structural information obtained for RNAs in solution accurately reflects their structure in ribonucleoprotein complexes. Furthermore, our identification of RNA–DEPENDENT RNA POLYMERASE6 substrates and conserved functional RNA domains within introns and both 5′ and 3′ untranslated regions (UTRs) of mRNAs using this approach strongly suggests that RNA molecules are correctly folded into their secondary structure in solution. Overall, our findings highlight the importance of base-paired RNAs in eukaryotes and present an approach that should be widely applicable for the analysis of this key structural feature of RNA.
Published in the journal:
. PLoS Genet 6(9): e32767. doi:10.1371/journal.pgen.1001141
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001141
Summary
The functional structure of all biologically active molecules is dependent on intra - and inter-molecular interactions. This is especially evident for RNA molecules whose functionality, maturation, and regulation require formation of correct secondary structure through encoded base-pairing interactions. Unfortunately, intra - and inter-molecular base-pairing information is lacking for most RNAs. Here, we marry classical nuclease-based structure mapping techniques with high-throughput sequencing technology to interrogate all base-paired RNA in Arabidopsis thaliana and identify ∼200 new small (sm)RNA–producing substrates of RNA–DEPENDENT RNA POLYMERASE6. Our comprehensive analysis of paired RNAs reveals conserved functionality within introns and both 5′ and 3′ untranslated regions (UTRs) of mRNAs, as well as a novel population of functional RNAs, many of which are the precursors of smRNAs. Finally, we identify intra-molecular base-pairing interactions to produce a genome-wide collection of RNA secondary structure models. Although our methodology reveals the pairing status of RNA molecules in the absence of cellular proteins, previous studies have demonstrated that structural information obtained for RNAs in solution accurately reflects their structure in ribonucleoprotein complexes. Furthermore, our identification of RNA–DEPENDENT RNA POLYMERASE6 substrates and conserved functional RNA domains within introns and both 5′ and 3′ untranslated regions (UTRs) of mRNAs using this approach strongly suggests that RNA molecules are correctly folded into their secondary structure in solution. Overall, our findings highlight the importance of base-paired RNAs in eukaryotes and present an approach that should be widely applicable for the analysis of this key structural feature of RNA.
Introduction
Recent discoveries reveal that RNAs perform a variety of tasks—ranging from the regulation of gene expression (e.g. small RNAs (smRNAs), and riboswitches) to catalytic activities (e.g. group I self-splicing introns)—and indicate that this functionality is intimately linked to their three-dimensional structure [1]–[5]. Correct secondary structure is also central to the proper regulation and maturation of RNA molecules [2], [3], [6], [7]. RNAs fold into their three-dimensional structures through specific base-pairing interactions (double-stranded RNA (dsRNA)) that are encoded within their sequence [2], [3], [6], [7]. These interactions can either be within (intra-molecular) or between (inter-molecular (heteroduplex)) RNA molecules. Although it is clear that secondary structure is abundantly important for the functionality and regulation of RNAs, comprehensive base-pairing interaction data are completely lacking for the majority of these molecules [3].
The recent discovery that RNA silencing pathways play a significant role in gene regulation has brought attention to a vast evolutionarily conserved post-transcriptional regulatory network dependent on self and foreign base-paired RNAs (dsRNAs) [8]–[10]. In RNA silencing, production of heteroduplex dsRNA or self-complementary fold-back structures gives rise to smRNAs through the activity of DICER or DICER-LIKE (DCL) RNase III-type ribonucleases [9]–[12]. In eukaryotes, smRNAs consist of microRNAs (miRNAs) and several classes of endogenous small interfering RNAs (siRNAs), which are differentiated from one another by their distinct biogenesis pathways and the classes of genomic loci from which they arise [8]. These smRNAs are the sequence-specific effectors of RNA silencing, and direct the negative regulation or control of genes, repetitive sequences, viruses, and mobile elements through inter-molecular base-pairing interactions [13], [14]. Overall, base-paired RNAs are at the core of both the biogenesis and function of all eukaryotic small silencing RNAs, emphasizing the importance of base-paired RNA in regulating gene expression.
In plants and several other organisms, there are numerous classes of endogenous and exogenous siRNAs that are processed from long dsRNA molecules synthesized by an RNA-dependent RNA polymerase (RDR) [8]–[10], [15]. The first RDR to be functionally identified as an RNA silencing pathway component in Arabidopsis thaliana, was RDR6 [16], [17]. RDR6 was initially uncovered due to its ability to utilize aberrant RNAs produced by transgenes as substrates for dsRNA synthesis [16]–[18]. These dsRNA molecules are subsequently converted by DCL4 into siRNAs that silence the transgenes [19]–[23]. More recently, RDR6 has been demonstrated to function in the biogenesis of endogenous smRNA populations [8], [20], [24]–[26]. One example is trans-acting siRNAs (tasiRNAs), which are processed from regions of non-coding RNAs known as TRANS-ACTING siRNA (TAS) transcripts [20], [25]–[27]. Biogenesis of tasiRNAs is initiated by siRNA or miRNA-mediated cleavage of the TAS transcript [20], [25]–[27]. The cleaved TAS transcript is then converted by RDR6 to dsRNA [20], [25]–[27], which is subsequently cleaved by DCL4 into phased 21 nucleotide (nt) siRNAs [20]–[23], [28].
Here, we describe a novel, genome-wide, high-throughput sequencing-based method, which we term dsRNA-seq, that can specifically interrogate base-paired (dsRNA) RNA molecules, and use this approach to identify and characterize ∼200 novel, smRNA-producing substrates of the dsRNA-synthesizing enzyme RDR6. Additionally, we find that mRNAs encoding proteins with functions in nucleic acid-based processes have a tendency to be highly structured. Making use of a seven-way comparative genomic approach, we demonstrate that the dsRNA-seq methodology can identify functionally conserved portions of UTRs (3′ and 5′), introns, transposable elements, as well as novel, structured RNA molecules throughout the Arabidopsis genome. Finally, we exploit the ability of dsRNA-seq to capture intra-molecular base-pairing interactions to produce mRNA secondary structural models on a genome-wide scale.
Results/Discussion
A novel approach to interrogate the dsRNA component of the Arabidopsis transcriptome
To obtain a transcriptome-wide view of base-paired RNA (dsRNA) in unopened flower buds of Arabidopsis thaliana Col-0 ecotype (hereafter referred to as wild-type Col-0), we married classical nuclease-based structure mapping techniques [29], [30] with high-throughput sequencing technology (see Figure S1A, and Materials and Methods for details). We characterized the dsRNA component of the Arabidopsis transcriptome after one round of ribosomal RNA (rRNA)-depletion, and obtained 15,499,789 raw reads representing 4,802,974 non-redundant (NR) sequences with an average clone-abundance of 3.2 (Accession #: GSE23439). (The size distributions for this dataset can be seen in Figure S3A.)
As expected, we found that the majority of our dsRNA sequencing reads corresponded to highly structured classes of RNA molecules (e.g., rRNA, tRNA, snoRNA, snRNA, etc.), smRNA-producing loci (e.g., miRNAs), and repetitive elements (e.g., transposons) (Figure 1A). We also found a large proportion of dsRNAs that correspond to protein-coding transcripts, which likely represent the self-complementary, base-pairing regions that form the secondary structure of mRNA molecules (Figure 1A). It is noteworthy that dsRNA-seq data mapped to all portions of protein-coding mRNAs, including introns, exons, and both (3′ and 5′) UTRs. Therefore, the dsRNA-seq methodology can identify base-paired regions within both mature and preprocessed mRNA molecules. (For this reason, we refer to protein-coding mRNAs within this manuscript as pre-mRNA.) Overall, our dsRNA-seq approach is robustly biased towards classes of RNA molecules that are highly base-paired in nature, which strongly suggests that this approach is interrogating the desired component of the transcriptome with a stringently estimated false discovery rate (FDR) of ≤0.067 (see Text S1).

The strand-specific nature of dsRNA-seq affords the opportunity to distinguish between intra-molecular fold-back dsRNAs (16.6% of total identified dsRNAs; example tRNA in Figure 1C) and inter-molecular heteroduplex molecules (83.4% of total identified dsRNAs; example in Figure 1D). To determine the strand bias for the different classes of RNAs captured by dsRNA-seq, we interrogated the ratio of sense versus anti-sense sequence reads. As indicated by the Log-odds (Lods) values of sense to antisense reads, the majority of RNA classes were strongly enriched for sense-strand reads, especially for the non-coding RNA classes (rRNA, tRNA, snoRNA, etc.) (Figure 1B). Specifically, functional RNAs (tRNA, miRNA, snoRNA, snRNA, and rRNA) were between 100–1000 fold enriched for the sense compared to the antisense-strand (Figure 1B). Conversely, we identified a strong anti-sense bias in our dsRNA-seq data for transposable element-derived sequences (Figure 1B). This may reflect an amplification of the antisense transposon sequence by an RDR to initiate production of siRNAs and subsequent RNA silencing of these mobile elements. For protein coding regions (exons) and 5′ UTRs of mRNAs, there was a significant sense-strand bias (∼16-fold), which was diluted for introns or 3′ UTRs of these RNA molecules. We suspect that the existence of many overlapping genes and non-coding RNAs (tRNAs, snRNAs, and snoRNAs) on the strand opposite to introns or 3′ UTRs is the confounding factor. This hypothesis is consistent with the stronger sense-strand bias in coding regions of mRNAs (Figure 1B), which have an extremely low probability of overlapping with expressed elements on the opposite strand. Additionally, there are numerous instances of 3′ end overlapping transcripts, as well as snRNA, snoRNA, and tRNA loci encoded within the introns and UTRs of protein coding mRNAs throughout the Arabidopsis genome. Taken together, these results suggest that by using dsRNA-seq we have identified the majority of base-paired RNA molecules (Figure S1B and S1C), which encompass a surprisingly large portion of the Arabidopsis genome (∼14.4% (17.3 Mb)).
As described above, dsRNA-seq captured both intra - and inter-molecular base-pairing interactions (Figure 1B–1D). In fact, we found that regions of tRNAs predicted to form intra-molecularly base-paired stems corresponded to higher levels of dsRNA-seq reads than the unpaired anti-codon loop and the amino acid acceptor stem as expected (Figure 1C). Furthermore, we observed dsRNAs that corresponded to both the Watson and Crick strands of the genome for a known substrate of the intermolecular dsRNA-synthesizing RDR6 (Figure 1D). Taken together, these results suggest that dsRNA-seq can be used to differentiate intra - from inter-molecular base-pairing interactions.
Genome-wide identification and characterization of Arabidopsis RDR6 smRNA–producing substrates
An ideal test to both validate and determine the utility of dsRNA-seq is to identify all known and novel substrates of Arabidopsis RDR6. Accordingly, we sequenced the full complement of base-paired RNA (using dsRNA-seq) and smRNA (using smRNA-seq) molecules from unopened flower buds of wild-type Col-0 and rdr6-11 mutant (referred to hereafter as rdr6) plants. For wild-type Col-0, we obtained the dsRNA-seq data described above, as well as 17,340,638 raw sequence reads representing 8,575,097 non-redundant smRNA sequences (the size distributions for this smRNA dataset can be seen in Figure S3B). Additionally, we generated a total of 18,345,980 and 18,850,891 raw sequence reads representing 9,725,315 and 9,860,471 non-redundant dsRNA and smRNA sequences for rdr6 mutant plants, respectively (the size distributions for these rdr6 datasets can be seen in Figure S3C and S3D, respectively).
To identify potential RDR6 substrates, we used a sliding-window analysis to select 1 kilobase (kb) regions of the genome that produced ≥2-fold more dsRNA in wild-type Col-0 than in rdr6 mutant plants with a p-value <0.001 (see Text S1). Using this approach, we identified 7,144 regions where dsRNAs are significantly depleted in rdr6 mutant compared to wild-type Col-0 plants (Figure 2A, positive Lods-ratio values). Within these molecules, we identified 7 of 8 previously characterized TAS transcripts (Figure 2A, Figure S2A and S2B, blue diamonds), while the eighth was represented by a single read in both (Col-0 and rdr6) dsRNA-seq libraries. Additionally, we found that the majority of RDR6-dependent dsRNAs are transposable elements (mostly MuDRs and Helitrons), mRNAs, intergenic RNAs (mostly centromeric tandem repeats), or tRNAs (Figure 2A and 2B (green bars), and Figure S2B). Taken together, these results suggest that RDR6 utilizes specific classes of repetitive elements, numerous categories of functional RNAs (e.g. tRNAs, snRNAs, snoRNAs, etc.), mRNAs, and intergenic transcripts as templates for dsRNA synthesis.
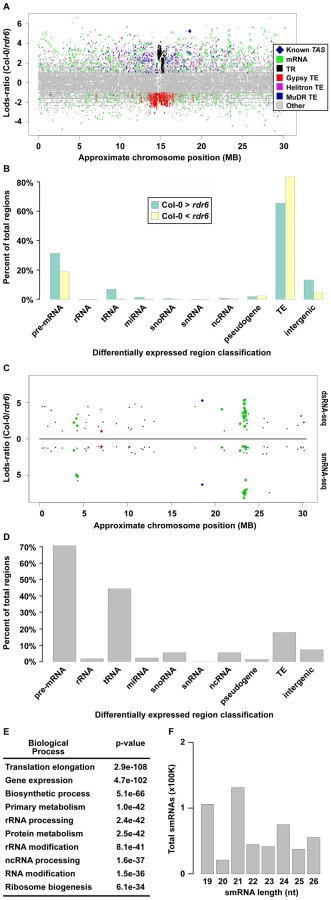
Our sliding window approach also identified 7,584 dsRNAs that are significantly stabilized in rdr6 mutant compared to wild-type Col-0 plants (Figure 2A, negative Lods-ratio values). The vast majority (>80%) of the molecules stabilized in rdr6 mutant plants are TEs (Figure 2B, yellow bars), most of which (∼95%) are pericentrometric Gypsy-like transposons (Figure 2A and 2B (yellow bars), and S2B). We also found a number of these dsRNAs correspond to mRNAs (∼15%) and intergenic transcripts (∼4%) (Figure 2B, yellow bars). Overall, the identification of dsRNA molecules that are stabilized in rdr6 mutant plants suggests a potential model where RDR6 antagonizes the action of other RDRs at some targets, especially at Gypsy-like transposons.
The consequence of dsRNA synthesis by RDR6 is often the subsequent formation of siRNAs [19]. Therefore, to identify those RDR6 dsRNA substrates that produce smRNAs, we identified regions that produce ≥2-fold more smRNAs in wild-type Col-0 than in rdr6 mutant plants. These sources of smRNA were then compared with the regions of the genome that produce more dsRNA in wild-type Col-0 than in rdr6 mutant plants, which identified 218 regions that met both criteria (Figure 2C and Figure 3A–3D; Table S1). These common regions include ∼50% (27 total) of the previously identified smRNA-producing RDR6 substrates, the majority of which were not known to be expressed in Arabidopsis unopened flower buds (Figure 2C and Figure S2C; Tables S1 and S2) [31]–[34]. The other 6,926 regions where dsRNAs, but not smRNAs, are significantly depleted in rdr6 mutant compared to wild-type Col-0 plants consist of mostly MuDR and Helitron transposable elements. These results suggest that the double-stranded MuDRs and Helitrons produced by RDR6 may only constitute an insignificant subset of the smRNA-producing population of these transposons. Conversely, RDR6 synthesized MuDR and Helitron dsRNAs may simply not be processed into smRNAs.

Our analysis also revealed that the majority of highly confident smRNA-producing RDR6 substrates are mRNAs with a variety of biological functions (Figure 2D and 2E) and, surprisingly, tRNAs (Figure 2D). As expected, the identified RDR6 substrates tend to produce 21 nt smRNAs (Figure 2F). It is noteworthy that RDR6-targeted mRNAs mostly encode proteins that function in nucleic acid-based biological functions (e.g. translation, RNA processing, etc.) and regulation of gene expression (Figure 2E). Taken together, these results suggest that an RDR6-dependent RNA silencing pathway regulates multiple stages of gene expression through siRNA production in Arabidopsis.
The identification of tRNAs as RDR6 substrates is intriguing because it was recently suggested that the mammalian telomerase reverse transcriptase catalytic subunit (Tert) functions as a smRNA-producing RDR that can also use tRNAs as substrates [15]. Taken together, these results suggest that plant RDR6 and animal Tert are functional orthologs that can use tRNAs as substrates for production of dsRNA precursors of smRNAs. Therefore, studies of RDR6 may be informative for gaining insight into the function of mammalian RDRs, and vice versa.
In order to validate and expand our characterization of new smRNA-producing RDR6 substrates, we turned to a quantitative reverse transcription polymerase chain reaction (qRT-PCR) approach. For these loci, RDR6 is required to produce a dsRNA precursor of siRNAs (see Figure 3A–3D). Therefore, if RDR6 is not active (rdr6 mutant plants), then the single-stranded transcripts may be stabilized. To test this hypothesis, we designed qRT-PCR primers to 14 (four known, 10 novel) identified smRNA-producing RDR6 substrates. We found that all fourteen tested loci, including the 10 newly identified RDR6 substrates (e.g. At1g20370 (Figure 3B), the intergenic region just upstream of At2g41490 (Figure 3C), and At3g19890 (Figure 3D)), had higher transcript levels in rdr6 mutant compared to wild-type Col-0 plants (Figure 3E). These results suggest that most, if not all of the 218 loci we identified using a combination of dsRNA-seq and smRNA-seq methodologies are true smRNA-producing RDR6 substrates; approximately 200 of these loci are novel (Tables S1 and S2).
Most previously identified endogenous RDR6 substrates produce phased 21 nt siRNAs [20]–[23], [28]. We found that 51 of the RDR6 substrates identified in this study also produce phased smRNAs (Table S2 and Figure S2D). This group includes 22 of the RDR6 substrates that have been previously reported [31]–[34], as well as the newly identified substrates, At1g20370 (Figure 3B and 3E), the intergenic region just upstream of At2g41490 (Figure 3C and 3E), and At5g02370 (Figure 3E; Tables S1 and S2). However, we found that >75% of all endogenous smRNA-producing RDR6 substrates (167) do not produce siRNAs with any recognizable phasing, including the newly identified At3g19890 (Figure 3D and 3E; Tables S1 and S2). These results suggest that there are multiple mechanisms by which transcripts become susceptible to RDR6-mediated silencing. In summary, our results suggest that the combination of dsRNA-seq and smRNA-seq is a highly sensitive method for identifying transcripts subject to RDR6-dependent silencing, and is likely to be useful for characterizing the substrates of other eukaryotic RDRs - such as mammalian Tert [15] - that have not been demonstrated to produce phased siRNAs.
Identification of dsRNA “hotspots” in the Arabidopsis genome
We next identified regions of the Arabidopsis genome that are significantly enriched for base-paired RNA using the dsRNA-seq data for wild-type Col-0. For this purpose, we used a geometric distribution-based approach to identify unusually long dsRNA molecules (dsRNA ‘hotspots’) based on the average size of dsRNAs computed for each chromosome independently. This analysis revealed 9,719 dsRNA ‘hotspots’ of varying lengths scattered along the entire length of all Arabidopsis chromosomes (Figure 4A and Figure S4A; Tables S3 and S4). In fact, we have identified the vast majority of highly base-paired RNA molecules in the Arabidopsis transcriptome (Figure S9). For example, the highly repetitive, transposon-rich pericentromeric regions of the Arabidopsis genome were found to be a rich source of dsRNA (Figure 4A and 4B, and Figure S4A). This is not surprising because cis transcriptional silencing of transposons and repetitive elements in the pericentromeric regions of Arabidopsis chromosomes is mediated by RDR2-dependent siRNAs [35]–[38]. These findings not only substantiate that dsRNA-seq interrogates the desired portion of the transcriptome, but also suggest that, as expected, Arabidopsis transposons and repetitive elements are highly enriched in dsRNA on a genome-wide scale.

A classification of Arabidopsis dsRNA ‘hotspots’ revealed that transposons and protein-coding mRNAs are the two most highly base-paired classes of RNA molecules (Figure 4B). In fact, we identified 1949 protein-coding mRNAs that contained dsRNA ‘hotspots’ (Figure 4B), so we interrogated over-represented molecular functions for these genes using Gene Ontology (GO) analysis. Ribulose-bisphosphate carboxylase was the most significantly over-represented protein in this analysis. However, the most highly over-represented group of genes were those involved in nucleic acid biology (e.g., translation, nucleic acid binding, etc.) (Figure 4C). Interestingly, genes involved in nucleic acid metabolism are also over-represented in dsRNA ‘hotspot’-containing transcripts of Drosophila melanogaster and Caenorhabditis elegans (Q.Z. and B.D.G., unpublished data). Thus, a propensity to form complex secondary structure (self base-pairing) may be a general feature of eukaryotic transcripts that encode proteins involved in processes involving nucleic acids. This may point to a feedback regulatory mechanism that is dependent on an interaction between the proteins encoded by these transcripts and highly structured RNA intermediates.
The biogenesis of all functional small silencing RNAs (e.g. miRNAs and siRNAs) requires a dsRNA intermediate. Therefore, we determined the propensity of highly base-paired regions (dsRNA ‘hotspots’) to be processed into smRNAs (Figure 4D) using corresponding smRNA-seq data (Figure 2C; see Figure S8 for smRNA data analysis). We found that the highly base-paired regions within 9 of 10 interrogated RNA categories were extremely likely to be processed into smRNAs, the exception being pre-mRNA molecules (Figure 4D). Although these results were expected for transposable elements and miRNAs - which are known to be smRNA biogenesis substrates - it was surprising that functional RNAs (e.g. rRNA, tRNA, snRNA, etc.) also have a high likelihood of being processed into smRNAs since intramolecular base-pairing interactions are intrinsic to their function.
The evidence that highly base-paired regions of RNA molecules are frequently processed into smRNA, suggests that this process may be important for regulating the abundance of functional RNAs in Arabidopsis cells. Our finding that any highly base-paired molecule can be processed into smRNAs, may provide an explanation for the restriction of the miRNA biogenesis machinery to specific sites within the plant nucleus (dicing bodies) [39], [40]. An intriguing hypothesis is that the sequestration of proteins involved in miRNA biogenesis and their MIRNA substrates to dicing bodies provides specificity to miRNA biogenesis, while protecting other structured RNAs (e.g. rRNA) from these proteins. Our findings suggest further studies of smRNA sources in eukaryotes will reveal additional siRNA-mediated regulatory pathways, as demonstrated, for example, by the analysis of tRNA-derived RNA fragments (tRFs) in human cells [41].
Comparative genomics of dsRNA “hotspots” reveals functionality within introns, both UTRs, and intergenic regions of the Arabidopsis genome
Regulation and maturation of eukaryotic pre-mRNA molecules is intimately linked to the proper formation of secondary structure [2], [3], [6], [7], which suggests that base-paired regions of these molecules are likely to be functionally conserved. To test this hypothesis, we employed a seven-way comparative genomics approach that determines an average conservation score (consScore) for all bases of dsRNA ‘hotspots’ and all other sequences (‘flanking regions’) within the four structural moieties (exons, introns, and both UTRs) of every mRNA. The consScores for dsRNA ‘hotspots’ and ‘flanking regions’ were then compared to determine if base-pairing mediates evolutionary conservation of mRNAs. Using this approach, we found that dsRNA ‘hotspots’ in exons are significantly less evolutionarily conserved than ‘flanking regions’ (Figure 5A), which suggests that intra - and/or intermolecular base-pairing interactions are disfavored in the protein-coding regions of plant mRNAs.

Our comparative genomic analysis of pre-mRNA data also demonstrated that dsRNA ‘hotspots’ are significantly more conserved than ‘flanking regions’ in 3′ UTRs (p = 0.0012) and introns (p = 1.73e–58) (Figure 5A), and that highly base-paired regions within 5′ UTRs (p = .072) were more evolutionarily conserved than ‘flanking regions’, but far less significantly than in 3′ UTRs and introns. This analysis suggests the ability to base-pair is functionally important, and has been selected during plant evolution. Just as selection for protein function maintains exonic sequences, base-pairing interactions may be important for conserving functionally important moieties in non-coding regions of mRNAs. These functions may include 1) providing appropriate structure for post-transcriptional and/or translational regulation, 2) maintaining mRNA stability, 3) providing cis-element sites for RNA binding proteins, and/or 4) forming the processed precursors of non-coding RNAs. Similar results have been obtained for Drosophila melanogaster and Caenorhabditis elegans (Q.Z. and B.D.G., unpublished data), suggesting that the ability to base pair is a critical feature of UTRs and introns in both plants and animals. An mRNA secondary structure prediction methodology (see below) was used to obtain a folded model of two highly conserved intronic dsRNAs (see Figure S5A and S5B for alignments), and suggested that these regions are almost entirely base-paired, and fold into unique, stable secondary structures (Figure 5C and 5D). Taken together, our results reveal that dsRNA-seq identifies functionally conserved regions of 5′ and 3′ UTRs and introns transcriptome-wide, and thus provides the critical first step towards understanding how such structural moieties affect the maturation and stability of transcripts in eukaryotic organisms.
We also noticed that a number of our dsRNA ‘hotspots’ are located in transposons and portions of the genome that do not contain any known genes. Comparative analysis revealed that dsRNA ‘hotspots’ in intergenic regions (p = 7.3e–5) and transposons (p = 9.1e–16) are significantly more conserved than their flanking regions (Figure 5B). In the case of transposons, this finding was quite surprising because the majority of these repetitive elements are selectively neutral, especially for ancestral repeats (ARs) [42], [43]. However, our findings demonstrate that the highly antisense-prone transposable element dsRNA ‘hotspots’ (Figure S4C and S4D) have been undergoing a significant purifying selection compared to their ‘flanking regions’, suggesting that these portions of TEs are not selectively neutral, but have important functions in plant cells. An intriguing hypothesis is that a class of smRNAs that are integral to initiate and/or maintain the transcriptional silencing of transposable elements are processed from these conserved highly-base paired regions. Overall, these results reveal functionally conserved portions of transposons, as well as novel, structured RNAs that have not been previously identified.
Identification and characterization of novel, highly base-paired RNAs with conserved functions in land plants
We identified a total of 1602 novel transcripts, ∼60% of which are unannotated transposable elements and/or simple repeats (Figure 6J; Tables S5 and S6). The other >700 transcripts represent newly identified RNAs. To determine the function of these 1602 transcripts we looked for the presence of these sequences in our flower bud smRNA dataset (see Figure S8 for smRNA analysis). 1437 (89.7%) of the novel RNAs overlapped regions of the genome that produce significant quantities of smRNAs (smRNA ‘hotspots’, Figure S8) (Figure 6 and Figure S6; Tables S5 and S6). Specifically, >98% of the unannotated transposable elements and/or simple repeats and ∼79% of the entirely novel RNAs produced smRNAs, respectively (Figure 6J). Most smRNAs from these transcripts were 24 nt in length (Figure 6K and 6L). In Arabidopsis, this size class is highly correlated with DNA methylation and heterochromatin formation [44], suggesting that these loci produce 24 nt smRNAs that direct transcriptional silencing.

To validate our sequencing data and further interrogate the newly identified transcription units, we characterized several of these RNAs by reverse transcription (RT) polymerase chain reaction (RT-PCR) in five different Arabidopsis tissues (leaf blade, leaf petiole, cauline leaves, stem, and unopened flower buds). We selected four loci that do (see Figure 6A and 6C; Figure S6A, S6C, and S6E; Table S5) and seven RNAs that do not (Figure 6B and 6D; Figure S6B, S6D, S6F, S6G, S6K, S6L, and S6M; Table S5) produce statistically significant amounts of smRNAs (11 total transcripts). As expected, all 11 of these RNAs are expressed in flower buds, the tissue used for the initial analysis of base-paired RNAs. Eight of these transcription units are expressed in all five tissues, and three are expressed only in unopened flower buds (Figure 6E–6I; Figure S6H, S6I, S6J, S6N, S6O, and S6P). Two of these latter transcripts are also the source of smRNAs (Figure 6A and Figure S6A; Table S5). Overall, our findings reveal a large collection of novel, structured RNAs in Arabidopsis flower buds, many of which have evolutionarily conserved functions in land plants (Figure 5B, intergenic).
Using dsRNA–seq data to produce models of mRNA secondary structure genome-wide
In principle, dsRNA-seq data should reveal the pairing status of all sequences within expressed mRNA molecules (Figure 1). If this is true, this approach can be used to generate and/or validate secondary structural predictions on a genome-wide scale. To test this hypothesis, we employed a novel methodology that produces structural models using sequence data obtained with a dsRNA-seq approach. For this analysis, we used sequence data obtained from samples that were processed using two rRNA-depletion steps (2X Ribominus approach (see Text S1; Figure S7)). We used this dataset because - although incredibly similar to the normal dsRNA-seq approach (see Text S1) - it is enriched for sense-strand mRNA sequences (Figure 7A and 7B, Figure S4D, and Figure S7), increasing the likelihood of generating useful secondary structure models. This mRNA secondary structure analysis revealed base-pairing differences between the structural models produced by the RNAfold program of the Vienna package (http://www.tbi.univie.ac.at/~ivo/RNA/) with and without dsRNA-seq constraints. Many regions that were predicted not to base-pair, but to form large loops and open regions by non-constrained RNAfold were more highly paired when constrained, and vice versa (see Figure 7C and 7D, http://tesla.pcbi.upenn.edu/annoj_at9/).

To test the ability of our structural modeling approach to predict highly base-paired regions, we characterized significantly paired regions of mRNAs (as determined by our methodology) (Figure 7C and 7D, see yellow regions) by reverse transcription (RT) polymerase chain reaction (RT-PCR) after digestion with a single-stranded or double-stranded RNase. We expected that the selected mRNA regions would be sufficiently intact for RT-PCR amplification after treatment with the single-stranded, but not the double-stranded RNase. As predicted, the regions of mRNA molecules determined to be highly base-paired were amplified following treatment with the ssRNase (Figure 7E). Conversely, we could not amplify these same regions after treatment with the dsRNase, which implies that they were completely degraded by this enzyme. These results demonstrated that dsRNA-seq reliably identifies base-paired portions of mRNAs. We also found that the models of secondary structure produced using dsRNA-seq data as constraints are predicted to be stable (Figure 7C, 7D, and 7F–7H, negative G values). In total, these results suggest that the constrained secondary structure models are accurate representations of folded RNAs in solution, providing valuable insight into the pairing status of RNA molecules genome-wide.
Finally, we used our mRNA secondary structure prediction methodology to produce folded models for the novel intergenic transcripts identified by the RNA-seq approach (Figure 6 and Figure S6). These structural models indicated that the new RNAs are highly base-paired, and are folded into a diverse array of stable (negative G values) secondary structures (Figure 7F–7H). Further evidence that these models are likely to be correct is provided by the observation that we obtained no dsRNA-reads for regions that are predicted to contain large loops by both dsRNA-seq data, as well as the RNAfold program of the Vienna package (http://www.tbi.univie.ac.at/~ivo/RNA/). We believe that these transcriptome-wide mRNA secondary structure models and corresponding web-based viewer (http://tesla.pcbi.upenn.edu/annoj_at9/) will be useful tools for elucidating the function of RNA folding in regulating gene expression and protein translation.
Conclusions
We describe in this report novel methodologies that produce a comprehensive genomic view of intra - and intermolecular base-paired RNAs at unprecedented resolution. We take advantage of the data from these approaches, which capture intra-molecular base-pairing interactions, to generate models of mRNA secondary structure in solution on a genome-wide scale (Figure 7). Although our methodology reveals the pairing status of RNA molecules in the absence of cellular proteins, previous studies have demonstrated that structural information obtained for RNAs in solution accurately reflects their structure in ribonucleoprotein complexes [3], [45]. Furthermore, our identification of conserved functional RNA domains using dsRNA-seq strongly suggests that RNA molecules are correctly folded into their secondary structure in solution (Figure 5). Overall, our results suggest we have produced highly informative models of mRNA secondary structure on a genome-wide scale for Arabidopsis, which can serve as a model for orthologous RNAs from other eukaryotic organisms.
As a resource for the larger community we have made available all sequencing data sets to NCBI Gene Expression OmniBus (GEO), and we have displayed them in a powerful and easy-to-use genome browser, Anno-J (http://tesla.pcbi.upenn.edu/annoj_at9/). Additionally, we have made the models of mRNA secondary structure freely available to the community through a structure viewer that has been incorporated into the dsRNA-seq Anno-J browser. Overall, the methods we have developed, as well as the highly informative sequencing data sets and models of RNA secondary structure that have resulted from this study will contribute positively to future work aimed at illuminating the numerous functions that RNA secondary structure has in regulating eukaryotic gene expression during developmental processes.
Materials and Methods
Text S1 information
Further details on the plant materials, experimental procedures, high-throughput sequencing, processing, mapping, and analysis of Illumina GA sequence reads are provided in Text S1. Primers used in this study are listed in Table S7.
dsRNA–seq library preparation
Briefly, total RNA is subjected to one (1X Ribominus) or two (2X Ribominus) rounds of rRNA depletion as per manufacturer's instructions (Ribominus, Invitrogen (Carlsbad, CA)). Next, these rRNA-depleted RNA samples are treated with a single-strand specific ribonuclease as per manufacturer's instructions (RNase One, Promega (Madison, WI)). The RNA sample is then used as the substrate for sequencing library construction using the Small RNA Sample Prep v1.5 kit (Illumina, San Diego, CA) as per manufacturer's instructions. For more detailed methodology see Text S1 and Figure S1A.
High-throughput sequencing
smRNA-seq and dsRNA-seq libraries were sequenced using the Illumina Genetic Analyzer II as per manufacturer's instructions (Illumina Inc., San Diego, CA).
Sequence read processing and mapping
Sequence information was extracted from the image files with the Illumina (San Diego, CA.) base calling software package (GAPipeline version 1.4). Prior to alignment, sequence reads were reduced to a list of only non-redundant (NR) sequences. NR sequences for which a 3′ adapter sequence was observed were truncated up to the junction with the adapter sequence, while sequences without recognizable 3′ adapters were also retained and processed independently. The dsRNA-seq and smRNA-seq reads were then aligned to the Arabidopsis genome (TAIR9 assembly). Finally, NR-sequences with their genomic coordinates were combined to form the final dataset. For more detailed methodology see Text S1.
Identification of dsRNA “hotspots” in the Arabidopsis genome
To identify dsRNA ‘hotspots’ in the Arabidopsis genome, we utilized a geometric distribution-based approach. For more detailed methodology see Text S1.
Gene Ontology (GO) enrichment of dsRNA “hotspot”-containing, protein-coding mRNAs
All protein-coding mRNAs overlapping identified dsRNA ‘hotspots’ were subjected to this analysis. Specifically, the GO enrichment analysis was carried out using the GOEAST web-based “Batch-Genes” tool [46].
Comparative genomics analysis of Arabidopsis dsRNA “hotspots”
The plant seven-way comparative genomics analysis was conducted as previously described. (http://genomewiki.ucsc.edu/index.php/Whole_genome_alignment_howto). For more detailed methodology see Text S1.
RNA structural models
We generated two computational structures for each annotated transcript. The unconstrained structure was obtained by folding with RNAfold v1.8.4 from the Vienna package with default parameters. The constrained structure was obtained with RNAfold using default parameters, but with structural constraints as additional input defined by reads from the dsRNA-seq approach. Specifically, any position covered by at least one mapped dsRNA read was constrained as paired (‘|’ in the structural constraint input); all other positions were left unconstrained (‘.’ in the structural constraint input).
Anno-J and RNA structure browser
The Anno-J Genome Browser is a REST-based genome annotation visualization program built using Web 2.0 technology. Licensing information and documentation are available at http://www.annoj.org.
We have developed a structure browser enhancement for Anno-J that enables visualization of the mRNA secondary structure models produced as described above. To do this, each predicted model was rendered as a SVG plot using Vienna (http://www.tbi.univie.ac.at/~ivo/RNA/) RNAplot. Reads and other features of interest such as UTR regions for mRNAs were then added to the SVG file. Read counts were normalized by the length of covered nucleotides (e.g. number of nucleotides covered by one or more reads). Users can visualize the structural model for an annotated transcript by selecting the corresponding genomic interval on Anno-J (RNA structures track) or by entering its accession number.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BrierleyI
PennellS
GilbertRJ
2007 Viral RNA pseudoknots: versatile motifs in gene expression and replication. Nat Rev Microbiol 5 598 610
2. CooperTA
WanL
DreyfussG
2009 RNA and disease. Cell 136 777 793
3. CruzJA
WesthofE
2009 The dynamic landscapes of RNA architecture. Cell 136 604 609
4. MendellJT
DietzHC
2001 When the message goes awry: disease-producing mutations that influence mRNA content and performance. Cell 107 411 414
5. MontangeRK
BateyRT
2008 Riboswitches: emerging themes in RNA structure and function. Annu Rev Biophys 37 117 133
6. BurattiE
MuroAF
GiombiM
GherbassiD
IaconcigA
2004 RNA folding affects the recruitment of SR proteins by mouse and human polypurinic enhancer elements in the fibronectin EDA exon. Mol Cell Biol 24 1387 1400
7. SharpPA
2009 The centrality of RNA. Cell 136 577 580
8. BaulcombeD
2004 RNA silencing in plants. Nature 431 356 363
9. CarthewRW
SontheimerEJ
2009 Origins and Mechanisms of miRNAs and siRNAs. Cell 136 642 655
10. BartelDP
2004 MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116 281 297
11. MeisterG
TuschlT
2004 Mechanisms of gene silencing by double-stranded RNA. Nature 431 343 349
12. Jones-RhoadesMW
BartelDP
BartelB
2006 MicroRNAS and their regulatory roles in plants. Annu Rev Plant Biol 57 19 53
13. AlmeidaR
AllshireRC
2005 RNA silencing and genome regulation. Trends Cell Biol 15 251 258
14. TomariY
ZamorePD
2005 Perspective: machines for RNAi. Genes Dev 19 517 529
15. MaidaY
YasukawaM
FuruuchiM
LassmannT
PossematoR
2009 An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA. Nature 461 230 235
16. DalmayT
HamiltonA
RuddS
AngellS
BaulcombeDC
2000 An RNA-dependent RNA polymerase gene in Arabidopsis is required for posttranscriptional gene silencing mediated by a transgene but not by a virus. Cell 101 543 553
17. MourrainP
BeclinC
ElmayanT
FeuerbachF
GodonC
2000 Arabidopsis SGS2 and SGS3 genes are required for posttranscriptional gene silencing and natural virus resistance. Cell 101 533 542
18. GazzaniS
LawrensonT
WoodwardC
HeadonD
SablowskiR
2004 A link between mRNA turnover and RNA interference in Arabidopsis. Science 306 1046 1048
19. VoinnetO
2008 Use, tolerance and avoidance of amplified RNA silencing by plants. Trends Plant Sci 13 317 328
20. AllenE
XieZ
GustafsonAM
CarringtonJC
2005 microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 121 207 221
21. AdenotX
ElmayanT
LauresserguesD
BoutetS
BoucheN
2006 DRB4-dependent TAS3 trans-acting siRNAs control leaf morphology through AGO7. Curr Biol 16 927 932
22. FahlgrenN
MontgomeryTA
HowellMD
AllenE
DvorakSK
2006 Regulation of AUXIN RESPONSE FACTOR3 by TAS3 ta-siRNA affects developmental timing and patterning in Arabidopsis. Curr Biol 16 939 944
23. GarciaD
CollierSA
ByrneME
MartienssenRA
2006 Specification of leaf polarity in Arabidopsis via the trans-acting siRNA pathway. Curr Biol 16 933 938
24. BorsaniO
ZhuJ
VersluesPE
SunkarR
ZhuJK
2005 Endogenous siRNAs derived from a pair of natural cis-antisense transcripts regulate salt tolerance in Arabidopsis. Cell 123 1279 1291
25. PeragineA
YoshikawaM
WuG
AlbrechtHL
PoethigRS
2004 SGS3 and SGS2/SDE1/RDR6 are required for juvenile development and the production of trans-acting siRNAs in Arabidopsis. Genes Dev 18 2368 2379
26. VazquezF
VaucheretH
RajagopalanR
LepersC
GasciolliV
2004 Endogenous trans-acting siRNAs regulate the accumulation of Arabidopsis mRNAs. Mol Cell 16 69 79
27. YoshikawaM
PeragineA
ParkMY
PoethigRS
2005 A pathway for the biogenesis of trans-acting siRNAs in Arabidopsis. Genes Dev 19 2164 2175
28. HunterC
WillmannMR
WuG
YoshikawaM
de la Luz Gutierrez-NavaM
2006 Trans-acting siRNA-mediated repression of ETTIN and ARF4 regulates heteroblasty in Arabidopsis. Development 133 2973 2981
29. WalkerTA
JohnsonKD
OlsenGJ
PetersMA
PaceNR
1982 Enzymatic and chemical structure mapping of mouse 28S ribosomal ribonucleic acid contacts in 5.8S ribosomal ribonucleic acid. Biochemistry 21 2320 2329
30. FischerRL
GoldbergRB
1982 Structure and flanking regions of soybean seed protein genes. Cell 29 651 660
31. AxtellMJ
JanC
RajagopalanR
BartelDP
2006 A two-hit trigger for siRNA biogenesis in plants. Cell 127 565 577
32. ChenHM
LiYH
WuSH
2007 Bioinformatic prediction and experimental validation of a microRNA-directed tandem trans-acting siRNA cascade in Arabidopsis. Proc Natl Acad Sci U S A 104 3318 3323
33. HowellMD
FahlgrenN
ChapmanEJ
CumbieJS
SullivanCM
2007 Genome-wide analysis of the RNA-DEPENDENT RNA POLYMERASE6/DICER-LIKE4 pathway in Arabidopsis reveals dependency on miRNA - and tasiRNA-directed targeting. Plant Cell 19 926 942
34. LuC
KulkarniK
SouretFF
MuthuValliappanR
TejSS
2006 MicroRNAs and other small RNAs enriched in the Arabidopsis RNA-dependent RNA polymerase-2 mutant. Genome Res 16 1276 1288
35. ChanSW
HendersonIR
JacobsenSE
2005 Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat Rev Genet 6 351 360
36. PontierD
YahubyanG
VegaD
BulskiA
Saez-VasquezJ
2005 Reinforcement of silencing at transposons and highly repeated sequences requires the concerted action of two distinct RNA polymerases IV in Arabidopsis. Genes Dev 19 2030 2040
37. QiY
HeX
WangXJ
KohanyO
JurkaJ
2006 Distinct catalytic and non-catalytic roles of ARGONAUTE4 in RNA-directed DNA methylation. Nature 443 1008 1012
38. ZhengX
ZhuJ
KapoorA
ZhuJK
2007 Role of Arabidopsis AGO6 in siRNA accumulation, DNA methylation and transcriptional gene silencing. EMBO J 26 1691 1701
39. FangY
SpectorDL
2007 Identification of nuclear dicing bodies containing proteins for microRNA biogenesis in living Arabidopsis plants. Curr Biol 17 818 823
40. SongL
HanMH
LesickaJ
FedoroffN
2007 Arabidopsis primary microRNA processing proteins HYL1 and DCL1 define a nuclear body distinct from the Cajal body. Proc Natl Acad Sci U S A 104 5437 5442
41. LeeYS
ShibataY
MalhotraA
DuttaA
2009 A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev 23 2639 2649
42. LunterG
PontingCP
HeinJ
2006 Genome-wide identification of human functional DNA using a neutral indel model. PLoS Comput Biol 2 e5 doi:10.1371/journal.pcbi.0020005
43. WaterstonRH
Lindblad-TohK
BirneyE
RogersJ
AbrilJF
2002 Initial sequencing and comparative analysis of the mouse genome. Nature 420 520 562
44. ListerR
O'MalleyRC
Tonti-FilippiniJ
GregoryBD
BerryCC
2008 Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133 523 536
45. DibrovSM
ParsonsJ
HermannT
2010 A model for the study of ligand binding to the ribosomal RNA helix h44. Nucleic Acids Res 38 4458 4465
46. ZhengQ
WangXJ
2008 GOEAST: a web-based software toolkit for Gene Ontology enrichment analysis. Nucleic Acids Res 36 W358 363
47. CannoneJJ
SubramanianS
SchnareMN
CollettJR
D'SouzaLM
2002 The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics 3 2
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 9
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Synthesizing and Salvaging NAD: Lessons Learned from
- Optimal Strategy for Competence Differentiation in Bacteria
- Long- and Short-Term Selective Forces on Malaria Parasite Genomes
- Identifying Signatures of Natural Selection in Tibetan and Andean Populations Using Dense Genome Scan Data
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy