
Actin Depolymerizing Factors Cofilin1 and Destrin Are Required for Ureteric Bud Branching Morphogenesis
The actin depolymerizing factors (ADFs) play important roles in several cellular processes that require cytoskeletal rearrangements, such as cell migration, but little is known about the in vivo functions of ADFs in developmental events like branching morphogenesis. While the molecular control of ureteric bud (UB) branching during kidney development has been extensively studied, the detailed cellular events underlying this process remain poorly understood. To gain insight into the role of actin cytoskeletal dynamics during renal branching morphogenesis, we studied the functional requirements for the closely related ADFs cofilin1 (Cfl1) and destrin (Dstn) during mouse development. Either deletion of Cfl1 in UB epithelium or an inactivating mutation in Dstn has no effect on renal morphogenesis, but simultaneous lack of both genes arrests branching morphogenesis at an early stage, revealing considerable functional overlap between cofilin1 and destrin. Lack of Cfl1 and Dstn in the UB causes accumulation of filamentous actin, disruption of normal epithelial organization, and defects in cell migration. Animals with less severe combinations of mutant Cfl1 and Dstn alleles, which retain one wild-type Cfl1 or Dstn allele, display abnormalities including ureter duplication, renal hypoplasia, and abnormal kidney shape. The results indicate that ADF activity, provided by either cofilin1 or destrin, is essential in UB epithelial cells for normal growth and branching.
Published in the journal:
. PLoS Genet 6(10): e32767. doi:10.1371/journal.pgen.1001176
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001176
Summary
The actin depolymerizing factors (ADFs) play important roles in several cellular processes that require cytoskeletal rearrangements, such as cell migration, but little is known about the in vivo functions of ADFs in developmental events like branching morphogenesis. While the molecular control of ureteric bud (UB) branching during kidney development has been extensively studied, the detailed cellular events underlying this process remain poorly understood. To gain insight into the role of actin cytoskeletal dynamics during renal branching morphogenesis, we studied the functional requirements for the closely related ADFs cofilin1 (Cfl1) and destrin (Dstn) during mouse development. Either deletion of Cfl1 in UB epithelium or an inactivating mutation in Dstn has no effect on renal morphogenesis, but simultaneous lack of both genes arrests branching morphogenesis at an early stage, revealing considerable functional overlap between cofilin1 and destrin. Lack of Cfl1 and Dstn in the UB causes accumulation of filamentous actin, disruption of normal epithelial organization, and defects in cell migration. Animals with less severe combinations of mutant Cfl1 and Dstn alleles, which retain one wild-type Cfl1 or Dstn allele, display abnormalities including ureter duplication, renal hypoplasia, and abnormal kidney shape. The results indicate that ADF activity, provided by either cofilin1 or destrin, is essential in UB epithelial cells for normal growth and branching.
Introduction
Depolymerization and severing of actin filaments produces new actin monomers and new free ends that facilitate dynamic changes in the actin cytoskeleton. These events are essential for several cellular processes including cell survival, shaping, cytokinesis, migration and chemotaxis [1]. For example, during migration and chemotaxis, cell protrusions are formed as a result of localized actin polymerization in the leading edge of a motile cell [2]. In dividing cells, actin depolymerization plays an important role in chromosome congression, cleavage plane orientation and furrow formation [3]. Three genes encode actin depolymerization factors (ADFs) in mammals: Cofilin1 (Cfl1, non-muscle Cofilin, n-Cofilin), Cofilin2 (Cfl2, muscle Cofilin) and Destrin (Dstn, also called ADF or Corn1).
Despite the vast amount of in vitro data on the functions of ADFs, remarkably little is known about their in vivo roles. ADF genes share overlapping expression patterns in many cell types, but the phenotypes of mouse mutants in either Cfl1 or Dstn suggest that they have somewhat distinct in vivo functions. Mice lacking Cfl1 are embryonic lethal at E11.5–12.5 and display defects in neural tube closure and neural crest cell migration [4], even though Dstn is highly expressed in the cranial neuroectoderm of these mutant embryos; thus, in this situation, Dstn seemed unable to compensate for the absence of Cfl1. No in vivo data on Cfl2 function are yet available. Dstn−/− homozygotes are viable but have corneal defects leading to eventual blindness in adult mice, whereas two alleles with inactivating point mutations (Dstncorn1 and Dstncorn1-2J) cause the same phenotype, indicating that they behave as null alleles [5], [6]. While Dstn−/− brains have a normal gross morphology, conditional deletion of Cfl1 in neuronal cells causes excessive differentiation, changes in cell proliferation, and migration defects, resulting in a lissencephaly phenotype [5].
While Cfl1 seems to be essential for the normal development of the neural tube and neuronal differentiation, the requirement for specific ADFs in other basic morphogenetic processes has not been addressed. Several organs including mammary and salivary gland, lung and kidney develop largely from an epithelial outgrowth that subsequently branches in an organ-specific manner. Kidney development, which serves as an excellent model to study epithelial branching, begins by the formation of the ureteric bud (UB) (at E10.5), which first arises as a thickening of the Wolffian (or nephric) duct. Subsequently, the UB elongates, invades the adjacent metanephric mesenchyme (MM) and begins to branch in a repeated and characteristic pattern, a process that continues until early postnatal life, and gives rise to the tree-like collecting duct system. The UB branching rate and pattern are tightly regulated by signals from the surrounding MM, while at the same time, the UB tips induce nephron progenitors in the MM to undergo mesenchyme-to-epithelium transformation, leading to formation of different nephron segments [7]–[9].
We became interested in potential role of ADF genes in renal epithelial branching for several reasons. First, evagination of the UB from the WD and its subsequent growth and branching require a number of cellular processes that involve the actin cytoskeleton, such as cell migration and proliferation [10], [11]. In vitro inhibition of ROCK, an upstream regulator of Cofilin1 activity, has controversial effects on renal development as it can either increase or decrease kidney size [12], [13]. Second, it was reported that Cfl1 gene expression is upregulated by the expression of activated forms of the Ret receptor tyrosine kinase in NIH3T3 cells [14]. Ret, which is expressed by UB cells, is the receptor for the secreted protein GDNF, which is produced by the MM. Ret and GDNF (as well as the GDNF co-receptor Gfrα1) play a critical role in UB branching morphogenesis, and their absence leads to renal agenesis in mice and humans [15]–[17].
Here we show that mice lacking Cfl1 in the ureteric epithelium, or those with an inactivating mutation in Dstn, develop mostly normal kidneys. However, simultaneous inactivation of both genes in UB epithelium causes a severe and early block in UB branching, indicating considerable functional overlap between cofilin1 and destrin in UB cells. Double mutant UB epithelial cells accumulate excess filamentous actin (F-actin), resulting in irregular epithelial organization and a defect in cell migration. The results show that actin depolymerization by either Cfl1 or Dstn is required for normal growth and branching of the UB epithelium during renal branching morphogenesis.
Results
Abnormal ureteric budding and kidney shape in Cfl1+/−;Dstn−/− compound mutants
Cfl1 and Dstn are both expressed in most or all cells of the developing kidney, while Cfl2 is apparently not expressed in kidney [18]. In order to investigate the requirement for ADF activity in ureteric bud morphogenesis, we studied the effects Dstn and Cfl1 mutations during renal development, initially using conventional loss-of function alleles. For Dstn, we used the Dstncorn1-2J mutant allele, which is phenotypically similar to the knockout allele [5], [6] and we therefore refer to Dstncorn1-2J heterozygotes and homozygotes as Dstn+/− and Dstn−/−. We observed no renal or ureteric abnormalities in Cfl1+/− or Dstn+/− heterozygotes (Figure 1A and 1B, and Figure 2A, 2C, 2E, 2G), or in Dstn+/−;Cfl1+/− compound heterozygotes (Table 1, column 3). Twenty-three percent of Dstn−/− embryos displayed a duplicated ureter, sometimes resulting in a duplex kidney (Table 1, column 1; Figure 1C, Figure S1A, S1B), but renal development appeared otherwise normal (data not shown). Surprisingly, none of the twenty-one Cfl1+/−;Dstn−/− embryos we examined displayed ureter duplications (Table 1, column 4), but instead 38% had kidneys that were abnormally shaped, elongated and uneven on the surface (Figure 1D, 1F), while an additional 29% had mildly hypoplastic kidneys (approximately 25% reduced in size, e.g., Figure 1H). Cultures of E12.5 Cfl1+/−;Dstn−/− renal explants sometimes showed a slight delay in branching, resulting in reduced UB tip numbers, but no obvious branching pattern abnormalities that might explain the abnormal kidney shapes were observed (Figure S1C, S1D, S1E, S1F). It is not clear why removing one Cfl1 allele would apparently rescue the ureteric duplications seen in some Dstn−/− mutants, but this may be a consequence of the mixed genetic background. Overall, however, the spectrum of ureteric and mild renal abnormalities observed in some Dstn−/− and Cfl1+/−;Dstn−/− kidneys suggests a role for actin depolymerization during renal branching morphogenesis.
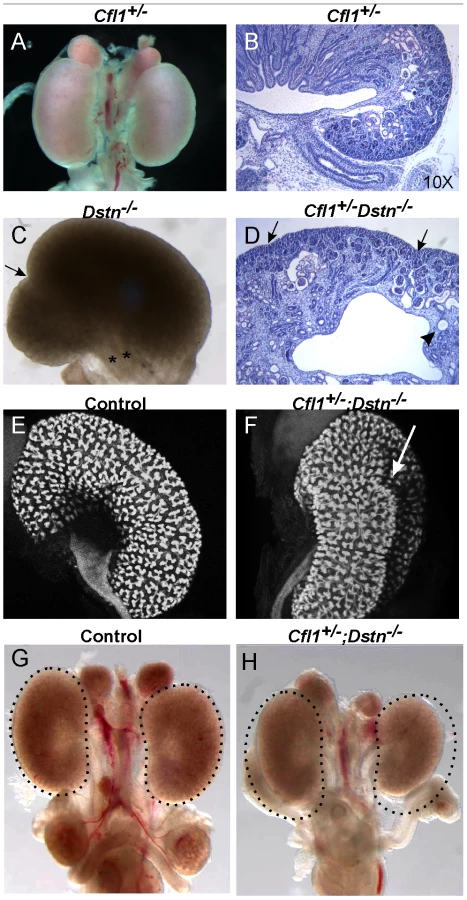


The gene dosage of Cfl1 and Dstn in the ureteric bud is important for normal branching patterns
Since Cfl1 and Dstn are expressed in most or all cell types in the kidney, the abnormal renal morphogenesis in some Cfl1+/−; Dstn−/− compound mutants could be due to either a direct effect in ureteric bud cells, or an indirect effect of faulty induction by the MM or stroma. To address this issue, and also to circumvent the early lethality in Cfl1−/− mice [4] we used a conditional knockout strategy to delete one or both Cfl1 alleles in the Wolffian duct and ureteric bud lineage. Mice carrying a floxed Cfl1 allele, Cfl1F [5] were crossed with Hoxb7/CreGFP, a transgenic line expressing Cre recombinase together with GFP in the Wolffian duct (from ∼E9.5) and UB [19]. Deletion of one or both Cfl1 alleles in a Dstn+/+ background (Hoxb7/CreGFP;Cfl1F/F or Hoxb7/CreGFP; Cfl1F/+) had no apparent effect on ureter or kidney development (data not shown). This suggested that Cfl1 is not required in the WD/UB lineage when Dstn is present at wild-type levels. However, deleting both Cfl1 alleles in the UB in a Dstn+/− background (Table 1, column 8), or deleting one Cfl1 allele in the UB in a Dstn−/− background (Table 1, column 6), caused occasional ureter duplications, and the same occasional renal hypoplasia or abnormal kidney shapes as seen in Cfl1+/−;Dstn−/− mice (Table 1, column 4). While the low frequency of animals with each of these specific genotypes precluded a quantitative comparison, the combined results indicate that normal cofilin1 levels in the UB are important, when Dstn is either reduced or absent, for normal renal and ureteric morphogenesis.
Homozygous deletion of Cfl1 in ureteric epithelium, in the absence of Dstn, abrogates ureteric bud branching and results in severe renal hypodysplasia
When both Cfl1 alleles were deleted in the UB, in the Dstn−/− background (Hoxb7/CreGFP; Cfl1F/F; Dstn−/−) kidney development failed almost completely (Figure 2A and 2B). Histological analysis revealed the presence of ureter and different nephron segments (glomeruli, proximal and distal tubules) but very little collecting duct epithelium (Figure 2D), and no renal pelvis or proper cortex-medulla compartmentalization. Hoxb7/CreGFP; Cfl1F/−; Dstn−/− kidneys (with one null and one floxed Cfl1 allele) appeared identical to Cfl1F/F; Dstn−/− (data not shown), and both of these genotypes will hereafter be called “double mutant”.
As double mutant kidneys contained virtually no UB derivatives at E18 (Figure 2D), we wanted to study the development of the UB at earlier stages. We used the GFP encoded by the Hoxb7/CreGFP transgene to visualize the UB in control and double mutant kidneys. The ureteric bud normally grows out from Wolffian duct at E10.5, by E11.5 it has branched once to form the “T-bud” shaped kidney, and by E12.5 it has branched several more times (Figure 2E). In all double mutant embryos examined at E11.5 (N = 6) or E12.5 (N = 8), the two UBs had successfully grown out from the Wolffian ducts and elongated, but none of them had branched within the kidney (Figure 2F). When cultured for 24 hours, the control kidneys continued to branch (Figure 2G), while the double mutant failed to branch (Figure 2H). Thus, deletion of Cfl1 using Hoxb7/CreGFP, in the absence of Dstn, did not prevent UB outgrowth, but completely blocked subsequent branching.
The finding that the UB always formed in Cfl1;Dstn double mutant kidneys, but failed to branch, raised the possibility that the activities of cofilin1 and destrin are required for UB branching, but not for Wolffian duct growth or initial UB formation. Alternatively, there might be a delay in the elimination of cofilin1 by Hoxb7/CreGFP, such that there is still sufficient cofilin1 at the time of UB outgrowth (E10.5) but not when branching initiates (E11.5). To study the timing and efficiency of cofilin1 elimination we stained mutant kidneys with anti-cofilin1 antibody. While the UB epithelium was clearly devoid of cofilin1 at E11.5 (data not shown) and E12.5 (Figure 3C–3D'), confirming the activity of Hoxb7/CreGFP, we found that cofilin1 protein levels at E10.5 were normal in the forming ureteric bud (Figure 3A–3B'). Thus, the ability of the UB to grow out in double mutants, but not to branch subsequently, is most likely due to residual cofilin1 expression at E10.5, which is eliminated by E11.5. A similar delay in cofilin1 protein elimination was observed when the floxed Cfl1 allele was deleted in the brain using nestinCre, a result that was attributed to the long half-life of the protein [5].

Exogenous GDNF is unable to rescue ureteric branching in Cfl1;Dstn double mutant kidneys
GDNF/Ret signaling is one of the most important pathways that promotes primary UB formation and subsequent branching [16]. To test if the UB branching defect in Cfl1;Dstn double mutants is due to insufficient GDNF/Ret signaling (as occurs in many other mutants with defective UB branching) [8], [15], we cultured double mutant kidneys with or without exogenous GDNF. When cultured without added GDNF, control kidneys with early T-shaped UBs at E11.5 developed several secondary branches over the next 48 h in culture (Figure 4A, 4B, 4I), while those cultured with GDNF showed a swelling of the UB tip and ectopic budding from Wolffian duct (Figure 4C, 4D, 4J), the typical response [20]. The Cfl1;Dstn double mutant UBs had not branched normally when dissected at E11.5 (Figure 4E) and when cultured for 48 hrs without added GDNF they elongated slightly but did not branch (Figure 4F, 4K). Added GDNF had no effect on UB morphogenesis in Cfl1;Dstn double mutant kidneys cultured from E11.5, and it was also unable to induce ectopic ureteric bud formation from the double mutant Wolffian duct (Figure 4G, 4H, 4L). However, all kidneys lacking two or three out of the four Cfl1;Dstn alleles responded like wild-type kidneys to GDNF treatment (data not shown).
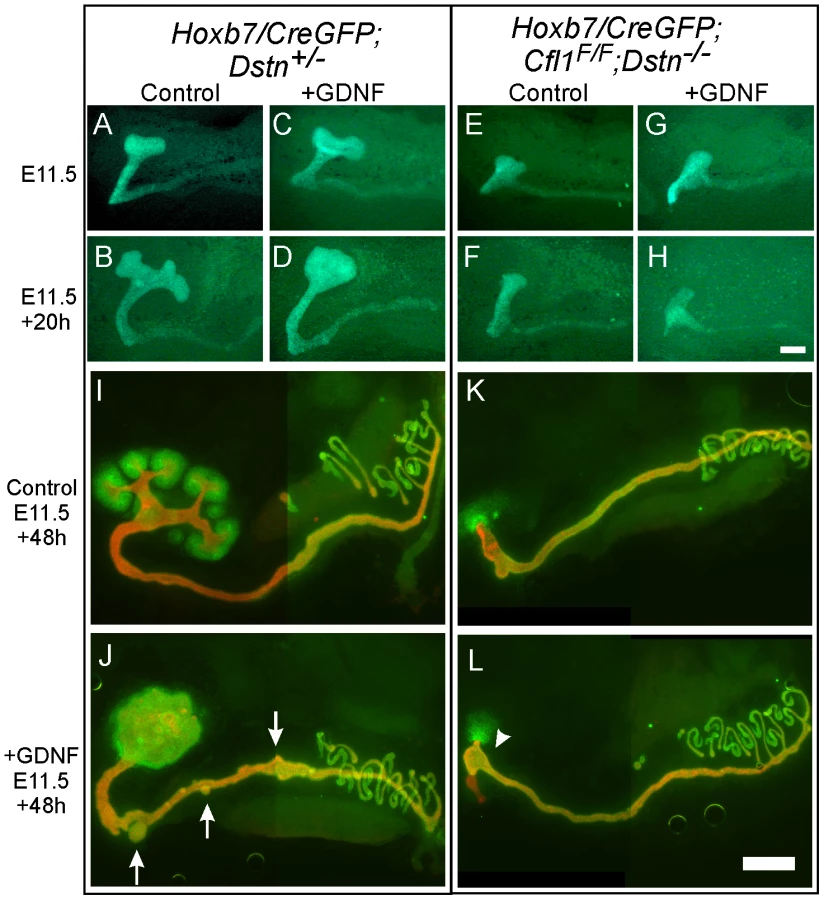
The finding that exogenous GDNF was unable to rescue UB branching in Cfl1;Dstn double mutant kidneys had at least two potential explanations. First we explored the possibility that lack of expression of the GDNF receptor Ret impairs the ability of ureteric epithelium to respond to GDNF signals. However, we found that Ret was expressed in a normal pattern (i.e., at the UB tip) and normal level in E11.5 double mutant kidneys (Figure S2A, S2B), but was somewhat reduced at a slightly later stage (Figure S2C, S2D). Like Ret, the Gdnf/Ret target gene Wnt11 [21] was expressed normally in the E11.5 UB tip (Figure S2E, S2F). Thus, a lack of Ret expression or Ret signaling in the UB seemed not to be the cause of the failure to branch. We next explored an alternative explanation, that defective branching was primarily due to cytoskeletal changes caused by the lack of ADF activity.
Lack of cofilin1 and destrin in UB epithelium causes F-actin accumulation and irregular cell shapes
Dynamic actin cytoskeleton rearrangements are involved in several cellular processes, such as apoptosis, proliferation and migration [1]. We took the advantage of the Hoxb7/myrVenus transgenic line, which expresses myristylated-Venus fluorescent protein at the cell membrane [22], to visualize epithelial cell shape and organization in the ureteric buds of Cfl1;Dstn double mutant mice. Confocal scanning of the UB epithelium at E12.5 revealed a variety of cell shapes in control kidneys, but the epithelium was well organized and cell outlines smooth and distinct (Figure 5A and 5B). In contrast, UB epithelial cells in Cfl1;Dstn mutant kidneys were disorganized, irregular in size and shape, and contained abnormal membranous (i.e., Venus-positive) bodies (Figure 5C and 5D). Thus, lack of both cofilin1 and destrin in the UB disrupts normal epithelial cell shape and organization.

In vivo, mutations in ADF genes have variable consequences on F-actin; they can cause accumulation of actin filaments [5], [6], [23] or a shortage of actin filaments [4], [23]. The latter suggests involvement of ADFs in actin nucleation, and is supported by in vitro studies [24], [25]. To understand how the absence of cofilin1 and destrin in UB epithelium affects the actin cytoskeleton, we stained sections of control and double mutant kidneys with phalloidin to visualize F-actin at E11.5 (data not shown) and E12.5 (Figure 5E–5H). As reported previously [12], the strongest phalloidin staining in control kidneys was observed at apical membranes of UB epithelial cells, but maximal projections of confocal images also revealed some actin filaments at the basolateral membranes (Figure 5E–5F). In accordance with the normal cofilin1 levels in UB epithelium of Cfl1;Dstn double mutant kidneys at E10.5 (Figure 3A–3B'), phalloidin staining was indistinguishable in double mutant and control kidneys at this stage (Figure S3). However, the epithelium of double mutant kidneys was full of phalloidin-positive inclusions at E11.5 (data not shown) and E12.5 (Figure 5G–5H). Accumulation of F-actin was strongest in the apical membranes but also obvious on the basolateral sides of mutant epithelial cells. These data suggest that cofilin1 and destrin are not required for actin nucleation in the UB epithelium but pivotal in its depolymerization and turnover.
Impaired actin depolymerization results in cell migration defect in UB epithelium of Cfl1;Dstn double mutant kidneys
Accumulation of actin filaments within the cells can impair their proliferation and migration [1]. No differences in the mitotic indices (% of phosphohistoneH3+ cells) of Cfl1;Dstn double mutant (1.6%±0.5, n = 4) and control (1.6%±0.3, n = 4) UB epithelium were detected at E11.5 (data not shown) suggesting that the primary cause for the branching defect in mutant mice is not a defect in cell proliferation. While the double mutant UBs did not branch, their continued elongation after E11.5 presumably reflects this continuing cell proliferation.
Primary cell cultures derived from the UB [26], allowed us to apply a scratch assay to non-immortalized primary cell cultures obtained from UBs of different Cfl1;Dstn genotypes. Briefly, individual UBs at E11.5 or E12.5 were separated from the surrounding mesenchyme and plated in fibronectin-coated wells, where the UB cells attached to the bottom and formed monolayers within the next 48 h. These primary epithelial cells survived for approximately two weeks without immortalization (for details, see Materials and Methods). The UB cells of all genotypes including double mutants remained quiescent as judged by lack of the proliferative marker Ki67 (Figure S4A and data not shown). The cells in such cultures were positive for the UB epithelial marker pan-cytokeratin, confirming their origin from the UB (Figure S4B). No differences in the capacity to adhere or form monolayers were observed between control cells (e.g., Dstn−/−) and those from Cfl1;Dstn double mutant UBs (Figure S4C, S4D).
Migration of epithelial cells was studied by introducing a scratch in the confluent cell monolayers at 48 h after plating the UB. Cultures were photographed after 3 h, 8 h and 24 h. The control cells migrated to completely fill in the gap by 24 h (n = 12, Figure 6A and 6B), while Cfl1;Dstn double mutant cells showed some movement but always failed to close the gap (n = 5, Figure 6C and 6D). Similarly to what we observed in the UB epithelium of E11.5 and E12.5 double mutant kidneys in vivo, the double mutant cells in culture were heterogeneous in size and morphology (Figure 6C and 6D) and huge F-actin accumulation was evident by phalloidin staining (data not shown), as it is in the intact UB (Figure 5).

As Cfl1;Dstn double mutant kidneys are delayed in their growth at E11.5 (Figure 4), we were concerned that, in addition to the abnormal cell morphology, the reduced number of cells in each UB might influence their ability to migrate in the scratch assay. To avoid this potential problem, we also measured the migration of primary UB cells isolated from Hoxb7/CreGFP;Cfl1F/F;Dstn+/− kidneys, which exhibit approximately similar numbers of UB branches as control kidneys at E12.5 (data not shown). The morphology of primary UB cells derived from Hoxb7/CreGFP;Cfl1F/F;Dstn+/− kidneys (Figure 7D–7F) was much better than that of double mutant cells (Figure 6C and 6D), and most of the time they had filled the gap by 24 h after the scratch was made (data not shown). However, their migration rate was slower than that of the wild-type cells: at 8 hr post-scratch, while wild-type cells had filled 61% of the gap, the mutant cells had filled only 31% of the gap (p<0.009) (Figure 7A–7G). Thus, even cells retaining one Dstn allele, in the absence of Cfl1, have a migration deficit. Altogether, these data suggest that loss of cofilin1 and Destrin in Cfl1;Dstn double mutants causes actin accumulation and defects in epithelial organization and cell migration, resulting in a failure of branching morphogenesis.

Cfl1 is not regulated by GDNF/Ret signaling in kidney
Cfl1 was identified as one of the potential Ret-induced genes in Ret-expressing NIH3T3 cells [14] and we were therefore interested in determining if Cfl1 could be induced by Ret signaling in the ureteric epithelium. Both gain - and loss-of-function strategies were used to analyze Cfl1 mRNA and protein regulation by GDNF/Ret signaling. Cfl1 is expressed in ureteric epithelium and metanephric mesenchyme of E11.5 kidneys cultured for 24 h (Figure 8A–8A'). While GDNF-soaked beads induced ectopic ureteric budding and local swelling of the UB tip, confirming the functionality of the protein, no change in Cfl1 mRNA expression was observed (Figure 8B–8B'). Two different Ret mutant mouse lines were used to study the effect of either reduced Ret signaling (Ret-hypomorphic mice) or lack of Ret signaling (Ret−/− mice) on cofilin1 protein levels. The Ret-hypomorphic mutant Rettm2(RET)Vpa has reduced UB branching [27] while UB formation fails in most of the Ret−/− kidneys [28]. Cofilin1 was present at normal levels in the early UB of E10.5 Ret−/− mutants (Figure 8C, 8D) as well as in E15 Ret-hypomorphic kidneys (data not shown). Furthermore, the distribution of F-actin appeared normal in both types of Ret-mutant kidneys (Figure 8E, 8F and data not shown). Therefore, although the failure of UB outgrowth in Ret−/− kidneys is phenotypically similar to the phenotype of Cfl1;Dstn double mutant UBs, this is apparently not due to reduced expression of Cfl1 in the Ret mutant.

Discussion
We examined the requirement for cofilin1 - and destrin-mediated cytoskeletal functions during UB branching. While animals retaining at least one wild type Cfl1 or Dstn allele exhibited either normal kidneys or low a frequency of renal/ureteric defects, double homozygotes lacking any cofilin1 or destrin in the UB epithelium had a severe branching defect at an early phase of kidney development. Characterization of cellular defects in Cfl1;Dstn double mutant animals revealed a huge accumulation of F-actin in UB cells, and a disorganized epithelium, at the same stage when the block in branching occurred. We found that primary UB epithelial cells isolated from Cfl1;Dstn double mutants were impaired in their ability to migrate, suggesting that the block in UB growth and branching in vivo is due, at least in part, to disruption in normal cell motility.
One important issue that our study addressed was the extent of functional overlap between cofilin1 and destrin, in vivo. The biochemical properties of these two proteins are highly similar, but significant functional differences have been observed in vitro: for example, destrin is more active in actin-depolymerization, while cofilin1 is a more potent nucleator of actinADP assembly [29]. These differences, as well as differences in expression patterns, led to the suggestion that the ADFs evolved to fulfill specific requirements for actin filament dynamics in different cell types [18]. During development, the ability of Cfl1 and Dstn to substitute for each other depends on two factors: their overlap in expression and their overlap in function. Cfl1 is more widely expressed than Dstn in the embryo [18], so the ability of Dstn−/− mice to develop normally (except for corneal defects) could be due to the ability of cofilin1 to replace the absent destrin in many cell types. In Cfl1−/− mutants, absence of cofilin1 throughout the embryo results in embryonic lethality at E11.5, with specific defects in the neural tube and neural crest cells, even though Dstn is strongly upregulated in the Cfl1−/− embryo [4]. This suggested a specific function for cofilin1. Similarly, the conditional knockout of Cfl1 in the brain (where Dstn is coexpressed) resulted in F-actin accumulation and defects in cell migration and cell cycle progression, indicating that cofilin1 performs roles that cannot be assumed by destrin [5].
In this study, a direct comparison of mice lacking Cfl1, Dstn or both genes in the UB allowed a clear test of redundancy. The lack of any cellular or developmental defects, or abnormal F-actin accumulation, in the kidneys of Dstn−/− or Hoxb7/CreGFP;Cfl1F/F mice suggests that either cofilin1 or destrin is sufficient for normal cellular functions. Furthermore, when both genes were deleted in the UB cells, there was a complete block to UB growth and branching. This clearly indicates that only in the absence of Cfl1 is Dstn required, while only in the absence of Dstn is Cfl1 required – in other words, there is high degree of functional overlap, at least in this cell lineage.
In embryos in which both Cfl1 alleles were deleted in the Wolffian duct/ureteric bud lineage, in a Dstn−/− background, UB growth was arrested shortly after outgrowth from the Wolffian duct, but before further branching. The timing of this developmental block was apparently a consequence of the slow turnover of Cofilin1 protein following deletion of the floxed gene: although the Hoxb7 promoter is active in the Wolffian duct at least as early as E9.5 [22], [30], and the Cfl1F alleles were presumably deleted in most or all cells by E10.5, cofilin1 protein was still present at normal levels in the UB at E10.5. Cofilin1 was not absent, nor did excess F-actin accumulate, until ∼E11.5, the approximate stage at which UB branching ceased. Thus, there is no reason to believe that ADF activity has a specific role in UB branching: it likely has a more general role in UB epithelial morphogenesis.
ADFs are important in various cellular processes involving the actin cytoskeleton [1], [31], [32]. We found that simultaneous lack of Cfl1;Dstn in UB epithelium does not impair the cells' ability to proliferate at normal rates. Therefore, the block in UB branching is not simply due to lack of cell division. Recently it has been demonstrated that cell movements in the Wolffian duct epithelium are important for primary UB outgrowth [10], and it is likely that similar cell movements continue to play a role during later UB growth and branching events. Therefore, the defects in motility of Cfl1;Dstn double mutant UB cells, which we observed in a scratch assay using primary UB cell cultures, provides one plausible explanation for the failure of UB growth. In addition to the migratory defects observed in cell cultures, the double mutant UB epithelium in vivo displayed unusual heterogeneity in cell size and shape, which is possibly a result of the abnormal accumulation of F-actin preventing the normal re-shaping of epithelial cells during branch formation.
Mice with intermediate numbers of mutant alleles (e.g., with one remaining wild type Dstn or Cfl1 allele) usually developed normal kidneys and ureters, revealing that a single Dstn or Cfl1 gene is usually sufficient, in the UB. But a fraction of these mice displayed visible defects, including double ureter, mild renal hypoplasia or irregular kidney shape. This effect of reduced gene dosage suggests that the level of total ADF expression (cofilin1 + destrin) in UB cells is important, which is consistent with the model, based on biochemical studies, that the various activities of ADFs (severing, stabilizing or nucleating actin filaments) are concentration-dependent [1]. UB cells with only one wild type Dstn allele and no Cfl1 (Hoxb7/CreGFP; Cfl1F/F; Dstn+/−) also showed a migration defect in the scratch assay, although less severe than the double-null cells; it is possible that this cellular defect contributes to the phenotypic defects observed in some mice of these genotypes.
Ureter duplications, which occur when the Wolffian duct gives rise to two UBs instead of one, were also seen in a fraction of Dstn−/− mice. Ureter duplication can result from excess GDNF/Ret signaling, ectopic GDNF expression, or reduced BMP4, an inhibitor of UB outgrowth [15], but the cause of this defect in ADF-deficient mice is not clear. It has also been observed in embryos lacking the chemokine SDF-1/CXCL12 or its receptor CXCR4 (F.C. and B. Lu, unpublished data), which play an important role in cell migration, and are also known to activate ADF proteins [33]. We therefore asked if Cxcr4 and Dstn would genetically interact in the control of ureteric bud formation, by examining Dstn−/−;Cxcr4−/− mutant embryos. These embryos displayed no increase in double UB formation compared to Dstn−/− alone, failing to support a synergistic role of CXCR4 signaling and ADF activity in this process.
Renal hypoplasia can result from reduced UB growth and branching; while we did not observe a consistent reduction in early UB branching in cultured E11.5 kidneys from embryos of the intermediate genotypes that sometimes cause renal hypoplasia, this may be due to the low penetrance of the defect. The irregular kidney shape sometimes observed in mice of several of the intermediate genotypes is an unusual phenotype, which has not been previously described, to our knowledge. It is likely that this also results from an abnormality in UB growth or elongation, as the collecting duct system is thought to be the main determinant of kidney shape [34]; however, no particular abnormality in early UB morphogenesis was observed in these kidneys, suggesting that the abnormal renal shape arises at a later stage of organogenesis, or else is not revealed in cultured kidneys, which flatten and lose their three dimensional shapes.
One of the most important pathways regulating UB branching is GDNF/Ret signaling, which was previously shown to induce Cfl1 expression in NIH3T3 cells [14]. The finding that Cfl1 expression is not regulated by GDNF/Ret signaling in developing kidney was therefore to some extent a surprise, but it is supported by a recent GDNF target screen performed in our laboratory [35], in which no changes in Cfl1 expression were detected in the UB. We cannot exclude the possibility that activation of Ret signaling would affect ADF protein activity, which is regulated by its phosphorylation/dephosphorylation status [1], but even if this was the case, Ret signaling apparently does not play major role in regulation of actin cytoskeleton dynamics in these cells, as the actin cytoskeleton appeared normal in Ret−/ − epithelium.
In summary, our results demonstrate that actin depolymerization by the cooperative function of cofilin1 and destrin is essential for normal UB branching, which is blocked due to F-actin accumulation in UB epithelial cells of double mutant Cfl1;Dstn mice. As renal abnormalities are common in newborns, this finding may help to understand the origins of certain congenital malformations in humans.
Materials and Methods
Ethics statement
All work on animals was conducted under PHS guidelines and approved by the relevant Institutional Animal Care and Use Committees.
Mouse strains
Cfl1 null and floxed alleles as well Ret−/− and Ret hypomorphic (Rettm2(RET)Vpa) mice and their genotyping by PCR have been described [4]–[6], [27], [28]. Dstncorn1-2J [6] mice were genotyped by amplifying the region of genomic DNA where the single mutation occurs with the primers 5′ TCC ACT GCA GCT GTC TTCAGACA 3′ and 5′ ATG ACA AAC CAA TGG ATC CCC AC 3′, then digesting with BanI, whose recognition site is mutated, resulting in a Pro106Ser substitution, in Dstncorn1-2J mice. All mice were on mixed genetic backgrounds (including strains C57BL6/J, FVB/N and 129/SvEv) except for Ret+/− mice, which were inbred 129/SvEv.
Organ cultures and bead experiments
E11.5 or E12.5 kidneys were isolated and cultured on Transwell filters (Fisher) in DMEM with 10% fetal calf serum, 1% Glutamax and 1% penicillin/streptomycin at 37°C and 5% CO2 for the indicated times. For GDNF beads, Affigel blue beads (100–200 mesh, Bio-Rad) were washed with PBS/0.1% BSA before incubating with 50 ng/µl recombinant GDNF (R&D) for 30 min at 37°C. Control beads were prepared similarly but incubated in 1% BSA. For organ cultures with GDNF in the culture medium, the concentration was 100 ng/ml.
Immunofluorescence and measuring mitotic index
Whole mount immunofluorescence staining with anti-Pax2 (1∶200, Zymed) and anti-pan-cytokeratin (1∶200, Sigma) antibodies (AB) was performed as previously described [36]. PFA-fixed 10 µm frozen sections were stained with anti-Calbindin AB (1∶200, Santa Cruz), phosphohistone-H3 (1∶100, Cell Signaling Technology) and phalloidin (1∶40, Molecular Probes). Antigen retrieval by 1 mg/ml pepsin (Sigma) digestion (10 min, 37°C) was performed for the samples stained with Cofilin1 AB [4].
For quantification of mitotic indexes, total epithelial cells and phosphohistone-H3+ epithelial cells were counted in sections through four E11.5 Dstn+/− and four Hoxb7/CreGFP; Cfl1F/F; Dstn−/− kidneys. For each specimen, the UB cells were counted in 15–19 serial sections. The percentage of pH3+ epithelial cells in mutant and control samples were compared using Student's t-test (two-tailed, equal variance).
In situ hybridization
Samples for whole mount in situ hybridization were dipped in ice-cold methanol, and fixed in 4% PFA overnight. Hybridization with digoxigenin-labeled Cfl1 [4] and Ret [37] riboprobes was performed according to Wilkinson [38].
Non-immortalized primary ureteric bud cultures and scratch assay
After an enzymatic treatment with collagenase (4 µg/µl, Gibco) ureteric buds were dissected free of metanephric mesenchyme and placed on fibronectin coated wells (BD Biosciences) containing DMEM supplemented with 10% FBS, 1% Glutamax, 1% penicillin/streptomycin, 5 ng/ml GDNF, 25 µg/ml FGF2 and 50 µg/ml HGF. Cultures were allowed to settle and form single cell layers for 48 h, before introducing the scratch, using standard 10 µl plastic pipette tips. For immunofluorescence staining, single cell layers were fixed in 4% PFA for 10 min, washed with PBS, and incubated with anti-pan-cytokeratin (1∶200, Sigma) and anti-Ki67 (1∶100, Abcam) antibodies or with phalloidin (1∶40, Molecular Probes). To quantify the cell migration, the width of the gap formed by the scratch was measured immediately after the scratch, and 3 h and 8 h later. The proportion of gap filled was calculated by dividing the width at each time by the initial gap width. All measurements were done using Image J program.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Van TroysM
HuyckL
LeymanS
DhaeseS
VandekerkhoveJ
2008 Ins and outs of ADF/cofilin activity and regulation. Eur J Cell Biol 87 649 667
2. InsallRH
MacheskyLM
2009 Actin dynamics at the leading edge: from simple machinery to complex networks. Dev Cell 17 310 322
3. LenartP
BacherCP
DaigleN
HandAR
EilsR
2005 A contractile nuclear actin network drives chromosome congression in oocytes. Nature 436 812 818
4. GurniakCB
PerlasE
WitkeW
2005 The actin depolymerizing factor n-cofilin is essential for neural tube morphogenesis and neural crest cell migration. Dev Biol 278 231 241
5. BellenchiGC
GurniakCB
PerlasE
MiddeiS
Ammassari-TeuleM
2007 N-cofilin is associated with neuronal migration disorders and cell cycle control in the cerebral cortex. Genes Dev 21 2347 2357
6. IkedaS
CunninghamLA
BoggessD
HawesN
HobsonCD
2003 Aberrant actin cytoskeleton leads to accelerated proliferation of corneal epithelial cells in mice deficient for destrin (actin depolymerizing factor). Hum Mol Genet 12 1029 1037
7. DresslerGR
2006 The cellular basis of kidney development. Annu Rev Cell Dev Biol 22 509 529
8. SchedlA
2007 Renal abnormalities and their developmental origin. Nat Rev Genet 8 791 802
9. SaxenL
1987 Organogenesis of the Kidney. Cambridge Cambridge University Press
10. ChiX
MichosO
ShakyaR
RiccioP
EnomotoH
2009 Ret-dependent cell rearrangements in the Wolffian duct epithelium initiate ureteric bud morphogenesis. Dev Cell 17 199 209
11. MichaelL
DaviesJA
2004 Pattern and regulation of cell proliferation during murine ureteric bud development. J Anat 204 241 255
12. MichaelL
SweeneyDE
DaviesJA
2005 A role for microfilament-based contraction in branching morphogenesis of the ureteric bud. Kidney Int 68 2010 2018
13. MeyerTN
SchwesingerC
SampognaRV
VaughnDA
StuartRO
2006 Rho kinase acts at separate steps in ureteric bud and metanephric mesenchyme morphogenesis during kidney development. Differentiation 74 638 647
14. WatanabeT
IchiharaM
HashimotoM
ShimonoK
ShimoyamaY
2002 Characterization of gene expression induced by RET with MEN2A or MEN2B mutation. Am J Pathol 161 249 256
15. CostantiniF
ShakyaR
2006 GDNF/Ret signaling and the development of the kidney. Bioessays 28 117 127
16. CostantiniF
2006 Renal branching morphogenesis: concepts, questions, and recent advances. Differentiation 74 402 421
17. SkinnerMA
LackeyKE
FreemermanAJ
2008 RET activation inhibits doxorubicin-induced apoptosis in SK-N-MC cells. Anticancer Res 28 2019 2025
18. VartiainenMK
MustonenT
MattilaPK
OjalaPJ
ThesleffI
2002 The three mouse actin-depolymerizing factor/cofilins evolved to fulfill cell-type-specific requirements for actin dynamics. Mol Biol Cell 13 183 194
19. ZhaoH
KeggH
GradyS
TruongHT
RobinsonML
2004 Role of fibroblast growth factor receptors 1 and 2 in the ureteric bud. Dev Biol 276 403 415
20. SainioK
SuvantoP
DaviesJ
WartiovaaraJ
WartiovaaraK
1997 Glial-cell-line-derived neurotrophic factor is required for bud initiation from ureteric epithelium. Development 124 4077 4087
21. PepicelliCV
KispertA
RowitchDH
McMahonAP
1997 GDNF induces branching and increased cell proliferation in the ureter of the mouse. Dev Biol 192 193 198
22. ChiX
HadjantonakisAK
WuZ
HyinkD
CostantiniF
2009 A transgenic mouse that reveals cell shape and arrangement during ureteric bud branching. Genesis 47 61 66
23. PhamH
YuH
LaskiFA
2008 Cofilin/ADF is required for retinal elongation and morphogenesis of the Drosophila rhabdomere. Dev Biol 318 82 91
24. AndrianantoandroE
PollardTD
2006 Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol Cell 24 13 23
25. van RheenenJ
CondeelisJ
GlogauerM
2009 A common cofilin activity cycle in invasive tumor cells and inflammatory cells. J Cell Sci 122 305 311
26. YeP
HabibSL
RiconoJM
KimNH
ChoudhuryGG
2004 Fibronectin induces ureteric bud cells branching and cellular cord and tubule formation. Kidney Int 66 1356 1364
27. de GraaffE
SrinivasS
KilkennyC
D'AgatiV
MankooBS
2001 Differential activities of the RET tyrosine kinase receptor isoforms during mammalian embryogenesis. Genes Dev 15 2433 2444
28. SchuchardtA
D'AgatiV
Larsson-BlombergL
CostantiniF
PachnisV
1994 Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 367 380 383
29. BamburgJR
BernsteinBW
2008 ADF/cofilin. Curr Biol 18 R273 275
30. SrinivasS
GoldbergMR
WatanabeT
D'AgatiV
al-AwqatiQ
1999 Expression of green fluorescent protein in the ureteric bud of transgenic mice: a new tool for the analysis of ureteric bud morphogenesis. Dev Genet 24 241 251
31. BernsteinBW
BamburgJR
2010 ADF/cofilin: a functional node in cell biology. Trends Cell Biol 20 187 195
32. WangW
EddyR
CondeelisJ
2007 The cofilin pathway in breast cancer invasion and metastasis. Nat Rev Cancer 7 429 440
33. YoderA
YuD
DongL
IyerSR
XuX
2008 HIV envelope-CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell 134 782 792
34. EkblomP
1992 Renal Development.
SeldinDW
GiebischG
The Kidney: Physiology and Pathophysiology. Second ed New York Raven Press 475 501
35. LuBC
CebrianC
ChiX
KuureS
KuoR
2009 Etv4 and Etv5 are required downstream of GDNF and Ret for kidney branching morphogenesis. Nat Genet 41 1295 1302
36. KuureS
SainioK
VuolteenahoR
IlvesM
WartiovaaraK
2005 Crosstalk between Jagged1 and GDNF/Ret/GFRalpha1 signalling regulates ureteric budding and branching. Mech Dev 122 765 780
37. PachnisV
MankooBS
CostantiniF
1993 Expression of the c-ret proto-oncogene during mouse embryogenesis. Development 119 1005 1017
38. WilkinsonDG
1992 Whole mount in situ hybridization of vertebrate embryos.
WilkinsonDG
In situ hybridization: a practical approach Oxford IRL Press 75 83
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Genome-Wide Identification of Targets and Function of Individual MicroRNAs in Mouse Embryonic Stem Cells
- Common Genetic Variants and Modification of Penetrance of -Associated Breast Cancer
- Allele-Specific Down-Regulation of Expression Induced by Retinoids Contributes to Climate Adaptations
- β-Actin and γ-Actin Are Each Dispensable for Auditory Hair Cell Development But Required for Stereocilia Maintenance
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy