
Ancient and Recent Adaptive Evolution of Primate Non-Homologous End Joining Genes
In human cells, DNA double-strand breaks are repaired primarily by the non-homologous end joining (NHEJ) pathway. Given their critical nature, we expected NHEJ proteins to be evolutionarily conserved, with relatively little sequence change over time. Here, we report that while critical domains of these proteins are conserved as expected, the sequence of NHEJ proteins has also been shaped by recurrent positive selection, leading to rapid sequence evolution in other protein domains. In order to characterize the molecular evolution of the human NHEJ pathway, we generated large simian primate sequence datasets for NHEJ genes. Codon-based models of gene evolution yielded statistical support for the recurrent positive selection of five NHEJ genes during primate evolution: XRCC4, NBS1, Artemis, POLλ, and CtIP. Analysis of human polymorphism data using the composite of multiple signals (CMS) test revealed that XRCC4 has also been subjected to positive selection in modern humans. Crystal structures are available for XRCC4, Nbs1, and Polλ; and residues under positive selection fall exclusively on the surfaces of these proteins. Despite the positive selection of such residues, biochemical experiments with variants of one positively selected site in Nbs1 confirm that functions necessary for DNA repair and checkpoint signaling have been conserved. However, many viruses interact with the proteins of the NHEJ pathway as part of their infectious lifecycle. We propose that an ongoing evolutionary arms race between viruses and NHEJ genes may be driving the surprisingly rapid evolution of these critical genes.
Published in the journal:
. PLoS Genet 6(10): e32767. doi:10.1371/journal.pgen.1001169
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001169
Summary
In human cells, DNA double-strand breaks are repaired primarily by the non-homologous end joining (NHEJ) pathway. Given their critical nature, we expected NHEJ proteins to be evolutionarily conserved, with relatively little sequence change over time. Here, we report that while critical domains of these proteins are conserved as expected, the sequence of NHEJ proteins has also been shaped by recurrent positive selection, leading to rapid sequence evolution in other protein domains. In order to characterize the molecular evolution of the human NHEJ pathway, we generated large simian primate sequence datasets for NHEJ genes. Codon-based models of gene evolution yielded statistical support for the recurrent positive selection of five NHEJ genes during primate evolution: XRCC4, NBS1, Artemis, POLλ, and CtIP. Analysis of human polymorphism data using the composite of multiple signals (CMS) test revealed that XRCC4 has also been subjected to positive selection in modern humans. Crystal structures are available for XRCC4, Nbs1, and Polλ; and residues under positive selection fall exclusively on the surfaces of these proteins. Despite the positive selection of such residues, biochemical experiments with variants of one positively selected site in Nbs1 confirm that functions necessary for DNA repair and checkpoint signaling have been conserved. However, many viruses interact with the proteins of the NHEJ pathway as part of their infectious lifecycle. We propose that an ongoing evolutionary arms race between viruses and NHEJ genes may be driving the surprisingly rapid evolution of these critical genes.
Introduction
DNA double-strand breaks are a particularly toxic form of DNA lesion. Such breaks are repaired through several pathways, the most well-studied being homologous recombination and non-homologous end joining (NHEJ; reviewed in [1]). NHEJ is also required for V(D)J recombination, which generates immunoglobulin and T cell receptor diversity. Accordingly, mutations in NHEJ genes have been linked to both cancer and immune deficiencies. Given the central importance of these processes, NHEJ genes are expected to have a low tolerance for mutations. Such a hypothesis would be supported if sequences of NHEJ genes are stable and relatively unchanging over evolutionary time.
In contrast to this expectation, a genome-wide analysis uncovered NHEJ as one of the two functional pathways most enriched for positive selection during Saccharomyces evolution [2]. Positive selection occurs when natural selection operates on an advantageous mutation, driving an increase in its prevalence over time, and sometimes leading to fixation of this mutation in the species in which it arose. Because advantageous mutations commonly involve a change in protein sequence, recurrent rounds of positive selection can lead to relatively rapid protein sequence evolution over time. Positive selection has been found to predominantly affect genes in three functional classes: reproduction, immunity, and environmental perception (smell, taste, etc), presumably because these processes are under strong selection for constant adaptive change [3]–[10]. The intriguing observation of positive selection in the NHEJ genes of Saccharomyces remains unexplained, but could potentially be attributed to the fact that NHEJ is not the major pathway for the repair of double-strand breaks in yeast [11]. Relaxation of evolutionary constraints on NHEJ genes in yeast species, due to their reliance predominantly on the homologous recombination pathway, could have made NHEJ genes vulnerable to competing evolutionary forces. In this study, we have analyzed the molecular evolution of NHEJ genes in primates, including humans, where NHEJ is the major pathway for DNA double-strand break repair.
NHEJ is activated upon detection of DNA double-strand breaks. After detection, NHEJ proteins enzymatically process broken DNA ends to allow for efficient end joining. Repair is then completed through the action of repair-specific DNA polymerases and the NHEJ ligation complex, which fill in and seal the break [1]. To analyze the selective pressures that have shaped the genes of the human NHEJ pathway, we generated sequence datasets of primate orthologs from twenty simian primate species. We find support for positive selection in five NHEJ genes: NBS1, CtIP, Artemis, XRCC4 and POLλ. Analysis of human polymorphism data indicates that positive selection has also operated on XRCC4 in modern humans. Crystal structures are available for the Nbs1, XRCC4, and Polλ proteins, and in all cases we find that amino acid sites targeted by positive selection fall on protein surfaces. It is well-established that rapidly evolving amino acid residues tend to be found on the surfaces of proteins [12]–[14]. In previous studies where the significance of these residues has been structurally or functionally investigated, it has been shown that they modulate protein-protein, protein-ligand, or protein-DNA interactions [15]–[24]. However, we demonstrate biochemically that positive selection in Nbs1 at one of the three residues identified has not affected its physical interactions with other DNA repair components. In the discussion, we propose that the positive selection of NHEJ genes may be explained by the diverse viruses and genetic parasites that interact with these proteins to promote their own lifecycle.
Results
Sliding window analysis of selective pressures shaping NHEJ genes
We utilized primate sequence datasets to study the evolutionary history of human NHEJ genes. With human population genetic data, evolutionary pressures can usually only be summarized for chromosomal regions larger than a single gene. However, with inter-species divergence data, resolution of evolutionary signatures can be increased to the level of a single gene, and it is sometimes possible to see the serial fixation of mutations in particular gene regions or even codons. The limitation in these studies is the number of available primate sequences. We first performed a preliminary survey of the selective pressures that have shaped all of the major genes of the NHEJ pathway (Figure 1A), so that we could generate appropriate primate datasets for candidate genes containing signatures suggestive of positive selection.

Five nearly complete primate genome projects are publicly available: human, chimpanzee, orangutan, rhesus macaque, and marmoset. Ten possible pairwise gene comparisons can be made between these five species, but three pairwise comparisons (human-orangutan, human-rhesus, and rhesus-marmoset) were chosen that maximize divergence and minimize phylogenetic re-sampling (Figure 1B). For each NHEJ gene, these three pairwise gene alignments were constructed and analyzed with a custom algorithm that calculates dN/dS in sliding windows along the length of each gene [25]. The dN/dS ratio captures the ratio of non-synonymous (dN; changing the encoded amino acid) to synonymous (dS; silent) DNA mutations that have accumulated since two genes last shared a common ancestor [26]. For most protein-encoding genes, the observed number of non-synonymous mutations is far less than the number of synonymous mutations observed (dN/dS<1) [5]. This is because mutations which cause an alteration in amino acid sequence are more likely to be detrimental to proper protein folding and function, and are therefore typically selected against (purifying selection). As expected in a typical gene, dN is less than dS (dN/dS<1) for all windows along the length of the NHEJ gene KU70 (Figure 1C).
Under positive selection, non-synonymous mutations are swept through populations more quickly than neutral or nearly-neutral synonymous mutations due to a selectable advantage that they convey. After many such rounds, such a regime gives rise to the dN>dS signature that is indicative of positive selection (dN/dS>1). Sliding window analysis of dN/dS is useful when making pairwise gene comparisons, as positive selection may be limited to specific regions that are buried within a gene that is otherwise conserved. In the case of XRCC4, sliding window analyses of human-rhesus and rhesus-marmoset pairwise alignments highlight the 3′ end of the gene as having signatures of dN/dS>1 (p<0.001 and p<0.005, respectively; Figure 1C). In this region, human and rhesus XRCC4 sequences differ by nine non-synonymous DNA mutations and zero synonymous mutations. In the human–orangutan comparison, a different region in the 5′ end of the gene shows a significant inflation of dN/dS above 1 (p<0.05). The different location of this signal may indicate a unique selective force that is operating specifically in the great apes.
Sliding window analyses have an inherent multiple testing problem that is difficult to correct because of the non-independence of tests (windows overlap) [27]. Nevertheless, we have successfully utilized sliding window analysis as a pre-screening tool in several previous studies [2], [28]. As an ad hoc method for eliminating some false positive signatures, we sought genes with regions of dN/dS significantly>1 in at least two out of three different pairwise primate comparisons made. All pairwise comparisons for each NHEJ gene are shown in Figure S1, and the maximum dN/dS value found in each comparison is summarized in Figure 1D. We find that five out of thirteen NHEJ genes bear significant regions of dN/dS>1 in at least two out of the three primate comparisons made (highlighted in gray in Figure 1D). Thus, we have identified preliminary signals of positive selection in five candidate NHEJ genes: NBS1, Artemis, CtIP, POLλ, and XRCC4.
Analysis of extended primate datasets for candidate genes
In order to verify positive selection with greater statistical rigor, larger sequence datasets are required. We sequenced all five candidate genes from 15 additional hominoid, old world monkey, and new world monkey species. Despite the fact that no significant windows of dN/dS>1 were observed in any of the pairwise comparisons of XLF (Figure 1D), we also included this gene because positive selection was previously reported in an analysis of mammalian XLF sequences [29]. In total, 90 primate genes were sequenced (6 genes, each from 15 species). We also re-sequenced all genes that were incomplete in the available primate genome projects (chimpanzee, orangutan, rhesus macaque, or marmoset). Details of primate cell lines, cell culture, mRNA extraction, cDNA library construction, and divergent-species PCR are given in the materials and methods section and in Tables S1, S2, S3. The resulting dataset for each gene is comprised of orthologs from 20 primate species that represent approximately 35 million years of primate evolution [30].
The multiple sequence alignment generated for each gene was analyzed for positive selection with the “codeml” program in PAML [31]. The codeml program provides a maximum likelihood framework for estimating dN/dS rates over the entire history of primate evolution by integrating over all ancestral gene sequences in the context of a phylogeny [32], [33]. This program offers several models for gene evolution, some where no codons are allowed to evolve with dN/dS>1 (NSsites models M1a, M7 and M8a), and others where positive selection of some codons is allowed (NSsites models M2a and M8). A likelihood ratio test allows comparison of positive selection models to null models. Results of all model comparisons for each gene are provided in Tables S4, S5, S6, S7, S8, S9, and the results of the M8a vs. M8 comparisons, using the f61 model of codon usage, are summarized in Table 1. The null model (M8a) is rejected (p<0.05) in favor of the model of positive selection (M8) in four of these six genes: CtIP, Artemis, XRCC4, and POLλ. For NBS1, the null model was very nearly rejected (p = 0.056). This analysis did not support a model of positive selection in primate XLF (p = 0.59). As mentioned above, sliding window analysis did not detect domains of positive selection in XLF. In conclusion, we find strong support for positive selection in four genes of the primate NHEJ pathway, a surprising finding given the critical role that these proteins play in DNA repair.

Analysis of the 20-species NBS1 dataset yielded marginal support for positive selection (p = 0.056; Table 1). However, we noticed that several amino acid positions in the NBS1 protein alignment had changed multiple times exclusively in hominoid species (humans, great apes, and gibbons). Based on this, we considered that positive selection of NBS1 may be specific to hominoids. Indeed, analysis of NBS1 from only the hominoid species resulted in improved statistical support for positive selection (p = 0.048; Table 1), despite the fact that the analysis of only eight sequences should greatly reduce statistical power. To formally test the hypothesis of hominoid-specific positive selection, we analyzed our datasets with “branch-site” models of evolution [34]. This test allowed us to determine whether there are codon positions evolving under positive selection specifically in the hominoid clade. NBS1 was the only one of the six NHEJ genes for which this hypothesis was supported (p<0.005; Table S10), and support is robust under all models of codon usage (Table S11). Because three total tests were performed on the NBS1 dataset, a Bonferroni-corrected p-value can be calculated for the rejection of the null hypothesis in the branch-sites test (p<0.015). Thus, hominoid-specific positive selection is supported in NBS1. Interestingly, the yeast ortholog of NBS1 (XRS2) was also identified as being under positive selection during Saccharomyces evolution [2].
Specific codon sites that have been the target of recurrent positive selection could be identified in the dataset for each NHEJ gene (Table 1). Posterior probabilities of codons included in the dN/dS>1 site class are commonly considered highly significant at cutoffs as low as P = 0.90, and potentially even lower [35]. The positions of these amino acid sites are summarized in Figure 2. Crystal structures have been solved for Polλ, XRCC4, and Nbs1, allowing us to further analyze the patterns of positive selection in these three proteins.
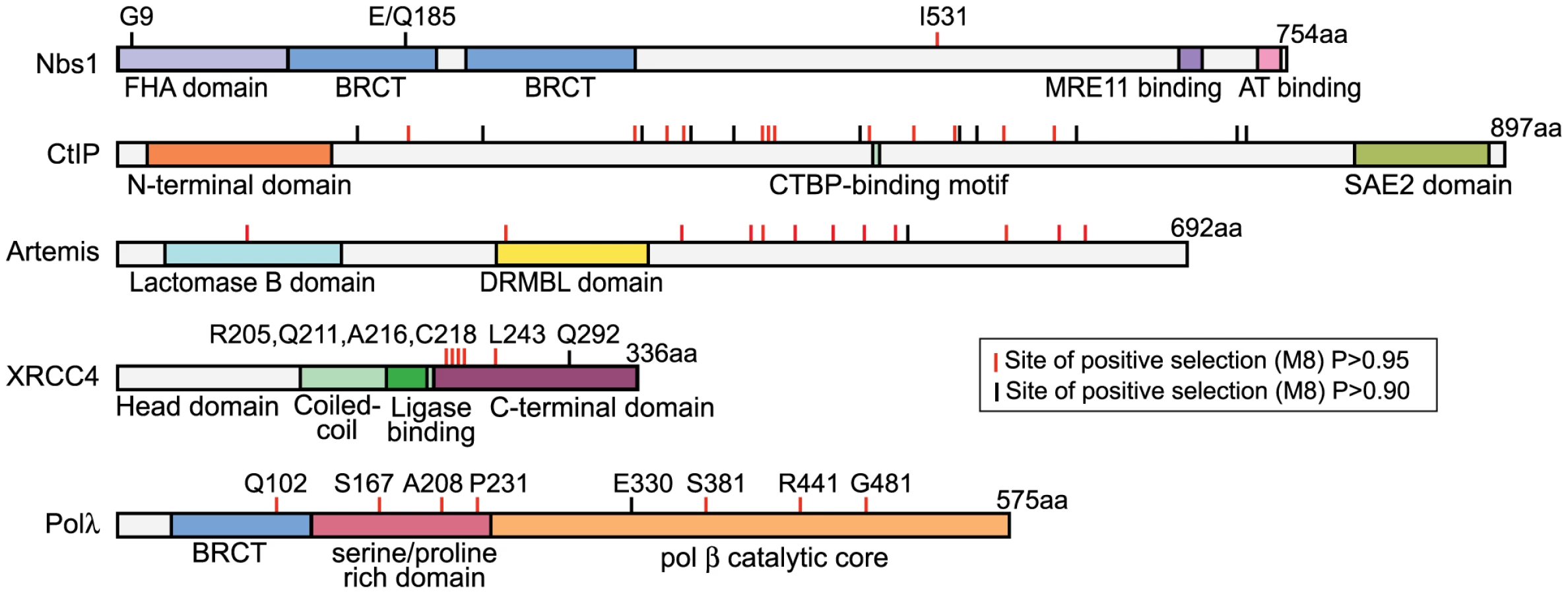
Positive selection of POLλ
Polλ is one of two DNA polymerases involved in the filling of gaps formed during NHEJ [36]. Approximately 5% of the codons in this gene were identified as evolving under positive selection, with an average dN/dS value of 3.2 (Table 1). Eight specific codons could be assigned to this class with high posterior probability (P>0.90), and these sites are scattered across the linear protein sequence (Figure 2). The crystal structure of the 39 kDa Polλ catalytic domain has been solved in complex with substrate DNA, and this catalytic core is comprised of the fingers, palm, thumb, and 8 kDa subdomains (Figure 3) [37]. Four of the eight amino acid sites identified as being positively selected are part of this catalytic core domain. All four (E330, S381, R441, and R484) map to the outer surface of the three-dimensional structure (red balls in Figure 3), with none of the sites being found within the enzyme active site. Thus, residues under recurrent positive selection fall on the protein surface, and mutations at these sites are not predicted to directly affect catalytic activity.

Positive selection of XRCC4 during primate evolution
The NHEJ-specific ligase complex is composed of DNA ligase IV (Lig4) along with the regulatory molecules XLF and XRCC4 [1]. The dN/dS>1 site class in XRCC4 is assigned a value of dN/dS = 15, nearly double the value seen for any other NHEJ gene (Table 1). Given the extreme value, only one codon, L243, can be supported as a member of this class with high posterior probability (P>0.99). To uncover more codons that may be evolving under positive selection, a secondary analysis was performed on the three XRCC4 structural domains: the N-terminal head domain, which is involved in DNA binding, the coiled-coil stalk domain, which includes the ligase binding domain, and the unstructured C-terminal domain (residues 204–336). Positive selection is supported only in the C-terminal domain (p<0.001; Table 1). Because four tests were performed on the XRCC4 dataset, the Bonferroni-corrected p-value for the observation of positive selection in the C-terminal domain is p<0.004. In this domain, six codon sites, including L243 identified previously, were identified as evolving under positive selection (P>0.90), with support for five of these being P>0.95. These codons were now collectively assigned a dN/dS value of 8.7. All of these codons were also identified, albeit with lower confidence, in the full-length XRCC4 analysis (Table S7).
The partial crystal structure of the XRCC4 dimer in complex with its binding partner, Lig4, has been solved [38]. All six of the identified codons map just downstream of the Lig4-binding domain (red dots in Figure 4A), in a region of the protein where the structure is predicted to transition from an alpha-helix to an unstructured domain. This unstructured domain is not included in the crystal structure, but has been represented in schematic form for illustration. Strikingly, of the five sites supported at the 95% confidence level, the first four (R205, Q211, A216, and C218) lie within a 14 amino acid stretch of the protein (4% of the length of the protein), and the fifth site (L243) lies just 25 residues downstream of this cluster. We assessed the significance of this clustering on the linear protein sequence by determining how many times a random sampling of five sites fell in a cluster equal to or smaller than the 39 amino acid region that contains the sites under positive selection. Comparing this observed distance to a null distribution (100,000 permutations) lends statistical support to the hypothesis that these positively selected sites are clustered (p = 0.0005). The functional significance of this “patch” of positive selection is unknown. A protein alignment of primate XRCC4 in this region is shown in Figure 4B. To the left, a cladogram shows the relationship of the twenty primate species used in this study. Amino acid positions evolving under positive selection are shown in the alignment in gray. This unstructured C-terminal domain has been shown to be dispensable for repair and V(D)J recombination [39], [40]. However, this domain also contains a number of regulatory sites including a SUMOylation site and several DNA-PKcs phosphorylation sites [41], [42], as well as a known cancer-linked mutation [43] (Figure 4B).

Positive selection of XRCC4 in modern humans
We investigated whether the NHEJ genes that have been subject to ancient recurrent positive selection in simian primates are also under recent local adaptation in humans. We examined the five genes POLλ, XRCC4, Artemis, NBS1, and CtIP for signals of selection in the HapMap Phase II [44] data using a recently published method, the Composite of Multiple Signals (CMS) [45]. By combining multiple tests, CMS increases resolution for localizing signals of selection by up to 100-fold, and has a lower false-positive rate than the component individual tests. We examined SNPs within and surrounding each gene of interest, with a window size of 100kb upstream and 100kb downstream of each gene (see Materials and Methods). In the European population, the CMS signal for XRCC4 is significant at a threshold that yields a 0.1% false positive rate in simulations, and is one of the top 60 strongest signals in the genome (Table S12). Applying CMS to fine-map the region, we localized the signal to 83kb entirely within the gene, suggesting that XRCC4 is a target of recent local adaptation (Figure 5). In the other four genes, we did not observe any signals significant at the same level as XRCC4, but we do observe suggestive signals by the individual tests (in the top 1–5% tail genome-wide) in POLλ and XRCC4 in the West African population, and Artemis in the European population (Table S12). As CMS is optimized to detect recent local adaptation in a single population, these signals by individual tests may reflect selective events outside of this model (e.g., selection on standing variation, or selection of the same allele in multiple populations). Indeed, a single allele of POLλ has previously been reported to be under positive selection in both Asian and Sub-Sahara African populations [46]. Thus we find that several of the genes that have been evolving under positive selection during primate evolution also show evidence suggestive of recent positive selection in human populations, with an especially strong signature identified in XRCC4.

Essential repair-related interactions are conserved despite positive selection of Nbs1
Nbs1 is part of the MRN complex, containing Mre11, Rad50, and Nbs1. This complex is involved in DNA break detection, end processing, and cellular signaling [47]. Mutations in NBS1 lead to the autosomal recessive disease, Nijmegen breakage syndrome, which is characterized by chromosomal instability. Three amino acid positions were identified as evolving under positive selection (Table 1). G9, Q185, and I531 are identified with P>0.90, with support for I531 being P>0.99. A partial Nbs1 structure is available [48], and two of the amino acid sites targeted by positive selection (residues 9 and 185) fall on the protein surface (Figure 6A). The third site, residue 531, is not included in this partial structure.
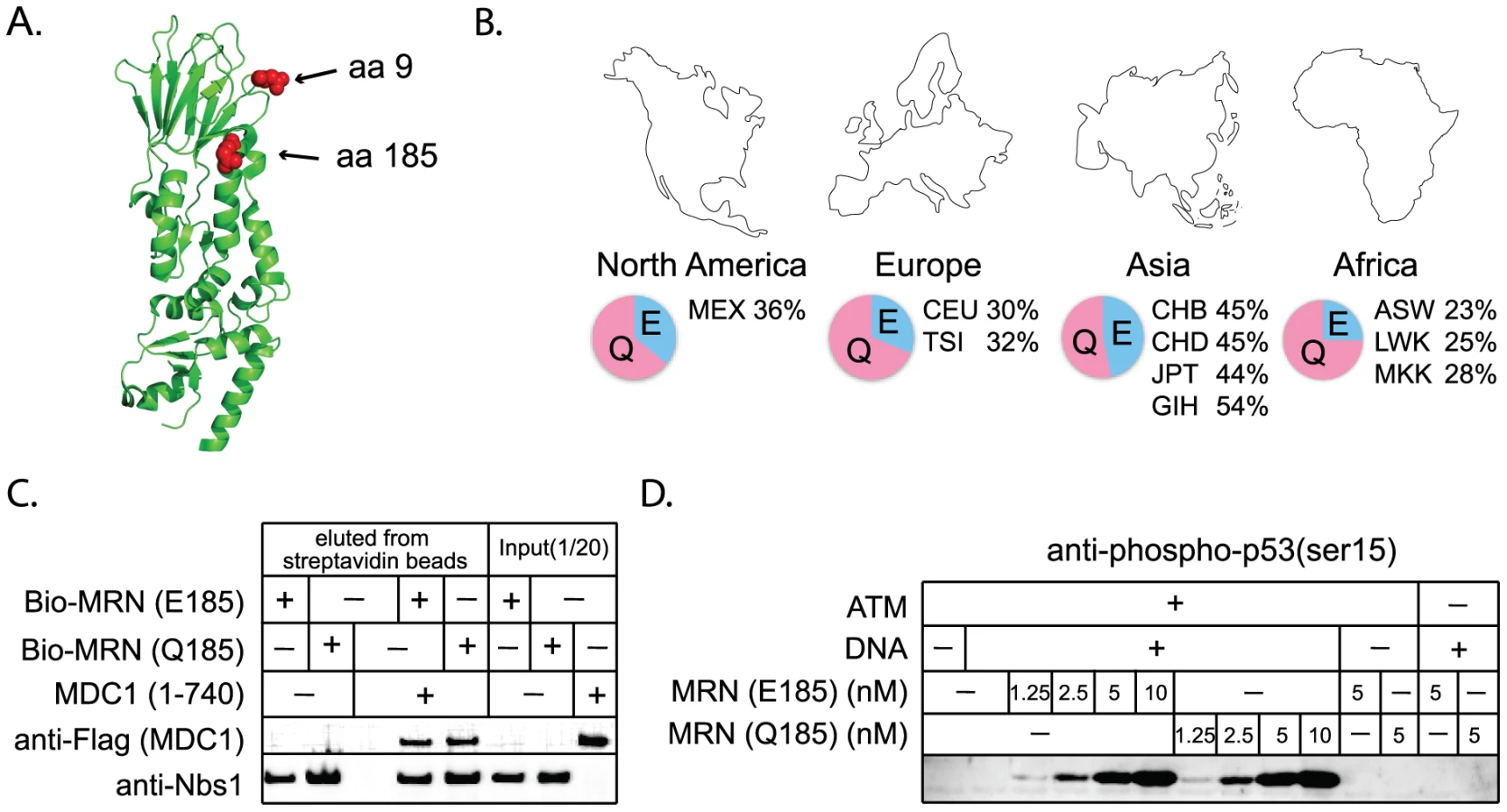
The positive selection of NHEJ genes suggests that certain mutations are providing a fitness advantage in an unknown context. While the essential DNA repair functions of these genes would be expected to remain conserved, there is a formal possibility that adaptive evolution of NHEJ genes could come at the cost of DNA repair. We wished to consider this hypothesis because a human SNP at a site of positive selection in NBS1 (Q185E; SNP ID rs1805794) has been linked to increased risk of renal, skin, and lung cancer in multiple association studies [49]–[52]. This SNP is found at high frequencies in human populations (Figure 6B). While Q185E has been linked to cancer, association studies are limited in that they may identify either a causal SNP, or a SNP that is linked to a causal SNP. We wished to test whether amino acid substitution in this codon changes the performance of Nbs1 in DNA repair, as the association with cancer might suggest.
We constructed NBS1 alleles encoding either an E or a Q at position 185, and expressed these proteins in insect cells using a baculovirus system. We then tested the effects of this mutation on several of the known activities of Nbs1. The Nbs1 N-terminus, including the BRCT domain in which this SNP is located, is known to bind to the checkpoint protein Mdc1 [53]–[56]. We produced and purified MRN complexes containing both versions of Nbs1 and find that both interact equally well with purified Mdc1 in an in vitro binding assay (Figure 6C). Thus the Nbs1 E/Q polymorphism is not expected to affect the association of MRN with Mdc1 at sites of DNA damage in vivo. The MRN complex is also required for the activation of the checkpoint protein ATM [57], [58]. We find that MRN complexes containing both versions of Nbs1 are equally efficient in stimulating ATM-dependent phosphorylation of one of the downstream targets of ATM, p53 (Figure 6D). Nbs1 is also known to bind XRCC4/Lig4 [59] and we find that both versions of Nbs1 interact equally well with this complex in vitro (data not shown). Therefore, we conclude that positive selection of this codon, regardless of what is driving it, has not affected the repair-related physical interactions of Nbs1. However, it should be noted that laboratory-based assays may not be sensitive enough to detect subtle defects that could cause a minor fitness effect in nature.
Discussion
The NHEJ pathway is over 3 billion years old, and is found in bacteria, archaea, and eukaryotes. Despite the ancient conservation of the pathway, we have identified five NHEJ genes that have evolved under positive selection during the evolution of simian primates: NBS1, CtIP, Artemis, XRCC4, and POLλ. An analysis of polymorphism data supports positive selection of XRCC4 in modern humans as well. Interestingly, the yeast ortholog of NBS1 (XRS2) was also identified as one of the two Saccharomyces NHEJ genes with the most extreme signatures of positive selection [2]. One hypothesis is that these signatures of positive selection are reflective of natural selection for more efficient DNA repair. As certain NHEJ components evolve, compensatory mutations may arise in other NHEJ components to re-optimize protein-protein interactions between the various components. We feel that this model is unlikely. In the absence of an antagonizing force, there is no reason that recurrent adaptive change should be required of any member of this pathway, which would then need to be followed by compensatory change. Four observations from our study additionally argue against this model. First, our biochemical experiments with Nbs1 suggest that positive selection of at least one of the three sites identified has not altered interactions with other repair proteins. Second, although there are several core complexes involved in NHEJ (the MRN complex and the Lig4/XRCC4/XLF complex), only one component of each of these was identified as evolving under positive selection. Third, the clustered sites of positive selection in XRCC4 fall within the C-terminal protein domain that is not essential for DNA repair. Fourth, the positive selection of the NHEJ pathway is not a primate specific phenomenon, but is also found in Saccharomyces yeast [2], arguing against a model where some novel role for DNA repair during primate evolution has driven this selection.
The finding of multiple primate NHEJ components evolving under positive selection, supported by parallel findings in Saccharomyces yeast, indicates a systematic perturbation of the NHEJ pathway. With positive selection observed in two highly divergent eukaryotic clades, a model for the cause of this rapid evolution must span such diverse species groups. We propose that NHEJ genes may be antagonized by genetic parasites, which in primates are comprised of viruses and retrotransposons.
Proteins of the NHEJ repair pathway have been shown to act as antiviral factors in the lifecycle of human adenovirus, a linear double-stranded DNA virus. Adenoviruses are a major cause of upper respiratory and other infections in humans. During infection, components of the NHEJ pathway join together viral genome ends, causing “dead-end” viral genome concatenation [60]. To counteract this antiviral tactic, adenovirus proteins (encoded by the E4 genes) sequester and target for degradation a number of components of the NHEJ pathway, including components of the Mre11/Rad50/Nbs1 and Lig4/XRCC4/XLF complexes [60]–[63]. CtIP has also been implicated in the adenovirus lifecycle through its interaction with the adenovirus early region 1A (AdE1A) protein [64]. If primate NHEJ genes are continually selected to encode variants that can evade interaction with these adenoviral antagonists, while the viral antagonists continually counter-evolve, this could drive positive selection of primate NHEJ genes. Adenovirus has been found in stool samples from great apes and macaques [65], indicating a possible long-standing co-evolution between this virus and primates.
Retroviruses like HIV may also provide the selective pressure that shapes the recurrent positive selection of NHEJ genes. There is abundant genetic evidence suggesting a role for NHEJ in the retroviral lifecycle [66]–[70]. Upon cellular entry, the retroviral RNA genome is reverse transcribed into double-stranded DNA. The ultimate destination for this retroviral cDNA is integration into the genome of the host, but it must first survive passage through the nucleus without being detected as broken DNA by the cell. NHEJ proteins have been found to physically associate with retroviral proteins, cDNA, and pre-integration complexes in vivo and in two-hybrid interactions [67], [71]–[74]. There are several models which have been proposed to explain this. In one model, NHEJ proteins are recruited by the viral complex to protect free viral cDNA ends from degradation or from triggering apoptosis. In another model, the viral complex recruits host NHEJ proteins to promote the repair of breaks created at sites of retroviral cDNA integration into the host genome. In a third model, NHEJ proteins act as antivirals, joining the two long-terminal repeat (LTR) ends of the viral cDNA into dead-end “2-LTR circles.” These 2-LTR circles are ubiquitously observed in the nuclei of infected cells [67]. Regardless of the model, allelic variants of NHEJ genes that result in lower infection rates would be selectively advantageous to the host. Should such alleles go to high frequency or fixation, retroviruses would be expected to counter-evolve, and the back-and-forth interplay would drive recurrent positive selection of NHEJ genes. Retroviruses and primates have co-evolved for tens of millions of years, as illustrated by the fact that all sequenced primate genomes contain the remnants of hundreds of thousands of integrated retroviruses [75].
It is unknown whether the positive selection observed in NHEJ genes represents a response to a single selective force, or whether multiple forces are shaping their evolution. At least eight additional viral families have been shown to evade or exploit the host DNA damage response [76]. Several NHEJ proteins include one or more “BRCT” domains, which have been linked to viral infection in multiple instances. The Epstein-Barr viral protein Zta has been shown to interact with the BRCT domains of 53BP1, a component of the DNA damage response, to prevent apoptosis that is activated in response to viral replication [77]. HIV-1 Tat has also been shown to interact with the BRCT domain of the human replication protein FCP1 [78]. In both Polλ and Nbs1, we find an amino acid position at the C-terminal end of the BRCT domain to be evolving under positive selection (Q185 in Nbs1 and Q102 in Polλ). The single site found to be under positive selection in Saccharomyces Xrs2 also falls near the end of the BRCT domain (site 298) [2]. BRCT domains could be a critical link in the interaction between viruses and the NHEJ pathway. Antagonism of host NHEJ proteins by genetic parasites may be a universal feature of cellular life, as yeast Ty retrotransposons also interact genetically and physically with NHEJ machinery [79], [80]. LINE-1 retrotransposons are major drivers of primate genome evolution, and LINE-1 retrotransposition rates are reduced in the absence of NHEJ genes [81]. The Corndog and Omega bacteriophages of mycobacteria have even incorporated the first gene in the bacterial NHEJ pathway, Ku, into their own genome [82]. This viral Ku now evolves under the selective pressures of the virus in order to recruit the bacterial NHEJ ligase, LigD, to circularize phage DNA.
In summary, we have documented abundant signatures of positive selection in genes of the NHEJ pathway, which is the major pathway for repairing double-strand chromosomal breaks in mammalian cells. We propose the hypothesis that these signatures result from the long-term co-evolution between NHEJ genes and genetic parasites. While it is well known that genetic parasites shape genome architecture through insertion and subsequent inter-element recombination, the present study may indicate that selective pressures imposed by genetic parasites can drive the evolution of protein sequence in critical human proteins.
Materials and Methods
Primate NHEJ gene sequences
Chimpanzee, orangutan, rhesus macaque, and marmoset gene sequences were obtained from the UCSC genome database (http://genome.ucsc.edu/) using the BLAT alignment tool [83]. NBS1, CtIP, Artemis, XRCC4, POLλ, and XLF were sequenced from 15 additional primate species, and poor-quality regions of chimpanzee, orangutan, rhesus and marmoset genes were also re-sequenced. Primary and immortalized primate cell lines (sources and individual primate identifiers are listed in Table S1) were grown in standard media supplemented with 15% fetal bovine serum at 37°C and in 5% CO2. Total RNA was harvested from cell lines using the AllPrep DNA/RNA kit (Qiagen). PCR was performed from total RNA and/or cDNA with OneStep RT-PCR kit (Qiagen) or PCR SuperMix High Fidelity (Invitrogen), respectively. Details of the PCR and sequencing strategy, along with primer sequences, can be found in Tables S2 and S3. Primate NHEJ gene sequences have been deposited in GenBank (accession numbers HM486750–HM486849).
Sliding window analysis
Alignments between orthologous gene pairs were performed using ClustalX2.0 [84]. Sliding-window dN/dS calculations for each alignment were performed with the SLIDERKK program [25]. Human-orangutan, human-rhesus and rhesus-marmoset alignments were analyzed with standard window sizes of 450bp, 306bp and 153bp, respectively, to reflect the increasing level of divergence in these species pairs (window size must be a multiple of nine in this program) [2], [28]. In order to generate confidence values for windows with dN/dS>1, the K-estimator program [85] was utilized to generate a null distribution through Monte Carlo simulation of randomly derived dN/dS values in the gene region of interest.
PAML analysis
Multiple alignments were created with ClustalX2.0 [84]. Maximum likelihood analysis was performed with codeml in the PAML 4.1 software package [31]. To detect selection, multiple alignments were fitted to the NSsites models M1a (neutral model, codon values of dN/dS are fit into two site classes, one with value between 0 and 1, and one fixed at dN/dS = 1), M2a (positive selection model, similar to M1a but with an extra class of dN/dS>1 allowed), M7 (neutral model, codon values of dN/dS fit to a beta distribution, dN/dS>1 disallowed), M8a (neutral model, similar to M7 except with a fixed codon class of at dN/dS = 1) and M8 (positive selection model, similar to M7 but with an extra class of dN/dS>1 allowed). Simulations were run with multiple seed values for dN/dS (ω) and assuming either the f61 or f3x4 model of codon frequencies. Likelihood ratio tests were performed to assess whether permitting codons to evolve under positive selection gives a significantly better fit to the data (model comparisons M1a vs. M2a, M7 vs. M8, M8a vs. M8). In situations where the null model could be rejected (p<0.05), posterior probabilities were assigned to individual codons belonging to the class of codons with dN/dS>1. Residues under positive selection were mapped onto existing crystal structures using MacPyMol (v.0.99; http://pymol.sourceforge.net/).
The branch-site test allows identification of positive selection that might be limited to a subset of codons along only a subset of the branches being analyzed [34]. To implement this test, multiple alignments were fitted to the branch-sites Model A (positive selection model, codon values of dN/dS along background branches are fit into two site classes, one (ω0) between 0 and 1 and one (ω1) equal to 1, on the foreground branches a third site class is allowed (ω2) with dN/dS>1), and Model A with fixed ω2 = 1 (null model, similar to Model A except the foreground ω2 value is fixed at 1). Hominoids were defined as the “foreground” clade, with all other branches in the tree being defined as background branches. The likelihood of Model A is compared to the likelihood of the null model with a likelihood ratio test. Simulations were run with multiple seed values for dN/dS and assuming either the f61 or f3x4 models of codon frequencies. The “Fequal” codon model was also tested in the branch-site analysis of NBS1.
Clustering analysis
To test the significance of clustering of the codons under positive selection in XRCC4, the statistical program R was utilized to perform a permutation test. The observed span of the positively selected codons on the primary sequence was compared with a null distribution created by calculating the span resulting from randomly generated sets of equivalent numbers of codons. We generated 100,000 random distances.
Population genetic tests
To examine evidence for recent positive selection in humans, we implemented a previously published method that combines multiple tests for selection, the Composite of Multiple Signals (CMS) [45]. We have adapted the method to detect genomic regions under selection by examining the fraction of high scores in 100kb sliding windows. To determine the significance threshold, we used the cosi coalescent simulator to simulate 1,000 1MB autosomal regions, evolving neutrally under a previously validated demographic model [86]. We set thresholds that yielded a 0.1% false positive rate in simulations. Two long-haplotype tests, XP-EHH and iHS, were used to examine evidence for selection in or around the genes of interest. iHS was calculated as described in [10] for all SNPs with a minor allele frequency greater than 5%. iHS was analyzed independently in the European (CEU), East Asian (JPT and CHB), and West African (Yoruban; YRI) populations. XP-EHH was calculated as in [9] for the each of the three populations. For each SNP, we found the maximum score of the comparisons with the two other populations. In each 100kb window along the gene regions, the fraction of SNPs with |iHS|>2 or the maximum XP-EHH score was used as the test statistic. To calculate empirical P-values for each window w, we calculated the test statistics for each 100kb window across the genome and found the fraction of genomic windows with values of the test statistic greater than that found for window w. The ancestral state for each SNP was determined by comparison to the chimpanzee genome. We calculated Fst for each SNP in the regions using the Weir-Cockerham estimator [87]. Three pairwise comparisons were made between the African (Yoruban), European, and East Asian populations. For each population, we compared the allele frequency in that population to the average frequency in the other two populations. For each 100kb window across the region, the maximum Fst was used as the test statistic. To generate the null distribution, we performed the same procedure on each 100kb window in the genome and derived an empirical p-value based on this distribution.
Plasmid constructs and expression
A biotinylated human MRN (E185) complex was expressed in a baculovirus system from the transfer vectors pTP11 (Rad50), pTP814 (Mre11), pTP1014 (Nbs1), and pTP1016 (BirA) as described earlier [88]. To make biotinylated human MRN (Q185) complex, the E to Q point mutation at Nbs1 position 185 was introduced into pTP994, whose bacmid form is pTP1014, by primer-based mutagenesis (QuikChange Kit, Invitrogen). Flag-tagged Mdc1 (amino acids 1–740) was expressed using bacmid construct pTP1188, which was made from the corresponding transfer vector pTP1187. Expression constructs for Flag-tagged and HA-tagged ATM were gifts from M. Kastan and R. Abraham. The E. coli expression construct for GST-p53 was described earlier [89].
Protein purification
Purification procedures for the biotinylated MRN complex were the same as for the non-biotinylated MRN complex as described earlier [90]. Dimeric ATM was made by transient transfection of expression constructs into 293T cells using calcium phosphate and purified as described earlier [91]. Mdc1 (aa 1–740) was expressed in Sf21 insect cells using the Bac-to-Bac system (Invitrogen) and was purified identically to 53BP1 as described earlier [88]. The GST-p53 was purified identically to the GST–Brca1 fragments as described earlier [92] and was further purified by separation on a Superdex 200 gel filtration column (GE) in buffer A (100 mM NaCl, 25 mM Tris pH8, 10% glycerol, and 1 mM DTT). Protein concentrations were determined by quantification of protein preparations with standards on colloidal Coomassie-stained SDS–PAGE gels using the Odyssey system (LiCor).
In vitro binding assay
20 nM biotinylated MRN complex was incubated with 45 nM Mdc1 (aa 1–740) in buffer A for 1 hour at 30°C in a final volume of 100 µl, then incubated with streptavidin-coated magnetic beads (Dynal) and 0.2% CHAPS (Sigma) while rotating at 4°C for 15 min. Beads with associated proteins were washed three times with buffer A containing 0.2% CHAPS, and bound proteins were eluted by boiling the beads in SDS loading buffer. Proteins were analyzed by SDS–PAGE and western blotting using antibodies directed against the Flag epitope (Sigma, F3165) and Nbs1 (Genetex, MSNBS10PX1).
Kinase assay
ATM kinase assays were performed with 0.2 nM dimeric ATM, 50 nM GST–p53 substrate, and varying amounts of MRN complex (concentrations of MRN = 1.25, 2.5, 5, and 10 nM). Kinase assays were performed in kinase buffer (50 mM HEPES, pH 7.5, 50 mM potassium chloride, 5 mM magnesium chloride, 10% glycerol, 1 mM ATP, 1 mM DTT, and 10 ng DNA) for 90 min at 30°C in a volume of 40 microliters as described earlier [91]. Phosphorylated p53 (ser15) was detected as described earlier [91] using phospho-specific antibody from Calbiochem (PC461).
Supporting Information
Zdroje
1. LieberMR
2008 The mechanism of human nonhomologous DNA end joining. J Biol Chem 283 1 5
2. SawyerSL
MalikHS
2006 Positive selection of yeast nonhomologous end-joining genes and a retrotransposon conflict hypothesis. P Natl Acad Sci USA 103 17614 17619
3. BarreiroLB
LavalG
QuachH
PatinE
Quintana-MurciL
2008 Natural selection has driven population differentiation in modern humans. Nat Genet 40 340 345
4. BustamanteCD
Fledel-AlonA
WilliamsonS
NielsenR
HubiszMT
2005 Natural selection on protein-coding genes in the human genome. Nature 437 1153 1157
5. MikkelsenTS
HillierLW
EichlerEE
ZodyMC
JaffeDB
2005 Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437 69 87
6. ClarkAG
GlanowskiS
NielsenR
ThomasPD
KejariwalA
2003 Inferring Nonneutral Evolution from Human-Chimp-Mouse Orthologous Gene Trios. Science 302 1960 1963
7. KosiolC
VinarT
da FonsecaRR
HubiszMJ
BustamanteCD
2008 Patterns of Positive Selection in Six Mammalian Genomes. PLoS Genet 4 e1000144 doi:10.1371/journal.pgen.1000144
8. SabetiPC
SchaffnerSF
FryB
LohmuellerJ
VarillyP
2006 Positive natural selection in the human lineage. Science 312 1614 1620
9. SabetiPC
VarillyP
FryB
LohmuellerJ
HostetterE
2007 Genome-wide detection and characterization of positive selection in human populations. Nature 449 913 918
10. VoightBF
KudaravalliS
WenXQ
PritchardJK
2006 A map of recent positive selection in the human genome. PLoS Biol 4 e72 doi:10.1371/journal.pbio.0040072
11. PâquesF
HaberJE
1999 Multiple Pathways of Recombination Induced by Double-Strand Breaks in Saccharomyces cerevisiae. Microbiol Mol Biol R 63 349 404
12. RothC
LiberlesDA
2006 A systematic search for positive selection in higher plants (Embryophytes). BMC Plant Biol 6
13. BloomJD
DrummondDA
ArnoldFH
WilkeCO
2006 Structural determinants of the rate of protein evolution in yeast. Mol Biol Evol 23 1751 1761
14. BustamanteCD
TownsendJP
HartlDL
2000 Solvent accessibility and purifying selection within proteins of Escherichia coli and Salmonella enterica. Mol Biol Evol 17 301 308
15. BishopJG
DeanAM
Mitchell-OldsT
1999 Rapid evolution in plant chitinases: Molecular targets of selection in plant-pathogen coevolution. P Natl Acad Sci USA 97 5322 5327
16. YangZ
SwansonWJ
2002 Codon-substitution models to detect adaptive evolution that account for heterogeneous selective pressures among site classes. Mol Biol Evol 19 49 57
17. BishopJG
RipollDR
BashirS
DamascenoCMB
SeedsJD
2005 Selection on glycine beta-1,3-endoglucanase genes differentially inhibited by a phytophthora glucanase inhibitor protein. Genetics 169 1009 1019
18. IvarssonY
MackeyAJ
EdalatM
PearsonWR
MannervikB
2003 Identification of residues in glutathione transferase capable of driving functional diversification in evolution - A novel approach to protein redesign. J Biol Chem 278 8733 8738
19. SawyerSL
WuLI
EmermanM
MalikHS
2005 Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. P Natl Acad Sci USA 102 2832 2837
20. ClarkNL
SwansonWJ
2005 Pervasive adaptive evolution in primate seminal proteins. PLoS Genet 1 e35 doi:10.1371/journal.pgen.0010035
21. EldeNC
ChildSJ
GeballeAP
MalikHS
2009 Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature 457 485 489
22. SwansonWJ
VacquierVD
1998 Concerted evolution in an egg receptor for a rapidly evolving abalone sperm protein. Science 281 710 712
23. OliverPL
GoodstadtL
BayesJJ
BirtleZ
RoachKC
2009 Accelerated Evolution of the Prdm9 Speciation Gene across Diverse Metazoan Taxa. PLoS Genet 5 e1000753 doi:10.1371/journal.pgen.1000753
24. MalikHS
HenikoffS
2001 Adaptive evolution of Cid, a centromere-specific histone in Drosophila. Genetics 157 1293 1298
25. ParmleyJL
HurstLD
2007 How common are intragene windows with KA>KS owing to purifying selection on synonymous mutations? J Mol Evol 64 646 655
26. HurstLD
2002 The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet 18 486 487
27. SchmidK
YangZ
2008 The trouble with sliding windows and the selective pressure in BRCA1. PLoS ONE 3 e3746 doi:10.1371/journal.pone.0003746
28. SawyerSL
EmermanM
MalikHS
2004 Ancient Adaptive Evolution of the Primate Antiviral DNA-Editing Enzyme APOBEC3G. PLoS Biol 2 e275 doi:10.1371/journal.pbio.0020275
29. PavlicekA
JurkaJ
2006 Positive selection on the nonhomologous end-joining factor Cernunnos-XLF in the human lineage. Biol Direct 2 15
30. PurvisA
1995 A composite estimate of primate phylogeny. Philos T Roy Soc B 348 405 421
31. YangZ
1997 PAML: A program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13 555 556
32. YangZH
2002 Inference of selection from multiple species alignments. Curr Opin Genet Dev 12 688 694
33. YangZ
BielawskiJP
2000 Statistical methods for detecting molecular adaptation. Trends Ecol Evol 15 496 503
34. ZhangJZ
NielsenR
YangZH
2005 Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol 22 2472 2479
35. AnisimovaM
BielawskiJP
YangZ
2002 Accuracy and power of bayes prediction of amino acid sites under positive selection. Mol Biol Evol 19 950 958
36. Garcia-DiazM
BebenekK
GaoGH
PedersenLC
LondonRE
2005 Structure-function studies of DNA polymerase lambda. DNA Repair 4 1358 1367
37. Garcia-DiazM
BebenekK
KrahnJM
PedersenLC
KunkelTA
2006 Structural analysis of strand misalignment during DNA synthesis by a human DNA polymerase. Cell 124 331 342
38. SibandaBL
CritchlowSE
BegunJ
PeiXY
JacksonSP
2001 Crystal structure of an Xrcc4-DNA ligase IV complex. Nat Struct Biol 8 1015 1019
39. GrawunderU
ZimmerD
KuleszaP
LieberMR
1998 Requirement for an interaction of XRCC4 with DNA ligase IV for wild-type V(D)J recombination and DNA double-strand break repair in vivo. J Biol Chem 273 24708 24714
40. MizutaR
ChengHL
GaoYJ
AltFW
1997 Molecular genetic characterization of XRCC4 function. Int Immunol 9 1607 1613
41. YurchenkoV
XueZ
SadofskyMJ
2006 SUMO modification of human XRCC4 regulates its localization and function in DNA double-strand break repair. Mol Cell Biol 26 1786 1794
42. LeeKJ
JovanovicM
UdayakumarD
BladenCL
DynanWS
2004 Identification of DNA-PKcs phosphorylation sites in XRCC4 and effects of mutations at these sites on DNA end joining in a cell-free system. DNA Repair 3 267 276
43. TsengHC
TsaiMH
ChiuCF
WangCH
ChangNW
2008 Association of XRCC4 codon 247 polymorphism with oral cancer susceptibility in Taiwan. Anticancer Res 28 1687 1691
44. The International HapMap Consortium 2007 A second generation human haplotype map of over 3.1 million SNPs. Nature 449 851 861
45. GrossmanSR
ShylakhterI
KarlssonEK
ByrneEH
MoralesS
2010 A Composite of Multiple Signals Distinguishes Causal Variants in Regions of Positive Selection. Science 327 883 886
46. KelleyJL
TurkheimerK
HaneyM
SwansonWJ
2009 Targeted resequencing of two genes, RAGE and POLL, confirms findings from a genome-wide scan for adaptive evolution and provides evidence for positive selection in additional populations. Hum Mol Genet 18 779 784
47. WilliamsRS
WilliamsJS
TainerJA
2007 Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem Cell Biol 85 509 520
48. WilliamsRS
DodsonGE
LimboO
YamadaY
WilliamsJS
2009 Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to Coordinate DNA Double-Strand Break Processing and Repair. Cell 139 87 99
49. LuMX
LuJC
YangXB
YangM
TanH
2009 Association between the NBS1 E185Q polymorphism and cancer risk: a meta-analysis. BMC Cancer 9
50. MedinaPP
AhrendtSA
PollanM
FernandezP
SidranskyD
2003 Screening of homologous recombination gene polymorphisms in lung cancer patients reveals an association of the NBS1-185GIn variant and p53 gene mutations. Cancer Epidem Biomar 12 699 704
51. MargulisV
LinJ
YangHS
WangW
WoodCG
2008 Genetic susceptibility to renal cell carcinoma: The role of DNA double-strand break repair pathway. Cancer Epidem Biomar 17 2366 2373
52. ThirumaranRK
BermejoJL
RudnaiP
GurzauE
KoppovaK
2006 Single nucleotide polymorphisms in DNA repair genes and basal cell carcinoma of skin. Carcinogenesis 27 1676 1681
53. ChapmanJR
JacksonSP
2008 Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep 9 795 801
54. SpycherC
MillerES
TownsendK
PavicL
MorriceNA
2008 Constitutive phosphorylation of MDC1 physically links the MRE11-RAD50-NBS1 complex to damaged chromatin. J Cell Biol 181 227 240
55. MelanderF
Bekker-JensenS
FalckJ
BartekJ
MailandN
2008 Phosphorylation of SDT repeats in the MDC1 N terminus triggers retention of NBS1 at the DNA damage-modified chromatin. J Cell Biol 181 213 226
56. WuLM
LuoKT
LouZK
ChenJJ
2008 MDC1 regulates intra-S-phase checkpoint by targeting NBS1 to DNA double-strand breaks. P Natl Acad Sci USA 105 11200 11205
57. LeeJH
PaullTT
2005 ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308 551 554
58. UzielT
LerenthalY
MoyalL
AndegekoY
MittelmanL
2003 Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 22 5612 5621
59. MatsuzakiK
ShinoharaA
ShinoharaM
2008 Forkhead-associated domain of yeast Xrs2, a homolog of human Nbs1, promotes nonhomologous end joining through interaction with a ligase IV partner protein, Lif1. Genetics 179 213 225
60. StrackerTH
CarsonCT
WeitzmanMD
2002 Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 418 348 352
61. WeitzmanMD
OrnellesDA
2005 Inactivating intracellular antiviral responses during adenovirus infection. Oncogene 24 7686 7696
62. EvansJD
HearingP
2005 Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J Virol 79 6207 6215
63. JayaramS
GilsonT
EhrlichES
YuXF
KetnerG
2008 E1B 55k-independent dissociation of the DNA ligase IV/XRCC4 complex by E4 34k during adenovirus infection. Virology 382 163 170
64. BrutonRK
RastiM
MappKL
YoungN
CarterRZ
2007 C-terminal-binding protein interacting protein binds directly to adenovirus early region 1A through its N-terminal region and conserved region 3. Oncogene 26 7467 7479
65. RoyS
VandenbergheLH
KryazhimskiyS
GrantR
CalcedoR
2009 Isolation and Characterization of Adenoviruses Persistently Shed from the Gastrointestinal Tract of Non-Human Primates. PLoS Pathog 5 e1000503 doi:10.1371/journal.ppat.1000503
66. SmithJA
DanielR
2006 Following the path of the virus: the exploitation of host DNA repair mechanisms by retroviruses. ACS Chem Biol 1 217 226
67. LiL
OlveraJM
YoderKE
MitchellRS
ButlerSL
2001 Role of the non-homologous DNA end joining pathway in the early steps of retroviral infection. EMBO J 20 3272 3281
68. KilzerJM
StrackerT
BeitzelB
MeekK
WeitzmanM
2003 Roles of host cell factors in circularization of retroviral DNA. Virology 314 460 467
69. DanielR
GregerJG
KatzRA
TaganovKD
WuX
2004 Evidence that stable retroviral transduction and cell survival following DNA integration depend on components of the nonhomologous end joining repair pathway. J Virol 78 8573 8581
70. DanielR
KatzRA
SkalkaAM
1999 A role for DNA-PK in retroviral DNA integration. Science 284 644 647
71. SmithJA
WangFX
ZhangH
WuKJ
WilliamsKJ
2008 Evidence that the Nijmegen breakage syndrome protein, an early sensor of double-strand DNA breaks (DSB), is involved in HIV-1 post-integration repair by recruiting the ataxia telangiectasia-mutated kinase in a process similar to, but distinct from, cellular DSB repair. Virol J 5
72. StudamireB
GoffSP
2008 Host proteins interacting with the Moloney murine leukemia virus integrase: multiple transcriptional regulators and chromatin binding factors. Retrovirology 5
73. LinC-W
EngelmanA
2003 The Barrier-to-Autointegration Factor Is a component of functional Human Immunodeficiency Virus type 1 preintegration complexes. J Virol 77 5030 5036
74. LauA
KanaarR
JacksonSP
O'ConnorMJ
2004 Suppression of retroviral infection by the RAD52 DNA repair protein. EMBO J 23 3421 3429
75. GibbsRA
RogersJ
KatzeMG
BumgarnerR
WeinstockGM
2007 Evolutionary and biomedical insights from the rhesus macaque genome. Science 316 222 234
76. LilleyCE
SchwartzRA
WeitzmanMD
2007 Using or abusing: viruses and the cellular DNA damage response. Trends Microbiol 15 119 126
77. BaileySG
VerrallE
SchelcherC
RhieA
DohertyAJ
2009 Functional Interaction between Epstein-Barr Virus Replication Protein Zta and Host DNA Damage Response Protein 53BP1. J Virol 83 11116 11122
78. AbbottKL
ArchambaultJ
XiaoH
NguyenBD
RoederRG
2005 Interactions of the HIV-1 tat and RAP74 proteins with the RNA polymerase IICTD phosphatase FCP1. Biochemistry 44 2716 2731
79. NyswanerKM
CheckleyMA
YiM
StephenstRM
GarfinkelDJ
2008 Chromatin-associated genes protect the yeast genome from Ty1 insertional mutagenesis. Genetics 178 197 214
80. DownsJA
JacksonSP
1999 Involvement of DNA end-binding protein Ku in Ty element retrotransposition. Mol Cell Biol 19 6260 6268
81. SuzukiJ
YamaguchiK
KajikawaM
IchiyanagiK
AdachiN
2009 Genetic evidence that the non-homologous end-joining repair pathway is involved in LINE retrotransposition. PLoS Genet 5 e1000461 doi:10.1371/journal.pgen.1000461
82. PitcherRS
TonkinLM
DaleyJM
PalmbosPL
GreenAJ
2006 Mycobacteriophage exploit NHEJ to facilitate genome circularization. Mol Cell 23 743 748
83. KentWJ
SugnetCW
FureyTS
RoskinKM
PringleTH
2002 The human genome browser at UCSC. Genome Res 12 996 1006
84. ThompsonJD
GibsonTJ
PlewniakF
JeanmouginF
HigginsDG
1997 The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25 4876 4882
85. ComeronJM
1999 K-Estimator: Calculation of the number of nucleotide substitutions per site and the confidence intervals. Bioinformatics 15 763 764
86. SchaffnerSF
FooC
GabrielS
ReichD
DalyMJ
2005 Calibrating a coalescent simulation of human genome sequence variation. Genome Res 15 1576 1583
87. CockerhamCC
WeirBS
1986 Estimation of inbreeding parameters in stratified populations. Ann Hum Genet 50 271 281
88. LeeJH
GoodarziAA
JeggoPA
PaullTT
2010 53BP1 promotes ATM activity through direct interactions with the MRN complex. EMBO J 29 574 585
89. LeeJH
PaullTT
2004 Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304 93 96
90. BhaskaraV
DupreA
LengsfeldB
HopkinsBB
ChanA
2007 Rad50 adenylate kinase activity regulates DNA tethering by Mre11/Rad50 complexes. Mol Cell 25 647 661
91. LeeJH
PaullTT
2006 Purification and biochemical characterization of ataxia-telangiectasia mutated and Mre11/Rad50/Nbs1. Method Enzymol 408 529 539
92. PaullTT
CortezD
BowersB
ElledgeSJ
GellertM
2001 Direct DNA binding by Brca1. P Natl Acad Sci USA 98 6086 6091
93. BrysonK
McGuffinLJ
MarsdenRL
WardJJ
SodhiJS
2005 Protein structure prediction servers at university college london. Nucleic Acids Res 33 W36 W38
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Genome-Wide Identification of Targets and Function of Individual MicroRNAs in Mouse Embryonic Stem Cells
- Common Genetic Variants and Modification of Penetrance of -Associated Breast Cancer
- Allele-Specific Down-Regulation of Expression Induced by Retinoids Contributes to Climate Adaptations
- β-Actin and γ-Actin Are Each Dispensable for Auditory Hair Cell Development But Required for Stereocilia Maintenance
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy