
Altering a Histone H3K4 Methylation Pathway in Glomerular Podocytes Promotes a Chronic Disease Phenotype
Methylation of specific lysine residues in core histone proteins is essential for embryonic development and can impart active and inactive epigenetic marks on chromatin domains. The ubiquitous nuclear protein PTIP is encoded by the Paxip1 gene and is an essential component of a histone H3 lysine 4 (H3K4) methyltransferase complex conserved in metazoans. In order to determine if PTIP and its associated complexes are necessary for maintaining stable gene expression patterns in a terminally differentiated, non-dividing cell, we conditionally deleted PTIP in glomerular podocytes in mice. Renal development and function were not impaired in young mice. However, older animals progressively exhibited proteinuria and podocyte ultra structural defects similar to chronic glomerular disease. Loss of PTIP resulted in subtle changes in gene expression patterns prior to the onset of a renal disease phenotype. Chromatin immunoprecipitation showed a loss of PTIP binding and lower H3K4 methylation at the Ntrk3 (neurotrophic tyrosine kinase receptor, type 3) locus, whose expression was significantly reduced and whose function may be essential for podocyte foot process patterning. These data demonstrate that alterations or mutations in an epigenetic regulatory pathway can alter the phenotypes of differentiated cells and lead to a chronic disease state.
Published in the journal:
. PLoS Genet 6(10): e32767. doi:10.1371/journal.pgen.1001142
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001142
Summary
Methylation of specific lysine residues in core histone proteins is essential for embryonic development and can impart active and inactive epigenetic marks on chromatin domains. The ubiquitous nuclear protein PTIP is encoded by the Paxip1 gene and is an essential component of a histone H3 lysine 4 (H3K4) methyltransferase complex conserved in metazoans. In order to determine if PTIP and its associated complexes are necessary for maintaining stable gene expression patterns in a terminally differentiated, non-dividing cell, we conditionally deleted PTIP in glomerular podocytes in mice. Renal development and function were not impaired in young mice. However, older animals progressively exhibited proteinuria and podocyte ultra structural defects similar to chronic glomerular disease. Loss of PTIP resulted in subtle changes in gene expression patterns prior to the onset of a renal disease phenotype. Chromatin immunoprecipitation showed a loss of PTIP binding and lower H3K4 methylation at the Ntrk3 (neurotrophic tyrosine kinase receptor, type 3) locus, whose expression was significantly reduced and whose function may be essential for podocyte foot process patterning. These data demonstrate that alterations or mutations in an epigenetic regulatory pathway can alter the phenotypes of differentiated cells and lead to a chronic disease state.
Introduction
The process of embryonic development determines the differentiated state of all cells by establishing unique gene expression patterns, or signatures, for individual cell types that define their phenotypes. Once a differentiated state is established, it is difficult to erase that epigenetic imprint and reprogram the cell towards a different cell lineage or phenotype. Although reprogramming can be forced by nuclear transplantation [1] or by the expression of Oct4 and accessory factors [2], [3], the low efficiency of these processes speaks to the inherent stability of a differentiated cell. Gene expression patterns must be established and maintained by compartmentalizing the genome into active and inactive regions, which is thought to occur through the covalent modifications of DNA and its associated nucleosomes. Such modifications include DNA methylation of CpG islands and methylation, acetylation, and ubiquitination of histone tails, all of which are thought to determine chromatin structure and accessibility [4], [5]. This epigenetic code is thus imprinted upon the primary genetic code during embryonic development to help establish cell lineages and restrict fate.
The genetics and biochemistry of histone modifications have been well studied in a variety of model organisms and developmental contexts. Genes of the Polycomb and Trithorax families encode proteins that are required for methylation of different histone lysine residues and often correlate with gene silencing or activation, respectively [6]–[9]. Many Trithorax group proteins, such as Drosophila TRX and human KMT2A (MLL), are histone H3 lysine 4 (H3K4) methyltransferases (KMTs) and are essential for maintaining gene expression patterns in diverse organisms. Recently, we discovered a novel co-factor, PTIP (Pax Transactivation-domain Interacting Protein), which is encoded by the Paxip1 gene. The PTIP protein co-purifies with the mammalian lysine methyltransferases KMT2B and KMT2C (formerly ALR and MLL3), is broadly expressed, and is essential for embryonic development [10]–[12]. At least in one case, PTIP is able to recruit the KMT2B complex to a developmental DNA binding protein in a locus specific manner [13]. Loss of PTIP function in the mouse results in gross developmental effects at gastrulation, with reduced levels of global H3K4 di - (me2) and trimethylation (me3) observed [13], [14]. In cultured mouse embryonic stem cells, PTIP is needed to maintain pluripotency, Oct4 expression, and normal levels of H3K4 trimethylation [15]. Similarly, in neuronal stem cells, differentiation is abrogated and levels of H3K4 methylation are reduced in tissue specific PTIP knockouts [13]. In mouse embryo fibroblasts, loss of PTIP blocks differentiation by inhibiting PPARγ and C/EPBα activation and H3K4 methylation at their respective promoters [16]. Similarly, the Drosophila homologue of PTIP is also essential for development, epigenetic control of gene expression, and global histone H3K4 methylation [17].
During cell division, patterns of histone methylation must be inherited by daughter cells such that the cellular phenotype is maintained. For repressive histone methylation marks, such as histone H3 lysine 27, the EED (Embryonic Ectodermal Development) protein is thought to bind and recruit the Polycomb Repressor Complex 2 to replicate and maintain gene silencing after mitotic cell division [18], [19]. For highly expressed genes, the KMT2A (MLL1) protein associates with promoter regions on condensed mitotic chromatin and is required to rapidly reactivate such genes after cell division [20]. These data suggest a model whereby histone methylation patterns are replicated during mitosis, but do not address the necessity for maintaining epigenetic modifications in terminally differentiated, non-dividing cells. Furthermore, changes in the expression of epigenetic regulatory genes have been reported in a variety of cancers [21] and disease states [22], but whether these are the cause or the result of disease remains to be determined.
To address the necessity of H3K4me3 in a stable non-dividing cell type, we utilized a Podocin-Cre transgenic driver to delete PTIP in the glomerular podocyte, a highly specialized and architecturally distinct cell that establishes the kidney filtration barrier. Podocytes are clinically relevant cells whose properties and expression profiles change in glomerular diseases and in older animals [23]. While the ubiquitous expression of PTIP, its role in H3K4 methylation, and its necessity in development and differentiation are all well established, whether PTIP deletion in terminally differentiated cells can induce changes in the pattern of H3K4me3 and gene expression has not been demonstrated. We show that loss of PTIP results in changes in the transcriptional profile of terminally differentiated podocyte cells, which ultimately leads to a chronic glomerular disease phenotype. Among the most affected is the neurotrophin receptor encoding gene Ntrk3, whose function had not been previously studied in podocytes. Our results demonstrate a maintenance function for PTIP-mediated H3K4 methylation and identify a novel role for Ntrk3 in podocyte foot process patterning.
Results
Generation of a Podocyte-Specific Paxip1 Deletion
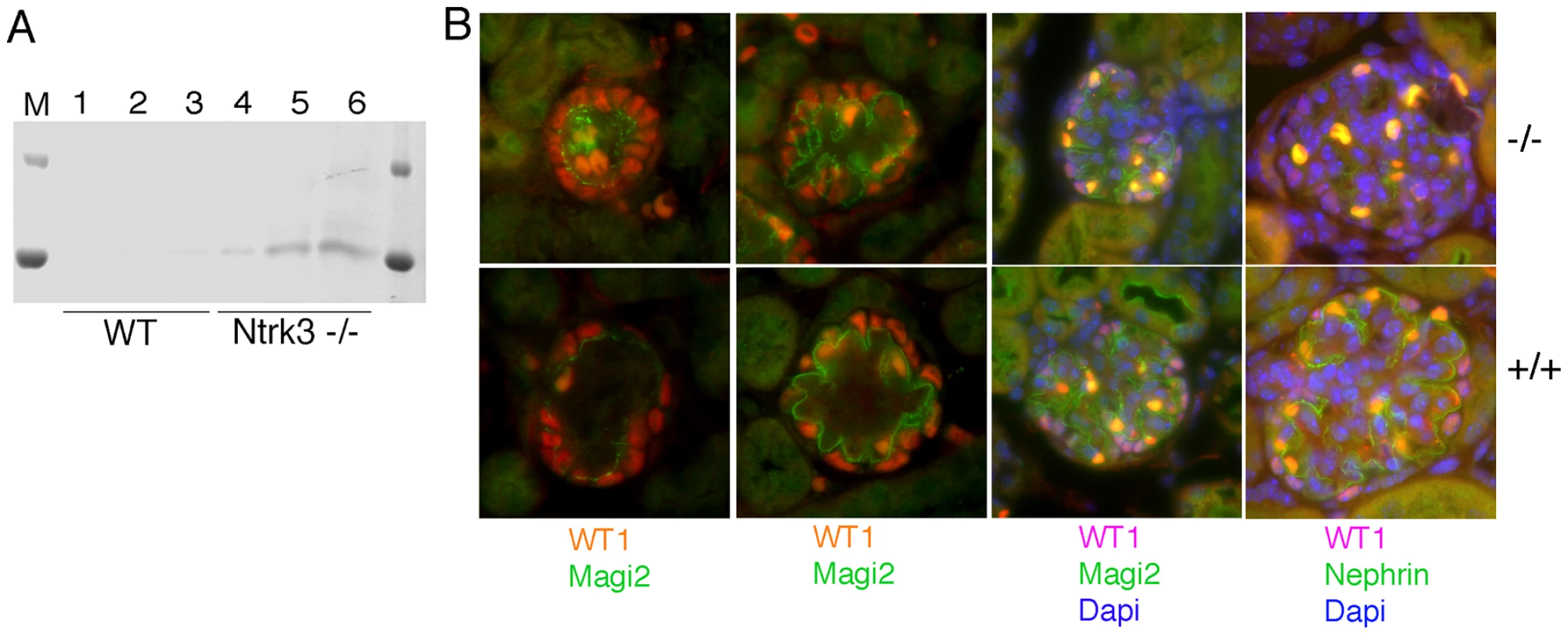
To specifically knockout PTIP protein in fully differentiated mouse podocytes, we utilized both floxed (fl) and conventional null (-) alleles of Paxip1 and a Cre driver strain specific for glomerular podocytes. The Paxip1fl/−:CreNPHS2 mice were crossed to Paxip1fl/fl animals to generate Paxip1fl/fl or Paxip1fl/− with or without CreNPHS2. The CreNPHS2 mice utilize the NPHS2 promoter to express Cre recombinase only in late developing and mature podocytes [24], [25]. The resulting progenies were born in the expected Mendelian ratios and did not show any gross kidney defects during the first 4 weeks of life (data not shown). For simplicity, we will refer to the mice as either PTIP − (Paxip1fl/−:CreNPHS2; Paxip1fl/fl:CreNPHS2) or PTIP+ (Paxip1−/fl, or Paxip1fl/fl). PCR analysis indicated that recombination occurred at the Paxip1 locus in DNAs isolated from kidneys but not in DNAs from tails (Figure 1A). Previous work established that the Paxip1fl allele produces normal levels of protein, but Cre-mediated excision of exon 1 and the promoter region results in complete absence of PTIP protein, essentially creating a null allele [13], [15]. The specificity of the Cre driver strain was confirmed by crossing CreNPHS2 mice to the Rosa26-LacZ reporter mice (Figure 1B). In 1 month old kidneys, lacZ expression was restricted to the glomerulus only, indicating efficient Cre mediated excision at this time. Immunostaining for PTIP and the podocyte marker WT1 also confirmed that PTIP protein levels were reduced only in the podocyte cells and not the mesangial or endothelial components of the glomerular tuft (Figure 1C). Previous work showed that a loss of PTIP function results in reduced levels of total H3K4me3 levels in embryos and cultured cells [13]–[17]. To test whether podocytes showed reduced H3K4me3, we stained kidney sections with antibodies specific for this modification (Figure 1D). Many podocytes were observed with reduced signal intensities. To quantitate this effect, images were analyzed for signal intensity by integrating a fixed area over the nuclei of both podocytes and other cell types (Figure 1E). Podocytes were co-stained with WT1 antibodies. The ratio of podocyte signal (WT1+) to other cell types (WT1−) was calculated by counting at least 6 cells of each type per glomerulus. The ratios from at least 8 glomeruli were averaged for each genotype and shown to decrease by more than 20% in PTIP − kidneys compared to PTIP+ controls (p<0.01). These data confirmed that the specific deletion of PTIP in the podocytes correlates with a reduction in H3K4me3 in this cell type.

Development of a Chronic Glomerular Disease Phenotype
Podocytes play a critical role in the establishment and maintenance of the glomerular filtration barrier. Interdigitated podocyte foot processes cover the glomerular basement membrane and form specialized junctions, called slit diaphragms, which create a highly selective barrier that filters small and negatively charged proteins and solutes from the blood to the urinary space. Damage to or loss of podocytes impairs the filtration barrier and results in increased rates of excretion of high molecular weight proteins, such as albumin, into the urine. Thus, we checked mice for proteinuria beginning at 1 month of age (Figure 2A). At 1 month, low levels of albumin were detected in the urine but these were not significantly different between PTIP+ and PTIP − animals. However, by 3 months of age the PTIP − mice showed significantly higher levels of albumin in the urine and these levels increased further at 6 and 12 months. The urine albumin to creatinine ratio (ACR) provides a quantitative assay that correlates with filtration barrier integrity. No significant differences were observed at 1 month (Figure 2B). However, by 3 and 12 months, ACR were 10 and 30 fold higher respectively in urines of PTIP − animals compared to PTIP+ mice. Mice that carried the CreNPHS2 transgene in a Paxip1+/+ or a Paxip1fl/+ genetic background did not show any renal abnormalities at 12 months (data not shown), consistent with many published reports that have used this particular Cre driver strain [25]–[28].

Renal pathology was characterized by light microscopy at 1, 3, and 12 months of age. Standard Masson's Trichrome and Periodic-Acid-Shiff stainings revealed significant sclerosis and matrix deposition in 12 month old glomeruli from PTIP − animals (Figure 2C). However, 3 month old kidneys did not show significant differences for most glomerular sections, at the light microscopy level, although evidence of limited matrix expansion could be observed in a small number of glomeruli of PTIP − kidneys. In 12 month old kidneys, significant interstitial fibrosis and protein filled cysts were also observed (Figure 2D). These are likely to be secondary effects due to the glomerular pathology.
Glomerular pathology and increased albuminuria can be the direct result of podocyte death [29]. Thus, we used a variety of markers to characterize the glomerular architecture and the numbers of podocyte cells at various ages to insure that the phenotype of the PTIP − mice was not just the result of early podocyte cell death. Immunostaining with WT1, Nephrin, and Podocin antibodies enabled us to determine the podocyte numbers, as average per mid-cross section, and to indirectly assess the integrity of the slit diaphragm (Figure 3). The number of WT1 positive podocytes was not significantly different between PTIP+ and PTIP − glomeruli at 1 or 3 months of age. At 6 months, PTIP − glomeruli had slightly fewer podocytes and by 12 months, the number of podocytes was half that of the PTIP+ littermates. Immunostainings for podocyte markers such as WT1, Nephrin, and Podocin did not reveal dramatic differences at 1 or 3 months, despite the increase in proteinuria, although some discontinuous staining could be seen with Podocin antibodies in PTIP − glomeruli (Figure 3B). Consistent with this data, TUNEL staining for apoptosis did not reveal differences between PTIP+ and PTIP − kidneys at 1 or 3 months of age (data not shown). Thus, the breakdown of the filtration barrier was not due to simple podocyte depletion at these early times. However by 12 months of age, the extensive network of Nephrin staining was partially depleted in PTIP − glomeruli (Figure 3B).

At the light microscopy level, the effects of PTIP loss on glomerular architecture seemed minimal at 3 months of age, yet the levels of albumin in the urine suggested significant functional defects. Thus, we utilized scanning and transmission electron microscopy to characterize the podocytes at the ultra structural level (Figure 4). Scanning electron micrographs revealed disorganized foot processes at 3 months. While PTIP+ podocytes had regularly arrayed tertiary foot-processes that were almost parallel (Figure 4A), the PTIP − podocyte foot processes were much more irregular and flattened. The parallel pattern of interdigitation was clearly different and resembled a jigsaw puzzle with random patterning (Figure 4B, 4C). Transmission electron micrographs at 3 months also revealed that the slit-diaphragms were not evenly spaced and fusion of foot processes was frequent (Figure 4D–4F). By 12 months, the remaining podocytes in the PTIP − kidneys were broader, flatter and displayed significant fusion or effacement (Figure 4G, 4H), consistent with the high levels of albumin detected in the urine. These data demonstrate that the initial glomerular phenotype in PTIP − kidneys is due primarily to differences in podocyte foot process morphology, which occurs prior to the loss of cell bodies.

Alteration of the Gene Expression Program Precedes the Disease Phenotype
Alterations in cellular phenotypes could be the result of changes in the transcriptional program of PTIP − podocytes. Thus, we prepared RNA from glomeruli enriched fractions at 1 month of age, prior to the onset of any significant phenotype, and assayed for gene expression changes by Affymetrix microarrays. We compared glomerular RNA preps from 10 independent PTIP − animals and 8 PTIP+ littermates at 1 month of age. The data were highly consistent and indicated both gain and loss of gene expression in the PTIP − kidneys (Table 1 and Table 2). The entire dataset can be accessed at the Gene expression Omnibus (GSE17709). Expression changes were confirmed by quantitative RT-PCR for selected genes (Figure 5). Among the genes increased was Protamine1 (Prm1), which is not normally expressed in podocytes or other somatic cells but is found only in spermatids where it is essential for chromatin condensation and fertility [30], [31]. The changes in RNA expression observed were surprising and did not correspond to any common pathways. In fact, the podocyte-specific genes that are known to function in cell viability and slit diaphragm integrity were largely unchanged (Table S1 and Figure 5C). The data suggest that loss of PTIP in podocytes alters the transcriptional program to affect a limited number of genes whose functions in the podocytes have not been previously characterized.



PTIP Deletion Affects Ntrk3 Expression and Histone Methylation
Among the most interesting genes whose expression was down regulated in PTIP − kidneys was the neurotrophic tyrosine kinase receptor type 3 (Ntrk3, formerly called TrkC), whose expression in podocytes had not been previously described. The Ntrk3 gene encodes two proteins that recognize neurotrophin 3 (NT-3) and functions in axon guidance and innervation and in cardiac development [32]–[34]. Ntrk3 promotes axon outgrowth and guidance, presumably through actin based extension and retraction of cellular processes [35]. Given that podocyte foot processes are also actin based and may require some type of guidance, we examined the role of Ntrk3 further. Quantitative RT-PCR confirmed that Ntrk3 expression was down approximately 10 fold in glomerular preps from PTIP − compared to PTIP+ animals (Figure 5A). We also examined Ntrk3 levels in kidneys by co-immunostaining kidney sections with Ntrk3, WT1 and Nephrin antibodies (Figure 6). At 3 months of age, Ntrk3 could be seen in glomeruli of PTIP+ kidneys, however the staining intensity in PTIP − kidneys was severely reduced in almost every glomerulus examined (Figure 6D, 6J). Some slight filamentous staining remained in the PTIP − glomeruli, but the overall intensity was markedly different. In PTIP+ glomeruli, Ntrk3 staining was remarkably similar to Nephrin (Figure 6G–6I). However, Nephrin staining intensity was unaffected in PTIP − glomeruli even though Ntrk3 was much lower (Figure 6J–6L). The Ntrk3 expression in glomerular preps and its decrease in the PTIP − kidneys suggested a function in foot process growth, guidance, and/or pattern formation.

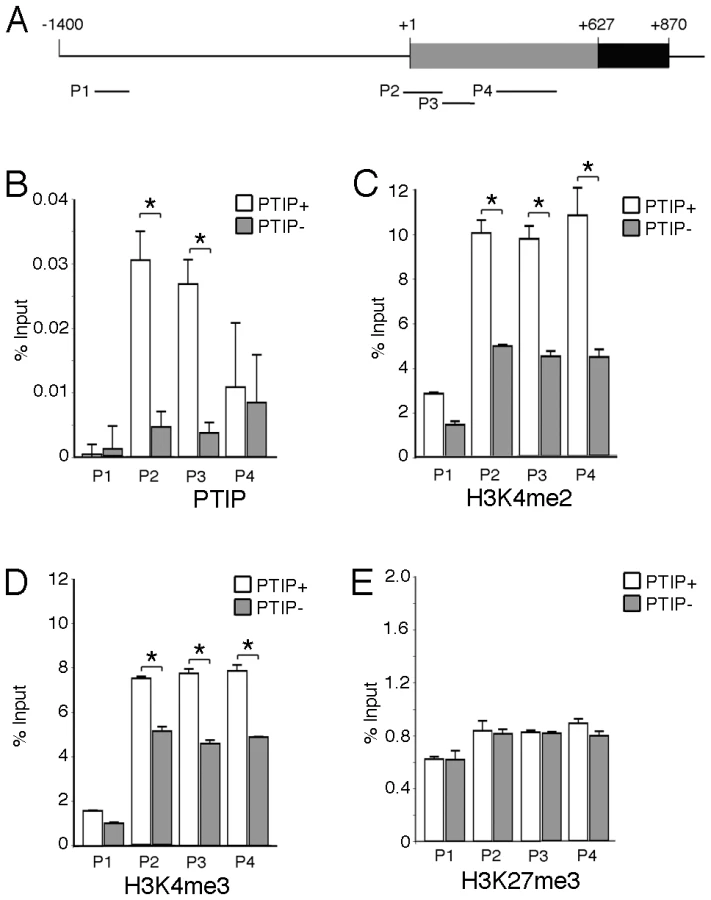
In order to more directly link PTIP to the Ntrk3 locus, we designed chromatin immunoprecipitation experiments to examine the presence of PTIP and the changes in histone methylation patterns around the transcription initiation site (+1) of Ntrk3 (Figure 7). Chromatin was prepared from whole glomerular preps from PTIP+ and PTIP − kidneys, which also included mesangial and endothelial cells. Despite the presence of other cell types in the glomerular chromatin, we were able to detect a 5–6 fold decrease in PTIP localization to sequences around the start site of Ntrk3 transcription when comparing PTIP+ to PTIP − chromatin (Figure 7B). No significant amount of PTIP was detected further upstream (−1200), nor did we see a significant difference, between PTIP+ and PTIP − chromatin, in PTIP localization within the 5′ UTR of exon 1 (Figure 7B, P4 site). Clear differences in H3K4me2 were also measured, with an approximately 50–60% decrease in PTIP − chromatin with primer pairs P2–P4, but not with P1 at −1200 (Figure 7C). Similarly, H3K4me3 levels were also decreased in PTIP − chromatin at P2–P4 but not at P1 (Figure 7D). We also examined changes in Polycomb mediated epigenetic silencing marks using an antibody against H3K27me3 (Figure 7E), which appeared unchanged at all sites examined. These data demonstrate recruitment of PTIP to the promoter region of Ntrk3 in normal glomeruli.
Ntrk3 Mutants Have Podocyte Foot Process Defects
In order to determine if the loss of Ntrk3 alone would impact normal glomerular patterning, we examined homozygous Ntrk3 mutant mice. The Ntrk3 mutants die shortly after birth due to cardiac and neuromuscular defects; however their kidneys had not been studied previously. Therefore, we collected urine and kidney tissue for light and electron microscopy from 3–4 day old Ntrk3 mutants and littermates. At three days post partum, Ntrk3 mutants were small and sickly. Higher levels of albumin could be observed in the urines of Ntrk3−/− pups (Figure 8A), compared to control littermates, although this could be due to delayed or arrested kidney development. Glomerular development was examined in kidney sections of 4 day old newborns (Figure 8B). At this time, nephrons are still undergoing development and glomeruli at the periphery are just beginning to form whereas cortical glomeruli closer to the medulla are already fully functional. The tight junction protein Magi2 specifically localizes to podocyte cell junctions and exhibited altered patterning in Ntrk3 mutant kidneys, with discontinuous staining and excessive looping of the developing tuft. In mature glomeruli, Nephrin staining was reduced and patchy in the Ntrk3 mutants. The number of podocytes did not seem affected in the Ntrk3−/− mice at this time.
Ultra structural analysis of Ntrk3 mutant kidneys revealed podocyte patterning defects both by scanning and transmission EM (Figure 9). At 4 days post-partum, we examined the most mature glomeruli, those located closest to the medullary zone. Podocyte foot processes from Ntrk3−/− mice exhibited disorganized secondary and tertiary processes that crisscrossed randomly over capillary vessels and were poorly interdigitated (Figure 9A′, 9B′). Few sections showed the characteristic spacing indicative of the slit diaphragms at the glomerular basement membranes (Figure 9D′). These data suggest a critical role for Ntrk3 in the fine patterning events of secondary and tertiary foot process formation and interdigitation.

Discussion
In this report, we utilized a conditional deletion to ask whether the PTIP dependent H3K4 methylation function is required in a terminally differentiated cell type, to maintain its differentiated state and its cell-type specific transcriptional program. Using the glomerular podocyte cell as a model, we show that deletion of PTIP results in subtle changes in gene expression patterns that ultimately lead to a slowly progressing disease state. These data support a model in which the gross stability of the differentiated state or podocyte cell survival, at least in the short term, does not depend on the PTIP/KMT complex, as many of the podocyte specific genes examined were unchanged in the absence of PTIP. Rather, the loss of PTIP was more subtle and revealed unexpected changes in a small number of genes and ultimately led to a chronic disease phenotype resembling glomerular sclerosis. Typical characteristics of chronic glomerular disease were present, including microalbuminuria, podocyte foot process fusion or effacement, remodeling of the filtration barrier, and increased extracellular matrix deposition.
Methylation of histone H3 at lysine 4 correlates with gene expression and is thought to regulate cellular identity by establishing and maintaining a stable epigenetic state. The PTIP protein is part of an H3K4 methyltransferase complex that includes the mammalian Trithorax homologues KMT2B and/or KMT2C [10], [11], [13], [16]. Previous studies in flies and mice demonstrated reduced H3K4 methylation in Paxip1 mutants and severe early lethal phenotypes. In the mouse, complete loss of PTIP protein results in developmental arrest just after gastrulation [14], a phenotype more severe than any individual mouse KMT2 family gene mutation [12], [36], [37], whereas a hypomorphic Paxip1 allele is lethal later in development [38]. In flies, maternal and zygotic ptip null embryos are embryonic lethal and fail to express many segmentation genes [17]. In mouse embryonic stem cells, PTIP protein is required for normal levels of H3K4 methylation and for maintaining pluripotency in cell culture [15], whereas in embryonic fibroblasts PTIP is required for adipocyte differentiation [16]. All of these findings suggest that a PTIP H3K4 methyltransferase complex is needed for differentiation of stem cells and progenitor cells in development. However in terminally differentiated cells, the requirement for active H3K4 methylation may be different and the lack of cell division may abrogate the need for de novo methylation. Our results suggest that PTIP must still function in some non-dividing cells, perhaps as part of a maintenance complex, as overall levels of H3K4 methylation were reduced and activation and suppression of a small number of genes was affected.
The mature podocyte is generally believed to be a non-dividing cell type, as classic cell BrdU labeling experiments do not mark this population over time [39]. However, more recent genetic lineage tracing experiments suggest that there is a population of parietal epithelial cells at the vascular pole of the Bowman's capsule that can replenish podocytes over time [40], [41]. This replacement of podocytes appears slow under normal conditions, but may be especially critical in cases of glomerular injury. In our animal model, we would expect any podocyte replacement to also delete the Paxip1 gene once expression of the Cre driver is activated. Given that we do not see significant loss of podocytes until at least 6 months of age, it may be that alterations in the transcriptional profile are not lethal. Rather, loss of podocytes may be the result of the damaged filtration barrier, the increase in the mesangium, and the general environment of the glomerulus in older mice. Alternatively, if podocyte replacement is accelerated in our model, it may be that by 6 months the ability of parietal cells to replenish the podocyte population is exhausted. In either case, the effects of manipulating the H3K4 methylation pathway is more apparent in older mice, suggesting a critical role for such epigenetic pathways in aging cells and tissues.
The changes in gene expression observed in response to PTIP deletion are surprising in that most of the well-characterized podocyte-specific genes appear unaffected. However, changes include both activation and suppression of previously uncharacterized genes in the podocytes. Activation of the Prm1 gene in PTIP − kidneys is unusual as this gene has only been associated with sperm maturation and is thought to encode a unique chromatin binding protein [31], [42]. Activation of the Padi4 gene could impact gene expression by deimination of arginines in the histone H3 tail, which prevents methylation [43]. The impact of increased Padi4 is likely to be complex as arginine methylation can correlate with gene activation or repression, depending on the context and specific residues.
The most compelling gene affected in PTIP − podocytes was Ntrk3, whose expression in the glomerulus had not been previously characterized. The reduction of Ntrk3 expression in PTIP − kidneys and the phenotype of Ntrk3−/− newborn kidneys suggest that this receptor is critical for tertiary foot process pattern formation. The podocyte is a highly specialized cell with a complex network of processes that cover the glomerular basement membrane. The large primary processes are microtubule containing structures, whereas the tertiary, interdigitated foot processes contain actin microfilaments [44]. Adjacent foot processes are connected through a specialized junctional complex, called the slit diaphragm, which is essential for maintaining a functional filtration pore. Some of the essential proteins in the slit-diaphragm, such as Nephrin, Podocin, and Neph1 are well characterized and mutations are associated with severe nephrotic syndromes [45]. Yet, how foot process outgrowth is regulated and maintained is not clear. Our data suggests that Ntrk3, and by inference its ligand NT–3, may be important for foot process growth and patterning. NT-3 is known to promote neuronal axon guidance by stimulating actin polymerization and lamellipodia formation [46], [47]. In cultured neuronal cells, NT-3 promotes localization of β-actin mRNA to the growth cones to stimulate motility and chemotaxis [48], [49]. Podocytes express many proteins known to function in neurite outgrowth, such as semaphorins, neuropilins, and ephrins. A recent report even describes the release and up-take of glutamate containing synaptic-like vesicles by podocytes [50]. Furthermore, foot processes are dynamic and can retract quickly in response to polyamines like protamine sulfate [51], [52]. This raises the possibility that sensing mechanisms are required for rapid actin dynamics; such mechanisms may be common to both podocytes and neurons. Still, reduction of Ntrk3 alone is unlikely to cause the phenotypic changes in PTIP − podocytes over time, as other genes whose functions are not well understood are also impacted.
Histone methylation by Trithorax or Polycomb complexes can imprint positive and negative epigenetic marks on chromatin during development. More recently, histone methyltransferases have been associated with cancer and other disease states. However, in many cases it is not clear whether changes in the expression of epigenetic modifiers are the cause or the result of disease progression. The results presented here suggest that mutations in an epigenetic pathway, which result in alterations of H3K4 methylation patterns, can lead to a chronic disease through subtle changes in gene expression patterns. This implies a direct function for HMTs in maintaining gene expression and the differentiated state in healthy organisms.
Methods
Animals
Mice carrying the Paxip1 null (Paxip1−) and floxed (Paxip1fl) alleles were previously described and genotyped as indicated [14], [53]. To obtain the specific deletion of the Paxip1fl allele in glomerular podocytes, these mice were crossed with the previously characterized 2.5P-Cre mice [24], [25], which express the Cre recombinase under the control of the human NPHS2 promoter (CreNPHS2). Among the next generations, mice carrying the Cre allele (Paxip1fl/fl:CreNPHS2 and Paxip1fl/−:CreNPHS2 mice) were considered as conditional null mutants (PTIP−), whereas littermates that did not express the Cre recombinase were used as controls (PTIP+). All animal procedures were approved by the University Committee on Use and Care of Animals (UCUCA) of the University of Michigan and performed in compliance with ULAM recommendations.
Antibodies
Rabbit polyclonal antibodies used to detect Nephrin (1∶1000) and Podocin (1∶500) were kindly provided by L.B. Holzman (University of Pennsylvania, Philadelphia, PA). Chicken anti-PTIP was described previously [54]. Additional antibodies were commercially available: mouse clone 6F-H2 anti-WT1 (1∶1000, DAKO, Carpinteria, CA), anti-H3K4me3 and anti-H3K27me3 (AbCam, Cambridge, MA), anti-Magi2 (Sigma-Aldrich, St. Louis, MO), anti-Ntrk3 (AF1404, R & D Systems, Minneapolis, MN), Alexa Fluor 488 F(ab′)2 fragment of goat anti-rabbit IgG, Alexa Fluor 568 F(ab′)2 fragment of goat anti-mouse IgG, Alexa Fluor 488 donkey anti-goat IgG (1∶500; Molecular Probes, Life Technologies, Carlsbad, CA).
Urine Collection and Analysis
Mice had access to a standard breeder chow (Purina 5008) and water ad libitum. Urine was collected early in the afternoon for three consecutive days from individual mice at 1, 3, 6 and 12 months of age and stored frozen until use. After thawing, 2 µL urine was run on a SDS-PAGE and stained with Coomassie Blue to test for the presence of proteins/albumin, using recombinant mouse albumin (Sigma-Aldrich, St. Louis, MO) as a control. Quantitative assessment of urine albumin and creatinine concentrations were determined by ELISA using the Albuwell M and Creatinine Companion kits (Exocell Inc., Philadelphia, PA).
Specimen Preparation for Microcopy Analyses
Mice at 1, 3, 6, and 12 months of age were sacrificed and their kidneys were perfused, fixed, and processed for histology, indirect fluorescence and electron microcopy analyses. Briefly, mice were anesthetized by intraperitoneal injection of 40 mg/kg sodium pentobarbital and prepared for systemic perfusion. A saline solution was first injected through the abdominal aorta to the entire mouse body at a pressure of approximately 70 mmHg as previously described [55]. As soon as the general bloodstream had been cleared, a solution of 4% paraformaldehyde in PBS was substituted. It was left to perfuse at the same flow conditions for approximately 10 minutes. Kidneys were removed, decapsulated, cut into pieces, and incubated for 2 additional hours in the appropriate fixative solution before being processed for histology, indirect immunofluorescence, and electron microscopy.
Histology and Indirect Immunofluorescence
Kidneys were fixed in 4% paraformaldehyde, embedded in paraffin, sectioned at 5 microns, and stained with Periodic Acid Shiff or Masson Trichrome. For immunofluorescence analyses with Nephrin, PTIP, WT1 and Magi2, sections were dewaxed, rehydrated, and microwaved for 10 minutes in a citric acid-based antigen unmasking solution (Vector Laboratories, Burlingame, CA). Sections were permeabilized with 0.3% Triton X-100 in PBS and blocked with 10% goat serum in PBS. Primary antibodies were incubated overnight at 4°C in PBS, 0.1% Triton, 2% goat serum. Sections were washed twice and incubated with the secondary fluorescent antibodies and DAPI in PBS, 0.1% Triton, 2% goat serum for 1 hour in the dark at room temperature. The sections were washed again and mounted in Mowiol. Stained and fluorescent-labeled sections were analyzed under a Nikon ES800 microscope. Micrographs were taken with a digital spot camera, using equivalent exposure times among sections. For Ntrk3 staining, fresh frozen sections were dried, fixed in methanol at −20°C and washed in PBS, 0.1% Tween 20 before incubation with anti-Ntrk3 antibodies at 1 µg/ml.
For quantitation of immunofluorescent signals, ImageJ 1.42 was utilized. H3K4me3 stained sections were digitally captured and light intensity measured by placing a fixed size circular area over the nuclei of cells and summing all pixels over the given area. At least 6 podocytes and 6 control cells, either mesangial or endothelial, were measured for each of 8 glomerular tufts (at least 48 podocytes and 48 other cells for each genotype). The average signal intensity was then expressed as a ratio of podocyte intensity to non-podocyte cell intensity for each of the glomerular micrographs taken.
For Cre activity detection, the Rosa26-lacZ reporter strain was used [56]. Mice carrying CreNPHS2 and Paxip1fl/fl were crossed to Rosa26-stop-lacZ:Paxip1fl/+ to generate Paxip1fl/fl:CreNPHS2:Rosa26-lacZ animals. Kidneys were excised at 1 month of age and stained for β-galactosidase activity as described [57].
Scanning and Transmission Electron Microscopy
Longitudinal slices of kidneys from PTIP+ and PTIP − mice fixed with 2.5% glutaraldehyde in 0.1M Sorensen's buffer (pH 7.2) for 2 hours at room temperature were processed for scanning electron microscopy following standard procedures. Briefly, after several washes with the Sorensen's buffer alone, the samples were dehydrated by successive washes in graded ethanol solutions, critical point dried, mounted on a stub, sputter coated with gold-palladium, and examined under an AMRAY 1910 field emission scanning electron microscope. Pieces of the kidney cortex (1 mm3), fixed with 2.5% glutaraldehyde in Sorenson's buffer for 2 hours at room temperature, were processed for transmission electron microscopy following standard procedures. They were embedded in PolyBed 812 resin (Polysciences Inc.), cut into 1-micron slices and stained with toluidine blue. Sample areas were selected based on the presence of glomeruli and cut into ultra-thin sections for analysis under a Philips CM-100 transmission electron microscope. The selected SEM and TEM images are representative of at least 10 different glomeruli per kidney.
Isolation of Mouse Glomeruli
Glomeruli were isolated from the kidneys of individual mice by sieving as described [58]. Briefly, 1 month-old mice were sacrificed by CO2 inhalation and kidneys were removed. After decapsulation, the kidneys were finely minced on ice and passed sequentially through nylon meshes of 90 and 41 microns (Sefar Filtration Inc., Depew, NY). The glomeruli-enriched fraction (GEF) was retained on top of the 41-micron mesh, while kidney tubules were flushed through. RNA was isolated directly from the mesh.
RNA Extraction and Reverse Transcription
Total RNA was extracted from the GEF of individual 1-month-old mice using the RNeasy Tissue Micro Kit (Qiagen, Valencia, CA) following the manufacturer's instructions. RNA concentration and purity were determined by nanodrop analysis on an Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA). Using the Ovation RNA Amplification System V2 (NuGEN Technologies, San Carlos, CA), 500 ng total RNA was reversed transcribed and linearly amplified into single-stranded cDNA, which concentration and purity were determined by nanodrop analysis on an Agilent Bioanalyzer 2100 (Agilent Technologies).
Microarray and Real-Time qPCR Analyses
Microarray analyses were done by the University of Michigan Comprehensive Cancer Center (UMCCC) Affymetrix and Microarray Core Facility. The FL-Ovation cDNA Biotin Module V2 kit (NuGEN Technologies, San Carlos, CA) was used to produce biotin-labeled cRNA, which was then fragmented and hybridized to a Mouse 430 2.0 Affymetrix GeneChip 3′ expression array (Affymetrix, Santa Clara, CA). Array hybridization, washes, staining, and scanning procedures were carried out according to standard Affymetrix protocols. Expression data were normalized by the robust multiarray average (RMA) method and fitted to weighted linear models in R, using the affy and limma packages of Bioconductor, respectively [59], [60]. Only probe sets with a variance over all samples superior to 0.1, a p-value inferior or equal to 0.05 after adjustment for multiplicity using the false discovery rate [61], and a minimum 2-fold difference in expression were selected for the analysis. The complete data set is available from the Gene Expression Omnibus database (accession number GSE17709).
Microarray data were confirmed by real-time quantitative PCR analysis. 25–50 ng single-stranded cDNA was amplified in triplicate in a 384-well plate, using the 7900HT Fast Real Time PCR system (Applied Biosystems, Foster City, CA) and expression levels of selected genes was determined by SYBR Green or TaqMan assays (Applied Biosystems). PCR primers pairs and TaqMan probes used in this study are presented in Table S2.
Chromatin Immunoprecipitation
Glomeruli were isolated from 6 PTIP+ and 6 PTIP − kidneys by sieving as described above. Glomeruli were resuspended in 1 ml PBS and cross linked with 1% formaldehyde for 10 minutes with rocking at room temperature. Chromatin preparation, immunoprecipitation, and PCR analysis was essentially as described previously [13]. Primers pairs for the Ntrk3 locus were as follows: P1, 5′ - CAATGTATTTTGCTTCCTTGCC, 5′ - AAGAAAGGGTTAGGGGAATCCG; P2, 5′ - AACCCGTGCGTTTCGTAAGG, 5′ - GGAGGAAGGAGGAGAAGGAAGATG; P3, 5′ - GCATCTTCCTTCTCCTCCTTCCTC, 5′ - AAGTCACCAAGTCCCACCTCCTAG; P4, 5′ - TTTGCCTTCCCACCGTCTGTTG, 5′ - TGCCTTTGAAACGCCGAAC.
Supporting Information
Zdroje
1. CampbellKH
McWhirJ
RitchieWA
WilmutI
1996 Sheep cloned by nuclear transfer from a cultured cell line. Nature 380 64 66
2. OkitaK
IchisakaT
YamanakaS
2007 Generation of germline-competent induced pluripotent stem cells. Nature 448 313 317
3. WernigM
MeissnerA
ForemanR
BrambrinkT
KuM
2007 In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 448 318 324
4. BergerSL
2007 The complex language of chromatin regulation during transcription. Nature 447 407 412
5. BernsteinBE
MeissnerA
LanderES
2007 The mammalian epigenome. Cell 128 669 681
6. RingroseL
ParoR
2004 Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu Rev Genet 38 413 443
7. SchuettengruberB
ChourroutD
VervoortM
LeblancB
CavalliG
2007 Genome regulation by polycomb and trithorax proteins. Cell 128 735 745
8. RingroseL
ParoR
2007 Polycomb/Trithorax response elements and epigenetic memory of cell identity. Development 134 223 232
9. SchwartzYB
PirrottaV
2007 Polycomb silencing mechanisms and the management of genomic programmes. Nat Rev Genet 8 9 22
10. IssaevaI
ZonisY
RozovskaiaT
OrlovskyK
CroceCM
2007 Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol Cell Biol 27 1889 1903
11. ChoYW
HongT
HongS
GuoH
YuH
2007 PTIP associates with MLL3 - and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem
12. LeeJ
SahaPK
YangQH
LeeS
ParkJY
2008 Targeted inactivation of MLL3 histone H3-Lys-4 methyltransferase activity in the mouse reveals vital roles for MLL3 in adipogenesis. Proc Natl Acad Sci U S A 105 19229 19234
13. PatelSR
KimD
LevitanI
DresslerGR
2007 The BRCT-Domain Containing Protein PTIP Links PAX2 to a Histone H3, Lysine 4 Methyltransferase Complex. Dev Cell 13 580 592
14. ChoEA
PrindleMJ
DresslerGR
2003 BRCT domain-containing protein PTIP is essential for progression through mitosis. Mol Cell Biol 23 1666 1673
15. KimD
PatelSR
XiaoH
DresslerGR
2009 The role of PTIP in maintaining embryonic stem cell pluripotency. Stem Cells 27 1516 1523
16. ChoYW
HongS
JinQ
WangL
LeeJE
2009 Histone methylation regulator PTIP is required for PPARgamma and C/EBPalpha expression and adipogenesis. Cell Metab 10 27 39
17. FangM
RenH
LiuJ
CadiganKM
PatelSR
2009 Drosophila ptip is essential for anterior/posterior patterning in development and interacts with the PcG and trxG pathways. Development 136 1929 1938
18. HansenKH
BrackenAP
PasiniD
DietrichN
GehaniSS
2008 A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol 10 1291 1300
19. MargueronR
JustinN
OhnoK
SharpeML
SonJ
2009 Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461 762 767
20. BlobelGA
KadaukeS
WangE
LauAW
ZuberJ
2009 A reconfigured pattern of MLL occupancy within mitotic chromatin promotes rapid transcriptional reactivation following mitotic exit. Mol Cell 36 970 983
21. ChiP
AllisCD
WangGG
Covalent histone modifications - miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer 10 457 469
22. GluckmanPD
HansonMA
BuklijasT
LowFM
BeedleAS
2009 Epigenetic mechanisms that underpin metabolic and cardiovascular diseases. Nat Rev Endocrinol 5 401 408
23. WigginsRC
2007 The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int 71 1205 1214
24. MoellerMJ
SandenSK
SoofiA
WigginsRC
HolzmanLB
2002 Two Gene Fragments that Direct Podocyte-Specific Expression in Transgenic Mice. J Am Soc Nephrol 13 1561 1567
25. MoellerMJ
SandenSK
SoofiA
WigginsRC
HolzmanLB
2003 Podocyte-specific expression of cre recombinase in transgenic mice. Genesis 35 39 42
26. HoJ
NgKH
RosenS
DostalA
GregoryRI
2008 Podocyte-specific loss of functional microRNAs leads to rapid glomerular and tubular injury. J Am Soc Nephrol 19 2069 2075
27. SuleimanH
HeudoblerD
RaschtaAS
ZhaoY
ZhaoQ
2007 The podocyte-specific inactivation of Lmx1b, Ldb1 and E2a yields new insight into a transcriptional network in podocytes. Dev Biol 304 701 712
28. El-AouniC
HerbachN
BlattnerSM
HengerA
RastaldiMP
2006 Podocyte-specific deletion of integrin-linked kinase results in severe glomerular basement membrane alterations and progressive glomerulosclerosis. J Am Soc Nephrol 17 1334 1344
29. WharramBL
GoyalM
WigginsJE
SandenSK
HussainS
2005 Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol 16 2941 2952
30. StegerK
PaulsK
KlonischT
FrankeFE
BergmannM
2000 Expression of protamine-1 and -2 mRNA during human spermiogenesis. Mol Hum Reprod 6 219 225
31. ChoC
WillisWD
GouldingEH
Jung-HaH
ChoiYC
2001 Haploinsufficiency of protamine-1 or -2 causes infertility in mice. Nat Genet 28 82 86
32. GencB
OzdinlerPH
MendozaAE
ErzurumluRS
2004 A chemoattractant role for NT-3 in proprioceptive axon guidance. PLoS Biol 2 e403 doi:10.1371/journal.pbio.0020403
33. DonovanMJ
HahnR
TessarolloL
HempsteadBL
1996 Identification of an essential nonneuronal function of neurotrophin 3 in mammalian cardiac development. Nat Genet 14 210 213
34. TessarolloL
TsoulfasP
DonovanMJ
PalkoME
Blair-FlynnJ
1997 Targeted deletion of all isoforms of the trkC gene suggests the use of alternate receptors by its ligand neurotrophin-3 in neuronal development and implicates trkC in normal cardiogenesis. Proc Natl Acad Sci U S A 94 14776 14781
35. PavesH
SaarmaM
1997 Neurotrophins as in vitro growth cone guidance molecules for embryonic sensory neurons. Cell Tissue Res 290 285 297
36. MilneTA
BriggsSD
BrockHW
MartinME
GibbsD
2002 MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell 10 1107 1117
37. GlaserS
SchaftJ
LubitzS
VinterstenK
van der HoevenF
2006 Multiple epigenetic maintenance factors implicated by the loss of Mll2 in mouse development. Development 133 1423 1432
38. MuW
WangW
SchimentiJC
2008 An allelic series uncovers novel roles of the BRCT domain-containing protein PTIP in mouse embryonic vascular development. Mol Cell Biol 28 6439 6451
39. PabstR
SterzelRB
1983 Cell renewal of glomerular cell types in normal rats. An autoradiographic analysis. Kidney Int 24 626 631
40. AppelD
KershawDB
SmeetsB
YuanG
FussA
2009 Recruitment of podocytes from glomerular parietal epithelial cells. J Am Soc Nephrol 20 333 343
41. RonconiE
SagrinatiC
AngelottiML
LazzeriE
MazzinghiB
2009 Regeneration of glomerular podocytes by human renal progenitors. J Am Soc Nephrol 20 322 332
42. WykesSM
KrawetzSA
2003 The structural organization of sperm chromatin. J Biol Chem 278 29471 29477
43. CuthbertGL
DaujatS
SnowdenAW
Erdjument-BromageH
HagiwaraT
2004 Histone deimination antagonizes arginine methylation. Cell 118 545 553
44. FaulC
AsanumaK
Yanagida-AsanumaE
KimK
MundelP
2007 Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol 17 428 437
45. PatrakkaJ
TryggvasonK
2007 Nephrin–a unique structural and signaling protein of the kidney filter. Trends Mol Med 13 396 403
46. CastellaniV
BolzJ
1999 Opposing roles for neurotrophin-3 in targeting and collateral formation of distinct sets of developing cortical neurons. Development 126 3335 3345
47. TessarolloL
CoppolaV
FritzschB
2004 NT-3 replacement with brain-derived neurotrophic factor redirects vestibular nerve fibers to the cochlea. J Neurosci 24 2575 2584
48. ZhangHL
EomT
OleynikovY
ShenoySM
LiebeltDA
2001 Neurotrophin-induced transport of a beta-actin mRNP complex increases beta-actin levels and stimulates growth cone motility. Neuron 31 261 275
49. ZhangHL
SingerRH
BassellGJ
1999 Neurotrophin regulation of beta-actin mRNA and protein localization within growth cones. J Cell Biol 147 59 70
50. RastaldiMP
ArmelloniS
BerraS
CalvaresiN
CorbelliA
2006 Glomerular podocytes contain neuron-like functional synaptic vesicles. Faseb J 20 976 978
51. KerjaschkiD
1978 Polycation-induced dislocation of slit diaphragms and formation of cell junctions in rat kidney glomeruli: the effects of low temperature, divalent cations, colchicine, and cytochalasin B. Lab Invest 39 430 440
52. KuriharaH
AndersonJM
KerjaschkiD
FarquharMG
1992 The altered glomerular filtration slits seen in puromycin aminonucleoside nephrosis and protamine sulfate-treated rats contain the tight junction protein ZO-1. Am J Pathol 141 805 816
53. KimD
WangM
CaiQ
BrooksH
DresslerGR
2007 Pax transactivation-domain interacting protein is required for urine concentration and osmotolerance in collecting duct epithelia. J Am Soc Nephrol 18 1458 1465
54. LechnerMS
LevitanI
DresslerGR
2000 PTIP, a novel BRCT domain-containing protein interacts with Pax2 and is associated with active chromatin. Nucleic Acids Res 28 2741 2751
55. VermaR
WharramB
KovariI
KunkelR
NihalaniD
2003 Fyn binds to and phosphorylates the kidney slit diaphragm component Nephrin. J Biol Chem 278 20716 20723
56. SorianoP
1999 Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21 70 71
57. KimD
DresslerGR
2005 Nephrogenic factors promote differentiation of mouse embryonic stem cells into renal epithelia. J Am Soc Nephrol 16 3527 3534
58. SalantDJ
DarbyC
CouserWG
1980 Experimental membranous glomerulonephritis in rats. Quantitative studies of glomerular immune deposit formation in isolated glomeruli and whole animals. J Clin Invest 66 71 81
59. IrizarryRA
WuZ
JaffeeHA
2006 Comparison of Affymetrix GeneChip expression measures. Bioinformatics 22 789 794
60. SmythGK
2004 Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3 Article3
61. BenjaminiY
HochbergY
1995 Controlling the false discovery rate: a practical approach to multiple testing. J Royal Stat Soc 57 289 300
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Genome-Wide Identification of Targets and Function of Individual MicroRNAs in Mouse Embryonic Stem Cells
- Common Genetic Variants and Modification of Penetrance of -Associated Breast Cancer
- Allele-Specific Down-Regulation of Expression Induced by Retinoids Contributes to Climate Adaptations
- β-Actin and γ-Actin Are Each Dispensable for Auditory Hair Cell Development But Required for Stereocilia Maintenance
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy