
Role for the Mammalian Swi5-Sfr1 Complex in DNA Strand Break Repair through Homologous Recombination
In fission yeast, the Swi5-Sfr1 complex plays an important role in homologous recombination (HR), a pathway crucial for the maintenance of genomic integrity. Here we identify and characterize mammalian Swi5 and Sfr1 homologues. Mouse Swi5 and Sfr1 are nuclear proteins that form a complex in vivo and in vitro. Swi5 interacts in vitro with Rad51, the DNA strand-exchange protein which functions during HR. By generating Swi5−/− and Sfr1−/− embryonic stem cell lines, we found that both proteins are mutually interdependent for their stability. Importantly, the Swi5-Sfr1 complex plays a role in HR when Rad51 function is perturbed in vivo by expression of a BRC peptide from BRCA2. Swi5−/− and Sfr1−/− cells are selectively sensitive to agents that cause DNA strand breaks, in particular ionizing radiation, camptothecin, and the Parp inhibitor olaparib. Consistent with a role in HR, sister chromatid exchange induced by Parp inhibition is attenuated in Swi5−/− and Sfr1−/− cells, and chromosome aberrations are increased. Thus, Swi5-Sfr1 is a newly identified complex required for genomic integrity in mammalian cells with a specific role in the repair of DNA strand breaks.
Published in the journal:
. PLoS Genet 6(10): e32767. doi:10.1371/journal.pgen.1001160
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001160
Summary
In fission yeast, the Swi5-Sfr1 complex plays an important role in homologous recombination (HR), a pathway crucial for the maintenance of genomic integrity. Here we identify and characterize mammalian Swi5 and Sfr1 homologues. Mouse Swi5 and Sfr1 are nuclear proteins that form a complex in vivo and in vitro. Swi5 interacts in vitro with Rad51, the DNA strand-exchange protein which functions during HR. By generating Swi5−/− and Sfr1−/− embryonic stem cell lines, we found that both proteins are mutually interdependent for their stability. Importantly, the Swi5-Sfr1 complex plays a role in HR when Rad51 function is perturbed in vivo by expression of a BRC peptide from BRCA2. Swi5−/− and Sfr1−/− cells are selectively sensitive to agents that cause DNA strand breaks, in particular ionizing radiation, camptothecin, and the Parp inhibitor olaparib. Consistent with a role in HR, sister chromatid exchange induced by Parp inhibition is attenuated in Swi5−/− and Sfr1−/− cells, and chromosome aberrations are increased. Thus, Swi5-Sfr1 is a newly identified complex required for genomic integrity in mammalian cells with a specific role in the repair of DNA strand breaks.
Introduction
Homologous recombination (HR) is a key pathway in mammalian cells for the repair of several types of lesions, including DNA strand breaks. Its importance is emphasized by the sensitivity of HR mutants to a variety of DNA damaging agents, as well as the loss of genomic integrity seen in these mutants arising from DNA damage. As a result, HR is a critical DNA repair pathway during development and for tumor suppression [1], [2].
Double-strand breaks (DSBs) arise in DNA as a result of both endogenous cellular processes and from exogenous sources [3], [4]. HR is a precise pathway for the repair of DSBs, during which homologous sequence information is copied from an intact donor template [1], [2], most frequently the sister chromatid during late S/G2 in mitotic cells [5]. A second key pathway for the repair of DSBs is nonhomologous end-joining (NHEJ), where two ends are joined with little or no sequence identity [6]. In addition to canonical two-ended DSBs, one-ended DSBs also arise in DNA [7]. These lesions form when a replication fork encounters a DNA single-strand break that is not repaired by base excision repair, for example, from a covalent topoisomerase I-DNA intermediate as a result of exposure to camptothecin [8], [9]. HR is the primary mechanism for the repair of one-ended DSBs, given that the joining of two unrelated one-ended DSBs by NHEJ would give rise to genomic rearrangements [7].
Many of the known HR factors in mammalian cells, including the central Rad51 protein, have been identified by their homology to yeast HR factors [2], [10], [11]. Rad51, the eukaryotic homologue of Eschericia coli RecA, binds to single-stranded DNA to form a nucleoprotein filament which catalyzes base pairing and strand exchange between homologous DNAs [12]–[14]. Single-stranded DNA is formed at DNA ends by resection [15]; although a substrate for Rad51 filament formation, single-stranded DNA is also bound by replication factor A (RPA), which binds at high affinity and removes secondary structure [16], [17]. While critical for the initiation of HR [18], RPA interferes with Rad51 loading onto single-stranded DNA. Several factors, referred to as “mediators”, are required to overcome the inhibition by RPA to facilitate Rad51 nucleoprotein filament formation [19]. Proposed mediators in yeast include the Rad51 paralogues, Rad55-Rad57, and Rad52 [20], [21]. Vertebrates have five Rad51 paralogues, of which a complex of two have been shown to have mediator activity in vitro [22]. Additionally, the breast cancer suppressor BRCA2, for which there is no homologue in budding or fission yeast, has been proposed to have mediator activity [23]. BRCA2 may also function to stabilize Rad51 filaments on single-stranded DNA, by inhibiting ATP hydrolysis while preventing the formation of non-productive filaments on double-stranded DNA [24].
A distinct complex that functions in fission yeast HR is Swi5-Sfr1. Mutation of either Swi5 or Sfr1 results in reduced HR in both mitotic and meiotic cells [25]–[28]. Like other HR mutants, Swi5 and Sfr1 mutants have elevated sensitivity to a number of DNA damaging agents, including ionizing radiation, UV, and methyl-methanesulfonate [29]. In vitro, the Swi5-Sfr1 complex binds to Rhp51 (the fission yeast Rad51 homologue) in an Sfr1-dependent manner [29], [30], and has been shown to possess mediator activity but importantly also to enhance strand exchange by Rhp51 [30]. While loss of either Swi5-Sfr1 or Rhp55-Rhp57 (fission yeast Rad55-Rad57 homologues) reduces HR, loss of both complexes complete abrogates Rhp51-dependent HR [26]. Both complexes are also required during meiotic recombination [31]. Budding yeast has a homologous complex to Swi5-Sfr1 termed Sae3-Mei5, although this complex is only expressed during meiosis where it plays a critical role in meiotic recombination [32]–[34].
Swi5 forms a second complex with an Sfr1-related protein, Swi2, which localizes to heterochromatin at the donor mating-type loci and promotes HR during switching [26], [29], [35]. In budding yeast, the function of Sae3-Mei5 appears to be limited to supporting the function of Dmc1, the meiosis-specific RecA homologue [33], [34].
Previous reports suggest that both Swi5/Sae3 and Sfr1/Mei5 are evolutionarily conserved, while Swi2 is only found in fission yeast [29], [34]. In this study, we isolated Swi5 and Sfr1 homologues from mice. Swi5 and Sfr1 form a complex in vivo and in vitro, and Rad51 binding to Swi5 is detected in vitro in GST-pull down assays, suggesting that the Swi5-Sfr1 complex has a conserved function in mouse. To investigate their function in vivo, we generated Swi5−/− and Sfr1−/− mouse embryonic stem (ES) cell lines. Although loss of either Swi5 or Sfr1 did not decrease HR frequency by itself, HR was perturbed to a greater extent in these cells by expression of a BRC peptide from BRCA2. Interestingly, Swi5−/− and Sfr1−/− cells were sensitive to ionizing radiation, camptothecin, and an inhibitor of poly(ADP-ribose) polymerase (Parp), all of which cause strand breaks. The induction of sister chromatid exchanges (SCE) by Parp inhibition was attenuated in the Swi5 and Sfr1-deficient cell lines; moreover, Parp inhibition resulted in increased chromatid breaks and radial chromosomes in Swi5−/− and Sfr1−/− cells. Thus, Swi5 and Sfr1 have an important role in the maintenance of genomic integrity in mammalian cells, in particular in the repair of DNA strand breaks.
Results
Cloning and structure of mammalian Swi5 and Sfr1
Based on amino acid conservation, putative mammalian homologues of Swi5/Sae3 and Sfr1/Mei5 have previously been reported [29],[34]. We cloned the mouse homologues, 2900010J23Rik for Swi5 and 6330577E15Rik for Sfr1, based on existing database information (http://www.informatics.jax.org/ and http://uswest.ensembl.org/index.html). The Sfr1 cDNA was successfully amplified by PCR following reverse transcription (RT-PCR) of RNA obtained from mouse ES cells. Sequence analysis of the Sfr1 cDNA revealed that the Sfr1 protein is 303 amino acids and is encoded by four exons (Figure 1A, Figure S1A and S1B). Ectopic expression of the cloned cDNAs complemented the phenotypes of Sfr1−/−cell lines (see below).

Swi5 cDNAs were also obtained by RT-PCR of RNA from mouse ES cells. Two differentially spliced forms were detected containing alternative first exons which encoded proteins of 89 and 121 amino acids (Figure S2A and S2C). When expressed in ES cells, we found that both forms migrated at a lower molecular weight than the endogenous protein (Figure S2B); attempts to clone a cDNA expressing a larger protein were unsuccessful, possibly because the 5′ end of the mRNA contains a structure which impedes amplification or a non-AUG initiation codon. Nevertheless, both forms complemented the phenotypes of Swi5−/− cells (see below and data not shown). In subsequent experiments, we used the Swi5 cDNA encoding the 89 amino acid protein (Figure 1A).
Overall, the sequence identities between mouse and fission yeast proteins were 28.6% (Swi5) and 20.9% (Sfr1). Significant variation was noted between the N-terminus of the various Sfr1 orthologues, even among mouse strains. Mouse Sfr1 has a proline-rich repeat of 16 amino acids at its N-terminus, which we named the RSfp motif (rodent Sfr1 proline rich motif) (Figure 1A and Figure S1B). In the mouse ES cells used in this study (E14) and in DBA/2J mice (Q3TI03), there are five repeats of the RSfp motif, whereas in C57BL/6J mice there are six repeats. The rat Sfr1 homologue (rCG57555) has two repeats. Repetition of the RSfp motif appears to be unique to rodents, as only a single RSfp motif is present in other mammals, including human, rabbit, dog and pig (Figure S1C). The RSfp motif is not present outside of mammals, although the downstream region is conserved (Figure S1D).
Mouse Swi5 and Sfr1 form a complex in vivo and in vitro
In fission yeast and in budding yeast, Swi5/Sae3 and Sfr1/Mei5 form a stable complex in vivo and in vitro. To determine whether mouse Swi5 and Sfr1 interact, we performed a yeast two-hybrid assay (Figure 1B). Swi5 fused to the Gal4 activation domain (AD) and Sfr1 fused to the Gal4 DNA binding domain (DBD) gave a positive interaction, suggesting a physical association between the two. The reverse test was uninformative as Swi5 fused to the Gal4DBD itself allowed growth on the test medium. In addition, Sfr1 showed self-association, which has also been observed in fission yeast [29].
We also tested complex formation with a GST pull-down assay using recombinant proteins expressed in E. coli. Unlike expression of yeast Sfr1/Mei5, which yields insoluble protein without co-expression of Swi5/Sae3 [30], , mouse His6-Sfr1 was soluble by itself (Figure 1C). The tagged Sfr1 migrated at a higher molecular weight (∼50 kDa) than the molecular weight calculated from the amino acid sequence (36 kDa). An unexpected lower mobility was seen with the endogenous Sfr1 protein (see below, Figure 2C). The E. coli extract expressing His6-Sfr1 was incubated with GST-Swi5 or GST alone immobilized on magnetic beads. Pull-down of GST-Swi5, but not GST, brought down His6-Sfr1 (Figure 1C), again indicating a physical association between Swi5 and Sfr1.
To determine their interacting domains, two-hybrid and GST pull-down assays were performed with N and C-terminal fragments from both proteins (Figure 1A). The N-terminal half of Swi5, but not the C-terminal half, interacted with Sfr1 in both assays (Figure 1D and 1F). Conversely, the C-terminal fragment of Sfr1, but not the N-terminal fragment, interacted with Swi5 (Figure 1E and 1G). The interacting fragments from both proteins contain coiled-coil motifs (Figure 1A), which may be responsible for the interaction. Consistent with their variability in different species, the RSfp motifs of Sfr1 did not appear to play a role in the interaction.
Co-immunoprecipitations were performed with mouse ES cell extracts to investigate the interaction in vivo. Using antibodies directed against the endogenous proteins, Swi5 co-precipitated Sfr1 and Sfr1 co-precipitated Swi5 (Figure 1H). Most of the Swi5 in the cell seems to be in a complex with Sfr1. Thus, despite the poor sequence conservation overall, Sfr1 is a major interacting partner for the Swi5 in the cell, consistent with the better conservation of the Sfr1 C-terminal portion, which interacts with Swi5.
Swi5 and Sfr1 are nuclear proteins that are interdependent for protein stability
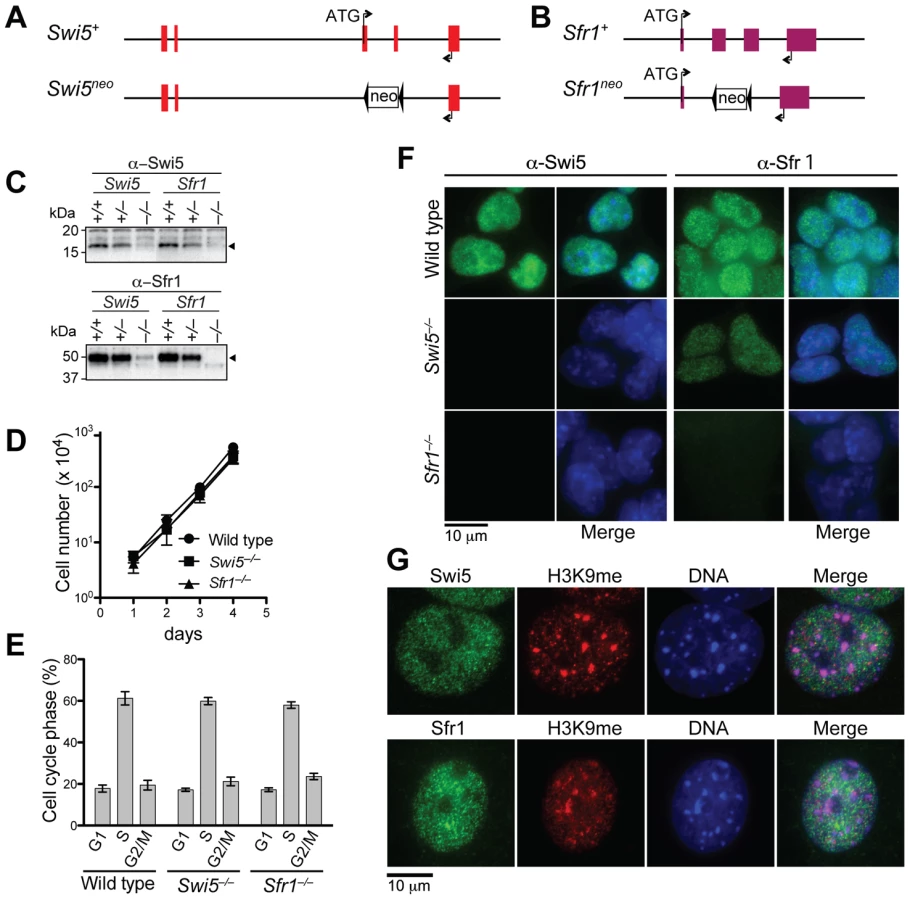
To investigate their cellular functions, we generated Swi5 and Sfr1-deficient mouse ES cell lines. The Swi5 targeting vector was designed to replace the exons 3 and 4 with a neomycin resistance gene (neo), resulting in deletion of most of the Swi5 coding sequence, including the sequence for Sfr1 interaction (Figure 2A and Figure S3A). In the Sfr1 targeting vector, the neo gene replaced exons 2 and 3, which encode amino acids 5 to 240, removing 78% of the coding region (Figure 2B and Figure S3B). Two rounds of gene targeting were performed, with an intervening step to delete the neo gene from the first targeted allele using Cre recombinase (Figure S3A and S3B). Successful gene targeting of both Swi5 and Sfr1 alleles in the respective cell lines was confirmed by Southern blotting (Figure S3A and S3B), and loss of protein was confirmed by Western blotting (Figure 2C) and immunofluorescence (Figure 2F). The Swi5−/− and Sfr1−/− cell lines (formally Swi5Δ/neo and Sfr1Δ/neo, respectively) exhibited similar proliferation kinetics and cell cycle distribution as wild-type cells (Figure 2D and 2E), indicating that Swi5 and Sfr1 are not essential for cell viability.
As Swi5 and Sfr1 form a complex, we determined whether loss of one affects the stability of the other by Western blotting (Figure 2C). Swi5 protein was not detectable in Sfr1−/− cells, indicating that the stability of Swi5 requires association with Sfr1. The level of Sfr1 in Swi5−/− cells was also diminished, although the protein was still detectable. These results provide further evidence for a physical association between Swi5 and Sfr1 in vivo.
To determine the sub-cellular localization of Swi5 and Sfr1, mouse ES cells and embryonic fibroblasts (MEFs) were examined by immunofluorescence. Swi5 and Sfr1 localized to the nucleus in both cell types (Figure 2F and 2G). Importantly, Swi5 was not detected in Sfr1−/− ES cells, providing further support that Swi5 is unstable without Sfr1; Sfr1 was detectable in Swi5−/− ES cells, albeit weakly (Figure 2F). In fission yeast, Swi5 localizes to heterochromatin as well as to euchromatin [26]. However, neither mouse protein specifically localized to heterochromatin, as marked by trimethyl-lysine 9 of histone H3 and intense DAPI staining (Figure 2G). Rather, both proteins had a more widespread nuclear distribution that was, nonetheless, somewhat granular.
Mouse Swi5 interacts with Rad51
The fission yeast Swi5-Sfr1 complex interacts with the Rad51 recombinase through the Sfr1 subunit [29], [30]. We tested whether Rad51 interaction would be conserved with the mouse proteins by co-immunoprecipitation from ES cell extracts. Neither Swi5 nor Sfr1 precipitated detectable amounts of Rad51 from either untreated (Figure 1H) or γ-irradiated cells (data not shown). To investigate this further, GST pull-down assays were performed with recombinant Rad51 expressed in E. coli (Figure 3A). Pull-down of Rad51 was detected with GST-Swi5, but not with GST-Sfr1 or GST alone (Figure 3A). Treatment of the extracts with ethidium bromide or DNase I did not affect the association between GST-Swi5 and Rad51 (Figure S4A). These results indicate a physical association between Swi5 and Rad51.

We tested whether Swi5 and Sfr1 co-localize with Rad51 in nuclear foci after X-irradiation. Unlike Rad51, Swi5 and Sfr1 were distributed throughout the nucleus, as in untreated cells, indicating that there was no specific recruitment of these proteins to DSB sites (Figure S4B). Further, Rad51 focus formation after X-irradiation was not noticeably affected in either Swi5−/− and Sfr1−/− cells (Figure S4C).
HR is reduced in Swi5−/− and Sfr1−/− cells when HR is compromised
The conservation of the protein complex and the interaction with Rad51 suggested that Swi5-Sfr1 could play a role in HR in mammalian cells. We examined HR levels in the Swi5−/− and Sfr1−/− ES cells using the DR-GFP assay [37] (Figure 3B). In this assay, a single DSB is introduced into the chromosomally integrated DR-GFP substrate by the I-SceI endonuclease; repair of the DSB by HR gives rise to cells expressing functional GFP. After I-SceI expression, Swi5−/− and Sfr1−/− cells gave similar levels of GFP positive cells (4.9% and 4.4%, respectively) as wild-type cells (5.2%; Figure 3C), indicating that Swi5 and Sfr1 are not essential for HR in mouse cells.
In fission yeast, the Swi5-Sfr1 complex stabilizes Rad51 filament formation on single-stranded DNA [38]. We hypothesized that if Rad51 nucleoprotein filaments were perturbed in mouse cells, a role for the Swi5-Sfr1 complex in HR might be uncovered. BRCA2 is a central HR protein in mammalian cells, binding Rad51 at a series of repeats ∼35 amino acids (BRC repeats); as an isolated peptide, the BRC repeat has been demonstrated to bind Rad51, to inhibit Rad51 focus formation [39]–[41] and, importantly, to decrease HR in mammalian cells [42]. Compared to cells transfected with an empty expression vector (5.8%) (Figure 3D), expression of BRC3 in wild-type cells resulted in a significantly reduced frequency of GFP positive cells (0.55%) and hence HR (Figure 3E), consistent with previous results. This inhibitory effect on HR was not observed with expression of the BRC3Δ peptide which is unable to bind Rad51 [43] (6.6%; Figure 3F) and, further, was rescued by Rad51 overexpression (data not shown).
With BRC3 expression, Swi5−/− and Sfr1−/− cells exhibited a 2.1-fold and 1.9-fold reduction of GFP positive cells (0.26% and 0.29%, respectively) compared to wild-type cells (Figure 3E), indicating that Swi5-Sfr1 plays a role in HR when it is compromised. Consistent with this interpretation, expression of the cognate cDNAs complemented the HR defect (Figure 3E). The defect in HR was dependent on the ability of the BRC3 repeat to bind Rad51, as a similar number of GFP positive cells were obtained with BRC3Δ expression (Figure 3F). These results indicate that the Swi5 and Sfr1 function in HR, but are not required for the process unless it is already compromised. Because BRC3 perturbs Rad51 focus formation, mouse Swi5-Sfr1 may play a role in stabilizing Rad51 filaments, as in fission yeast.
Swi5−/− and Sfr1−/− cells are sensitive to agents that cause strand breaks
Given the HR phenotype associated with these cells, we next examined the sensitivity of Swi5−/− and Sfr1−/− cells to DNA damaging agents. In these assays, Brca2lex1/lex2 cells were included for comparison, as they are known to be defective in HR [44]. Swi5−/− and Sfr1−/− cells were found to be more sensitive to X-rays than wild-type cells, although their sensitivity was less pronounced than that of Brca2lex1/lex2 cells (Figure 4A). Expression of the Swi5 or Sfr1 cDNA in the respective mutant cells restored survival to the level observed in wild-type cells, demonstrating that the sensitivity was specifically due to the deletion of Swi5 or Sfr1.

Cells were also exposed to topoisomerase poisons, which like X-rays lead to strand breaks. Both Swi5−/− and Sfr1−/− cells exhibited sensitivity to the type I topoisomerase poison camptothecin, although not as severely as Brca2lex1/lex2 cells (Figure 4B). Interestingly, Sfr1−/− cells were somewhat more sensitive to camptothecin than Swi5−/− cells. Sfr1−/− cells were also sensitive to the type II topoisomerase poison etoposide. The two mutants again showed differential sensitivity, with Sfr1−/− cells showing a more severe phenotype. In this case, Sfr1−/− cells were even more sensitive than Brca2lex1/lex2 cells, whereas Swi5−/− cells were no more sensitive than wild-type cells (Figure 4C). These results suggest that Swi5 and Sfr1 have a function in repairing DNA strand breaks, the primary lesions from X-irradiation and topoisomerase poisons. Given the greater sensitivity observed in Sfr1−/− cells, they also indicate that the roles of Swi5 and Sfr1 are not equivalent in the cell.
Cells with defective DNA damage checkpoints often exhibit sensitivity to DNA damaging agents. Chk1 and Chk2 are two proteins that are phosphorylated upon X-irradiation [45], [46]. After X-irradiation, Swi5−/− and Sfr1−/− cells were proficient at phosphorylation of both proteins and showed similar kinetics (Figure 4D). Checkpoint-proficient cells also arrest after DNA damage rather than proceed into mitosis. Mitotic populations were reduced to a similar extent in Swi5−/− and Sfr1−/− cells as in wild-type cells (Figure 4E). These results point to intact DNA damage checkpoints in both mutants.
We also tested the sensitivity of Swi5−/− and Sfr1−/− cells to a variety of other DNA damaging agents. HR mutants are typically sensitive to interstrand crosslinking agents [47]–[49], yet we observed that Swi5−/− and Sfr1−/− cells were not any more sensitive to either mitomycin C or cisplatin than wild-type or complemented cells (Figure S5A and S5B). In addition, cells were not sensitive to the replication inhibitor hydroxyurea (Figure S5C), implying that the camptothecin sensitivity is specifically related to strand breaks generated by this agent rather than indirectly to problems with replication per se. Finally, neither mutant was sensitive to ultraviolet light (Figure S5D), indicating that the proteins do not play a role in nucleotide excision repair. Interestingly, Brca2lex1/lex2 cells were found to be sensitive, suggesting a role for HR repair of UVC lesions.
Swi5−/− and Sfr1−/− cells are sensitive to Parp inhibition
Poly(ADP-ribose) polymerase (Parp) plays an important role in the repair of DNA single-strand breaks, such that inhibition of Parp activity leads to the accumulation of the unrepaired single-strand breaks that turn into DSBs when encountered by replication forks. Since the repair of DSBs arising during replication largely depends on the HR pathway, cells deficient in HR are extremely sensitive to Parp inhibitors [50], [51]. To further investigate the effects of Swi5 and Sfr1 deficiency on the repair of DNA strand breaks, Swi5−/− and Sfr1−/− cells were exposed to the Parp inhibitor olaparib. Consistent with previous reports, Brca2 mutant cells were exquisitely sensitive to olaparib; by contrast, the NHEJ mutant Ku70−/− was not (Figure 5A). Swi5−/− and Sfr1−/− cells were also significantly more sensitive to olaparib than wild-type cells, although not as sensitive as Brca2lex1/lex2 cells (Figure 5A). This sensitivity was suppressed by introducing the cognate cDNAs into the Swi5−/− and Sfr1−/− cells (Figure 5A). Sensitivity of the cell lines to Parp inhibition further implicates Swi5 and Sfr1 in the repair of DNA strand breaks.

To further examine the effect of Parp inhibition on Swi5 and Sfr1-deficient cells, chromosomes were examined for aberrations in metaphase spreads. In Swi5−/− and Sfr1−/− cells, chromatid breaks were elevated 30 and 20-fold, respectively, after exposure to olaparib compared with untreated cells, significantly more than that observed in wild-type cells (9-fold; Figure 5B). Radial chromosomes, which were not observed in untreated cells, were also induced in Swi5−/− and Sfr1−/− cells. Both of these types of aberrations typically arise from problems encountered during DNA replication. Brca2lex1/lex2 cells showed a substantial number of chromatid breaks even without olaparib, but chromatid breaks increased and radial chromosomes were observed upon olaparib treatment. The level of aberrations in olaparib-treated Brca2lex1/lex2 cells was similar to that found in the treated Swi5−/− and Sfr1−/− cells, but aberrations may be underestimated if the G2/M checkpoint was activated. The observation of increased chromatid breaks and radial chromosomes in Swi5−/− and Sfr1−/− cells suggest that unrepaired DSBs accumulate, which may be responsible for the toxicity observed with Parp inhibition in these cells.
The accumulation of chromatid breaks induced by Parp inhibition may be the result of HR deficiency. To test this, we examined sister-chromatid exchange (SCE), which is one of outcome of HR (Figure 5C). The spontaneous SCE frequency was similar among wild-type, Swi5−/− and Sfr1−/− cells (9.3, 9.3 and 9.7 SCEs per metaphase, respectively), while Brca2lex1/lex2 cells showed a lower frequency of SCE (7.1 SCEs per metaphase). With Parp inhibition, SCEs were significantly induced in wild-type cells (41.1 SCEs per metaphase) as well as in Brca2lex1/lex2 cells, although the overall level was lower (30.1 SCEs per metaphase). In Swi5−/− and Sfr1−/− cells, the overall level of SCEs was reduced compared with wild type (35.0 and 35.4 SCEs per metaphase, respectively). These results indicate that SCE induction by Parp inhibition is partially dependent on Swi5 and Sfr1.
Discussion
In this study, we identified Swi5 and Sfr1 orthologues in mammalian cells and determined that they have critical roles in the repair of DNA strand breaks. Despite their low conservation with the respective yeast proteins, we found that mouse Swi5 and Sfr1 form a complex in vivo and in vitro, as do fission yeast Swi5 and Sfr1 and budding yeast Sae3 and Mei5 [29], [30], [34]. The integral nature of the protein-protein interactions is emphasized by the mutual interdependence of the Swi5 and Sfr1 for stability, and by the finding that Sfr1 co-immunoprecipitates Swi5 to a similar extent as immunoprecipitation of Swi5 itself. Although the budding yeast complex is only expressed during meiosis [32], [34], mouse Swi5-Sfr1 is expressed in mitotically dividing cells, making it more akin to the fission yeast complex.
We found that Swi5 or Sfr1-deficient mammalian cells are sensitive to agents that cause DNA strand breaks, including X-rays, camptothecin, and the Parp inhibitor olaparib. Consistent with a DNA damage repair defect in Swi5−/− and Sfr1−/− cells, chromosome aberrations are increased compared to wild-type when cells are challenged with olaparib. For the most part, the sensitivities of Swi5−/− and Sfr1−/− cells are similar to each other, although unlike Swi5−/− cells, Sfr1−/− cells are also sensitive to etoposide. In contrast to Swi5, the stability of Sfr1 is not fully compromised when its partner protein is absent, consistent with Sfr1 functions that are independent of Swi5 in some contexts, as is the case with fission yeast [52], [53]. While fission yeast Swi5 acts independent of Sfr1 during mating-type switching, mouse Swi5 is unlikely to have Sfr1-independent functions, given its instability in the absence of Sfr1.
Sensitivity to camptothecin and olaparib is consistent with a defect in the ability to repair DNA damage by HR. That Swi5 interacts with Rad51 in vitro, the critical strand exchange protein for HR reactions, supports a role for the mammalian Swi5-Sfr1 complex in HR, like the cognate complexes in fission and budding yeast [26], [33], [34]. Further, DNA damage-induced SCEs are reduced in Swi5−/− and Sfr1−/− cells compared with wild-type cells. Moreover, although direct assay of DSB-induced HR in these cells did not reveal an intrinsic HR defect, a more severe defect in HR is observed in both the Swi5−/− and Sfr1−/− cells when HR is compromised by interfering with Rad51 function.
Unlike typical mammalian HR mutants, however, Swi5−/− and Sfr1−/− cells are not sensitive to interstrand crosslinking agents or the replication inhibitor hydroxyurea. Although both agents lead to DSBs during S phase and induce HR, DSBs are detected by pulse field gel electrophoresis only after prolonged incubation with these agents and require the structure-specific nuclease Mus81 for their formation [54], [55]. By contrast, when a replication fork encounters a single-strand break, a one-ended DSB is generated with fast kinetics, as DSBs appear within 30 min after camptothecin exposure during S phase [56]. In fission yeast, evidence points to a role for Swi5-Sfr1 (or Swi5-Swi2) acting specifically at one end of a DSB or at the one-ended DSBs at the mat locus during either mating-type switching or sister chromatid recombination in donorless strains [26], [57], [58]. Taken together, we propose that Swi5-Sfr1 is an evolutionarily conserved complex that acts at specific types of lesions, specifically at one-ended DSBs.
These experiments reveal a role for the mammalian Swi5-Sfr1 complex in HR. Although Swi5-Sfr1 are required for repair when the DNA damage load is high, the role of the complex appears to be more restricted than that of BRCA2 and the Rad51 paralogues, given the more severe phenotype seen when these other proteins are deficient [37], [44], [59]. In fission yeast, which does not have a BRCA2 orthologue, both Swi5-Sfr1 and the Rad51 paralogue complex Rhp55-Rhp57 are required for high levels of HR [26]. Thus, a shift in dependence on the Swi5-Sfr1 complex may have occurred during evolution. How might Swi5-Sfr1 function in HR? In vitro, the fission yeast Swi5-Sfr1 complex has mediator activity [30]. Moreover, the fission yeast Swi5-Sfr1 complex stabilizes the Rad51 filament on single-stranded DNA [38]. We hypothesize that the mammalian complex plays a similar role, given the reduced recombination in Swi5−/− and Sfr1−/− cells in the presence of the BRC3 repeat, which is known to perturb Rad51 focus formation [40], [41].
It is noteworthy, however, that the interaction of the Swi5-Sfr1 complex with Rad51 is through Swi5, in contrast to fission yeast where the interaction with Rhp51 is through Sfr1 [29], [30]. In both mouse cells and fission yeast, the interaction between Swi5-Sfr1 and Rad51 is detected in vitro, but not in vivo, as co-precipitation of the endogenous proteins has been unsuccessful, even under DNA damaging conditions [29]. Thus, Swi5-Sfr1 and Rad51 may interact weakly or transiently in cells. In fission and budding yeast, Swi5-Sfr1 and Sae3-Mei5, respectively, bind and promote the activity of Dmc1 [30], [33], [34], the meiosis-specific strand exchange protein, which is also critical for mouse meiosis [60], [61]. Whether Swi5-Sfr1 plays a similar role in mammalian cells awaits mouse knockout studies of the complex, although notably we have detected high level of expression of the complex in the testis, including a testis-specific isoform of Swi5 (Y.A. and M.J., unpublished results).
In summary, we have characterized a novel complex critical for DNA strand break repair in mammalian cells. The importance of strand break repair is well recognized, as defective repair is associated with various neurodegenerative diseases [62]. Moreover, therapeutic approaches to some cancers are being developed which increase the cellular load of DNA strand breaks through Parp inhibition [63]. The identification of Swi5-Sfr1 as being important for cellular resistance to agents like olaparib therefore has potential clinical as well as biological relevance.
Materials and Methods
Swi5 and Sfr1 cDNA cloning
Primers for Swi5 and Sfr1 cDNA cloning were designed based on annotated transcripts from the Ensembl database. For Swi5, forward-reverse primer pairs, YA110 (5′ATACCCACCCCTCCCAATAC)-YA113 (5′AGTTTAAGCCCACCCCACTC) and YA532 (5′ATTATTGTCGACATGGGAAGCAGGGGCGGAAC)-YA127 (5′GCCGGCGGCCGCTTACTATCAGTCATTCAGGTTTAGATC), were designed based on annotated transcripts ENSMUSG00000044627 and ENSMUST00000113400, respectively. For Sfr1, the forward-reverse primer pair, YA114 (5′GGCTGTGTGTACGGTGTGTC)-YA115 (5′CCTCCCTCTAAGCCACAACA), was designed based on annotated transcript ENSMUST00000099353. The genomic structures presented in Figures S1A and S2A were derived by comparing the amplified cDNA sequences to the genomic structures in Ensembl.
GST-pull down assay
The full length and truncated Swi5 cDNAs were cloned into the GST expression vector pGEX6P-1 (GE healthcare). GST and GST-Swi5 proteins were expressed in E. coli (UT481). The cell lysates were obtained by sonication of cells in R-buffer (20 mM Tris-HCl, pH 7.6/1 mM EDTA/100 mM NaCl/0.1% Triton X-100/1 mM DTT/10% Glycerol) followed by centrifugation at 15000 ×g for 20 min. The expressed GST and GST-Swi5 protein in the lysates were immobilized to the MagneGST (Promega). The His6-Sfr1 and Rad51 proteins were expressed in E. coli BL21-CodonPlus (DE3) from plasmids pET15b or pET21d (Novagen) respectively. The lysates (40 µg of proteins) obtained in R-buffer with sonication followed by centrifugation were mixed with 10 µl of the GST or GST-Swi5 protein immobilized to MagneGST, and incubated three hours at 4°C. The precipitates were then washed three times with R - buffer and eluted by boiling in SDS-PAGE sample buffer. The co-precipitations were subjected to SDS-PAGE with Coomassie Brilliant Blue (CBB) staining and to Western blotting.
Yeast two-hybrid assay
The full length and truncated Swi5 and Sfr1 cDNAs were cloned to pGADT7 or pGBKT7 expression vectors to fuse to the Gal4 activation domain (AD) or the Gal4 DBA-binding domain (DBD). The experiments were performed according to the manufacturer's instructions (Matchmaker Two-Hybrid system 3 from Clontech).
Generation of mouse Swi5 and Sfr1 antibodies
The full length of Swi5 and Sfr1 cDNAs were cloned in pET15b vector (Novagen). The His6-tagged Swi5 and Sfr1 proteins, as immunogens, were expressed in E. coli (BL21-Codonplus DE3) and purified using affinity to TALON (Clontech). Polyclonal antisera against Swi5 and Sfr1 were generated by Covance. Each antiserum was affinity purified against the respective protein.
Immunoprecipitataion
Protein extracts from mouse ES cells were obtained by lysing cells in L-buffer (50 mM Tris-HCl, pH 8.0/2 mM EDTA/125 mM NaCl/1% NP-40/Complete protease inhibitor cocktail from Roche/Halt phosphatase inhibitor mixture from Pierce) on ice for 20 min followed by centrifugation at 15000 ×g for 20 min. The antibodies were added to the protein extract (200 µg of protein) and then incubated for 2 hours at 4°C. Protein G Dynabeads (Invitrogen) were added and the mixtures were incubated for an additional hour. The precipitates were washed six times with W-buffer (50 mM Tris-HCl, pH 8.0/2 mM EDTA/200 mM NaCl/1% NP-40) and subsequently eluted by boiling in SDS-PAGE sample buffer. Immunoprecipitated proteins were analyzed by Western blotting.
Immunofluorescence
The cytospin slide centrifuge was used to spread ES cells on glass slides. MEF cells were grown directly on cover slips. Cells were fixed with 4% paraformaldehyde in PBS for 10 min and then permeabilized in PBS containing 0.25% Triton X for 10 min. Following incubation with blocking buffer (10% FBS in PBS) for 1 hour, cells were incubated with the indicated primary antibodies (diluted in blocking buffer) for 16 hours at 4°C followed by Alexa 488 or 594 conjugated secondary antibodies (Invitrogen) for 2 hours at RT and washed with PBS before mounting in ProLong antifade reagent with DAPI (Invitrogen).
Targeting vector construction
To create pYA163, the beta-actin promoter driven diphtheria toxin A (DTA) fragment from pBADT3-BSKII (a gift of Dr. Valter Agosti) and the loxP-Neo-loxP cassette from pEGFPKT1loxneo (a gift from Dr Willie Mark) was cloned into Sma I/Xba I and Cla I/Hind III sites respectively in pBluescript SK+. The targeting arms were PCR amplified from mouse genomic DNA, using primer sets; YA217 (5′TATAGTCGACTCTTTCCTTTCTCAGACATGGGTTC) and YA218 (5′ATATCTCGAGAACATTACAGATCAGAGTCTATGAATAT) for the Swi5 long targeting arm; YA189 (5′GGTCTTGGAGTTTACTCCTTATC) and YA191 (5′GGCCCTCTGAAGATAAGATTTGT) for the Swi5 short targeting arm; YA266 (5′ATAAACAATCAGCCAGATAACCAGA) and YA267 (5′TGAGACAGAAAGAGGGTGGATCT) for the Sfr1 short targeting arm; YA270 (5′TAATGTCGACCATTTCCAACATCCAGCATTCCT) and YA271 (5′TATACTCGAGACGCGAATGATAATCAAATTATCTC) for the Sfr1 long targeting arm. PCR products were confirmed by sequencing. The long targeting arms and the short targeting arms were cloned to pYA163 at Sal I and at Eco RV, respectively, to generate the Swi5 targeting vector (pYA186) and the Sfr1 targeting vector (pYA249).
Cell lines
The E14 DR-GFP mouse ES cell line was established previously [37]. For gene targeting of Swi5 and Sfr1, 10 µg of Sal I-linearized targeting vectors were electroporated into 1×107 E14 DR-GFP cells suspended in OPTI-MEM by pulsing cells at 250 V, 500 µF. After 24 hours of incubation, G418 was added at final concentration of 300 µg/ml. The medium was changed every other day. After 7 days, colonies were isolated and the gene targeting was confirmed by PCR and Southern blotting. To remove the neo gene, the pCAGGS-Cre vector (10 µg) was transiently transfected into cells by electroporation. Colonies were grown without G418 treatment, and clones were examined by PCR and Southern blotting. To create constructs which complement Swi5 and Sfr1 deficient cell lines, cDNAs encoding Swi5 and Sfr1 were cloned into the mammalian expression vector pCAGGS that was modified to contain a hygromycin resistance gene (Hyg) at Hind III. These constructs were electroporated into Swi5−/− and Sfr1−/− cells, and cells were grown in hygromycin to select stable clones. Swi5 and Sfr1 expressing cells were confirmed by Western blotting.
DR-GFP assay
This assay has been described previously [64]. Briefly, 30 µg of I-SceI expression vector, pCBASce was electroporated into ES cells suspended in 650 µl of OPTI-MEM (Invitrogen) at 250 V, 950 µF in a 0.4 cm cuvette. In the experiments with the BRC3 peptide, 30 µg of BRC3, BRC3Δ, or the empty expression vector [42] were additionally added. BRC3Δ contains a 7 amino acid deletion, abrogating the interaction with Rad51 [42], [43]. To measure HR frequency, GFP-positive cells were scored by flow cytometry at 48 hours following electroporation.
Survival assay
For the clonogenic survival assay, 500 cells were seeded onto a 6 cm dish and incubated for 24 hours to allow cells to attach to the bottom. For X-ray sensitivity assays, cells were irradiated with the indicated doses. For camptothecin or etoposide sensitivity assays, cells were exposed to the indicated concentrations of drug continuously for 9 days. Then colonies were fixed with methanol and stained with Giemsa. To examine olaparib sensitivity, 500 cells were seeded per well of a 24-well plate. After 24 hours incubation, olaparib was added at the indicated concentration, and cells were continuously exposed to olaparib for 7 days before fixing and staining with Giemsa.
Chromosome analysis
To prepare metaphase spreads, ES cells were treated with 0.03 µg/ml of colcemid for 30 min. Cells were collected and incubated in hypotonic solution (0.56% KCl) for 20 min. Subsequently cells were fixed in methanol: acetic acid (3∶1) and washed. The cell suspensions in fixative were spotted to slides and air-dried. To measure chromatid aberrations, the slides were stained with 2% Giemsa/Sorensen's buffer for 5 min. After washing with water, the spreads were mounted in Permount. To visualize SCE, ES cells were incubated with 10 µM BrdU during two cell cycles. After metaphase spreads were prepared, the slides were treated with 1 µg/ml Hoechst 33258 in Sorensen's buffer and rinsed with 2× SSC. The slides were exposed to black light for 20 min, incubated at 60°C for 2 hours, and stained in 2% Giemsa/Sorensen's buffer. The excess staining was washed with water and the slides were mounted in Permount.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. MoynahanME
JasinM
2010 Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol 11 196 207
2. San FilippoJ
SungP
KleinH
2008 Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 77 229 257
3. LindahlT
WoodRD
1999 Quality control by DNA repair. Science 286 1897 1905
4. van GentDC
HoeijmakersJH
KanaarR
2001 Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet 2 196 206
5. JohnsonRD
JasinM
2000 Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J 19 3398 3407
6. LieberMR
2010 The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 79 181 211
7. HelledayT
LoJ
van GentDC
EngelwardBP
2007 DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair (Amst) 6 923 935
8. StrumbergD
PilonAA
SmithM
HickeyR
MalkasL
2000 Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol Cell Biol 20 3977 3987
9. Saleh-GohariN
BryantHE
SchultzN
ParkerKM
CasselTN
2005 Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol Cell Biol 25 7158 7169
10. ShinoharaA
OgawaH
MatsudaY
UshioN
IkeoK
1993 Cloning of human, mouse and fission yeast recombination genes homologous to RAD51 and recA. Nat Genet 4 239 243
11. YoshimuraY
MoritaT
YamamotoA
MatsushiroA
1993 Cloning and sequence of the human RecA-like gene cDNA. Nucleic Acids Res 21 1665
12. SungP
RobbersonDL
1995 DNA strand exchange mediated by a RAD51-ssDNA nucleoprotein filament with polarity opposite to that of RecA. Cell 82 453 461
13. OgawaT
YuX
ShinoharaA
EgelmanEH
1993 Similarity of the yeast RAD51 filament to the bacterial RecA filament. Science 259 1896 1899
14. YuX
JacobsSA
WestSC
OgawaT
EgelmanEH
2001 Domain structure and dynamics in the helical filaments formed by RecA and Rad51 on DNA. Proc Natl Acad Sci U S A 98 8419 8424
15. MimitouEP
SymingtonLS
2009 Nucleases and helicases take center stage in homologous recombination. Trends Biochem Sci 34 264 272
16. BrillSJ
StillmanB
1989 Yeast replication factor-A functions in the unwinding of the SV40 origin of DNA replication. Nature 342 92 95
17. SugiyamaT
ZaitsevaEM
KowalczykowskiSC
1997 A single-stranded DNA-binding protein is needed for efficient presynaptic complex formation by the Saccharomyces cerevisiae Rad51 protein. J Biol Chem 272 7940 7945
18. SungP
1994 Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science 265 1241 1243
19. SungP
KrejciL
Van KomenS
SehornMG
2003 Rad51 recombinase and recombination mediators. J Biol Chem 278 42729 42732
20. SungP
1997 Yeast Rad55 and Rad57 proteins form a heterodimer that functions with replication protein A to promote DNA strand exchange by Rad51 recombinase. Genes Dev 11 1111 1121
21. SungP
1997 Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J Biol Chem 272 28194 28197
22. SigurdssonS
Van KomenS
BussenW
SchildD
AlbalaJS
2001 Mediator function of the human Rad51B-Rad51C complex in Rad51/RPA-catalyzed DNA strand exchange. Genes Dev 15 3308 3318
23. San FilippoJ
ChiP
SehornMG
EtchinJ
KrejciL
2006 Recombination mediator and Rad51 targeting activities of a human BRCA2 polypeptide. J Biol Chem 281 11649 11657
24. CarreiraA
HilarioJ
AmitaniI
BaskinRJ
ShivjiMK
2009 The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell 136 1032 1043
25. KhasanovFK
SalakhovaAF
KhasanovaOS
GrishchukAL
ChepurnajaOV
2008 Genetic analysis reveals different roles of Schizosaccharomyces pombe sfr1/dds20 in meiotic and mitotic DNA recombination and repair. Curr Genet 54 197 211
26. AkamatsuY
TsutsuiY
MorishitaT
SiddiqueMS
KurokawaY
2007 Fission yeast Swi5/Sfr1 and Rhp55/Rhp57 differentially regulate Rhp51-dependent recombination outcomes. EMBO J 26 1352 1362
27. EllermeierC
SchmidtH
SmithGR
2004 Swi5 acts in meiotic DNA joint molecule formation in Schizosaccharomyces pombe. Genetics 168 1891 1898
28. SchmidtH
Kapitza-FeckeP
StephenER
GutzH
1989 Some of the swi genes of Schizosaccharomyces pombe also have a function in the repair of radiation damage. Curr Genet 16 89 94
29. AkamatsuY
DziadkowiecD
IkeguchiM
ShinagawaH
IwasakiH
2003 Two different Swi5-containing protein complexes are involved in mating-type switching and recombination repair in fission yeast. Proc Natl Acad Sci U S A 100 15770 15775
30. HarutaN
KurokawaY
MurayamaY
AkamatsuY
UnzaiS
2006 The Swi5-Sfr1 complex stimulates Rhp51/Rad51 - and Dmc1-mediated DNA strand exchange in vitro. Nat Struct Mol Biol 13 823 830
31. HyppaRW
SmithGR
2010 Crossover Invariance Determined by Partner Choice for Meiotic DNA Break Repair. Cell 142 243 255
32. McKeeAH
KlecknerN
1997 Mutations in Saccharomyces cerevisiae that block meiotic prophase chromosome metabolism and confer cell cycle arrest at pachytene identify two new meiosis-specific genes SAE1 and SAE3. Genetics 146 817 834
33. TsubouchiH
RoederGS
2004 The budding yeast mei5 and sae3 proteins act together with dmc1 during meiotic recombination. Genetics 168 1219 1230
34. HayaseA
TakagiM
MiyazakiT
OshiumiH
ShinoharaM
2004 A protein complex containing Mei5 and Sae3 promotes the assembly of the meiosis-specific RecA homolog Dmc1. Cell 119 927 940
35. JiaS
YamadaT
GrewalSI
2004 Heterochromatin regulates cell type-specific long-range chromatin interactions essential for directed recombination. Cell 119 469 480
36. FerrariSR
GrubbJ
BishopDK
2009 The Mei5-Sae3 protein complex mediates Dmc1 activity in Saccharomyces cerevisiae. J Biol Chem 284 11766 11770
37. PierceAJ
JohnsonRD
ThompsonLH
JasinM
1999 XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 13 2633 2638
38. KurokawaY
MurayamaY
Haruta-TakahashiN
UrabeI
IwasakiH
2008 Reconstitution of DNA strand exchange mediated by Rhp51 recombinase and two mediators. PLoS Biol 6 e88 doi:10.1371/journal.pbio.0060088
39. GalkinVE
EsashiF
YuX
YangS
WestSC
2005 BRCA2 BRC motifs bind RAD51-DNA filaments. Proc Natl Acad Sci U S A 102 8537 8542
40. ChenCF
ChenPL
ZhongQ
SharpZD
LeeWH
1999 Expression of BRC repeats in breast cancer cells disrupts the BRCA2-Rad51 complex and leads to radiation hypersensitivity and loss of G(2)/M checkpoint control. J Biol Chem 274 32931 32935
41. YuanSS
LeeSY
ChenG
SongM
TomlinsonGE
1999 BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res 59 3547 3551
42. StarkJM
HuP
PierceAJ
MoynahanME
EllisN
2002 ATP hydrolysis by mammalian RAD51 has a key role during homology-directed DNA repair. J Biol Chem 277 20185 20194
43. DaviesAA
MassonJY
McIlwraithMJ
StasiakAZ
StasiakA
2001 Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol Cell 7 273 282
44. MoynahanME
PierceAJ
JasinM
2001 BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell 7 263 272
45. HiraoA
KongYY
MatsuokaS
WakehamA
RulandJ
2000 DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 287 1824 1827
46. LiuQ
GuntukuS
CuiXS
MatsuokaS
CortezD
2000 Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 14 1448 1459
47. LiuN
LamerdinJE
TebbsRS
SchildD
TuckerJD
1998 XRCC2 and XRCC3, new human Rad51-family members, promote chromosome stability and protect against DNA cross-links and other damages. Mol Cell 1 783 793
48. TakataM
SasakiMS
TachiiriS
FukushimaT
SonodaE
2001 Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol Cell Biol 21 2858 2866
49. YuVP
KoehlerM
SteinleinC
SchmidM
HanakahiLA
2000 Gross chromosomal rearrangements and genetic exchange between nonhomologous chromosomes following BRCA2 inactivation. Genes Dev 14 1400 1406
50. BryantHE
SchultzN
ThomasHD
ParkerKM
FlowerD
2005 Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434 913 917
51. FarmerH
McCabeN
LordCJ
TuttAN
JohnsonDA
2005 Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434 917 921
52. WillisN
RhindN
2010 The fission yeast Rad32(Mre11)-Rad50-Nbs1 complex acts both upstream and downstream of checkpoint signaling in the S-phase DNA damage checkpoint. Genetics 184 887 897
53. WillisN
RhindN
2009 Mus81, Rhp51(Rad51), and Rqh1 form an epistatic pathway required for the S-phase DNA damage checkpoint. Mol Biol Cell 20 819 833
54. HanadaK
BudzowskaM
ModestiM
MaasA
WymanC
2006 The structure-specific endonuclease Mus81-Eme1 promotes conversion of interstrand DNA crosslinks into double-strands breaks. EMBO J 25 4921 4932
55. HanadaK
BudzowskaM
DaviesSL
van DrunenE
OnizawaH
2007 The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat Struct Mol Biol 14 1096 1104
56. ArnaudeauC
LundinC
HelledayT
2001 DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol 307 1235 1245
57. RoseaulinL
YamadaY
TsutsuiY
RussellP
IwasakiH
2008 Mus81 is essential for sister chromatid recombination at broken replication forks. EMBO J 27 1378 1387
58. ArcangioliB
de LahondesR
2000 Fission yeast switches mating type by a replication-recombination coupled process. EMBO J 19 1389 1396
59. JohnsonRD
LiuN
JasinM
1999 Mammalian XRCC2 promotes the repair of DNA double-strand breaks by homologous recombination. Nature 401 397 399
60. PittmanDL
CobbJ
SchimentiKJ
WilsonLA
CooperDM
1998 Meiotic prophase arrest with failure of chromosome synapsis in mice deficient for Dmc1, a germline-specific RecA homolog. Mol Cell 1 697 705
61. YoshidaK
KondohG
MatsudaY
HabuT
NishimuneY
1998 The mouse RecA-like gene Dmc1 is required for homologous chromosome synapsis during meiosis. Mol Cell 1 707 718
62. CaldecottKW
2008 Single-strand break repair and genetic disease. Nat Rev Genet 9 619 631
63. RouleauM
PatelA
HendzelMJ
KaufmannSH
PoirierGG
2010 PARP inhibition: PARP1 and beyond. Nat Rev Cancer 10 293 301
64. PierceAJ
JasinM
2005 Measuring recombination proficiency in mouse embryonic stem cells. Methods Mol Biol 291 373 384
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Genome-Wide Identification of Targets and Function of Individual MicroRNAs in Mouse Embryonic Stem Cells
- Common Genetic Variants and Modification of Penetrance of -Associated Breast Cancer
- Allele-Specific Down-Regulation of Expression Induced by Retinoids Contributes to Climate Adaptations
- β-Actin and γ-Actin Are Each Dispensable for Auditory Hair Cell Development But Required for Stereocilia Maintenance
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy