
DSIF and RNA Polymerase II CTD Phosphorylation Coordinate the Recruitment of Rpd3S to Actively Transcribed Genes
Histone deacetylase Rpd3 is part of two distinct complexes:
the large (Rpd3L) and small (Rpd3S) complexes. While Rpd3L targets specific promoters for gene repression, Rpd3S is recruited to ORFs to deacetylate histones in the wake of RNA polymerase II, to prevent cryptic initiation within genes. Methylation of histone H3 at lysine 36 by the Set2 methyltransferase is thought to mediate the recruitment of Rpd3S. Here, we confirm by ChIP–Chip that Rpd3S binds active ORFs. Surprisingly, however, Rpd3S is not recruited to all active genes, and its recruitment is Set2-independent. However, Rpd3S complexes recruited in the absence of H3K36 methylation appear to be inactive. Finally, we present evidence implicating the yeast DSIF complex (Spt4/5) and RNA polymerase II phosphorylation by Kin28 and Ctk1 in the recruitment of Rpd3S to active genes. Taken together, our data support a model where Set2-dependent histone H3 methylation is required for the activation of Rpd3S following its recruitment to the RNA polymerase II C-terminal domain.
Published in the journal:
. PLoS Genet 6(10): e32767. doi:10.1371/journal.pgen.1001173
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001173
Summary
Histone deacetylase Rpd3 is part of two distinct complexes:
the large (Rpd3L) and small (Rpd3S) complexes. While Rpd3L targets specific promoters for gene repression, Rpd3S is recruited to ORFs to deacetylate histones in the wake of RNA polymerase II, to prevent cryptic initiation within genes. Methylation of histone H3 at lysine 36 by the Set2 methyltransferase is thought to mediate the recruitment of Rpd3S. Here, we confirm by ChIP–Chip that Rpd3S binds active ORFs. Surprisingly, however, Rpd3S is not recruited to all active genes, and its recruitment is Set2-independent. However, Rpd3S complexes recruited in the absence of H3K36 methylation appear to be inactive. Finally, we present evidence implicating the yeast DSIF complex (Spt4/5) and RNA polymerase II phosphorylation by Kin28 and Ctk1 in the recruitment of Rpd3S to active genes. Taken together, our data support a model where Set2-dependent histone H3 methylation is required for the activation of Rpd3S following its recruitment to the RNA polymerase II C-terminal domain.
Introduction
Histone acetylation was the first covalent histone modification shown to be involved in transcription regulation. Indeed, histones at the promoter of active genes tend to be hyper-acetylated while repressed genes have promoters with hypo-acetylated nucleosomes. It is now well established that this is due to the recruitment of histone acetyltransferases (HATs) and histone deacetylases (HDACs) by transcriptional activators and repressors, respectively [1]. The first described and best characterized HDAC is Rpd3. The repressive effect of yeast Rpd3 on transcription has been well studied over the last 15 years, paving the way for the characterization of its mammalian orthologs [2]. Yeast Rpd3 is recruited to the promoters of specific genes by DNA-binding repressors, leading to the repression of many important pathways such as stress response, meiosis, the cell cycle and others [3]–[19]. Moreover, Rpd3 also plays roles in silencing [20]–[27], DNA replication [28]–[31] and recombination [32], [33].
Recent proteomic studies have determined that Rpd3 is found in two distinct complexes: the large (Rpd3L) and the small (Rpd3S) complex [34], [35]. Both complexes share a core composed of Rpd3, Sin3 and Ume1. The large complex is composed of 11 additional proteins whereas the small complex contains only two additional subunits, namely Rco1 and Eaf3. While Rpd3L is likely responsible for the repressive function of Rpd3, the function of Rpd3S remains far less understood. Recent work by several groups has shown that Rpd3S is involved in the suppression of cryptic transcription [34], [36], [37] and that its activity is linked to the Set2 histone methyltransferase (HMT). Furthermore, in vitro studies have shown that Rpd3S is recruited to H3K36-methylated nucleosomes and that its Rco1 and Eaf3 subunits are essential for this recruitment [34], [35], [37], [38]. Rco1 mediates interactions with histones in a modification-independent manner through a PHD zinc finger domain, while Eaf3 contains a methyl-lysine binding chromodomain (CHD) that is essential for recognition of H3K36-trimethylated (H3K36me3) nucleosomes in vitro. These studies also indicate that genome-wide histone acetylation levels on promoters and coding regions are altered when either H3K36 methylation or Rpd3S is disrupted.
Taken together, these data lead to a model where Rpd3S is recruited to coding regions through the interaction of Eaf3 with H3K36me3 in order to deacetylate nucleosomes after the passage of the transcriptional machinery [39]. Deacetylation would allow chromatin disrupted by elongating RNA polymerase II (RNAPII) to return to a more ordered and compact structure, thereby restoring an environment hostile to cryptic transcription initiation within the coding region. However, this model relies heavily on the in vitro observation that the Eaf3 CHD binds preferentially to H3K36me3 peptides or nucleosomes [34], [35], [37], [38]. It was never formally demonstrated that this interaction is required for the targeting of Rpd3S to coding regions in vivo. In addition, whether Rpd3S is recruited to all transcribed genes or to only a subset of them has never been assessed. In order to address these questions, we performed genome-wide ChIP-chip experiments looking at both Rpd3S - and Rpd3L-specific subunits in wild type cells and in various mutants, including set2Δ and H3K36A (Table S1 and S2).
Quite interestingly, our data show that Rpd3S specifically binds to the coding region of actively transcribed genes whose promoters are also bound by Rpd3L. Surprisingly, the binding of Rpd3S to active genes is not dependant on Set2-mediated H3K36 methylation. However, methylation by Set2 is required for the activity of Rpd3S, as assayed by histone acetylation and RNAPII levels. We also provide in vivo evidence that Rpd3S is recruited to active genes via the phosphorylation of the RNAPII C-terminal domain (CTD). Finally, we show that the yeast DSIF transcription elongation factor negatively regulates Rpd3S recruitment. Based on these results, we propose that the recruitment and activity of Rpd3S on ORFs depend on a two steps mechanism: an initial recruitment to the elongation complex -coordinated by DSIF and RNAPII phosphorylation-, followed by the H3K36me3-dependant modulation of Rpd3S activity through the Eaf3 chromodomain.
Results
Rpd3S is not recruited to all transcribed genes
While it is generally accepted that Rpd3L negatively regulates specific sets of genes, the specific function of Rpd3S remains largely unaddressed. However, it is expected to be ubiquitously recruited to active genes since it interacts with methylated H3K36. In order to address the specificity of Rpd3S and Rpd3L in a systematic manner, we performed ChIP-chip experiments on myc-tagged Rpd3, Rco1 and Sds3, the last two being specific subunits of Rpd3S and Rpd3L, respectively. As shown in Figure 1A, Rpd3 binds to a large subset of promoters (Figure 1A, clusters 1 and 2). In addition, a subset of these genes also exhibit Rpd3 binding on their coding regions (Figure 1A, cluster 2). Figure 1B and 1C show the average signal of Rpd3, Rco1 and Sds3 over the genes from cluster 1 and cluster 2, respectively. The data for cluster 3, representing the genes not bound by Rpd3, are also shown. Cluster 1, which is enriched for genes previously demonstrated to be repressed by Rpd3 (genes involved in M phase (p-value 10−8), cell cycle (p-value 10−9), etc.), shows binding of both Rpd3 and Sds3 to the promoter. The presence of Rco1 is not detectable on these genes. This cluster therefore represents genes repressed by Rpd3L, which is consistent with the fact that these genes have low level of RNAPII (Figure S1A). Cluster 2, however, shows evidence for the presence of both Rpd3L and Rpd3S since all three subunits tested for are detected. Rco1 is restricted to the coding region of these genes, consistent with the fact that they are actively transcribed (Figure S1A). Sds3 is present at the promoter, which is expected since Rpd3 binds to these promoters. More surprisingly, however, some level of Sds3 is also detected on the coding region of these genes. These data - also observed with another subunit of Rpd3L (Rxt2; Figure S1B) - suggest that the large complex may play some role during transcriptional elongation, perhaps in conjunction with the small complex (see Discussion). Nevertheless, the data presented here clearly show that Rpd3S binds to the coding region of active genes.

Strikingly, these experiments also show that Rpd3S preferentially associates with genes that are also bound by Rpd3L. In fact, we found no clusters of genes where Rpd3 binds in the coding region but not in the promoter. This suggests that Rpd3S, contrary to what has been expected, does not ubiquitously bind to active genes but rather targets some of them, namely a subset of those that are bound by Rpd3L at their promoter. In order to test if Rpd3S ubiquitously binds active genes, we performed ChIP-chip experiments of RNAPII and H3K36me3, two proxies for active gene expression. Figure 1D (cluster 4) clearly shows that many genes, despite having strong enrichment for RNAPII and H3K36me3, show no evidence of Rpd3S binding. Three conclusions can be drawn from these results; firstly, they confirm that the recruitment of Rpd3S is not a general phenomenon occurring on all transcribed genes; secondly, they suggest that the methylation of H3K36 by Set2 is not providing the specificity for the recruitment of Rpd3S; and finally they suggest that the large complex may play a role in the recruitment of the small complex as Rpd3S appears to target primarily genes also bound by Rpd3L.
Set2 is not required for Rpd3S binding to ORFs in vivo
The idea that H3K36me3 recruits Rpd3S through the Eaf3 subunit is well established in the literature [39]. Work done in vitro by several groups has clearly shown this using peptides and nucleosomal substrates [34], [35], [37], [38]. Our ChIP-chip experiments, however, clearly demonstrate that many genes harboring high levels of H3K36me3 are free of Rpd3S. We have therefore endeavored to examine whether the well characterized interaction between the Eaf3 CHD and H3K36me3 is responsible for the targeting of Rpd3S in vivo.
First, we looked at Rco1 binding in a set2Δ mutant. Since this mutant cannot methylate H3K36, the current model predicts that Rpd3S should not bind to ORFs under these conditions. To our considerable surprise, Rco1 enrichment on ORFs in this mutant is not significantly altered for about two-thirds of Rpd3S target genes (Figure 2A, cluster 5). For other genes, Rpd3S occupancy is decreased significantly, although not completely abolished (Figure 2A, cluster 6). We next repeated these experiments in a H3K36A mutant (where lysine 36 is mutated into an alanine) with similar results (Figure 2A). The deletion of the Rco1 PHD domain had no effect on Rpd3S occupancy (despite the fact that it destabilizes the Rco1 protein; Figure 2E), while deletion of the Eaf3 CHD domain phenocopied set2Δ and H3K36A. Interestingly, a set1Δ/set2Δ/dot1Δ triple mutant (abolishing all histone methylation activity in yeast) was similar to wild type, suggesting that deletion of SET1 and/or DOT1 can partially suppress the set2Δ phenotype. Taken together, these data demonstrate that H3K36 methylation is not required for the recruitment of Rpd3S to most genes.
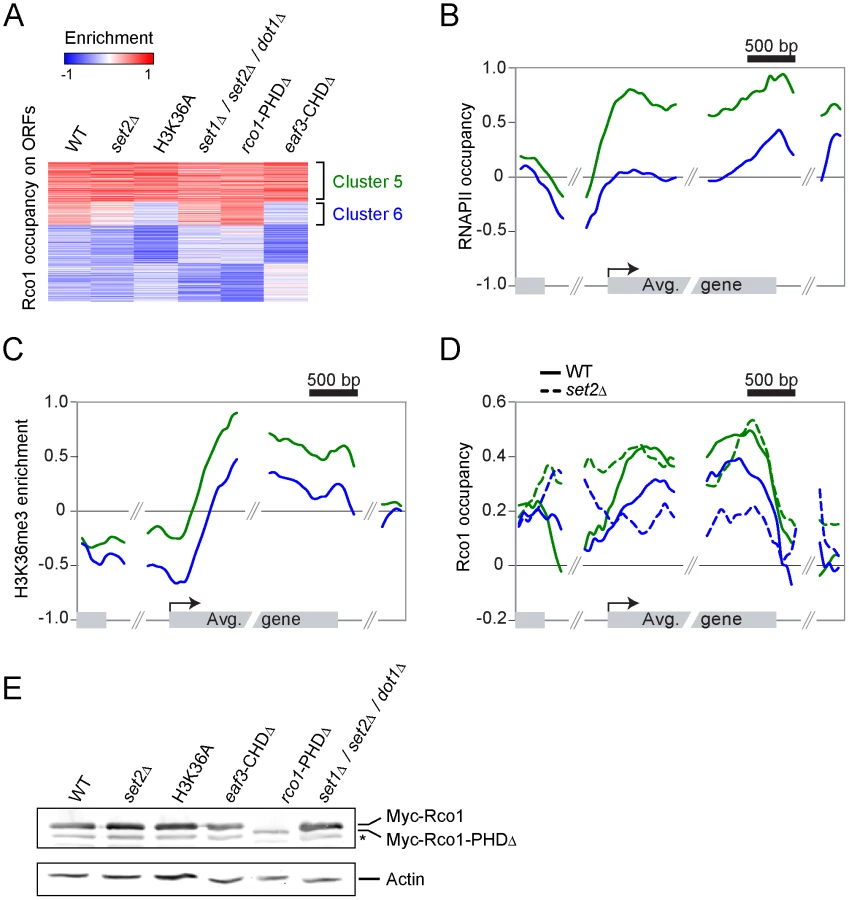
Since Rpd3S occupancy seems to be more dependent on H3 methylation at some genes than others, we looked more closely at clusters 5 and 6. As shown in Figure 2B and 2C, clusters 5 and 6 are markedly different with regards to transcription levels. While cluster 5 is highly transcribed (as shown by the presence of high levels of both RNAPII and H3K36me3), cluster 6 is less so. Next we looked at the distribution of Rco1 on genes contained within these clusters in all strains shown in Figure 2A. As expected from data shown in Figure 2A, Rco1 occupancy on ORFs is not (or only slightly) affected in these mutants for the cluster 5 genes, but it is reduced for the genes from cluster 6 (Figure 2D and Figure S2). In addition, for all Rpd3S-bound genes, a redistribution of Rco1 to the promoter region was observed in all mutants tested. This redistribution towards the promoter remains unexplained but correlates with our observation that histone acetylation is decreased at promoters in these same mutants (Figure S3). Collectively, these data clearly demonstrate that H3K36 methylation has no impact on Rpd3S occupancy at highly transcribed genes (genes from cluster 5). However, the methyl mark, or the ability to recognize it through the Eaf3 chromodomain, is important for optimal Rpd3S association to ORFs with lower levels of RNAPII (genes from cluster 6).
Set2 and Rpd3S regulate the distribution of RNAPII and histone acetylation
It is known that set2Δ mutants, along with null mutants of Rpd3S subunits Eaf3 or Rco1, exhibit a cryptic transcription phenotype [34], [36], [37]. This phenotype is thought to be due to the improper deacetylation of transcribed ORFs by Rpd3S after each round of transcription [39] because of a lack of Rpd3S recruitment. Other groups that have characterized acetylation levels on coding regions in Set2 and Rpd3S mutants either looked at bulk chromatin by western blotting [37], or at specific genes by ChIP [34], [35], [38], [40] and have come to the conclusion that acetylation levels increase on ORFs when Set2 or Rpd3S is disrupted.
Since we—quite surprisingly—observed, however, that Rpd3S binding to genes is mostly independent of histone methylation by Set2, we decided to test whether the activity of Rpd3S requires methylation of H3K36 by Set2. To do so, we looked at H4K5 acetylation (H4K5ac) by ChIP-chip. We used H4K5 acetylation to score for Rpd3S activity because it was shown previously to be a robust read out for Rpd3 activity in ChIP-chip assays [41]. Similar to other groups [34], [35], [38], [40], we observed decreased acetylation on promoters in set2Δ, H3K36A or Rpd3S mutants (Figure S3). Histone acetylation is also dramatically affected across ORFs in these mutants. As shown in Figure 3A, we observed a loss of acetylation for normally highly acetylated ORFs, and a gain in acetylation for ORFs that exhibit low levels in the wild type. Because Set2 and Rpd3S are both known to prevent cryptic initiation within ORFs, we repeated the same analyses on RNAPII ChIP-chip results, and found a similar pattern to that observed for H4K5ac, namely that ORFs with high RNAPII enrichment show decreased RNAPII levels in the absence of Set2 or Rpd3S, whereas ORFs with low RNAPII tend to display higher levels of polymerase (Figure 3B). These results clearly show that the activity of the Rpd3S complex requires methylation of H3K36 by Set2.

The effect of the loss of Rpd3S activity on histone acetylation and RNAPII distribution on ORFs is more complex than previously described. Our data indeed suggest that both histone acetylation and RNAPII occupancy are redistributed in a more even manner across the genome than expected. A plausible explanation of the genome-wide averaging of RNAPII and histone acetylation levels in Set2 and Rpd3S mutants would entail aberrant recruitment of the transcriptional apparatus to low-expression genes whose coding regions were not reset properly by a, now inactive, Rpd3S complex. Assuming a limited pool of transcription machinery in a cell, this would result in a lower abundance of RNAPII at the more active ORFs, and aberrant genomic acetylation levels.
These results, combined with the Rpd3S occupancy profiles in rco1Δ, rco1-PHDΔ and eaf3-CHDΔ mutants, suggest that the loss of histone H3 methylation at lysine 36 affects ORF identity through a modulation of Rpd3S deacetylase activity rather than through altered recruitment of the Rpd3S complex on coding regions as previously thought.
Spt4 is required for the proper localization of Rpd3S
Since the interaction between the Eaf3 CHD and H3K36me3 does not account for the initial recruitment of Rpd3S to active genes, we decided to look for a factor that would fulfill that role. Quan and Hartzog have shown genetic interactions between H3K36 methylation and Rpd3S with Spt5 [42]. Their data suggest that Rpd3S opposes the function of the elongation factor Spt4/5, which is the yeast ortholog of the human elongation factor DSIF [43]. DSIF negatively regulates elongation in its non-phosphorylated form, but is turned into a positive elongation factor upon phosphorylation by P-TEFb (Bur1 in yeast) [44]–[46]. The genetic interaction between Rpd3S and Spt5 led us to test whether Spt4/Spt5 is involved in the recruitment of Rpd3S. We therefore performed ChIP-chip experiments of Rco1 in spt4Δ cells (the deletion of SPT5 is lethal). As shown in Figure 4A, deletion of SPT4 leads to massive changes in Rco1 binding across the genome. Notably, the effect is far more dramatic compared to the deletion of SET2 (Figure 4A). Importantly, the level of RNAPII observed on these genes is not significantly affected in the mutant, ruling out the possibility that the effect is solely due to a reshuffling of the transcriptome (Pearson correlation = 0.94, Figure S4A). The deletion of SPT4 causes a decrease of Rco1 binding at some transcribed genes normally strongly associated with Rco1 (Figure 4A, cluster 7), as well as an increase at others where Rco1 is otherwise only found at low levels (cluster 8). Deletion of SPT4 even causes a slight increase of Rco1 occupancy at genes where it is normally undetectable (cluster 9). Overall, these effects lead to a distribution of Rpd3S that correlates better with RNAPII occupancy than in wild type cells (Figure 4B). To test the possibility that, in the absence of Spt4, Rpd3S is recruited via H3K36 methylation, we profiled Rco1 binding in a spt4Δ/set2Δ double mutant. As shown in Figure 4A, deleting both SPT4 and SET2 leads to a similar Rpd3S localization phenotype compared to the single spt4Δ mutant, giving further evidence that Set2 does not play a large role in Rpd3S recruitment, even in the absence of Spt4.

Spt4 negatively regulates the association of Rco1 with transcribed genes
To distinguish the direct effect of the loss of SPT4 from eventual indirect effects of the mutation, we localized Spt4 in wild type cells by ChIP-chip using a strain carrying a myc-tagged SPT4 gene. Spt4 associates with genes in a manner that correlates with levels of RNAPII (Pearson correlation = 0.84) suggesting that DSIF acts as a general elongation factor. Moreover, it is present across the whole ORF, indicating that it travels with RNAPII, but is further enriched in the 3′ end of genes (Figure S4B), suggesting that it may also regulate the elongation-termination transition, as shown by others [47]–[49]. This is also consistent with the fact that Spt5 interacts with components of the capping and termination machineries [50]. Even more interestingly, the more a gene is occupied by Spt4 in wild type cells, the more Rco1 we detect in the spt4Δ mutant (Figure 4C), suggesting that the direct effect of the loss of Spt4 is an increase in Rpd3S binding (as observed for cluster 8). Consequently, the decrease in occupancy observed in cluster 7 is most likely indirect since Spt4 is barely detectable at these genes (Figure 4A). Similarly, the level of Rco1 increases dramatically in the spt4Δ mutant on cluster 4 genes (from Figure 1D), representing transcribed genes highly occupied by Spt4 where Rco1 is absent in wild type cells (Figure S4C). In general, genes with a higher Spt4/RNAPII ratio tend to have less Rco1 than genes with lower Spt4/RNAPII ratios (Pearson correlation = −0.42, Figure S4D). Taken together, these data suggest that Spt4 acts as a negative regulator of Rpd3S binding and that its presence prevents the HDAC from freely associating with transcribed genes. This model is in agreement with genetic data showing that Rco1 opposes the function of Spt4/5 [42].
We then looked at the distribution of Rpd3S on transcribed genes where Spt4 is also bound (cluster 8) and found strong differences between the wild type and spt4Δ mutants. While Rco1 occupies the whole ORF at a constant level in wild type cells, it accumulates to abnormally high levels towards the 3′ end of the gene in spt4Δ cells (Figure 4D, dashed line). This binding pattern is also found in the spt4Δ/set2Δ double mutant (compare dashed and dotted lines in Figure 4D). The occupancy profile of Rco1 in a spt4Δ mutant shows a strong similarity to the occupancy profile of RNAPII with a CTD phosphorylated at Ser2 (Figure 4D, red solid line). This led us to hypothesize that CTD phosphorylation by Ctk1 (the major serine 2 kinase) might be implicated in Rpd3S recruitment in the absence of Spt4/5. To test this hypothesis, we profiled Rco1 occupancy in a spt4Δ/ctk1Δ background. Surprisingly, ctk1Δ partially suppressed the spt4Δ Rpd3S binding pattern phenotype (Figure 4A) leading to an intermediate binding profile between wild type and spt4Δ. Looking at it more closely, we observed that short genes fail to accumulate Rco1 in spt4Δ/ctk1Δ cells (Figure 4E), whereas Rco1 is observed at genes irrespective of their length in wild type (Figure S5) or spt4Δ cells (Figure 4F), suggesting that Rpd3S accumulates more slowly in spt4Δ/ctk1Δ cells. We therefore conclude that in the absence of Spt4, Rpd3S binds to the RNAPII CTD and that the phosphorylation of the CTD at serine 2 contributes to that phenomenon.
Phosphorylation of the RNAPII CTD triggers the association of Rpd3S to active genes
The data presented above demonstrate that Spt4 negatively regulates the association of Rpd3S to highly transcribed genes. The data also suggest that phosphorylation of the RNAPII CTD, notably at serine 2, plays some role in the recruitment of Rpd3S to active genes in the absence of Spt4. We next tested whether the phosphorylation of the CTD is implicated in the association of Rpd3S with transcribed genes in wild type cells and performed ChIP-chip experiments of Rco1 in mutants for the known CTD kinases. As shown in Figure 5A, deletion of CTK1, the major serine 2 kinase, has a clear effect on Rco1 occupancy (compare solid line with dashed line). To test the effect of serine 5 and 7 phosphorylation, we used a strain carrying ATP-analog-sensitive alleles of KIN28 since the deletion of the KIN28 gene is lethal. As shown in Figure 5A (dotted line), inhibition of Kin28 has a dramatic effect on Rco1 occupancy. Indeed, no Rco1 can be detected on ORFs in that mutant. This clearly demonstrates that phosphorylation of serine 5 and/or 7 by Kin28 is a major element in the recruitment of Rpd3S to active genes. These results are supported by data from the Hinnebusch lab who have shown that Rpd3S interacts with the phosphorylated form of RNAPII and has high affinity for doubly phosphorylated (serine 2/5) CTD peptides in vitro [51]. Interestingly, CTD peptides carrying a single phosphate group on serine 5 or serine 2 respectively have a much weaker or no affinity to Rpd3S compared to doubly phosphorylated CTD peptides. Also noteworthy is the fact that Rpd3S is redistributed to promoter regions when Kin28 is inactive (Figure 5A). As we will be discuss below, this might be due to a decrease in H3K36 methylation in that mutant, most likely due to a defective Bur1/2 recruitment, as suggested by [52] and [53], leading to a defect in Rad6 phosphorylation. Our genome-wide data, together with these in vitro experiments, suggest that phosphorylation of the RNAPII CTD stimulates the recruitment of Rpd3S to transcribed genes while the elongation factor DSIF counteracts this recruitment.

Phosphorylation of Spt5 by Bur1 negatively regulates the activity of DSIF on Rpd3S recruitment
In mammalian cells, DSIF is phosphorylated by Cdk9, a cyclin-dependent kinase associated with the elongation factor P-TEFb [54]. Cdk9 also phosphorylates the RNAPII CTD on serine 2 as well as other proteins including NELF. In yeast, the function of Cdk9 is fulfilled by two distinct kinases. Ctk1 mainly phosphorylates the RNAPII CTD and Bur1 targets Spt5 and Rad6 [55]–[57]. Inactivation of Bur1 has modest effects on phosphorylation of the RNAPII CTD, as shown by western blot and by ChIP-chip experiments (data not shown; see also [52], [57], [58]). Because Bur1 phosphorylates Spt5, the partner of Spt4, we tested the effect of inactivating Bur1 activity on Rco1 recruitment. Not surprisingly, inhibiting Bur1 using an ATP-analog-sensitive strategy (bur1AS) has a profound effect on Rco1 occupancy. In the absence of a functional Bur1, Rco1 is depleted from the coding region and redistributed to promoter regions (Figure 5B, dashed line) as was observed in the Kin28 mutant while the RNAPII level is mostly unchanged (data not shown). Deleting the CTD of Spt5 (spt5ΔC), the region phosphorylated by Bur1, caused a similar, although milder, phenotype (Figure 5B, dotted line). The stronger effect observed in the Bur1 inactivation experiment, compared to truncation of Spt5, is most likely due to the fact that Bur1 has additional targets. For example, Bur1 phosphorylates Rad6, an event that is required for the methylation of H3K4 and K79 by Set1 and Dot1 respectively [59]–[61]. In a triple mutant for Set1, Set2 and Dot1, we observed a shift of Rpd3S toward intergenic regions (Figure S2B), suggesting that the redistribution of Rpd3S to promoter regions in Bur1-impaired cells is at least in part a consequence of Bur1's activity on Rad6. Nevertheless, on the coding region, where our spt4Δ data showed that DSIF negatively regulates Rpd3S binding, we observed a clear decrease in Rco1 occupancy in both bur1AS and spt5ΔC. These data strongly suggest that phosphorylation of Spt5 by Bur1 negatively regulates the activity of DSIF on Rpd3S recruitment. While we cannot completely rule out the possibility that some of the effect observed in the bur1AS strain is due to an effect of Bur1 on the RNAPII CTD, our data on spt5ΔC rather suggest that Bur1 regulates Rpd3S recruitment by regulating DSIF. Interestingly, phosphorylation of Spt5 by Bur1 was previously shown to stimulate its activity as an elongation factor while our data suggest that it inhibits its activity as a negative regulator of Rpd3S recruitment. It will therefore be interesting to see whether these activities are linked.
Discussion
The Rpd3S complex is recruited to the coding region of transcribed genes where it represses cryptic transcription by deacetylating histones in the wake of the elongating polymerase, therefore resetting chromatin to its pre-transcriptional state. The recruitment of Rpd3S to transcribed regions was thought to be mediated by the interaction between the Eaf3 CHD and H3K36me. However, the interaction of Rpd3S on the coding regions of actively transcribed genes has never been directly demonstrated in vivo. Here, we build on that model and show that: 1) Rpd3S targets a subset of transcribed genes; 2) Rpd3L is present at the promoter of genes where Rpd3S is bound; 3) The activity of Rpd3S requires methylation of H3K36 by Set2 but its association with active genes does not; 4) The recruitment of Rpd3S on ORFs requires the phosphorylation of the RNAPII CTD by Kin28 and Ctk1; and 5) The DSIF elongation complex counteracts the recruitment of Rpd3S to transcribed genes, a phenomenon that is regulated by the phosphorylation of Spt5 by Bur1. We therefore propose a model where the opposing effects of CTD phosphorylation and DSIF on Rpd3S recruitment together allow for the complex occupancy profile of that HDAC in vivo (Figure 6).

The distribution of Rpd3S along the genome is more complex than expected
Because H3K36 methylation correlates with transcription, it is generally accepted to be present at all transcribed genes; Rpd3S was therefore expected to follow the same pattern. Our finding that many transcribed genes do not show any signs of Rpd3S binding despite the presence of H3K36me3 was therefore a considerable surprise. This has important implications since it suggests that not all genes are equally protected against cryptic initiation. Other mechanisms exist that prevent cryptic transcription [62], so it will be interesting to investigate how these various activities share the labor of protecting the genome from aberrant transcription.
Does Rpd3L play a role in the targeting of Rpd3S?
Another peculiarity of Rpd3S is that it is preferentially enriched on the coding regions of genes where Rpd3L is found on the promoter: we rarely find Rpd3S on genes where Rpd3L is not present. The presence of Rpd3L on the promoter of active genes is counterintuitive given its role as a co-repressor but it has nevertheless being observed before [17]. The co-occurrence of Rpd3L at promoter and Rpd3S on the ORFs of the same genes suggests that Rpd3L may play a role in targeting Rpd3S. One possibility would be that Rpd3S emerges from Rpd3L during the transition from initiation to elongation. The transition to elongation may trigger the exchange of subunits to transform Rpd3L into Rpd3S. Our finding that DSIF and RNAPII CTD phosphorylation are involved in the proper recruitment of Rpd3S to chromatin suggests that they may play a role in that process.
Unexpectedly, we also observe Rpd3L on the coding regions of the genes where Rpd3S is also present. This last result was surprising in that it does not agree with the generally accepted function of Rpd3L as a promoter-recruited co-repressor. These data argue for a new model where subunits of both Rpd3L and Rpd3S coexist on actively transcribed coding regions. According to the model described above, the presence of Rpd3L subunits may reflect an imperfect exchange of subunits during the transition from initiation to elongation. Alternatively, it may reflect the presence of an Rpd3 “super large” complex that contains subunits of both Rpd3S and Rpd3L. In such a model, Rpd3S (as we normally see it) would only exist in solution and would represent a module that joins Rpd3L after initiation. This hypothesis is supported by data provided by Collins et al. [63] who combined and re-analyzed previously published mass spectrometry data [64], [65] to generate a high-accuracy yeast protein interaction dataset. They found that several subunits of Rpd3L can be co-purified using Rco1 as bait. Reciprocally, subunits of Rpd3S can be co-purified with Rpd3L subunits used as baits. These data argue that some interactions between Rpd3S and Rpd3L do exist in the cell. Since the complexes analyzed in these studies were purified from the soluble cellular fraction, the relative low abundance of these inter-complex interactions may be explained by the fact that these complexes normally exist together only on chromatin. The recent development of techniques to purify protein complexes from chromatin [66], [67] may help testing these intriguing possibilities.
The role of the Eaf3 CHD in Rpd3S function
We show that the recruitment of Rpd3S to active genes does not require H3K36 methylation, Set2 or the Eaf3 chromodomain. Interestingly, Rco1 binding is not abolished in a triple deletion mutant for all the known yeast HMTs (Set1, Set2 and Dot1), ruling out the possibility that the previously described interaction between H3K4me3 and Eaf3 [38] (or an eventual interaction with methylated H3K79) is compensating for the loss of H3K36 methylation in set2Δ and H3K36A mutants. Moreover, although the methylation state of H3K36 does not affect Rpd3S binding at highly transcribed genes, it has an effect on the occupancy of Rpd3S at ORFs showing lower transcription levels. This suggest that, despite not providing the main recruitment signal, H3K36 methylation provides some stabilizing effect on the association of Rpd3S with chromatin. While Rpd3S recruitment is largely independent on H3K36 methylation, the activity of Rpd3S, however, depends on the integrity of the Set2/Rpd3S pathway, namely Set2-dependant H3K36 methylation, the Rco1 PHD and the Eaf3 CHD. We therefore propose that the main function of H3K36 methylation is to “activate” Rpd3S after it has been recruited by virtue of its association with the RNAPII CTD. This may involve the anchoring of Rpd3S on chromatin via the Eaf3-H3K36me interaction.
RNAPII phosphorylation by Kin28 and Ctk1 is required for the recruitment of Rpd3S
Our data clearly show that phosphorylation by Kin28 is absolutely required for the association of Rpd3S with the coding region of transcribed genes. This suggests that phosphorylation of serine 5 provides the signal for the association of Rpd3S with early elongating RNA polymerases II molecules. Since Kin28 was recently shown to phosphorylate serine 7 in addition to serine 5, we cannot rule out the possibility that serine 7 phosphorylation also contributes to the binding of Rpd3S to RNAPII. Using a ctk1Δ strain, we also show that phosphorylation of serine 2 also contributes -although to a lesser extend than serine 5 - to the association of Rpd3S with transcribed genes. These results are in perfect agreement with a recent paper from the Hinnebusch group who showed that RNAPII CTD peptides harboring a phosphate group on both serines 5 and 2 have a high affinity for Rpd3S in vitro while a single phosphate on serine 5 has a reduced affinity [51]. More importantly, they were not able to detect any interaction of Rpd3S with non-phosphorylated CTD peptides, and peptides carrying a single phosphate on serine 2 barely show any affinity with Rpd3S [51]. We therefore propose that Rpd3S is recruited to active genes via interaction with the phosphorylated RNAPII CTD. However, this complex remains inactive until it has been “anchored” on chromatin via H3K36 methylation. As discussed above, this phenomenon does not appear to occur equally on all genes. Negative regulation of these interactions by the DSIF elongation complex modulates association of Rpd3S with genes, therefore creating situations where active genes with high level of Spt4 carry much less Rpd3S than expected from their transcriptional level.
DSIF is a negative regulator of Rpd3S recruitment
Interestingly, we found that deletion of SPT4, a subunit of DSIF, has a profound effect on Rpd3S occupancy in vivo. In the absence of Spt4, Rpd3S associates with the genome in a manner that correlates better with transcription than in wild type cells. This suggests that DSIF is involved in regulating the amount of Rpd3S on transcribed genes. DSIF appears to prevent the association of Rpd3S to a subset of transcribed genes, and even at genes occupied by Rpd3S, DSIF also plays a role since it prevents the hyper-accumulation of the HDAC in the 3′end of the gene. How exactly an elongation factor can regulate the association of a HDAC with elongating RNAPII remains obscure but we envision several mechanisms by which it may operate: 1) Rpd3S may directly or indirectly interact with DSIF; 2) The association of DSIF with the elongation complex may prevent the association of the HDAC; 3) DSIF may modulate the speed of the elongation complex in a way that makes it less favorable for Rpd3S binding; 4) DSIF may also impinge on the phosphorylation of the RNAPII CTD. More complex mechanisms may also be envisioned. For instance, Pin1, a proline isomerase that may modify the RNAPII CTD, binds to both, phosphorylated DSIF [68] and Rpd3/Sin3 [69]. CTD isomerisation may therefore be involved in the regulation of the recruitment of Rpd3S.
Bur1 downregulates the activity of DSIF
In human and yeast, DSIF is regulated by phosphorylation of the CTD of its Spt5 subunit. In yeast, this phosphorylation is mediated by the Bur1 cyclin-dependent kinase [56], [57]. We therefore tested whether phosphorylation of Spt5 by Bur1 affects Rpd3S occupancy in vivo. As expected, both the catalytic inactivation of Bur1 and the removal of its substrate (by deleting the CTD of Spt5) have a dramatic impact on Rpd3S occupancy. Both these mutants indeed show a decreased in Rpd3S occupancy along genes. These data suggest that phosphorylation of Spt5 by Bur1 negatively regulate the activity of DSIF on Rpd3S recruitment. In addition, the inactivation of Bur1 also causes Rpd3S to redistribute to promoter regions, a phenomenon that we also observed in set2Δ cells. Since Bur1 phosphorylates Rad6, which is also required for H3K36 methylation by Set2, it appears likely that this redistribution in Bur1 mutants is due to its effect on H3K36 methylation via Rad6. Why a lack in H3K36 methylation leads to an association of Rpd3S with promoters remains unknown, but is in agreement with the previous observation that histone acetylation decreases at promoters in these mutants.
Taken together, our in-depth analysis of Rpd3S genomic occupancy has revealed several key insights about its recruitment to genes in vivo. Our study highlights a complex network of protein-protein interactions mediated by phosphorylation of several substrates by at least three kinases. Interestingly, there is previous evidence in the literature linking these kinases to the function of DSIF. First, P-TEFb (Cdk9) and Bur1 can phosphorylate DSIF [56], [57], [70]. Second, Kin28, Ctk1 and Bur1 exhibit synthetic genetic interactions with Spt4 and Spt5 [71]. And third, the recruitment of Bur1 is stimulated by Kin28 [52], [72]. Finally, both DSIF and Kin28 have been shown to stimulate the recruitment of the Paf1 complex to the elongation complex [57], [73]. A lot more work will be required before we completely understand the interplay between these factors and Rpd3S.
Materials and Methods
Strains, cell growth, and crosslinking conditions
All strains used in this study are listed in Table S1. All strains were grown in 50mL of YPD to an OD600 of 0.6–0.8 before crosslinking, unless otherwise indicated. For ChIP-chip, most strains were crosslinked with 1% formaldehyde for 30 min at room temperature on a wheel. The Rco1-9myc strains were crosslinked with 1% paraformaldehyde for 30 min at room temperature followed by 90 min at 4°C on a wheel. ATP analog-sensitive strains were treated with 6 µM of NAPP1 for 15 minutes prior to crosslinking.
Chromatin immunoprecipitation and antibodies
ChIP experiments were performed as per [74], with minor modifications. For myc-tag ChIP, we used 5µg of 9E11 antibody coupled to 2×107 pan-mouse IgG DynaBeads (Invitrogen) per sample. For histone H4K5 acetylation ChIP, we used 4µL of a rabbit serum (Upstate 07-327) coupled to 2×107 protein G DynaBeads per sample. Histone H3K36me3 was immunoprecipitated with 4µL of antibody (Abcam Ab9050) coupled to 2×107 protein G DynaBeads per sample. Histone H4 levels were assayed with 2µL of an antibody raised against recombinant yeast histone H4 (a gift from Alain Verreault) coupled to 2×107 protein G DynaBeads per sample. RNAPII ChIPs were done using 2µL of 8WG16 antibody coupled to 2×107 pan-mouse IgG DynaBeads per sample. Note that in our ChIP-chip assays, 8WG16 generates profiles that are nearly identical as using a tagged RNAPII (data not shown). 8WG16 is therefore used here to measure total RNAPII levels on genes. RNAPII CTD serine 2 phosphorylation was assayed using 5µL of H5 antibody (Covance Research MMS-129R-200) coupled to 2×107 protein G DynaBeads per sample.
The microarrays used for location analysis were purchased from Agilent Technologies (Palo Alto, California, United States) and contain a total of 44,290 Tm-adjusted 60-mer probes covering the entire genome for an average density of one probe every 275 bp (±100 bp) within the probed regions (catalog # G4486A and G4493A). Myc-tag ChIPs were hybridized against ChIPs from isogenic strains that did not contain the tag as controls. Acetylation, RNAPII and histone H4 ChIPs were hybridized against a sample derived from 400ng of input (non-immunoprecipitated) DNA. Acetylation levels were normalized to histone H4 levels by subtracting Log2 (Histone H4/input) from Log2 (H4K5 acetyl/input). All microarray experiments described in this work are listed in Table S2, the processed data are available in Datasets S1, S2, S3, S4, and the raw data have been deposited into the GEO database (Accession # GSE22636).
Data analysis
The data were normalized and biological replicates were combined using a weighted average method as described previously [74]. The log2 ratio of each spot of combined datasets was then converted to Z-score, similar to Hogan et al. [75], to circumvent the large differences in the immunoprecipitation efficiencies of the different factors. Visual inspection of the Z-scores was carried out on the UCSC Genome Browser (http://genome.ucsc.edu/). All data analyses described here were done using data from protein-coding genes longer than or equal to 500 bp. Median Z-score values for promoter and complete length of each annotation from SGD (version Feb. 02 2008) were calculated without interpolation (Dataset S5) and used in our clustering and Pearson correlation analyses. Promoters are defined as the shortest of either 250bp or half the intergenic region (half-IG) relative to the reference gene's 5′ boundary. Self-organizing map (SOM) clustering was done with the Cluster software [76] and visualized with Java Treeview [77]. Only genes with no missing value were used for clustering.
Gene mapping was performed as in Rufiange et al. [78] on selected groups of genes described in the text. Briefly, data were mapped onto the 5′ and 3′ boundaries in 50 bp windows for each half-gene and adjacent half-IG regions. A sliding window of 300 bp was then applied to the Z-scores to smooth the curve.
Supporting Information
Zdroje
1. ShahbazianMD
GrunsteinM
2007 Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem 76 75 100
2. YangXJ
SetoE
2008 The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol 9 206 218
3. WangH
CareyLB
CaiY
WijnenH
FutcherB
2009 Recruitment of Cln3 cyclin to promoters controls cell cycle entry via histone deacetylase and other targets. PLoS Biol 7 e1000189
4. HuangD
KaluarachchiS
Van DykD
FriesenH
SopkoR
2009 Dual regulation by pairs of cyclin-dependent protein kinases and histone deacetylases controls G1 transcription in budding yeast. PLoS Biol 7 e1000188 doi:10.1371/journal.pbio.1000188
5. KremerSB
GrossDS
2009 The SAGA and Rpd3 chromatin modification complexes dynamically regulate heat shock gene structure and expression. J Biol Chem
6. TakahataS
YuY
StillmanDJ
2009 The E2F functional analogue SBF recruits the Rpd3(L) HDAC, via Whi5 and Stb1, and the FACT chromatin reorganizer, to yeast G1 cyclin promoters. EMBO J 28 3378 3389
7. VeisJ
KlugH
KorandaM
AmmererG
2007 Activation of the G2/M-specific gene CLB2 requires multiple cell cycle signals. Mol Cell Biol 27 8364 8373
8. SharmaVM
TomarRS
DempseyAE
ReeseJC
2007 Histone deacetylases RPD3 and HOS2 regulate the transcriptional activation of DNA damage-inducible genes. Mol Cell Biol 27 3199 3210
9. InaiT
YukawaM
TsuchiyaE
2007 Interplay between chromatin and trans-acting factors on the IME2 promoter upon induction of the gene at the onset of meiosis. Mol Cell Biol 27 1254 1263
10. RobertF
PokholokDK
HannettNM
RinaldiNJ
ChandyM
2004 Global position and recruitment of HATs and HDACs in the yeast genome. Mol Cell 16 199 209
11. De NadalE
ZapaterM
AlepuzPM
SumoyL
MasG
2004 The MAPK Hog1 recruits Rpd3 histone deacetylase to activate osmoresponsive genes. Nature 427 370 374
12. MalloryMJ
StrichR
2003 Ume1p represses meiotic gene transcription in Saccharomyces cerevisiae through interaction with the histone deacetylase Rpd3p. J Biol Chem 278 44727 44734
13. WashburnBK
EspositoRE
2001 Identification of the Sin3-binding site in Ume6 defines a two-step process for conversion of Ume6 from a transcriptional repressor to an activator in yeast. Mol Cell Biol 21 2057 2069
14. KadoshD
StruhlK
1998 Targeted recruitment of the Sin3-Rpd3 histone deacetylase complex generates a highly localized domain of repressed chromatin in vivo. Mol Cell Biol 18 5121 5127
15. RundlettSE
CarmenAA
SukaN
TurnerBM
GrunsteinM
1998 Transcriptional repression by UME6 involves deacetylation of lysine 5 of histone H4 by RPD3. Nature 392 831 835
16. KadoshD
StruhlK
1997 Repression by Ume6 involves recruitment of a complex containing Sin3 corepressor and Rpd3 histone deacetylase to target promoters. Cell 89 365 371
17. KurdistaniSK
RobyrD
TavazoieS
GrunsteinM
2002 Genome-wide binding map of the histone deacetylase Rpd3 in yeast. Nat Genet 31 248 54
18. FazzioTG
KooperbergC
GoldmarkJP
NealC
BasomR
2001 Widespread collaboration of Isw2 and Sin3-Rpd3 chromatin remodeling complexes in transcriptional repression. Mol Cell Biol 21 6450 6460
19. KadoshD
StruhlK
1998 Histone deacetylase activity of Rpd3 is important for transcriptional repression in vivo. Genes Dev 12 797 805
20. ZhouJ
ZhouBO
LenzmeierBA
ZhouJQ
2009 Histone deacetylase Rpd3 antagonizes Sir2-dependent silent chromatin propagation. Nucleic Acids Res 37 3699 3713
21. LoewithR
SmithJS
MeijerM
WilliamsTJ
BachmanN
2001 Pho23 is associated with the Rpd3 histone deacetylase and is required for its normal function in regulation of gene expression and silencing in Saccharomyces cerevisiae. J Biol Chem 276 24068 24074
22. SunZW
HampseyM
1999 A general requirement for the Sin3-Rpd3 histone deacetylase complex in regulating silencing in Saccharomyces cerevisiae. Genetics 152 921 932
23. RundlettSE
CarmenAA
KobayashiR
BavykinS
TurnerBM
1996 HDA1 and RPD3 are members of distinct yeast histone deacetylase complexes that regulate silencing and transcription. Proc Natl Acad Sci U S A 93 14503 14508
24. VannierD
BalderesD
ShoreD
1996 Evidence that the transcriptional regulators SIN3 and RPD3, and a novel gene (SDS3) with similar functions, are involved in transcriptional silencing in S. cerevisiae. Genetics 144 1343 1353
25. SandmeierJJ
FrenchS
OsheimY
CheungWL
GalloCM
2002 RPD3 is required for the inactivation of yeast ribosomal DNA genes in stationary phase. EMBO J 21 4959 4968
26. SmithJS
CaputoE
BoekeJD
1999 A genetic screen for ribosomal DNA silencing defects identifies multiple DNA replication and chromatin-modulating factors. Mol Cell Biol 19 3184 3197
27. OakesML
SiddiqiI
FrenchSL
VuL
SatoM
2006 Role of histone deacetylase Rpd3 in regulating rRNA gene transcription and nucleolar structure in yeast. Mol Cell Biol 26 3889 3901
28. KnottSR
ViggianiCJ
TavareS
AparicioOM
2009 Genome-wide replication profiles indicate an expansive role for Rpd3L in regulating replication initiation timing or efficiency, and reveal genomic loci of Rpd3 function in Saccharomyces cerevisiae. Genes Dev 23 1077 1090
29. VogelauerM
RubbiL
LucasI
BrewerBJ
GrunsteinM
2002 Histone acetylation regulates the time of replication origin firing. Mol Cell 10 1223 1233
30. LambTM
MitchellAP
2001 Coupling of Saccharomyces cerevisiae early meiotic gene expression to DNA replication depends upon RPD3 and SIN3. Genetics 157 545 556
31. AparicioJG
ViggianiCJ
GibsonDG
AparicioOM
2004 The Rpd3-Sin3 histone deacetylase regulates replication timing and enables intra-S origin control in Saccharomyces cerevisiae. Mol Cell Biol 24 4769 4780
32. MerkerJD
DominskaM
GreenwellPW
RinellaE
BouckDC
2008 The histone methylase Set2p and the histone deacetylase Rpd3p repress meiotic recombination at the HIS4 meiotic recombination hotspot in Saccharomyces cerevisiae. DNA Repair (Amst) 7 1298 1308
33. DoraEG
RudinN
MartellJR
EspositoMS
RamirezRM
1999 RPD3 (REC3) mutations affect mitotic recombination in Saccharomyces cerevisiae. Curr Genet 35 68 76
34. CarrozzaMJ
LiB
FlorensL
SuganumaT
SwansonSK
2005 Histone H3 Methylation by Set2 Directs Deacetylation of Coding Regions by Rpd3S to Suppress Spurious Intragenic Transcription. Cell 123 581 592
35. KeoghMC
KurdistaniSK
MorrisSA
AhnSH
PodolnyV
2005 Cotranscriptional set2 methylation of histone h3 lysine 36 recruits a repressive rpd3 complex. Cell 123 593 605
36. LickwarCR
RaoB
ShabalinAA
NobelAB
StrahlBD
2009 The Set2/Rpd3S pathway suppresses cryptic transcription without regard to gene length or transcription frequency. PLoS One 4 e4886 doi:10.1371/journal.pone.0004886
37. LiB
GogolM
CareyM
PattendenSG
SeidelC
2007 Infrequently transcribed long genes depend on the Set2/Rpd3S pathway for accurate transcription. Genes Dev 21 1422 1430
38. JoshiAA
StruhlK
2005 Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to Pol II elongation. Mol Cell 20 971 978
39. LeeJS
ShilatifardA
2007 A site to remember: H3K36 methylation a mark for histone deacetylation. Mutat Res 618 130 134
40. BiswasD
TakahataS
StillmanDJ
2008 Different genetic functions for the Rpd3(L) and Rpd3(S) complexes suggest competition between NuA4 and Rpd3(S). Mol Cell Biol 28 4445 4458
41. RobyrD
SukaY
XenariosI
KurdistaniS
WangA
2002 Microarray deacetylation maps determine genome-wide functions for yeast histone deacetylases. Cell 109 437 446
42. QuanTK
HartzogGA
2010 Histone H3K4 and K36 methylation, Chd1 and Rpd3S oppose the functions of Saccharomyces cerevisiae Spt4-Spt5 in transcription. Genetics 184 321 334
43. WadaT
TakagiT
YamaguchiY
FerdousA
ImaiT
1998 DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev 12 343 356
44. PriceDH
2008 Poised polymerases: on your mark…get set…go! Mol Cell 30 7 10
45. SaundersA
CoreLJ
LisJT
2006 Breaking barriers to transcription elongation. Nat Rev Mol Cell Biol 7 557 567
46. SimsRJIII
BelotserkovskayaR
ReinbergD
2004 Elongation by RNA polymerase II: the short and long of it. Genes Dev 18 2437 2468
47. CuiY
DenisCL
2003 In vivo evidence that defects in the transcriptional elongation factors RPB2, TFIIS, and SPT5 enhance upstream poly(A) site utilization. Mol Cell Biol 23 7887 7901
48. BucheliME
BuratowskiS
2005 Npl3 is an antagonist of mRNA 3′ end formation by RNA polymerase II. EMBO J 24 2150 2160
49. KaplanCD
HollandMJ
WinstonF
2005 Interaction between transcription elongation factors and mRNA 3′-end formation at the Saccharomyces cerevisiae GAL10-GAL7 locus. J Biol Chem 280 913 922
50. LindstromDL
SquazzoSL
MusterN
BurckinTA
WachterKC
2003 Dual roles for Spt5 in pre-mRNA processing and transcription elongation revealed by identification of Spt5-associated proteins. Mol Cell Biol 23 1368 1378
51. GovindCK
QiuH
GinsburgDS
RuanC
HofmeyerK
2010 Phosphorylated Pol II CTD recruits multiple HDACs, including Rpd3C(S), for methylation-dependent deacetylation of ORF nucleosomes. Mol Cell 39 234 246
52. QiuH
HuC
HinnebuschAG
2009 Phosphorylation of the Pol II CTD by KIN28 enhances BUR1/BUR2 recruitment and Ser2 CTD phosphorylation near promoters. Mol Cell 33 752 762
53. ChuY
SuttonA
SternglanzR
PrelichG
2006 The BUR1 cyclin-dependent protein kinase is required for the normal pattern of histone methylation by SET2. Mol Cell Biol 26 3029 3038
54. PeterlinBM
PriceDH
2006 Controlling the elongation phase of transcription with P-TEFb. Mol Cell 23 297 305
55. WoodA
SchneiderJ
DoverJ
JohnstonM
ShilatifardA
2005 The Bur1/Bur2 complex is required for histone H2B monoubiquitination by Rad6/Bre1 and histone methylation by COMPASS. Mol Cell 20 589 599
56. ZhouK
KuoWH
FillinghamJ
GreenblattJF
2009 Control of transcriptional elongation and cotranscriptional histone modification by the yeast BUR kinase substrate Spt5. Proc Natl Acad Sci U S A
57. LiuY
WarfieldL
ZhangC
LuoJ
AllenJ
2009 Phosphorylation of the transcription elongation factor Spt5 by yeast Bur1 kinase stimulates recruitment of the PAF complex. Mol Cell Biol 29 4852 4863
58. KeoghMC
PodolnyV
BuratowskiS
2003 Bur1 kinase is required for efficient transcription elongation by RNA polymerase II. Mol Cell Biol 23 7005 7018
59. SunZW
AllisCD
2002 Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature 418 104 108
60. NgHH
XuRM
ZhangY
StruhlK
2002 Ubiquitination of histone H2B by Rad6 is required for efficient Dot1-mediated methylation of histone H3 lysine 79. J Biol Chem 277 34655 7
61. NakanishiS
LeeJS
GardnerKE
GardnerJM
TakahashiYH
2009 Histone H2BK123 monoubiquitination is the critical determinant for H3K4 and H3K79 trimethylation by COMPASS and Dot1. J Cell Biol 186 371 377
62. BerrettaJ
MorillonA
2009 Pervasive transcription constitutes a new level of eukaryotic genome regulation. EMBO Rep 10 973 982
63. CollinsSR
MillerKM
MaasNL
RoguevA
FillinghamJ
2007 Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature 446 806 810
64. GavinAC
AloyP
GrandiP
KrauseR
BoescheM
2006 Proteome survey reveals modularity of the yeast cell machinery. Nature 440 631 636
65. KroganNJ
CagneyG
YuH
ZhongG
GuoX
2006 Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440 637 643
66. AygunO
SvejstrupJ
LiuY
2008 A RECQ5-RNA polymerase II association identified by targeted proteomic analysis of human chromatin. Proc Natl Acad Sci U S A 105 8580 8584
67. LambertJP
MitchellL
RudnerA
BaetzK
FigeysD
2009 A novel proteomics approach for the discovery of chromatin-associated protein networks. Mol Cell Proteomics 8 870 882
68. LavoieSB
AlbertAL
HandaH
VincentM
BensaudeO
2001 The peptidyl-prolyl isomerase Pin1 interacts with hSpt5 phosphorylated by Cdk9. J Mol Biol 312 675 685
69. Arevalo-RodriguezM
CardenasME
WuX
HanesSD
HeitmanJ
2000 Cyclophilin A and Ess1 interact with and regulate silencing by the Sin3-Rpd3 histone deacetylase. EMBO J 19 3739 3749
70. WadaT
TakagiT
YamaguchiY
WatanabeD
HandaH
1998 Evidence that P-TEFb alleviates the negative effect of DSIF on RNA polymerase II-dependent transcription in vitro. EMBO J 17 7395 7403
71. LindstromDL
HartzogGA
2001 Genetic interactions of Spt4-Spt5 and TFIIS with the RNA polymerase II CTD and CTD modifying enzymes in Saccharomyces cerevisiae. Genetics 159 487 497
72. ViladevallL
St AmourCV
RosebrockA
SchneiderS
ZhangC
2009 TFIIH and P-TEFb coordinate transcription with capping enzyme recruitment at specific genes in fission yeast. Mol Cell 33 738 751
73. QiuH
HuC
WongCM
HinnebuschAG
2006 The Spt4p subunit of yeast DSIF stimulates association of the Paf1 complex with elongating RNA polymerase II. Mol Cell Biol 26 3135 3148
74. RenB
RobertF
WyrickJJ
AparicioO
JenningsEG
2000 Genome-wide location and function of DNA binding proteins. Science 290 2306 2309
75. HoganGJ
LeeCK
LiebJD
2006 Cell cycle-specified fluctuation of nucleosome occupancy at gene promoters. PLoS Genet 2 e158 doi:10.1371/journal.pgen.0020158
76. EisenMB
SpellmanPT
BrownPO
BotsteinD
1998 Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A 95 14863 8
77. SaldanhaAJ
2004 Java Treeview–extensible visualization of microarray data. Bioinformatics 20 3246 3248
78. RufiangeA
JacquesPE
BhatW
RobertF
NouraniA
2007 Genome-wide replication-independent histone H3 exchange occurs predominantly at promoters and implicates H3 K56 acetylation and Asf1. Mol Cell 27 393 405
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Genome-Wide Identification of Targets and Function of Individual MicroRNAs in Mouse Embryonic Stem Cells
- Common Genetic Variants and Modification of Penetrance of -Associated Breast Cancer
- Allele-Specific Down-Regulation of Expression Induced by Retinoids Contributes to Climate Adaptations
- β-Actin and γ-Actin Are Each Dispensable for Auditory Hair Cell Development But Required for Stereocilia Maintenance
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy