
Tight Regulation of the Gene of the KplE1 Prophage: A New Paradigm for Integrase Gene Regulation
Temperate phages have the ability to maintain their genome in their host, a process called lysogeny. For most, passive replication of the phage genome relies on integration into the host's chromosome and becoming a prophage. Prophages remain silent in the absence of stress and replicate passively within their host genome. However, when stressful conditions occur, a prophage excises itself and resumes the viral cycle. Integration and excision of phage genomes are mediated by regulated site-specific recombination catalyzed by tyrosine and serine recombinases. In the KplE1 prophage, site-specific recombination is mediated by the IntS integrase and the TorI recombination directionality factor (RDF). We previously described a sub-family of temperate phages that is characterized by an unusual organization of the recombination module. Consequently, the attL recombination region overlaps with the integrase promoter, and the integrase and RDF genes do not share a common activated promoter upon lytic induction as in the lambda prophage. In this study, we show that the intS gene is tightly regulated by its own product as well as by the TorI RDF protein. In silico analysis revealed that overlap of the attL region with the integrase promoter is widely encountered in prophages present in prokaryotic genomes, suggesting a general occurrence of negatively autoregulated integrase genes. The prediction that these integrase genes are negatively autoregulated was biologically assessed by studying the regulation of several integrase genes from two different Escherichia coli strains. Our results suggest that the majority of tRNA-associated integrase genes in prokaryotic genomes could be autoregulated and that this might be correlated with the recombination efficiency as in KplE1. The consequences of this unprecedented regulation for excisive recombination are discussed.
Published in the journal:
. PLoS Genet 6(10): e32767. doi:10.1371/journal.pgen.1001149
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001149
Summary
Temperate phages have the ability to maintain their genome in their host, a process called lysogeny. For most, passive replication of the phage genome relies on integration into the host's chromosome and becoming a prophage. Prophages remain silent in the absence of stress and replicate passively within their host genome. However, when stressful conditions occur, a prophage excises itself and resumes the viral cycle. Integration and excision of phage genomes are mediated by regulated site-specific recombination catalyzed by tyrosine and serine recombinases. In the KplE1 prophage, site-specific recombination is mediated by the IntS integrase and the TorI recombination directionality factor (RDF). We previously described a sub-family of temperate phages that is characterized by an unusual organization of the recombination module. Consequently, the attL recombination region overlaps with the integrase promoter, and the integrase and RDF genes do not share a common activated promoter upon lytic induction as in the lambda prophage. In this study, we show that the intS gene is tightly regulated by its own product as well as by the TorI RDF protein. In silico analysis revealed that overlap of the attL region with the integrase promoter is widely encountered in prophages present in prokaryotic genomes, suggesting a general occurrence of negatively autoregulated integrase genes. The prediction that these integrase genes are negatively autoregulated was biologically assessed by studying the regulation of several integrase genes from two different Escherichia coli strains. Our results suggest that the majority of tRNA-associated integrase genes in prokaryotic genomes could be autoregulated and that this might be correlated with the recombination efficiency as in KplE1. The consequences of this unprecedented regulation for excisive recombination are discussed.
Introduction
Temperate bacteriophages are characterized by their ability to maintain their genome into the host, a process called lysogeny. Most temperate phages integrate their genome into the host's chromosome, becoming prophages. Alternatively, circularized phage genomes are maintained as episomes. Once integrated, the now so-called prophage is stable and replicates passively with its host genome. This situation can continue as long as outside conditions do not become threatening for the host, and therefore for the virus. Prophages are indeed able to detect many stressful signals, such as DNA damage, excessive heat or pressure [1]–[3]. By “listening” and hijacking the host's response to various stresses, prophages behave like perfect stress biosensors. Once the prophage is induced, the process of lysogeny escape is engaged, and the phage enters a lytic mode of development [1]. A crucial event in this process is the excision of the prophage from the host's chromosome. Replication of the viral genome follows, as well as the synthesis and the assembly of the virion proteins. Thus, excisive recombination is a highly regulated process that relies on two different levels of regulation: (i) protein activity, through the control of directionality by a recombination directionality factor (RDF), and (ii) protein synthesis via the coordinated expression of the integrase and RDF genes.
Temperate bacteriophages use site-specific recombination to integrate into and excise their genomes out of the host genomes. Integration consists of a strand exchange between the recombination region attP on the phage genome and attB on the bacterial chromosome leading to the formation of the recombined halves attL and attR at the junctions between the bacterial chromosome and the integrated phage genome (Figure 1). Lambda phage integrase has been extensively studied for its role in site-specific recombination and is essential for lysogeny establishment as well as for the transition to productive lytic development (reviewed in [4], [5]). The Int tyrosine recombinase catalyzes integrative and excisive recombination [6], [7]. Xis acts as a recombination directionality factor (RDF) as it bears no catalytic activity but rather directs the Int-driven reaction toward excision [8]. Xis plays an architectural role in the formation of the excisive intasome by binding and bending DNA, and prevents reintegration of the excised phage genome [9]–[11]. Precise stoechiometry of Int and Xis proteins is required for the correct assembly of the intasome nucleoprotein complex [12]. Since the organization of the protein binding sites of the att regions is not conserved, this suggests that the intasome architecture may vary according to the number and orientation of the recombination protein binding sites [13]. The phage-encoded integrase is a hetero-bivalent DNA binding protein in which the N - and C-terminal domains bind to different DNA substrates. The C-terminal domain, where the catalytic activity takes place, binds to and recombines the identical core-type sequences present in attP and attB, or in attL and attR, depending on the direction of the reaction considered [14]–[16]. The N-terminal domain binds to arm-type sequences [17], and this binding allows the assembly of the intasome, the nucleoprotein complex for site-specific recombination. Host-encoded proteins are also involved in this process, including IHF and Fis that bind and bend DNA in order to assist intasome formation [9], [18]–[20]. Recombination occurs through pair-wise exchange of four DNA strands between two att substrates. A four-way Holliday junction is formed upon the exchange of one pair of strands and then resolved after the DNA cleavage activity is switched from one pair of strands to another [21]–[24]. In all temperate phages, site-specific recombination events are believed to be identical; however, the organization of the att regions varies from one family of phages to another according to the number and orientation of the recombination protein binding sites. This suggests that the assembly and final composition of the intasome might follow different paths to eventually end with the same recombination reaction.

The KplE1 prophage (also named CPS-53) is a defective prophage integrated into the argW tRNA gene in E. coli K12 (Figure 1). The prophage's remaining genome (10.2 kb) contains 16 open reading frames (ORF) bordered by a duplicated core sequence of 16 nucleotides (CTGCAGGGGACACCAT). None of these ORFs seems to encode a repressor consistent with the finding that KplE1 is not SOS-inducible (M. Ansaldi, unpublished observation). Despite the small remnant genome, the KplE1 prophage can be excised in vivo [13], [25]. The KplE1 recombination module has been analyzed, and indeed it contains all the elements required for site-specific recombination to occur, including RDF and integrase genes as well as the attL and attR recombination regions [26]. This recombination module is highly conserved in several enterobacteria phage genomes such as CUS-3 and HK620 that infect E. coli strains K1 RS218 and TD2158, respectively, and Sf6, which infects Shigella flexneri, as well as in prophages present in E. coli strains APEC-O1 and UTI89 [27]–[32]. One advantage of studying the KplE1 prophage is that we can dissect the excisive recombination and its regulation in vivo independently of prophage induction since the CI regulator module is missing in KplE1. Directionality of the site-specific recombination has been studied using KplE1 DNA substrates as well as HK620 substrates and requires the RDF protein TorI to direct the recombination reaction towards excision [26]. One prominent feature of the KplE1 recombination is the orientation of the intS gene relative to the attL region (Figure 1). Indeed, the intS gene is transcribed from a dedicated promoter that overlaps with the attL region. In λ, int gene expression depends on the activity of two promoters PI and PL [1], [5]. While lysogeny is established, int expression relies on the PI promoter located in the xis gene and allows transcription of int independently of xis. Therefore, this promoter is used to establish lysogeny and ensures that more Int than Xis is being made [33]. During the escape from lysogeny, xis and int are co-transcribed as a consequence of PL promoter activation and N antitermination (Figure 1). The differential expression of Int by these two promoters depends upon a site (sib) located distal to the int gene. Thus, lower amounts of Int are made, and Xis production is not affected by this element [34].
Based on the localization and orientation of the intS promoter that overlaps the attL recombination region (Figure 1), we performed preliminary experiments that led us to conclude that the intS gene is negatively autoregulated and poorly expressed during the exponential growth phase [26]. In this study, we further investigate the regulation of the intS gene in relation to the recombination efficiency. We provide in silico evidence that a majority of integrase genes associated with tRNA inserted prophages are predicted to negatively autoregulate. This prediction was subsequently confirmed in vivo with several examples. As a consequence, the integrase gene appears constantly expressed at a low level in KplE1, and the control of excisive recombination seems to rely only on the RDF expression rather than on a coordinate expression of the integrase and RDF genes.
Results
Experimental determination of the intS transcription start site
Previous work described the PintS promoter based on sequence analysis of the region upstream from the ATG starting codon [26]. This allowed the identification of putative −10 and −35 sequences close to the consensus sequences recognized by the σ70-RNA polymerase holoenzyme (TAaAAa and TTGACA, respectively) (Figure 2C). To show that the RNA polymerase actually utilizes this promoter to start intS transcription, we experimentally determined the intS transcription start site. Primer extension analysis was performed using total RNAs extracted from a wild-type as well as an intS strain, annealed with a labeled primer hybridizing downstream from the intS ATG (see Materials and Methods for details). In the presence of IntS (Figure 2A, lane 1), extension products were scarcely apparent. However, in the intS background (Figure 2A, lane 2) we observed two main extension products, indicating that transcription started at T and A residues at positions 2464536 and 2464537 on the E. coli chromosome, respectively. These transcription start sites are correctly located relative to the σ70-RNA polymerase holoenzyme binding sites, and the A at position 2464537 is perfectly positioned relative to the −10 box [35]. This latter transcription start site was also detected in a genome-scale analysis of transcription in E. coli [36]. Altogether, these experiments confirmed the previous localization of the intS promoter and the downregulation of the intS gene by its own product.

Expression of the PintS promoter in vivo
The intS promoter, due to its location, obviously overlaps with the attL recombination region, and thus overlaps with IntS and TorI binding sites as previously characterized [26] (Figure 2C). In that study, we showed that the intS transcript originating at the chromosomal Pints promoter was five-fold more abundant in an intS background than in a wild-type strain. To study the influence of each protein binding site on PintS regulation in vivo, an accurate method was needed to quantify gene expression that would allow easy mutagenesis of the protein binding sites. We chose to use a gfp fusion-based vector (pUA66) that contains a sc101 replication origin, which leads to a low copy number (3 to 4 copies in the logarithmic growth phase) of the plasmid in vivo to avoid titration of the regulators [37]. The experiment was calibrated by cloning the entire attL region (positions 2464344 to 2464630 on the E. coli chromosome) in the pUA66 vector in order to measure pattL-gfp expression in various genetic backgrounds. Primer extension was used to control that transcription initiation occurred at the same site in this construct rather than in the chromosome (Figure 2A, lane 3). Indeed, the transcription start sites proved to be identical to those characterized on the chromosome when expressing the PintS promoter from a plasmid. Using this construct, we observed an increased transcription level of the PintS promoter compared to the chromosomal expression. This was likely due to a combination of two effects: the plasmid copy number and the fact that total RNAs were extracted from the LCB1019 strain that lacks the entire KplE1 prophage, and therefore the intS gene. Another explanation could be that this increase in transcription is linked to an increase in translation of the fusion. However, this is probably not the case because although integrase genes often contain rare codons that may slow down translation, a particular rare codon (AGA) is also present in the gfp gene.
We measured the fusion expression with two different methods: direct fluorescence measurement (Figure 3), which gave a whole population measurement, and microscopic counting (Figure S1), which estimated the homogeneity of the fluorescent population. As indicated in Figure 3B, the attL-gfp wild-type fusion was expressed at a high level in the absence of IntS (6368±914 Units) and was repressed in the presence of IntS (1270±208 Units), leading to a repression ratio of ∼5 when the control ratio of placZ-gfp expression was close to 1 in the same conditions. This ratio of ∼5 is in complete agreement with the values we obtained by measuring intS expression from the chromosomal gene with quantitative RT-PCR [26], indicating that the fusion expression from several copies did not modify the regulatory ratio. Expression of the fusion was homogenous under all conditions, and the most resolved peaks were observed for cells producing TorI or IntS and therefore emitting little fluorescence (Figure S1). Thus, the results measured in the whole population (Figure 3) reflect homogenous expression of the fusion.
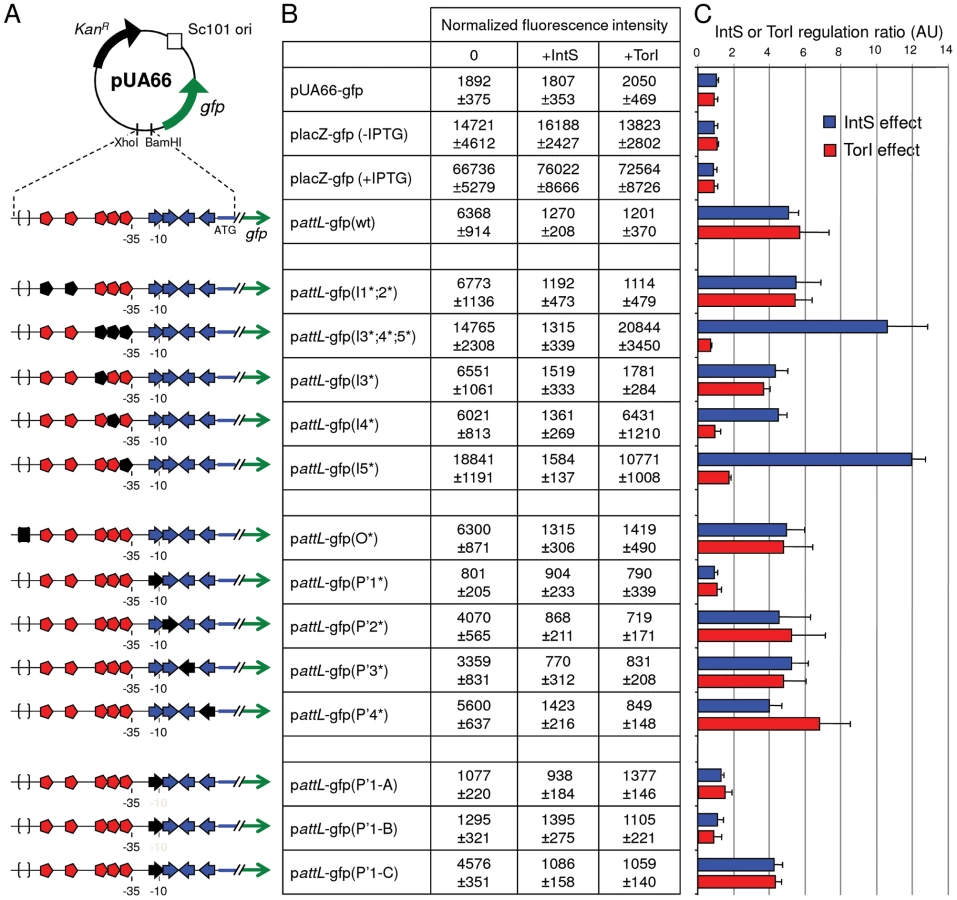
Looking at the recombination protein binding sites identified on attL (Figure 2C), it was obvious that some of the TorI RDF binding sites were also near the −35 sequence. We thus looked at a possible effect of TorI on intS expression. As indicated in Figure 3B, overexpression of TorI in an intS background led to a strong decrease in expression of the pattL-gfp(wt) fusion (compare 6368±914 with 1201±370 units). Taken together, these results show that intS expression is under the negative control of both TorI and IntS.
Identification of the recombination protein binding sites involved in intS downregulation
The attL region contains five TorI binding sites organized in two blocks (Figure 2C, red symbols). The first one (I1;2), encompasses sites I1 and I2 that are separated by 12 nucleotides (positions 2464409 to 2464436). The second block is composed of three binding sites (I3;4;5) separated by 2 nucleotides (positions 2464472 to 2464499). We mutated each site by changing the sequences GTTCG, GATCG, GTCCG into CAAGC. When both sites of block I1;2 were mutated we did not observe any effect on the TorI mediated downregulation of intS (pattL-gfp(I1*;2*)) with a repression ratio of 5.4 (Figure 3). In contrast when the sites of the second block were changed, pattL-gfp(I3*;4*;5*), TorI was no longer able to repress the expression of the fusion, meaning that at least one of these sites was important for repression. We thus measured the effect of each site independently. If the mutation of site I3 had little effect on the repression ratio (4.7), the mutations of sites I4 and I5 led to expression of the fusion independent of the presence of TorI (repression ratios of 1.0 and 1.7, respectively). These two sites are the closest to the −35 sequence and are therefore appropriate candidates for mediating TorI repressor activity. We observed increased basal expression of the fusion (from 6,400 to 15,000–19,000 units) when the I5 site was mutated. This effect is probably due to the change in the nucleotides adjacent to the −35 sequence that results in a promoter-up phenotype.
We then studied the implication of the arm-type binding sites (P' sites, blue symbols in Figure 2B and 2C) of the integrase. For the P' sites the conserved motif TAAA present in all P' sites was changed into its complement ATTT. Interestingly, none of the individual mutations led to derepression of the fusion; indeed, in all cases (except for the P'1*, see below), the repression ratio ranged from 4.0 to 5.2 (Figure 3B and 3C). The P'1 site's influence was more difficult to study since it overlapped with the −10 sequence (Figure 2C). Thus, any mutation of the conserved motif led to an inactive promoter whose measured fluorescence did not exceed that of the promoter-less fusion (Figure 3, compare pUA66-gfp with pattL-gfp(P'1*)). Additional constructs were made to avoid this effect on the promoter activity; however, any change we made that altered IntS binding also affected promoter activity (Figure 3, constructs pattL-gfp(P'1-A and B)), and in the case the latter was not affected (pattL-gfp(P'1-C)), neither was the down regulation of intS. In a control experiment, we mutated the core site, which is the binding site for the catalytic domain of the integrase, and this construct showed an unaltered repression phenotype (Figure 3) as well as IntS binding similar to that observed with the wild-type sequence (data not shown and [13]). Altogether, these data demonstrate that both TorI and IntS negatively regulates the intS gene in vivo and point to the TorI and IntS sites located near the −35 and −10 sequences as being responsible for the downregulation of intS gene expression. These results also show that the intS gene is tightly regulated and is thus expressed at a low level under all tested growth conditions.
A critical IntS concentration is required for efficient excisive recombination
One could ask about the “raison d'être” of this atypical integrase gene regulation compared with the lambda int gene. For that purpose, we measured the efficiency of the excisive recombination reaction in vitro as a function of the integrase concentration. Briefly, 32 nM of attL and attR linear substrates were incubated at 37°C for 1 h in the presence of constant concentrations of TorI and IHF (1.6 µM and 0.25 µM, respectively) and increasing concentrations of IntS (0.02 to 6.7 µM). The attP product was quantified by Q-PCR and the efficiency of the reaction was calculated as the percentage of substrates transformed into products. As the concentration of IntS increased, the efficiency of the reaction increased until a maximum level of ∼80% was achieved for an IntS concentration around 1 µM (Figure 4). However, when the IntS concentration exceeded 1.2 µM, we rapidly observed an inhibitory effect of IntS on the excisive reaction. Subsequently, the concentration range for which the efficiency of the reaction reached more than 50% was very narrow (0.8 µM up to 1.2 µM). These results show that to obtain the maximum efficiency in excisive recombination a precise integrase concentration is required.

The effect of IntS overloading was then analyzed in vivo. Strain LCB6005 contains a Km resistance cassette in the tail fiber encoding gene (tfaS) of the KplE1 prophage, thus allowing an in vivo excision assay to be performed without any effect on the site-specific recombination process. This strain was transformed with the pJFi plasmid that contains the torI gene under the control of an IPTG inducible promoter as well as with the pBAD33 vector containing or not the intS gene under the control of an arabinose inducible promoter (pBADintS). Colonies were counted after the different cultures induced with IPTG were plated on LB medium containing ampicillin or kanamycin (see the Material and Methods section). ApR colonies are representative of the total number of cells since all contain the pJFi plasmid (ApR), whereas KmR colonies originate from cells that have kept the tfaS::kan marker, and thus the KplE1 prophage. As shown before [25], expressing torI from a multicopy plasmid (pJFi) is sufficient to promote excisive recombination. Indeed, in the strain containing the low copy vector alone (pBAD33), the maximal level of excision was achieved in the presence of TorI as revealed by a high ApR/KmR ratio (Figure 5A), and the addition of the arabinose inducer did not impede the reaction's efficiency. However, in the presence of the pBintS plasmid, even without adding the arabinose inducer, we observed dramatically decreased recombination activity (Figure 5A, compare bars 1 and 3). It is striking that, even at a concentration of integrase that could not be detected on a Western blot (Figure 5B, lane 3), i.e., in the absence of an inducer, the efficiency of the reaction underwent a 50-fold decrease. We explain this effect by the leakage of the pBAD promoter in the absence of glucose. Indeed, this promoter is induced in the presence of arabinose and repressed in the presence of glucose [38]. Since we do not use glucose in the medium, the pBAD promoter is not repressed, and some integrase is being made, although not sufficiently to immunodetect it. We therefore consider the empty vector as the actual negative control. Adding arabinose to the medium, which led to overproduction of IntS (Figure 5B, lanes 4 to 8), amplified this negative effect on the in vivo excision reaction. As a result, the in vivo recombination efficiency was negatively correlated with the increasing integrase concentration, thus confirming the results we obtained in vitro.

Occurrence of predicted self-regulated integrase genes in prokaryotic genomes
To address the general relevance of the negative autoregulation of the intS gene, a large-scale in silico analysis of tRNA-associated integrase genes was performed on the complete prokaryotic genomes available at that time. The in silico outline is described in the “Materials and Methods” section. Experimentally well-characterized integrases such as λInt and KplE1IntS contain at least one of the three functional domains, Phage_integrase, Phage_integ_N, and Phage_integr_N, referred to as PF00589, PF09003 and PF02899 in the Pfam database, respectively. By using these functional domains as queries, we detected 8368 protein homologs within 1014 complete prokaryotic genomes, and 1273 of the corresponding integrase genes (15% of the total) are adjacent to a tRNA gene. These couples of tRNA-integrase genes (called InTr shape) constitute the primary data set used in this study (Table S1). InTr shapes were classified according to their gene coding orientation, leading to four different types of InTr shapes (Figure 6A): STI (Same orientation and T precedes I), SIT (Same orientation and I precedes T), OC (Opposite and Convergent orientation) and OD (Opposite and Divergent orientation). We then analyzed the distribution of the InTr copy-number per organism (Figure 6B) as well as the distribution of InTr shapes over the prokaryotic phylum (Figure 6C). A detailed analysis of these data is available in the Text S1. Overall analysis shows that the majority of the InTr shapes exhibits STI and OC shapes with 736 and 438 representatives, respectively. The other two classes (SIT and OD) occur relatively rarely (less than 8% in total) in the analyzed genomes. Therefore, the high occurrence of STI and OC shapes within the prokaryotes may highlight the functional importance of these shapes in microbial organisms.

Relationship between the InTr shapes and integrase regulation
To study a possible correlation between the prevalence of InTr shapes and the autoregulation of the integrase genes as demonstrated for the intS gene, the number of putative autoregulated integrase genes was determined. Based on our experimental model, we proposed that STI and OD shapes should be subjected to autoregulation, since in these cases the integrase gene promoter overlaps with the recombination region, whereas SIT and OC shapes should show integrase gene expression independent of the integrase protein. Our in silico results indicated that InTr shapes containing Asn, Cys, Gln, Gly, Leu, Phe, SelC, and Ser tRNA genes were mainly predicted to autoregulated (Table S2). In contrast, the opposite conclusion can be drawn for InTr shapes containing Ile, Lys and Tyr tRNA genes, which is consistent with the observation that prophages are preferentially inserted in poorly expressed tRNA genes, probably to avoid a deleterious effect on cell fitness ([39]–[41]. A detailed analysis of the distribution of InTr shapes with respect to tRNAs in prokaryotic genomes is available in Figure S2 and Table S3. Out of the 1273 InTr shapes analyzed, 61.5% were detected as potentially autoregulated, most encoded within the Proteobacteria, Cyanobacteria, Bacteroidetes and Crenarcheota genomes (Figure 6C and Table S2). Thus, a situation that has rarely been described and studied in the literature is actually predominant in the sequenced prokaryotic genomes.
We next addressed whether a relationship exists between the length of the intergenic region (IR, Figure 6A) and the fact that an integrase gene is predicted to be autoregulated. Therefore, the IR length was determined for each InTr shape, and the distribution of the obtained values was analyzed as a function of autoregulated and non-autoregulated InTr shapes (Figure 7). The lower values of the IR lengths are statistically associated with predicted non-autoregulated InTr shapes as the 95% confidence intervals of the mean IR length values are [157.5–158.4] for non-autoregulated InTr and [208.3–227.9] for predicted autoregulated InTr. These results clearly indicate that autoregulated InTr shapes are linked to large IRs. Our prediction is that autoregulation of the integrase mostly correlates with STI and OD shapes, and therefore the IR should be large enough to contain an entire attL region. As mentioned above, the average distance observed for predicted autoregulated InTr shapes [208.3–227.9] is perfectly compatible with the presence of an average attL region of 80–170 nucleotides.

Biological validation of the autoregulation of integrase genes involved in STI and OD InTr shapes.
To validate the in silico predictions, we chose to study the expression of several integrase promoters from E. coli strains K12 MG1655 and O157:H7 EDL933. The promoters of the integrase genes were cloned into the pUA66 vector upstream of the gfp gene and the cognate integrase coding sequences were cloned into the pJF119EH vector (see plasmid list in Table 1). Regarding the InTr shapes, in addition to the well-characterized STI argW-intS, we studied 2 STI shape argW-intC (the argW-intS homologous shape in EDL933) and selC-intL, 2 OC shapes argU-intD (MG1655) and thrW-intH (EDL933) and 1 OD ptwF-intF (MG1655). Of these integrase genes, 3 are predicted to be autoregulated (intC, intF, and intL), and 2 should not exhibit autoregulation (intD and intH). To avoid the influence of the chromosomal copies of MG1655 integrase genes, we transformed both kinds of plasmids (the empty vector and the integrase encoding vector) in the appropriate deletion mutant, and when applicable, we used the MG1655 mutant for the EDL933 equivalent. As indicated in Figure 8, none of the OC shape associated integrase genes showed self-regulation, and the STI and OD shape integrase genes were negatively autoregulated. However, different regulation ratios were observed depending on the integrase gene considered. Interestingly, the pIntS-gfp fusion was repressed almost 15 times during the exponential growth phase (time point ∼2 h) whereas a repression ratio of 6 was measured during the stationary phase (∼4.5 h), which is consistent with the data shown in Figure 2 and Figure 3. A similar expression pattern was obtained with the pPintC-gfp fusion for which the repression ratios were higher than for pPintS (28 in the exponential phase and 10 in the stationary phase). A high regulatory ratio was observed with the pPintF-gfp fusion whose expression was decreased around 23 times in the presence of the pIntF plasmid in exponential as well as in stationary growth phases, without any induction of the ptac promoter, indicating that the leak of the promoter allowed sufficient IntF production to produce a negative effect on the fusion expression. In contrast, the pPintL-gfp fusion was only down-regulated by a factor of 4 in the exponential growth phase, and this occurred in the presence of 0.1 mM of IPTG. Thus, the level of IntL required to lead to a negative effect on the fusion expression is probably higher than that necessary for the IntF integrase. One possible explanation is that integrase genes from E. coli MG1655 interfere with the downregulation of EDL933 genes. This hypothesis is strengthened by the fact that the regulatory ratios measured with the pPintC fusion were higher in an intS background than in a WT MG1655 background (data not shown). Together, these results supported the in silico prediction that STI and OD shape associated integrase genes should be negatively autoregulated. However, this prediction could be associated with promoter and recombination region sequence analysis to ensure that the two overlap.


Discussion
In temperate phages, site-specific recombination is a highly regulated process; indeed, both the activity and integrase gene expression are controlled. Little is known about integrase gene expression in general, except for the lambda phage integrase for which extensive studies have been available for almost 40 years. Integrase gene expression has been detected in natural environment samples induced with mitomycin C, which promotes productive growth. Therefore, integrase gene expression is used as a marker of (pro)phage presence [42].
In the case of the well-characterized lambda integrase, little integrase is made during lysogeny as none of the promoters is activated (PL and PI). Under lytic conditions, the int gene is transcribed together with the xis gene from the PL promoter due to the antitermination role of the N protein [1]. In KplE1, and other phages related by their recombination module such as HK620, Sf6 and CUS-3, transcription of the integrase and RDF genes is clearly uncoupled. Uncoupling of the integrase and RDF gene transcription has been described in P2 and 186 phages, where the int gene is expressed from the lysogenic transcript, and the RDF is the first gene on the lytic transcript [43]. The intS promoter, according to its orientation, overlaps with the attL region, where recombination proteins, including IntS itself, bind. We measured intS transcription during the E. coli exponential growth phase, and as expected, the intS transcript could be detected by RT-QPCR at the different time points of the growth ([26] and data not shown). In this work, we show that IntS as well as the RDF TorI negatively regulate intS expression. A similar situation is found in the P4 satellite phage, although the regulatory mechanism might be slightly different. In P4, Piazzolla and co-workers showed that the integrase and the RDF protein Vis negatively regulate int gene expression [44]. Although integrase self-regulation occurs through direct DNA binding at similar positions relative to the int transcription start site compared to those we described for IntS, the authors suggest that Vis binds to the int mRNA and therefore may inhibit translation [44]. In Figure 3, we show that TorI binds to DNA at positions favorable for transcription inhibition. In both cases, the RDF protein eventually promotes a lower integrase amount in the cell although Vis binds to RNA and TorI to DNA. These are the only documented cases of such a double down-regulation of integrase gene expression by the integrase itself and its cognate RDF protein. However, we can speculate that this regulatory process will be present in all cases where the integrase promoter overlaps the attL recombination region as long as the att region and integrase promoter overlap.
We then asked about the biological significance of such tight regulation of the integrase gene in KplE1. We measured the excisive reaction efficiency using fixed TorI and IHF concentrations, and variable IntS concentrations (Figure 4). Interestingly, the IntS concentration range that led to more than 50% efficiency was narrow, indicating that excisive recombination occurs at a precise integrase concentration. Moreover, when IntS was artificially overexpressed in vivo, the excision efficiency dropped rapidly as the IntS concentration increased (Figure 4). According to these results, a tight regulation of the integrase gene appears crucial for the recombination event to take place, as described earlier in vivo for lambda prophage [45]. The regulatory scenario characterized in KplE1 is dual. First, the integrase itself regulates its own expression by directly binding to the promoter sequence close to the −10 box. Negative autoregulatory loops are widespread in all organisms. For example, in mammals and photosynthetic bacteria, circadian oscillations are generated by a set of genes forming a transcriptional autoregulatory feedback loop [46], [47]. Feedback regulation plays a crucial role in the robust control of many cellular systems and is a way of stabilizing and maintaining the concentration of gene products. Recent models of feedback loops suggest that the strength of a feedback loop controls the oscillations of a regulatory path [48]. Therefore, one must consider the synthesis rate together with the degradation rate of the feedback regulator. In the case of IntS, the protein was very stable in the conditions we examined, suggesting that the loop is controlled only by the synthesis rate of the integrase. However, under certain conditions the integrase might undergo degradation by a yet unknown mechanism.
The second component of intS gene downregulation involves the TorI RDF (Figure 3). As mentioned above, this negative regulation involves the I4 and I5 sites located near the −35 sequence (Figure 2C). It is therefore likely that TorI prevents the binding of the RNA polymerase holoenzyme to the −35 region. This is a way to control the ratio integrase/RDF in order to obtain optimal excision conditions. In the lambda phage, the coupling of xis and int transcription upon lytic induction together with the presence of the sib untranslated region allow the accumulation of higher amounts of Xis than Int under lytic conditions [1], [34]. We therefore propose that the downregulation of intS by TorI is a different method of achieving a similar pattern in recombination protein concentrations. Indeed, when the prophage undergoes excision of its genome under lytic conditions, the RDF is needed in higher amounts than the integrase because of its dual role, directing the reaction towards excision and preventing the re-integration of the newly replicated phage genomes. As a consequence, in the absence of transcriptional coupling, as is the case in P4, KplE1 and other related (pro)phages, the RDF protein may directly control the appropriate integrase/RDF ratio through negative regulation of the integrase gene. Several lines of evidence support this statement; in particular, we showed in previous papers that the chromosomal intS gene was transcribed at a low level during the exponential growth phase [26] and that expressing the torI gene from a multicopy plasmid was sufficient to promote in vivo excision [25]. Altogether, our results show that the integrase gene is permanently expressed at a low level due to a strong negative control by the integrase itself and by the RDF. However, the gene is expressed at a sufficient level to allow prophage excision as soon as the RDF is produced [25]. Therefore, we propose that the main control of prophage excision targets the RDF gene when the integrase promoter is not coupled to the lytic promoter.
The narrow optimum Int concentration for recombination is probably the consequence of the strict stoechiometry required for the correct assembly of the intasome [12]. The role of the RDF protein is often restricted to a helper function as a DNA bender required to position the integrase molecules. However, the strict dependency of the lambda and KplE1 recombination systems on their respective RDF may suggest a more active role. In KplE1, the efficiency of the excisive recombination is dependent on the IntS/TorI protein ratio [13]. We speculate that, in this case, keeping a constant rate of integrase synthesis allows control of this ratio, and therefore the intasome forms only through the RDF production. Alternatively, researchers recently suggested that the alternative sigma factor (σH) in Staphylococcus aureus participates in maintaining prophages by controlling integrase expression to ensure that more integrase than excisionase is made thus avoiding undesired excision [49]. Although xis transcription is strictly repressed by CI in the lambda prophage during lysogeny, one could imagine that transcriptional leakage is possible; thus, a moderate expression of the int gene could maintain the right balance of Int/Xis during lysogeny.
To address the general relevance of the negative autoregulation of the intS integrase gene, we performed a large-scale study of tRNA inserted prophages on complete prokaryotic genomes. The first step consisted of identifying of the InTr shapes. Current computational methods (Phage_Finder, Prophage Finder, DRAD) detect prophages in genomes by identifying possible essential proteins such as integrases, a region containing proteins similar to those occurring in prophages, or by dinucleotide relative abundance difference (DRAD) [40], [50], [51]. While these programs have been shown independently to give reliable results, comparative analysis of prophages identified by these methods showed high heterogeneity with low overlapping results probably arising from the mosaic nature of the prophages [51]. Therefore, we preferred identification without any a priori, based on the presence of the essential integrase gene. Moreover, our procedure can likely identify complete integrated elements and defective prophage regions encountered within prokaryotic genomes as long as they contain an integrase gene. The obtained data combined with tRNA searches gave 92.6% of STI InTr shapes in which the integrases have only a «Phage integrase» domain, and therefore, this procedure avoids many false positive results. Thus, without any a priori on the data, the most frequently observed STI shapes were with proteins likely to be similar to IntS, indicating a clear tRNA sublocation preference with this integrase subfamily. Several genome analysis studies showed that a vast majority of prophages are inserted in or adjacent to tRNA genes [39]–[41], [52], [53]. Williams revealed that tRNA sequence sites are preferred for prophage integration sites [39]. This analysis also demonstrated that for 34 cases out of 58 (59%) the attB sequence is in a tRNA or tmRNA and that some of the prophages are flanked by tRNA genes. A bias was also noted for the selenocysteinyl tRNA (tRNA SelC), tRNA Arg, tRNA Met, and tRNA Ala genes (Figure S2 and Table S3). The same conclusion was drawn by Fouts as his analysis of 285 putative attachment sites (from 302 complete bacterial genomes) revealed that tRNAs are the most frequently used targets (33%) for integration, and that the most popular tRNA targets are Arg, Leu, Ser and Thr [40]. Our integrase identification procedure, combined with the fact that we were working on >1000 organisms (compared to 302 bacterial genomes analyzed by Fouts [40]) may explain the difference observed (15% vs 33%) of the prophages that used tRNA as target sites. In E. coli and Shigella genomes, comparative genomic analysis also showed that tRNA loci are preferentially used as an insertion site for integrative elements, with the majority of tRNA genes remaining intact after insertion [52], [53]. Finally, Boyd and colleagues' analysis of island-encoded integrases revealed that half of the available tRNA genes were used as integration sites, in particular among members of the γ-Proteobacteria [41]. The vast majority of these integrase genes were adjacent to the tRNA loci. However, in the mentioned studies, less is done about functional relationships between the integrase and the proximal tRNA gene. We therefore focused on this particular couple of integration shapes, as some benefits could be expected by the genetic element from its association with a tRNA gene. As suggested by Swenson et al. a possible benefit could be the transcriptional coupling of the integrated element and the tRNA gene, as tRNA promoters are typically regulated by the growth rate [54]. Non-regularity in the orientation of prophages to tRNA genes has been observed, and researchers have suggested that the tRNA gene setting might directly affect integrase function or the directionality of recombination in a way that is beneficial for genetic elements.
The main focus of the in silico analysis was to study the occurrence of a regulatory path similar to the one we described for the intS gene. To our surprise, we found that the majority of tRNA associated integrase genes (61.5%) exhibited a promoter that overlapped with the attL recombination region (STI and OD shapes). As a consequence, and given the results we obtained with the intS promoter, we were able to predict that these genes may undergo negative autoregulation, which was confirmed in vivo for several genes (Figure 8). This prediction can be expanded to any locus containing an STI or OD InTr shape, as long as the recombination protein binding sites and RNA polymerase binding sites somehow overlap. For example, at the tRNA Ser locus in Vibrio cholera, the integrase gene associated to the genomic island VPI-2 should be autoregulated which may have some implication for the maintenance of this pathogenicity island.
Concluding remarks
The regulatory switch leading to the controlled expression of the integrase and RDF proteins that allows the excision of the lambda prophage and therefore permits productive growth to resume has long been the paradigm for all temperate phages [1], [5]. In this study, we show that the particular organization we identified for the KplE1 attL recombination region and related (pro)phages is widespread among the tRNA inserted prophages. The fact that the attL region overlaps the integrase promoter has several consequences: (i) the integrase gene is likely down-regulated by itself and the RDF, as long as the recombination protein and the RNA polymerase binding sites overlap sufficiently, (ii) the transcription of the integrase and RDF genes are uncoupled, and (iii) the regulatory switch that permits prophage excision relies on RDF gene expression. Full understanding of prophage excision control will require focusing on the expression of the RDF genes that are uncoupled to the integrase gene transcription.
Materials and Methods
Bacterial strains, plasmids, media, and growth conditions
Bacterial strains and plasmids are listed in Table 1. Strains were grown in LB medium and, when necessary, ampicillin (50 µg mL−1), chloramphenicol (25 µg mL−1), kanamycin (25 µg mL−1) or IPTG (0.1–1 mM) were added.
Strain construction
Strains LCB6005, LCB6006, LCB6035 and LCB6036 are derivatives of ENZ1734 (MG1655 ΔlacIZ) [55] obtained by P1 transduction of the tfaS::KanR (JW5383), intS::KanR (JW2345), intD::KanR (JW0525) and intF::KanR (JW0275) markers [56], respectively, into ENZ1734. The kan gene was then removed from strains LCB6006, LCB6035 and LCB6036 by using the pCP20 plasmid [57] to generate strains LCB6007 (intS, KanS), LCB6037 (intD, KanS), LCB6038 (intF, KanS). Strains are described in Table 1.
Plasmid construction
To construct plasmid pBintS, the intS coding sequence was PCR-amplified using MG1655 chromosomal DNA as a template with appropriate primers. After enzymatic hydrolysis, the PCR product was cloned into the KpnI/HindIII sites of the pBAD33 vector [38]. Plasmid pattL-gfp was constructed by the insertion of the attL region (220 bp, Figure 2C) into the XhoI and BamHI sites of the pUA66 vector [37]. A similar procedure was used to clone the promoter regions of intD, intH, intL intF into the pUA66 vector. Positions of the cloned sequences are indicated in Table 1, and primer sequences are available upon request from the authors. The sequence accuracy of the cloned inserts was checked by sequencing.
Primer extension
Total RNAs extracted from strains MC4100 and LCB1024 (ΔintS), and strain LCB1019 (ΔKplE1) containing pattL-gfp were hybridized with a primer complementary to the positions +40 to +64 relative to the ATG of intS (attL-ter). attL-ter was 32P labeled by using [γ32P]ATP and T4 polynucleotide kinase (Biolabs). A total of 12 µg of ARNs and 4 ng of labeled primer were incubated together with 200 units of Superscript III reverse transcriptase (Invitrogen) for 50 minutes at 50°C, followed by 10 minutes at 70°C to inactivate the enzyme. The sequencing ladder was PCR amplified with the same labeled primer and 5′ primer hybridizing to positions −196 to −173 relative to the ATG of intS (attL-Kpn). The sequencing reaction was performed using the Thermo Sequenase Cycle Sequencing Kit (USB Corporation). Extension and sequencing products were separated onto a 6 M urea 8% acrylamide (19∶1) gel.
Site-directed mutagenesis of attL
Mutations in the recombination protein binding sites were generated by an overlapping PCR procedure [58]. Mutated primers were used to amplify the protein binding sites whereas the wild-type primers attL-pro-XhoI and attL-ter-BamHI delimit the attL region. After enzymatic hydrolysis, mutated attL were cloned into pUA66. Mutations in the IntS and TorI binding site are summarized in Figure 2C. All primer sequences used for mutagenesis are available upon request.
Protein purifications
IntS, TorI and IHF proteins were overproduced and purified near homogeneity as described [26], [59], [60]. All proteins were dialyzed in 40 mM Tris-HCl buffer (pH 7.6) containing 50 mM KCl and 10% glycerol. Denaturing polyacrylamide gel electrophoresis (SDS-PAGE) was used to estimate the protein purity, and the Lowry method was used to estimate protein concentrations.
In vivo excision assay
Strain LCB6005 (KanR gene inserted in the tfaS gene of KplE1) carrying plasmids pJFi and pBAD33 (control) or pJFi and pBintS were grown in LB medium supplemented by increasing amounts of arabinose as indicated in Figure 5 legend. When the A600 reached 0.5 units (0.5×109 cells mL−1), IPTG (1 mM) was added and the culture resumed for 2 h at 37°C under agitation. Culture dilutions were prepared and plated onto rich medium containing either ampicillin (pJFi) or kanamycin (tfaS::kan). Numeration of the colonies plated on both antibiotics was performed and the ratio of ampicillin-resistant/kanamycin-resistant (ApR/KnR) colonies was calculated. This value is close to one when the excision rate is low and the tfaS::kan marker is present, and increases when excision efficiency increases and the cells no longer contain the KplE1 prophage. The values represent the average of at least three independent determinations. The IntS relative amount in crude extracts was analyzed after 12% SDS-PAGE with Western blot using a polyclonal IntS antiserum.
In vitro excision assay
Purified IHF, IntS and TorI were used in all experiments. All reaction mixtures (25 µl) included 32 nM of linear attL (attL-SpeI/attL-KpnI primers) and attR (attR-XbaI/attR-IHF2 primers) in buffer containing 30 mM Tris-HCl (pH 7.6), 10 mM spermidine, 5 mM EDTA, 1 mg.mL−1 bovine serum albumin, 34 mM KCl and 5% glycerol. IHF (0.25 µM) and TorI (1.6 µM) were added in all samples in the presence of a range of IntS concentrations (0.02 to 8 µM). The reactions were carried out in optimized conditions: 37°C for 1 h. The best efficiency was obtained for IntS concentrations ranging from 0.8 to 1.2 µM, leading to an IHF:IntS:TorI protein ratio of 1∶4∶6.
Real-time PCR analysis (Q-PCR)
The abundance of attP formed during in vitro excision assays was quantified by real-time PCR, using a known concentration of PCR amplified attP as a reference standard. The real-time PCR quantifications were performed with an Eppendorf Mastercycler ep realplex instrument and the SYBR Premix Ex Taq (TaKaRa) according to the manufacturer specifications. Serial dilutions of each in vitro reaction were mixed with 1.5 µM of primers and 6 µL of master mix in a 14 µL final volume. The primer pair used to quantify attP was attR-IHF2/attL-SpeI. PCR parameters were as follows: one cycle at 95°C for 2 min followed by 40 cycles at 95°C for 5 s, 55°C for 15 s and 72°C for 10 s. Excision efficiency was calculated as the percentage of the initial substrate (32 nM) transformed into product.
GFP transcriptional fusion measurement
GFP fluorescence was measured on whole cells after an overnight aerobic growth at 37°C in LB medium supplemented by IPTG (0.1–1 mM) for TorI and/or integrase induction (Figure 3). The pJF119EH empty vector was used as a negative control and to ensure that the growth conditions (presence of ampicillin) were identical for all strains. After centrifugation, bacteria were washed, resuspended and diluted in 0.25X M9 medium. Cells (150 µL) were loaded on an Optilux black/clear Bottom Microtest 96-well assay plate (Falcon). Alternatively, fluorescence intensity was measured on bacterial cultures over time. Precultures of the various strains were diluted in fresh LB medium containing the appropriate antibiotics and IPTG (0.1 mM) when indicated. Each strain was assayed in quadruplet. The incubation protocol included an initial 5-min shake (double orbital, 1.5 mm diameter, normal speed), followed by 85 cycles consisting of the following actions: a 1-sec measurement (see below), a 6-min shake and a 1-min standing. The time course was performed at 37°C for approximately 10 h. A600 and fluorescence measurements were performed using the Infinit M200 instrument (Tecan) and the Tecan i-control 1.3 application (488 nm excitation wavelength, 521 nm emission wavelength, 160 gain, 20 µs integration time and 25 reads per sample). The value of blank (0.25X M9 or LB) was withdrawn and normalized fluorescence intensities (emission at 521 nm/A600) were calculated. The values represent the averages of at least four independent measurements. Microscopic analysis was performed using an automated and inverted epifluorescence microscope TE2000-E-PFS (Nikon, France) and adequate filters (excitation 480±15 nm, emission 535±20 nm). Images were recorded with a CoolSNAP HQ 2 (Roper Scientific, Roper Scientific SARL, France) and a 40x/0.75 DLL “Plan-Apochromat” or a 100x/1.4 DLL objective; image analysis was conducted with MetaMorph 7.5 software (Molecular Devices). For each cell preparation, a total of 25 images were taken randomly on different optical fields, and the average intensity of each cell was calculated (Figure S1).
Bioinformatic analyses
The complete genomes of 1014 prokaryotic (946 bacterial and 68 archaeal) organisms available in December 2009 were downloaded from the NCBI ftp site (ftp:/ftp.ncbi.nih.gov/genomes/Bacteria/) and constitute the primary data source. To identify integrase promoters overlapping the integration site, the analysis was restricted to prophage insertion targeted to tRNA sites. The HMMER-3 package [61] and self-written Perl scripts were then used to search for protein integrase homologs (with phage λ int and E. coli intS as reference seed proteins) in the complete genomes. The presence of one of these functional domains (from Pfam 24.0 [62]), Phage_integrase (PF00589), Phage_integ_N (PF09003) or Phage_integr_N (PF02899), was a requisite. Alignments with a score higher than the Pfam gathering thresholds were considered significant. Note that homologs with protein sizes lower than 140 amino acids (corresponding to 80% of the Phage_integrase profile length) were removed from the data. The obtained sequences were subsequently analyzed with the same software in order to locate additional known functional domains. In-house Perl scripts were used to define the domain organization. The search for tRNA genes, located in the region between the integrase gene and the downstream/upstream neighboring gene was performed by using the tRNAscan-SE program [63]. Finally, protein integrase homologs were filtered by the presence of an adjacent tRNA gene (downstream or upstream of the integrase gene), leading to the final set of integrase homologs used in this study. We then computed the IR length as the distance in nucleotides between a given integrase gene and the immediately adjacent tRNA gene.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. PtashneM
2004 A genetic switch: phage lambda revisited. Cold Spring Harbor Cold Spring Harbor Laboratory Press
2. AertsenA
VanHR
VanoirbeekK
MichielsCW
2004 An SOS response induced by high pressure in Escherichia coli. J Bacteriol 186 6133 6141 10.1128/JB.186.18.6133-6141.2004 [doi];186/18/6133 [pii]
3. RokneyA
KobilerO
AmirA
CourtDL
StavansJ
2008 Host responses influence on the induction of lambda prophage. Mol Microbiol 68 29 36 MMI6119 [pii];10.1111/j.1365-2958.2008.06119.x [doi]
4. Van DuyneGD
2005 Lambda integrase: armed for recombination. Curr Biol 15 R658 R660
5. CourtDL
OppenheimAB
AdhyaS
2006 A New Look at Bacteriophage Lambda Genetic Networks. J Bacteriol 189 298 304
6. KittsP
RichetE
NashHA
1984 Lambda integrative recombination: supercoiling, synapsis, and strand exchange. Cold Spring Harb Symp Quant Biol 49 735 744
7. WarrenD
LeeSY
LandyA
2005 Mutations in the amino-terminal domain of lambda-integrase have differential effects on integrative and excisive recombination. Mol Microbiol 55 1104 1112
8. LewisJA
HatfullGF
2001 Control of directionality in integrase-mediated recombination: examination of recombination directionality factors (RDFs) including Xis and Cox proteins. Nucleic Acids Res 29 2205 2216
9. MillerHI
FriedmanDI
1980 An E. coli gene product required for lambda site-specific recombination. Cell 20 711 719
10. AbremskiK
GottesmanS
1982 Purification of the bacteriophage lambda xis gene product required for lambda excisive recombination. J Biol Chem 257 9658 9662
11. NumrychTE
GumportRI
GardnerJF
1992 Characterization of the bacteriophage lambda excisionase (Xis) protein: the C-terminus is required for Xis-integrase cooperativity but not for DNA binding. EMBO J 11 3797 3806
12. MummJP
LandyA
GellesJ
2006 Viewing single lambda site-specific recombination events from start to finish. EMBO J 25 4586 4595
13. PanisG
DuvergerY
ChampS
AnsaldiM
2010 Protein binding sites involved in the assembly of the KplE1 prophage intasome. Virology 404 41 50
14. RossW
LandyA
KikuchiY
NashH
1979 Interaction of int protein with specific sites on lambda att DNA. Cell 18 297 307
15. TirumalaiRS
KwonHJ
CardenteEH
EllenbergerT
LandyA
1998 Recognition of core-type DNA sites by lambda integrase. J Mol Biol 279 513 527
16. DorgaiL
SloanS
WeisbergRA
1998 Recognition of core binding sites by bacteriophage integrases. J Mol Biol 277 1059 1070
17. RossW
LandyA
1982 Bacteriophage lambda int protein recognizes two classes of sequence in the phage att site: characterization of arm-type sites. Proc Natl Acad Sci U S A 79 7724 7728
18. GardnerJF
NashHA
1986 Role of Escherichia coli IHF protein in lambda site-specific recombination. A mutational analysis of binding sites. J Mol Biol 191 181 189
19. NumrychTE
GumportRI
GardnerJF
1991 A genetic analysis of Xis and FIS interactions with their binding sites in bacteriophage lambda. J Bacteriol 173 5954 5963
20. GoodmanSD
NicholsonSC
NashHA
1992 Deformation of DNA during site-specific recombination of bacteriophage lambda: replacement of IHF protein by HU protein or sequence-directed bends. Proc Natl Acad Sci U S A 89 11910 11914
21. BauerCE
GardnerJF
GumportRI
WeisbergRA
1989 The effect of attachment site mutations on strand exchange in bacteriophage lambda site-specific recombination. Genetics 122 727 736
22. LandyA
HsuPL
RossW
BuraczynskaM
1980 Site-specific recombination in bacteriophage lambda: structural analyses of reactive DNA sequences. Am J Trop Med Hyg 29 1099 1106
23. NashH
1996 Site-Specific Recombination: Integration, Excision, Resolution, and Inversion of Defined DNA Segments.
NeidhardtFC
Escherichia coli and Salmonella: cellular and molecular biology Washington ASM Press 2363 2376
24. BiswasT
AiharaH
Radman-LivajaM
FilmanD
LandyA
2005 A structural basis for allosteric control of DNA recombination by lambda integrase. Nature 435 1059 1066
25. ElAntakL
AnsaldiM
GuerlesquinF
MéjeanV
MorelliX
2005 Structural and genetic analyses reveal a key role in prophage excision for the TorI response regulator inhibitor. J Biol Chem 280 36802 36808
26. PanisG
MéjeanV
AnsaldiM
2007 Control and regulation of KplE1 prophage site-specific recombination: a new recombination module analyzed. J Biol Chem 282 21798 21809
27. DhillonTS
PoonAP
ChanD
ClarkAJ
1998 General transducing phages like Salmonella phage P22 isolated using a smooth strain of Escherichia coli as host. FEMS Microbiol Lett 161 129 133
28. ClarkAJ
InwoodW
CloutierT
DhillonTS
2001 Nucleotide sequence of coliphage HK620 and the evolution of lambdoid phages. J Mol Biol 311 657 679
29. CasjensS
Winn-StapleyDA
GilcreaseEB
MoronaR
KuhleweinC
2004 The chromosome of Shigella flexneri bacteriophage Sf6: complete nucleotide sequence, genetic mosaicism, and DNA packaging. J Mol Biol 339 379 394
30. ChenSL
HungCS
XuJ
ReigstadCS
MagriniV
2006 Identification of genes subject to positive selection in uropathogenic strains of Escherichia coli: a comparative genomics approach. Proc Natl Acad Sci U S A 103 5977 5982 0600938103 [pii];10.1073/pnas.0600938103 [doi]
31. KingMR
VimrRP
SteenbergenSM
SpanjaardL
PlunkettG
III
2007 Escherichia coli K1-specific bacteriophage CUS-3 distribution and function in phase-variable capsular polysialic acid O acetylation. J Bacteriol 189 6447 6456 JB.00657-07 [pii];10.1128/JB.00657-07 [doi]
32. JohnsonTJ
KariyawasamS
WannemuehlerY
MangiameleP
JohnsonSJ
2007 The genome sequence of avian pathogenic Escherichia coli strain O1:K1:H7 shares strong similarities with human extraintestinal pathogenic E. coli genomes. J Bacteriol 189 3228 3236 JB.01726-06 [pii];10.1128/JB.01726-06 [doi]
33. BenedikM
MascarenhasD
CampbellA
1982 Probing cII and himA action at the integrase promoter pi of bacteriophage lambda. Gene 19 303 311
34. SchmeissnerU
McKenneyK
RosenbergM
CourtD
1984 Transcription terminator involved in the expression of the int gene of phage lambda. Gene 28 343 350
35. HarleyCB
ReynoldsRP
1987 Analysis of E. coli promoter sequences. Nucleic Acids Res 15 2343 2361
36. ChoBK
ZenglerK
QiuY
ParkYS
KnightEM
2009 The transcription unit architecture of the Escherichia coli genome. Nat Biotechnol 27 1043 1049 nbt.1582 [pii];10.1038/nbt.1582 [doi]
37. ZaslaverA
BrenA
RonenM
ItzkovitzS
KikoinI
2006 A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat Methods 3 623 628
38. GuzmanLM
BelinD
CarsonMJ
BeckwithJ
1995 Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177 4121 4130
39. WilliamsKP
2002 Integration sites for genetic elements in prokaryotic tRNA and tmRNA genes: sublocation preference of integrase subfamilies. Nucleic Acids Res 30 866 875
40. FoutsDE
2006 Phage_Finder: automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res 34 5839 5851 gkl732 [pii];10.1093/nar/gkl732 [doi]
41. BoydEF
Almagro-MorenoS
ParentMA
2009 Genomic islands are dynamic, ancient integrative elements in bacterial evolution. Trends Microbiol 17 47 53 S0966-842X(09)00003-1 [pii];10.1016/j.tim.2008.11.003 [doi]
42. McDanielL
BreitbartM
MobberleyJ
LongA
HaynesM
2008 Metagenomic analysis of lysogeny in tampa bay: implications for prophage gene expression. PLoS ONE 3 e3263 doi:10.1371/journal.pone.0003263
43. NilssonAS
Haggard-LjungquistE
2006 The P2-like bacteriophages.
CalendarR
The Bacteriophages Oxford Oxford University Press 365 390
44. PiazzollaD
CaliS
SpoldiE
FortiF
SalaC
2006 Expression of phage P4 integrase is regulated negatively by both Int and Vis. J Gen Virol 87 2423 2431
45. EnquistLW
KikuchiA
WeisbergRA
1979 The role of lambda integrase in integration and excision. Cold Spring Harb Symp Quant Biol 43 Pt 2 1115 1120
46. IshiuraM
KutsunaS
AokiS
IwasakiH
AnderssonCR
1998 Expression of a gene cluster kaiABC as a circadian feedback process in cyanobacteria. Science 281 1519 1523
47. SiepkaSM
YooSH
ParkJ
LeeC
TakahashiJS
2007 Genetics and neurobiology of circadian clocks in mammals. Cold Spring Harb Symp Quant Biol 72 251 259 10.1101/sqb.2007.72.052 [doi];10.1101/sqb.2007.72.052 [pii]
48. NguyenLK
KulasiriD
2009 On the functional diversity of dynamical behaviour in genetic and metabolic feedback systems. BMC Syst Biol 3 51 1752-0509-3-51 [pii];10.1186/1752-0509-3-51 [doi]
49. TaoL
WuX
SunB
2010 Alternative sigma factor sigmaH modulates prophage integration and excision in Staphylococcus aureus. PLoS Pathog 6 e1000888 doi:10.1371/journal.ppat.1000888
50. BoseM
BarberRD
2006 Prophage Finder: a prophage loci prediction tool for prokaryotic genome sequences. In Silico Biol 6 223 227 2006060020 [pii]
51. SrividhyaKV
AlagurajV
PoornimaG
KumarD
SinghGP
2007 Identification of prophages in bacterial genomes by dinucleotide relative abundance difference. PLoS ONE 2 e1193 doi:10.1371/journal.pone.0001193
52. OuHY
ChenLL
LonnenJ
ChaudhuriRR
ThaniAB
2006 A novel strategy for the identification of genomic islands by comparative analysis of the contents and contexts of tRNA sites in closely related bacteria. Nucleic Acids Res 34 e3 34/1/e3 [pii];10.1093/nar/gnj005 [doi]
53. GermonP
RocheD
MeloS
Mignon-GrasteauS
DobrindtU
2007 tDNA locus polymorphism and ecto-chromosomal DNA insertion hot-spots are related to the phylogenetic group of Escherichia coli strains. Microbiology 153 826 837 153/3/826 [pii];10.1099/mic.0.2006/001958-0 [doi]
54. SwensonDL
KimKJ
SixEW
CleggS
1994 The gene fimU affects expression of Salmonella typhimurium type 1 fimbriae and is related to the Escherichia coli tRNA gene argU. Mol Gen Genet 244 216 218
55. MoreauPL
2007 The Lysine Decarboxylase CadA Protects Escherichia coli Starved of Phosphate against Fermentation Acids. J Bacteriol 189 2249 2261
56. BabaT
AraT
HasegawaM
TakaiY
OkumuraY
2006 Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2 2006
57. DatsenkoKA
WannerBL
2000 One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97 6640 6645
58. HoSN
HuntHD
HortonRM
PullenJK
PeaseLR
1989 Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77 51 59
59. AnsaldiM
ThéraulazL
MéjeanV
2004 TorI, a response regulator inhibitor of phage origin in Escherichia coli. Proc Natl Acad Sci U S A 101 9423 9428
60. MurtinC
EngelhornM
GeiselmannJ
BoccardF
1998 A quantitative UV laser footprinting analysis of the interaction of IHF with specific binding sites: re-evaluation of the effective concentration of IHF in the cell. J Mol Biol 284 949 961
61. DurbinR
EddyS
KroghA
MichisonG
1998 Biological sequence analysis: probabilistic models of proteins and nucleic acids. Cambridge University Press
62. FinnRD
MistryJ
TateJ
CoggillP
HegerA
2010 The Pfam protein families database. Nucleic Acids Res 38 D211 D222 gkp985 [pii];10.1093/nar/gkp985 [doi]
63. LoweTM
EddySR
1997 tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25 955 964 gka184 [pii]
64. FursteJP
PansegrauW
FrankR
BlockerH
ScholzP
1986 Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene 48 119 131
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 10
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Genome-Wide Identification of Targets and Function of Individual MicroRNAs in Mouse Embryonic Stem Cells
- Common Genetic Variants and Modification of Penetrance of -Associated Breast Cancer
- Allele-Specific Down-Regulation of Expression Induced by Retinoids Contributes to Climate Adaptations
- β-Actin and γ-Actin Are Each Dispensable for Auditory Hair Cell Development But Required for Stereocilia Maintenance
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy