
Nové poznatky o patogenezi folikulárního lymfomu a koncepty cílené léčby
Novel Findings in Follicular Lymphoma Pathogenesis and the Concepts of Targeted Therapy
The molecular pathogenesis of follicular lymphoma (FL) was partially revealed by the discovery of BCL2 translocations to the region encoding the immunoglobulin heavy chain, which accompany the vast majority of cases. This aberration leads to the ectopic and constitutive expression of anti-apoptotic BCL2 protein in B-cells. Nevertheless, the aberration alone is not sufficient for FL development, which suggests necessity of further genetic aberrations acquisition for neoplastic transformation to FL. Their discovery has been enabled by recent progress in the field of massive parallel sequencing (next generation sequencing), which revealed high number of genetic aberrations connected with onset and progression of FL. The occurrence of many of these aberrations in the early stages of the disease, and the fact that they are shared by the majority of patients with FL, fundamentally changed our former understanding of the disease onset. Furthermore, in a large fraction of patients, FL undergoes histological transformation to a more aggressive lymphoma, which is also associated with specific genetic alterations. In this review, we summarize the current knowledge of molecular pathways connected with FL biology and discuss their role in the context of normal B-cell development. Understanding of FL biology is essential for the development of new targeted therapies and the stratification of patients, and potentially also for the selection of treatment for specific patients who share the same genetic aberrations.
Key words:
follicular lymphoma – mutation – aberration – apoptosis – epigenetic regulators – microRNA
This research was carried out under the project CEITEC 2020 (LQ1601) with financial support from the Ministry of Education, Youth and Sports of the Czech Republic under the National Sustain ability Programme II.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted:
28. 1. 2017
Accepted:
5. 3. 2017
Autoři:
J. Deván 1; K. Musilová 1; A. Janíková 1,2; M. Mráz 1,2
Působiště autorů:
CEITEC – Středoevropský technologický institut, MU, Brno
1; Interní hematologická a onkologická klinika LF MU a FN Brno
2
Vyšlo v časopise:
Klin Onkol 2017; 30(4): 247-257
Kategorie:
Přehled
doi:
https://doi.org/10.14735/amko2017247
Souhrn
Molekulární podstata folikulárního lymfomu (FL) byla částečně poodhalena již poměrně dávno zjištěním, že téměř ve všech případech je v maligních B lymfocytech přítomna translokace genu BCL2 do oblasti kódující těžký řetězec imunoglobulinů. Tato aberace vede k ektopické a konstitutivní expresi antiapoptotického BCL2 v B lymfocytech. Samotná však zřejmě není pro vznik onemocnění postačující, což naznačují jak výsledky získané na myším modelu, tak i častý výskyt translokace BCL2 v B lymfocytech zdravých jedinců. Předpokládalo se, že musí existovat další aberace, jejichž výskyt je spojen se vznikem FL. K jejich odhalení přispěl nedávno rozvoj metod masivního paralelního sekvenování (sekvenování nové generace), díky kterému bylo popsáno množství genetických aberací, které provázejí vznik a progresi této malignity. Skutečnost, že některé z nich se vyskytují již v raných stadiích FL a jsou sdílené většinou pacientů, zásadně koriguje předešlou představu o vzniku tohoto onemocnění. FL navíc u nezanedbatelné části pacientů podléhá histologické transformaci do agresivnějšího onemocnění, což s sebou nese další specifické aberace. V tomto přehledovém článku shrnujeme nejnovější poznatky o molekulárních drahách zapojených v biologii FL a jejich význam v kontextu přirozeného vývoje B lymfocytů. Pochopení molekulární patogeneze FL je základem pro vývoj nových cílených léčiv, stratifikaci pacientů a možná i volbu léčby pro skupiny pacientů dle specifických aberací.
Klíčová slova:
folikulární lymfom – mutace – aberace – apoptóza – epigenetické regulátory – mikroRNA
Úvod
Folikulární lymfom (FL) je nejčastějším typem indolentního nehodgkinského lymfomu (NHL), který představuje ~20 % všech NHL s incidencí asi 3–4/100 000 za rok [1]. Toto nádorové onemocnění patří historicky k prvním, u nichž byla identifikována jednotící genomická aberace, tj. translokace t (14; 18) (q32; 21) (~95 % případů) [2,3]. Klinicky jde o pomalu rostoucí nádor projevující se obvykle nebolestivou lymfadenopatií, která může v čase narůstat i ustupovat. FL je dobře senzitivní na radio-i chemoterapii, avšak léčebné odpovědi jsou u většiny pacientů dočasné, následované téměř pravidelně relapsy. U části pacientů dochází v průběhu choroby k transformaci do agresivního lymfomu (nejčastěji do difuzního velkobuněčného B lymfomu (diffuse large B-cell lymphoma – DLBCL)). Osud konkrétního pacienta s nově diagnostikovaným FL lze prozatím jen nedostatečně odhadnout. K tomu slouží řada prognostických skóre (FLIPI, FLIPI2, POD24-PI) postavených zejména na základních klinicko-laboratorních parametrech (věk, klinické stadium, laboratorní aktivita choroby vyjádřená obvykle hodnotami LDH, hemoglobinem, beta-2-mikroglobulinem atd.) nebo na klinickém chování v čase [4]. Nedostatkem těchto skóre je jejich malá individuální přesnost. V současné době však bylo díky metodám masivního paralelního sekvenování (sekvenování nové generace – NGS) popsáno mnoho aberací, které provázejí vznik a progresi FL. Bylo zjištěno, že existuje několik aberací, které jsou přítomny již v raných stadiích onemocnění a jsou sdílené většinou pacientů, což podstatně změnilo pohled na vznik onemocnění. V posledních 2 letech je navíc snaha začlenit některé časté aberace do nových klinicko-genetických prognostických skóre. Příkladem takového skórovacího systému je M7-FLIPI, které je založené na hodnocení mutačního statusu sedmi genů (EZH2, ARID1A, MEF2B, EP300, FOXO1, CREBBP a CARD11) v kombinaci s klinickými faktory (FLIPI, ECOG škála) u pacientů s FL [5,6]. Prognosticky zajímavá je zejména výrazná asociace mutovaného ARID1A a EZH2 s dobrou prognózou [5,6]. Takový přístup představuje významný posun v stratifikaci pacientů do prognostických skupin. Nicméně pro vývoj cílené léčby a event. navrhování specifické léčby pro subtypy FL je důležité detailní pochopení molekulární biologie tohoto nádorového onemocnění. V následujícím textu se proto věnujeme jednotlivým genetickým aberacím a změnám v genové expresi charakteristickým pro buňky FL a diskutujeme jejich úlohu v kontextu fyziologického vývoje B lymfocytů a evoluce onemocnění. Shrnujeme poznatky, které vedly od představy jednotící aberace (tj. translokace BCL2) ke komplexnější integraci deregulace epigenomu, aberantní signalizace z B bu-něčného receptoru a změn v mikroprostředí do představ o vývoji FL. Změna v chápání biologie FL, k níž v posledních letech došlo, je určitým pozitivním posunem paradigmatu a lze předpokládat, že proběhne i u dalších onkologických onemocnění. Důležité je, že toto pochopení také vede k testování nové cílené léčby a potenciálně k navrhování specifické léčby pro subtypy FL.
První kroky ve vzniku FL – translokace BCL2
Pro ochranu organizmu proti patogenům musí být B lymfocyty schopné vytvářet imunoglobuliny (Ig) s obrovskou variabilitou v antigen-rozpoznávajících regionech. B lymfocyty disponují sofistikovaným mechanizmem, který ve třech navzájem nezávislých krocích (tj. VDJ/VJ rekombinace, somatické hypermutace a izotypový přesmyk Ig) umožňuje produkci protilátek s téměř nelimitovanou specificitou. Navzdory nenahraditelné roli pro imunitní systém představují tyto reakce zároveň vysoce rizikové procesy, neboť vyžadují tvorbu zlomů a mutací v DNA. Chyby v těchto procesech jsou mimo jiné zdrojem translokací umísťujících proto-onkogeny (jako je BCL2) pod vliv vysoce aktivních zesilovačů promotorů imunoglobulinů a představují tak první krok ve vzniku FL, ke kterému dochází v kostní dřeni (obr. 1) [2].
Za normálních okolností opustí funkční naivní (IgM+, IgD+) B lymfocyty kostní dřeň a dostávají se krevním řečištěm do niche sekundárních lymfatických orgánů. Tato migrace je významným krokem ve vývoji B lymfocytů a je řízena expresí specifických cytokinových a chemokinových receptorů a adhezivních molekul. Sekundární lymfatické orgány jsou místem afinitní maturace a podmínky v jejich germinálních centrech (GC) umožňují rychlou proliferaci B lymfocytů a zároveň toleranci genetických změn (somatické hypermutace, přesmyk Ig). Nicméně genový expresní profil B lymfocytů v GC je spíše pro-apoptotický, což je důležitou podmínkou umožňující jejich fyziologickou selekci. Selekce B lymfocytů je založena na jejich schopnosti, resp. neschopnosti iniciovat interakce s buňkami mikroprostředí GC. Ta je fyziologicky podmíněna dostatečně silnou vazbou k antigenu pomocí B buněčného receptoru (BCR), čímž je spouštěna signalizace zabezpečující přežívání buňky a její proliferaci. Pokud však premaligní B lymfocyty dokáží účinně inhibovat apoptózu (např. díky translokaci BCL2), přestávají podléhat selekci a mohou akumulovat genomické aberace, které jim umožňují dát vznik plně malignímu klonu lymfomových buněk (obr. 1). Samotný výskyt translokace BCL2 není zřejmě pro vznik FL dostačující, což naznačují jak výsledky pokusů na myším modelu, tak relativně častý výskyt této aberace v B lymfocytech zdravých jedinců (v nízkých frekvencích až u 50 % populace) [7–9]. U myší s BCL2 aberací obdobnou té, která se vyskytuje u lidského FL, dochází sice k rozvoji folikulárních hyperplazií nebo (po dlouhé době latence) agresivních lymfomů, ne však FL [8]. Bylo však popsáno, že výskyt BCL2 translokace časteji než v 1 : 104 lymfocytů periferní krve je asociován s více než 20násobným zvýšením rizika vzniku FL v následujících 15 letech [9]. U některých zdravých jedinců se t (14; 18) vyskytuje i v populaci centrocytů (B lymfocyty světlé zóny GC) určitých lymfatických folikulů, avšak bez zasahování do zbývající části lymfatické uzliny (~2–3 %). Tento jev byl popsán jako in situ FL a u značné části těchto jedinců dochází během několika let k rozvoji FL [10]. Retrospektivní studie ale ve většině případů dokumentují nízkou nebo žádnou míru klonality mezi populacemi FL a in situ FL. Tyto výsledky ukazují na divergentní způsob evoluce, anebo dokonce nezávislý vznik těchto jevů. Předpokládá se proto, že první signály umožňující proliferaci t (14; 18) + centrocytů jsou podobně jako u normálních centrocytů podmíněné afinitou BCR k antigenu. Až akumulací aberací vedoucích k deregulaci epigenomu a aberantně posílené BCR signalizaci buňky FL částečně svou závislost na afinitě k antigenu ztrácejí. Nicméně i po získání mnoha genomických aberací zůstávají maligní buňky FL závislé na interakcích s faktory mikroprostředí a FL si zachovává základní strukturu GC (shrnuto v Janíková et al [11]). Význam mikroprosředí demonstruje i výrazná míra apoptózy, kterou vykazují primární buňky FL při kultivaci in vitro [12]. Předpokládá se, že mikroprostředí hraje podstatnou roli v průběhu celého vývoje onemocnění od raných fází, kdy chrání buňky před apoptózou, než dojde ke kumulaci genomických aberací, až po event. histologickou transformaci. Nicméně je zřejmé, že změny v mikroprostředí jsou do značné míry (ne-li zcela) iniciovány aberacemi postihujícími B lymfocyty. U B lymfocytů FL se totiž vyskytují přímo mutace v genech kódujících cytokiny a některé povrchové molekuly zodpovědné za interakce s komponentami imunitních niche (viz níže) [13,14].

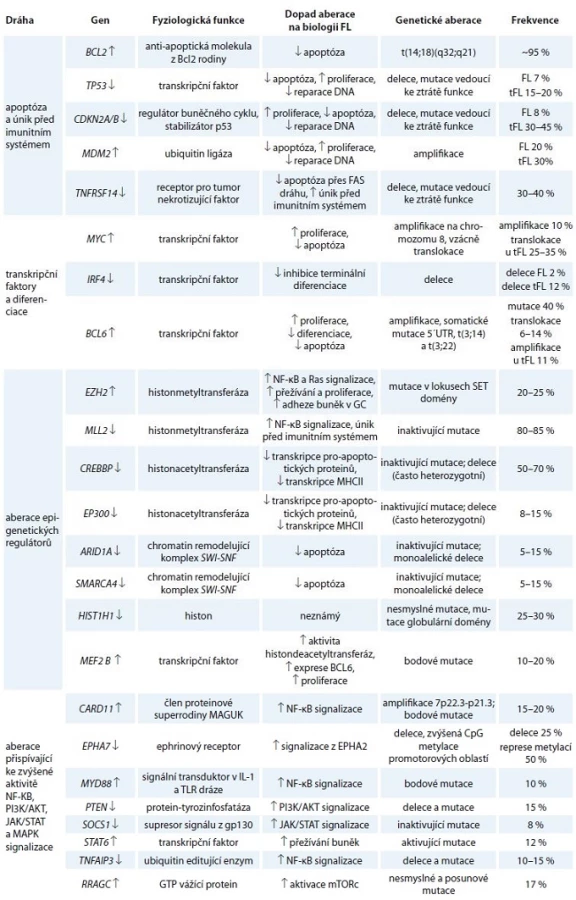
Kromě translokace BCL2 bylo u FL popsáno i mnoho dalších mutací a genomických aberací. U drtivé většiny případů je přítomna aberace některého z epigenetických regulátorů z kategorie histonmetyltransferáz, histonacetyltransferáz či komponent chromatin remodelujících komplexů. Předpokládá se, že aberace epigenetických regulátorů jsou relativně brzkou událostí ve vývoji maligního klonu FL buněk. V popředí stojí především mutace histonmetyltransferázy MLL2, které jsou je díky svému mimořádně častému výskytu (80 % případů) jedním z důležitých faktorů v patogenezi FL [14,15]. Časté aberace v raných fázích FL jsou (podobně jako u DLBCL) mutace v drahách vedoucích k aktivaci B lymfocytů, a to zejména v BCR signalizační dráze, CD40 a TLR signalizaci [14–17]. Ty vedou ke zvýšené aktivitě kináz spouštějících signální kaskády, které aktivují transkripční faktory chránící buňky před apoptózou a umožňující jejich proliferaci (viz níže). Aberace vedoucí k úniku před imunitním systémem a deregulace nekódujících RNA, tzv. mikroRNA (miRNA), mají také své důležité postavení v biologii tohoto onemocnění [14,15,18]. Navzdory vysoké frekvenci výskytu některých aberací se žádná z nich nevyskytuje ve všech případech FL, dokonce ani translokace BCL2 (graf 1, tab. 1). Důležité je povšimnout si, že deregulace těchto genů a proteinů probíhá na podkladu preexistujících molekulárních drah, které fyziologicky řídí osud normálních B lymfocytů v GC lymfatických folikulů. V následujícím textu se proto jednotlivě věnujeme zmíněným genetickým aberacím a kontextu jejich přítomnosti právě u FL.
![Přibližná frekvence aberací vybraných genů zahrnutých v patogenezi FL [14–17,44].
↓ označuje aberaci vedoucí ke ztrátě funkce, ↑ označuje aberaci vedoucí ke zvýšené funkci/expresi](https://pl-master.mdcdn.cz/media/image/546d30be91bf880ee29af8d0656644fc.jpg?version=1537794030)
Interakce v mikroprostředí, BCR signalizace a aktivace transkripčních faktorů
Osud B lymfocytů je v sekundárních lymfatických orgánech spojený se signalizací z povrchových receptorů, jako je BCR, TLR a CD40. Tyto dráhy nejen spouštějí signalizační kaskády vedoucí k aktivaci transkripčních faktorů jako NF-κB, STAT nebo MYC, ale také zabezpečují aktivaci buněčných komponent mikroprostředí, a tím produkci cytokinů a kontakt mezi buňkami [19]. V odpovědi na tyto signály spouštějí membránově vázané receptory na B lymfocytech kaskády navzájem propojených reakcí umožňujících tak přežívání a proliferaci B lymfocytů FL i v případě, že jejich BCR vykazují nízkou nebo žádnou afinitu k antigenu. Aberace v těchto drahách se vyskytují již v raných stadiích onemocnění (CARD11, SOCS1, STAT6, PTEN, EPHA7), přičemž frekvence některých vzrůstá s jeho progresí (CARD11, PTEN, MYB). Jiné jsou spojovány spíše s histologickou transformací FL (MYD88, TNFAIP3) (tab. 1) [14,15,17,20].
Jednou z klíčových drah v patogenezi FL je NF-κB signalizace, což je zřejmé i z vysoké frekvence aberací jejích regulátorů. Aktivita transkripčního faktoru NF-κB je u B lymfocytů GC přirozená v populaci centrocytů, kde souvisí především s vazbou antigenu a BCR, resp. CD40/CD40L signalizací. Zvýšení aktivity NF-κB u FL vede k posílení schopnosti proliferace a k inhibici apoptózy (schematicky zobrazeno na obr. 2). NF-κB je transkripční faktor schopný odpovědět na mnoho signálů a v závislosti na nich ovlivnit expresi tisíců genů. Aktivita této molekuly je kromě BCR úzce propojena také s PI3K/AKT dráhou. V souvislosti s aktivací NF-κB přes BCR jsou prominentní zejména deregulace genů CARD11, CD79A/B a změny v expresi miRNA miR-155 (obr. 2) [14,17,21]. CARD11 kóduje membránovou kinázu s důležitou pozicí v reakční kaskádě vedoucí k NF-κB aktivaci (obr. 2). CD79A/B je intracelulární částí BCR receptoru a její fosforylace je prvním krokem v reakční kaskádě amplifikace BCR signalizace.

Důležitou roli v NF-κB aktivaci hraje kromě BCR také signalizace z jiných povrchových receptorů, jako jsou TLR nebo CD40 (obr. 2). V buňkách FL dochází k aktivaci NF-κB vlivem zvýšené exprese těchto povrchových molekul, vyšších hladin jejich ligandů v mikroprostředí a aberací postihující komponenty signálních kaskád spouštěných těmito receptory. Signalizace z TLR nebo CD40 aktivuje NF-κB přes proteiny rodiny TRAF (obr. 2) a je transduktorem signálu z Tfh lymfocytů (pomocné folikulární T lymfocyty), které napomáhají přežívání buněk FL. Zvýšená aktivita této signální dráhy je spojována s transformací FL a může být způsobena také inaktivací TNFAIP3 (~15 % případů) a mutacemi stabilizujícími MYD88 (10 % případů) (graf 1, obr. 2) [17]. NF-κB signalizace je navíc u jisté frakce FL zřejmě posílená v důsledku aberantní signalizace přes receptory NOTCH (tab. 1) [22]. Signalizace přes TRAF, resp. NOTCH zvyšuje také aktivitu dalších důležitých molekul jako MAPK a transkripčních faktorů z rodiny STAT. STAT signalizace je u FL také posílena aktivujícími mutacemi STAT6 (12 % případů), inaktivujícími mutacemi SOCS1 (8 % případů) a zvýšenými hladinami ligandů (jako IL-4 a IL-6) (tab. 1, graf 1, obr. 2) [13,14,23–25]. Ke zvýšené aktivitě těchto drah také zřejmě napomáhá nadměrná aktivita ephrinového receptoru 2 (EPHA2) způsobená absencí jeho negativního regulátoru EPHA7 (tab. 1, graf 1, obr. 2) [14,20]. Navíc je známo, že transkripční faktory kooperují a jejich interakce mohou iniciovat expresi či zvyšovat aktivitu svých vlastních aktivátorů, což dále posiluje deregulaci spuštěnou genetickými aberacemi.
Zvýšená BCR/PI3K/AKT signalizace a NF-κB aktivita způsobují deregulaci exprese mnoha genů vedoucí ke zvýšení odolnosti buněk FL vůči apoptóze a k jejich proliferaci. Inhibice BCR signalizace (a sekundárně NF-κB, STAT a MYC) pomocí nízkomolekulárních léčiv, jako je idelalisib (cílí PI3Kδ) či ibrutinib (cílí BTK) představuje rozvíjející se oblast cílené léčby B buněčných malignit [26,27]. V současné době probíhá množství studií, které testují tyto inhibitory samostatně či častěji v kombinaci s dalšími léčivy u FL.
Aktivita AID a mutace BCL6
Po migraci z kostní dřeně do sekundárních lymfatických orgánů recirkulují normální B lymfocyty mezi zónami GC, což umožňuje jejich selekci a afinitní maturaci. Chování recirkulujících buněk je řízeno cyklickou změnou exprese mnoha genů, přičemž jedněmi z klíčových jsou geny kódující aktivací indukovanou cytidin deaminázu (AID) a transkripční represor BCL6. AID je zodpovědná za vnášení somatických hypermutací do variabilních řetězců Ig, ale v případě FL indukuje mutace také v jiných genech, čímž přispívá k vývoji onemocnění a transformaci FL do DLBCL. Jedním z nejčastějších cílů těchto aberantních somatických mutací je BCL6, u kterého se somatické mutace 5‘nekódujícího regionu vyskytují u ~10 % případů FL a u > 70 % případů DLBCL [14]. Tyto aberace patrně brání autoregulaci BCL6, čímž umožňují jeho zvýšenou expresi, která je normálně v GC typická jenom pro masivně proliferující centroblasty [28]. Zvýšený výskyt translokací BCL6 u transformovaného FL (tFL), častější mutace způsobené aktivitou AID a identifikace zvýšených hladin AID jako prediktoru histologické transformace FL naznačují podstatnou úlohu těchto dvou molekul v evoluci onemocnění [15,16,29].
Recirkulace v GC a zejména mutační aktivita AID je mechanizmus, kterým mohou buňky FL získat aberace umožňující jim přežít a proliferovat. Pro buňky FL je tedy adheze v GC výhodná a aberace zajišťující, aby k ní docházelo, jsou důležitou součástí patogeneze FL. Významná v tomto směru je zvýšená aktivita BCL6 a mutace histonmetyltransferázy EZH2 (viz níže) (tab. 1) [30]. B lymfocyty FL tak mohou zůstat v GC, kde podstupují další a další cykly proliferace a somatických hypermutací působením AID.
Aktivita MYC
Exprese genu pro transkripční faktor MYC má zásadní vliv na intracelulární procesy u B lymfocytů. Od jeho objevení před 30 lety bylo publikováno mnoho prací popisujících jeho úlohu při kontrole exprese > 10 % lidských genů. V GC je MYC za normálních okolností aktivní pouze v subpopulaci B lymfocytů, které jej opouštějí a nacházejí se na hranici mezi světlou a vnější zónou [31]. Jeho prvořadým úkolem je zde zřejmě represe negativního regulátoru buněčného cyklu p27, a tím posílení proliferace této populace centrocytů (podobně jako BCL6 umožňuje proliferaci centroblastů, viz výše) [32].
Přirozená aktivace MYC v důsledku BCR signalizace má nezastupitelnou roli pro správný vývoj centrocytů. Zvýšené hladiny MYC hrají ale také důležitou roli v biologii FL tím, že ovlivňují expresi genů zahrnutých v regulaci buněčného cyklu. Mimo jiné byla v důsledku zvýšené aktivity MYC pozorována snížená aktivita některých miRNA, vč. miR-150 a miR-34a, které jsou negativními regulátory pro-proliferativních proteinů FOXP1 a GAB1 [33,34]. Naopak MYC přímo aktivuje transkripci klastru miR-17-92, který represí specifické skupiny genů významně přispívá k MYC navozeným onkogenním vlastnostem B lymfocytů [35,36]. Genomické aberace MYC jsou však u FL poměrně vzácné, a jeho aktivita je proto ve většině případů důsledkem inaktivujících mutací jeho negativních regulátorů (ARID1A, SMARCA4, SMAD6) či zvýšené aktivity pozitivních regulátorů (aktivace BCR, RAS a MAPK) (graf 1, obr. 2) [14,15]. Případy FL s genomickými aberacemi MYC a BCL2 jsou označovány jako FL se dvěma zásahy a jsou spojeny zpravidla s velice krátkou dobou přežití pacientů [37]. Navzdory onkogenním vlastnostem MYC není ale samotné zvýšení jeho exprese pro rozvoj lymfomu dostačující. Vysoká hladina MYC totiž vede buňky k senescenci, která musí být překonána dalšími aberacemi molekulárních drah (např. mutacemi TP53 nebo aberantní aktivitou klastru miR-17-92). Stojí za povšimnutí, že 30–50 % případů transformace FL do DLBCL je spojeno s translokací, mutací nebo amplifikací genu MYC [16].
Mutace histonmetyltransferáz
Rozhodnutí, zda bude gen exprimován, podléhá vysoce sofistikované síti regulačních mechanizmů, které řídí epigenetické modifikace chromatinu. Není tedy až tak překvapivé, že mutace genů kódujících tyto regulátory mají význam i v procesech vzniku a progrese FL. Histonmetyltransferázy (HMT) jsou epigenetické regulátory, které katalyzují přenos jedné nebo více metylových skupin na argininová nebo lyzinová rezidua histonových proteinů. U FL dochází především k deregulaci dvou HMT – MLL2 a EZH2 [14]. Oba tyto proteiny metylují lyzinová rezidua, avšak v rozličných pozicích a s rozličným výsledným efektem. Zatímco metylace H3K4 zprostředkovaná MLL2 proteinem vede k aktivaci transkripce, metylace H3K27 řízená EZH2 vede k její represi. Inaktivující aberace MLL2 se v buňkách FL vyskytují s téměř stejnou frekvencí jako translokace BCL2 (tab. 1). Funkce MLL2 v B lymfocytech však zůstává do dnešních dnů poměrně málo známá a není plně popsán ani efekt jeho deregulace na buňky FL. Údaje získané delecí myšího homologu k MLL2 naznačují, že absence MLL2 přispívá k lymfomagenezi především indukcí aktivity NF-κB a zvyšováním exprese AID a mnoha genů hrajících roli v regulaci buněčného cyklu a v imunitní odpovědi [38]. Naopak fyziologická funkce i dopad mutací EZH2 jsou popsány poměrně dobře. Mutace EZH2 kódujícího protein, který je součástí polycomb represorového komplexu 2 (PRC2), se vyskytují zhruba u 25 % případů FL (graf 1, tab. 1) [14,15]. Většina mutací se nachází v lokusech, které ovlivňují substrátovou specifitu tohoto proteinu, a zvyšují tak jeho schopnost trimetylace H3K27. Bylo prokázáno, že zvýšená exprese mutovaného EZH2 vede k posílení reakce GC a akumulaci hyperproliferativních B lymfocytů GC [39], což je zřejmě důsledkem represe exprese genů podílejících se na diferenciaci B lymfocytů, jako jsou IRF4, resp. PRDM1 [16,40]. Neméně důležitou funkcí EZH2 je schopnost stimulovat angiogenezi, přičemž induktor angiogeneze VEGF je zase schopen přispívat k zvýšené aktivitě EZH2 [41].
Díky pleiotropním funkcím EZH2 se tato metyltransferáza stala potenciálním terčem cílené terapie. V nedávné době bylo vyvinuto několik selektivních inhibitorů EZH2, které přinášející slibné výsledky už i v klinických testech [42,43].
Mutace histonacetyltransferáz a histonových proteinů
Histonacetyltransferázy (HAT) mají zásadní roli v regulaci genové exprese, neboť neutralizací lyzinových reziduí dekondenzují chromatin, čímž umožňují přístup transkripčních komplexů k promotorům genů. U B lymfocytů FL jsou často již v době diagnózy detekovány mutace (55–85 % případů) způsobující ztrátu funkce dvou HAT – CBP nebo p300 (graf 1, tab. 1) [14,44]. Mutace obou jsou ve většině případů heterozygotní, což naznačuje haploinsuficienci ve vztahu k acetylaci jejich substrátů, a jen zcela vzácně jsou přítomny ve stejných buňkách, tj. vzájemně se vylučují. Předpokládá se, že k tomu dochází kvůli jejich rovnocennému vlivu na patogenezi, bez rozdílu ve ztrátě funkce jednoho nebo obou genů, nebo je zachování funkce u alespoň jednoho z nich nezbytnou podmínkou pro přežití B lymfocytů.
Mutace vedoucí ke ztrátě funkce u CBP vedou překvapivě také ke snížení exprese MHCII molekul, díky snížené schopnosti CBP asociovat s CIITA, který je hlavním koaktivátorem MHCII [45,46]. Snížení exprese MHCII vede k menší T lymfocytární infiltraci, čímž mutace CREBBP přispívají k úniku před imunitním systémem [47]. Bylo také prokázáno, že mutace CREBBP ovlivňují acetylaci tumor-supresoru p53 a proteinu BCL6 [44]. U p53 je acetylace nezbytná pro zajištění jeho transkripční aktivity a u BCL6 vede acetylace k inaktivaci jeho funkce transkripčního represoru. Inaktivace CREBBP tak vytváří u B lymfocytů FL charakteristiky podobné centroblastům, tj. nízká exprese MHCII, zvýšená aktivita BCL6 a nízká aktivita p53.
CBP a p300 acetylují H3K27, ale tato rezidua jsou také ovlivněna inhibiční metylací způsobenou PRC2. Je známa antagonistická aktivita těchto dvou proteinových skupin a nutnost zabránit acetylaci H3K27, aby byla umožněna PRC2 zprostředkovaná represe transkripce [48]. V této souvislosti je zajímavé, že jak HAT, tak HDAC využívají stejné vazebné místo na proteinech rodiny MEF2 [49]. MEF2 rodina je tvořena čtyřmi transkripčními faktory, u kterých se předpokládalo, že se všechny váží jak k HAT, tak i k HDAC [49]. Výsledky současných studií ale ukazují, že jeden člen této rodiny – MEF2B – je schopen vazby pouze s HDAC. Aktivující mutace tohoto genu se vyskytují u FL ve zhruba 15 % případů a přispívají ke snížené acetylaci H3K27, čímž umožňují inhibiční metylaci histonů zprostředkovanou např. EZH2 (graf 1) [50,51].
Pro epigenetickou regulaci transkripce genů jsou nepostradatelné i další jaderné proteiny ovlivňující strukturu chromatinu. Rekurentní mutace v genech pro komponenty na ATP závislých chromatin remodelujících komplexů (ARID1A a SMARCA4) a mutace postihující C konec histonu H1 patří mezi ty nejčastější (tab. 1) [14,15]. Histon H1 představuje proteinovou rodinu, jejíž členové stabilizují chromatinové vlákno vazbou na nukleozom a zároveň na DNA spojující nukleozomy. Mutace jsou ve většině případů lokalizovány v 5’konci a vedou k snížené interakci proteinu s DNA-metyltranferázami DNMT1 a DNMT3B, což ovlivňuje expresi mnoha genů [52]. S četností aberací téměř 30 % patří mutace genů pro HIST1H1 mezi nejčastější aberace u FL (tab. 1) [14]. Do dnešních dní zůstává velké množství nezodpovězených otázek týkajících se fyziologických funkcí chromatinových modifikátorů, a je tedy těžké jich využít v cílené léčbě FL. Nicméně deaktivace HDAC pomocí jejich syntetických inhibitorů, jako je abexinostat nebo vorinostat, představuje potenciální terapeutický přístup, který by mohl být úspěšný alespoň pro jistou skupinu pacientů s FL [53,54].
„Únik“ před imunitním systémem
Pro nádorové buňky je obecně typická snaha o uniknutí kontrole ze strany imunitního systému. Jednou z možností je absence charakteristických povrchových molekul na nádorových buňkách schopných zprostředkovat vazbu s efektorovými imunitními buňkami. U FL se vyskytují mutace vedoucí ke ztrátě funkce receptoru pro tumor nekrotizující faktor ze superrodiny 14 (TNFRSF14) (30–40 % případů FL). TNFRSF14 kóduje transmembránový protein, který zprostředkovává interakce s T lymfocyty a jeho stimulace posiluje aktivaci apoptózy přes FAS a protinádorovou imunitu. Tyto mutace se opakovaně vyskytují již v buňkách tzv. společné progenitorové populace, jež dává vznik jednotlivým maligním klonům B lymfocytů vytvářejících tumor. V případech, kdy FL vzniká ze společné progenitorové populace, která sdílí jenom minimum aberací, dochází k selekci těchto mutací nezávisle v jednotlivých klonech. Takový konvergentní způsob evoluce jednotlivých klonů FL implikuje zásadní význam těchto aberací v relativně raných stadiích jeho patogeneze [14]. Kromě uvedených mutací se u buněk FL často vyskytuje delece druhé alely pro TNFRSF14 nebo uniparentální dizomie stejné oblasti, které vedou ke ztrátě tohoto proteinu [14,15]. Z hlediska inaktivace vnější apoptotické dráhy hrají asi v 15 % případů FL důležitou roli také delece dalšího člena rodiny TNF receptorů – FAS (tab. 1) [15].
V kontextu imunitní odpovědi je třeba zmínit aberace TP53 a jeho regulátorů. Molekula p53 sice není povrchovou strukturou, je ale transkripčním aktivátorem mnoha pro-apoptotických proteinů vč. povrchových molekul z rodiny TNF. Inaktivace p53 je často spojována s histologickou transformací FL. U tFL jsou genetické aberace TP53 přítomny u téměř 20 % případů, což je asi 2× častěji než u FL [15,44]. Avšak ještě častější než aberace postihující gen pro p53 jsou delece jeho pozitivního regulátoru CDKN2A/B (kóduje p14-ARF, p16-INK4A a p15-INK4B), které jsou přítomny téměř u poloviny tFL, a amplifikace jeho negativních regulátorů MDM2 a MDM4 v třetině případů tFL (tab. 1, obr. 2) [16].
Jedním z nejčastěji popisovaných způsobů pro únik imunitnímu systému u solidních tumorů vůbec je exprese povrchové molekuly PD-L1, která přes vazbu na PD-1 na T lymfocytech tlumí jejich cytotoxické funkce. Buňky folikulárního lymfomu typicky neexprimují ligandy PD-1, ale FL může obsahovat PD-L1+ histiocyty (v T bohatých oblastech folikulů) a přispívat tak k „vyčerpání“ a utlumení funkce PD1+ T lymfocytů infiltrujících FL. Lze tedy předpokládat, že inhibice PD-1/PD-L1 signalizace by mohla být využita terapeuticky, což bylo částečně potvrzeno v prvních klinických studiích [55].
Jinou „nádorovou strategií“ jsou aberace vedoucí ke ztrátě funkce beta-2-mikroglobulinu (B2M) a CD58 (> 5 % případů) [14]. Simultánní ztráta funkce těchto molekul způsobuje, že buňky nejsou rozpoznávány cytotoxickými lymfocyty a ani NK buňkami. Několikanásobně častější výskyt delecí B2M u tFL naznačuje význam tohoto mechanizmu v procesu transformace [16]. V raných fázích vývoje FL může podobnou roli zastávat výše popsaná inaktivace histonacetyltransferázy CBP, která snižuje expresi genů nutných pro aktivaci T lymfocytů [44].
Aberace umožňující toleranci genetických změn v B lymfocytech FL, ať už v důsledku zabezpečení jejich „neviditelnosti“ buňkami imunitního systému, nebo poruchou vnitřních kontrolních mechanizmů, významně přispívají k jejich přežívání. Frekvence těchto aberací navíc často stoupá s progresí FL. Navození citlivosti buněk FL k imunitnímu systému pomocí léčiv jako je lenalidomid [56] (obnovuje synapse mezi T lymfocyty a B lymfocyty FL) nebo inhibitory PD1-/PD-L1 signalizace [55] představuje proto slibný terapeutický přístup.
MikroRNA
Během posledních dvou dekád byla popsána nová důležitá skupina post-transkripčních regulátorů genové exprese, tzv. miRNA. Doposud bylo po-psáno > 2 500 lidských miRNA zapojených do regulace většiny buněčných drah, vč. těch spojených s patogenezí B buněčných malignit [57]. MiRNA hrají důležitou roli v precizně regulovaném procesu hematopoézy a jejich zásadní význam pro patogenezi B buněčných malignit byl poprvé prokázán u chronické lymfatické leukemie (CLL), kde delece části chromozomu 13 vede specificky ke ztrátě miR-15a a miR-16 [58]. Opakovaně se vyskytující deregulace některých miRNA v buňkách FL, stejně jako jejich popsaný vliv na přežívání pacientů s DLBCL či CLL, naznačují jejich důležitost v biologii FL. Ukazuje se, že miRNA mají zřejmě podstatnou roli také v histologické transformaci FL. V nedávno publikové práci Thompson et al byly identifikované některé miRNA jako prediktory histologické transformace FL [18].
Opakovaně je u lymfomů popisována zvýšená aktivita klastru miR-17-92, který snižuje expresi genů BIM a PTEN, a tím posiluje aktivitu BCR/PI3K/AKT signalizace a brání apoptóze (graf 1) [15,59]. Vyšší hladiny těchto miRNA u tFL, stejně jako výrazně vyšší frekvence jejich amplifikací, naznačují jejich důležitost v transformačním procesu. Ke zvýšené aktivitě PI3K/AKT signalizace přispívá i deregulace miR-155, miR-150 a miR-21 (viz výše a obr. 2) [21,34,57,60]. Především vysoké hladiny miR-155 umožňují silnější aktivaci BCR snížením hladin PIK3R1 a SHIP1 [21,61] a snižují také expresi SMAD5 a HGAL, čímž zvyšují odolnost buněk vůči apoptóze a jejich motilitu [21,62]. Vysoká exprese miR-155 byla popsána u mnoha B buněčných malignit a u transgenních myší vyvolává vznik lymfomů [63]. Důležitou vlastností miR-155 je, že také reguluje proces, při kterém B lymfocyty opouštějí GC, tím, že reprimuje expresi genu pro PU.1. Takto miR-155 na jedné straně zvyšuje BCR signalizaci a na druhé brání diferenciaci B lymfocytů (která je silnou BCR signalizací přirozeně indukována).
MiRNA jsou také potenciálním terčem cílené terapie s využitím syntetických anti-miR či miR-mimic. Terapeutickými kandidáty jsou především onkogenní miRNA jako miR-155 či miR-21, jejichž inhibice vedla k regresi lymfomů v myších modelech. Cheng et al dosáhli chemickou modifikací miRNA dobrého průniku do tkání a úspěšně inhibovali funkce miR-155 in vivo v myším modelu, což významně zpomalilo růst lymfomů [64]. V současné době jsou připravovány klinické studie s inhibitory miR-155 u pacientů s B buněčnými malignitami.
Závěr
FL vzniká z B lymfocytů GC, které již během svého vývoje v kostní dřeni získaly výhodu v přežívání díky translokaci a konstitutivní expresi BCL2. Protein BCL2 sice brání indukci apoptózy, na druhou stranu se ale vyznačuje anti-proliferačními účinky a nedokáže chránit buňky před mechanizmy buněčné smrti zprostředkované efektorovými buňkami imunitního systému. Pro vznik FL je proto třeba více aberací umožňujících lymfocytům nekontrolovanou proliferaci, únik před imunitním systémem a získání nezbytných signálů od buněk mikroprostředí. Ve většině případů je toho dosáhnuto deregulací genů, jejichž exprese řídí i osud normálních B lymfocytů GC. Nezastupitelnou roli v patogenezi tohoto onemocnění hraje deregulace epigenetických modifikátorů vedoucí k ovlivnění exprese celých skupin genů a vyskytující se téměř ve všech případech FL. Komplexnost biologie FL je umocněna důležitostí aberací postihujících mikroprostředí jak z hlediska jeho složení, tak i z hlediska aktivity jednotlivých komponent. Navzdory lepšímu pochopení patogeneze zůstává FL nevyléčitelným onemocněním, ale lze očekávat, že získané poznatky o rekurentních mutacích u FL povedou v budoucnu k lepší stratifikaci pacientů do prognostických skupin a možná i k volbě terapie na základě specifických aberací. V současnosti se již pro stanovení prognózy využívá mutační status sedmi genů v tzv. M7-FLIPI skóre. Lze také očekávat, že tato schémata budou v budoucnu obsahovat také stanovení exprese některých miRNA, které se zdají být velmi spolehlivými biomarkery. Snaha o vývoj cílené léčby dospěla k zavedení inhibitoru PI3Kδ idelalisibu do léčby relabujících pacientů. Do praxe by v nejbližších letech mohly být také zavedeny inhibitory BTK jako ibrutinib, PI3Kδ/γ inhibitor duvelisib či inhibitory histonmetyltransferázy EZH2, které přinášejí slibné výsledky v klinických testech. Je důležité, že vzrůstající poznání biologie FL nyní vede k testování nových potenciálních cílených léčiv, která mohou být velmi účinná u skupin pacientů s deregulací specifické molekulární dráhy.
Výsledky tohoto výzkumu byly získány v rámci projektu CEITEC 2020 (LQ1601) za finančního přispění Ministerstva školství, mládeže a tělovýchovy České republiky v rámci účelové podpory z prostředků Národního programu udržitelnosti II.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
doc. MUDr. Mgr. Marek Mráz, Ph.D.
CEITEC – Středoevropský technologický institut, MU
Kamenice 5
625 00 Brno
e-mail: marek.mraz@email.cz
Obdrženo: 28. 1. 2017
Přijato: 5. 3. 2017
Zdroje
1. Smith A, Crouch S, Lax S et al. Lymphoma incidence, survival and prevalence 2004–2014: sub-type analyses from the UK’s Haematological Malignancy Research Network. Br J Cancer 2015; 112 (9): 1575–1584. doi: 10.1038/bjc.2015.94.
2. Tsujimoto Y, Cossman J, Jaffe E et al. Involvement of the bcl-2 gene in human follicular lymphoma. Science 1985; 228 (4706): 1440–1443.
3. Tsujimoto Y, Gorham J, Cossman J et al. The t (14; 18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science 1985; 229 (4720): 1390–1393.
4. Casulo C, Byrtek M, Dawson KL et al. Early Relapse of Follicular Lymphoma After Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone De-fines Patients at High Risk for Death: An Analysis From the National LymphoCare Study. J Clin Oncol 2015; 33 (23): 2516–2522. doi: 10.1200/JCO.2014.59.7534.
5. Pastore A, Jurinovic V, Kridel R et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol 2015; 16 (9): 1111–1122. doi: 10.1016/S1470-2045 (15) 00169-2.
6. Jurinovic V, Kridel R, Staiger AM et al. Clinicogenetic risk models predict early progression of follicular lymphoma after first-line immunochemotherapy. Blood 2016; 128 (8): 1112–1120. doi: 10.1182/blood-2016-05-717355.
7. Liu Y, Hernandez AM, Shibata D et al. BCL2 translocation frequency rises with age in humans. Proc Natl Acad Sci U S A 1994; 91 (19): 8910–8914.
8. McDonnell TJ, Korsmeyer SJ. Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t (14; 18). Nature 1991; 349 (6306): 254–256.
9. Roulland S, Kelly RS, Morgado E et al. t (14; 18) Translocation: a predictive blood biomarker for follicular lymphoma. J Clin Oncol 2014; 32 (13): 1347–1355. doi: 10.1200/JCO.2013.52.8190.
10. Cong P, Raffeld M, Teruya-Feldstein J et al. In situ localization of follicular lymphoma: description and analysis by laser capture microdissection. Blood 2002; 99 (9): 3376–3382.
11. Janíková A, Michalka J, Tichý B et al. Folikulární lymfom a význam nádorového mikroprostředí. Transfuze Hematol Dnes 2010; 16 (3): 150–157.
12. Mraz M, Zent CS, Church AK et al. Bone marrow stromal cells protect lymphoma B-cells from rituximab-induced apoptosis and targeting integrin α-4-β-1 (VLA-4) with natalizumab can overcome this resistance. Br J Haematol 2011; 155 (1): 53–64. doi: 10.1111/j.13652141.2011.08794.x.
13. Labidi SI, Ménétrier-Caux C, Chabaud S et al. Serum cytokines in follicular lymphoma. Correlation of TGF-β and VEGF with survival. Ann Hematol 2010; 89 (1): 25–33. doi: 10.1007/s00277-009-0777-8.
14. Okosun J, Bödör C, Wang J et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet 2014; 46 (2): 176–181. doi: 10.1038/ng.2856.
15. Bouska A, McKeithan TW, Deffenbacher KE et al. Genome-wide copy-number analyses reveal genomic abnormalities involved in transformation of follicular lymphoma. Blood 2014; 123 (11): 1681–1690. doi: 10.1182/blood-2013-05-500595.
16. Pasqualucci L, Khiabanian H, Fangazio M et al. Genetics of follicular lymphoma transformation. Cell Rep 2014; 6 (1): 130–140. doi: 10.1016/j.celrep.2013.12.027.
17. Okosun J, Wolfson RL, Wang J et al. Recurrent mTORC1-activating RRAGC mutations in follicular lymphoma. Nat Genet 2016; 48 (2): 183–188. doi: 10.1038/ng.3473.
18. Thompson MA, Edmonds MD, Liang S et al. miR-31 and miR-17-5p levels change during transformation of follicular lymphoma. Hum Pathol 2016; 50 : 118–126. doi: 10.1016/j.humpath.2015.11.011.
19. Dave SS, Wright G, Tan B et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N Engl J Med 2004; 351 (21): 2159–2169.
20. Oricchio E, Nanjangud G, Wolfe AL et al. The Eph-receptor A7 is a soluble tumor suppressor for follicular lymphoma. Cell 2011; 147 (3): 554–564. doi: 10.1016/j.cell. 2011.09.035.
21. Huang X, Shen Y, Liu M et al. Quantitative proteomics reveals that miR-155 regulates the PI3K-AKT pathway in diffuse large B-cell lymphoma. Am J Pathol 2012; 181 (1): 26–33. doi: 10.1016/j.ajpath.2012.03.013.
22. Karube K, Martínez D, Royo C et al. Recurrent mutations of NOTCH genes in follicular lymphoma identify a distinctive subset of tumours. J Pathol 2014; 234 (3): 423–430. doi: 10.1002/path.4428.
23. Calvo KR, Dabir B, Kovach A et al. IL-4 protein expression and basal activation of Erk in vivo in follicular lymphoma. Blood 2008; 112 (9): 3818–3826. doi: 10.1182/blood-2008-02-138933.
24. Yildiz M, Li H, Bernard D et al. Activating STAT6 mutations in follicular lymphoma. Blood 2015; 125 (4): 668–679. doi: 10.1182/blood-2014-06-582650.
25. Mottok A, Renné C, Seifert M et al. Inactivating SOCS1 mutations are caused by aberrant somatic hypermutation and restricted to a subset of B-cell lymphoma entities. Blood 2009; 114 (20): 4503–4506. doi: 10.1182/blood-2009-06-225839.
26. Maffei R, Fiorcari S, Martinelli S et al. Targeting neoplastic B cells and harnessing microenvironment: the “double face” of ibrutinib and idelalisib. J Hematol Oncol 2015; 8 : 60. doi: 10.1186/s13045-015-0157-x.
27. Mráz M, Doubek M, Mayer J. Inhibition of B Cell Receptor Signaling: a First Targeted Therapeutic Approach for Chronic Lymphocytic Leukemia and Other B Cell Lymphomas. Klin Onkol 2013; 26 (3): 179–185.
28. Pasqualucci L, Migliazza A, Basso K et al. Mutations of the BCL6 proto-oncogene disrupt its negative autoregulation in diffuse large B-cell lymphoma. Blood 2003; 101 (8): 2914–2923.
29. Correia C, Schneider PA, Dai H et al. BCL2 mutations are associated with increased risk of transformation and shortened survival in follicular lymphoma. Blood 2015; 125 (4): 658–667. doi: 10.1182/blood-2014-04-571786.
30. Béguelin W, Popovic R, Teater M et al. EZH2 is re-quired for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 2013; 23 (5): 677–692. doi: 10.1016/j.ccr.2013.04.011.
31. Klein U, Tu Y, Stolovitzky GA et al. Transcriptional analysis of the B cell germinal center reaction. Proc Natl Acad Sci U S A 2003; 100 (5): 2639–2644.
32. Pérez-Roger I, Solomon DL, Sewing A et al. Myc activation of cyclin E/Cdk2 kinase involves induction of cyclin E gene transcription and inhibition of p27 (Kip1) binding to newly formed complexes. Oncogene 1997; 14 (20): 2373–2381.
33. Rao DS, O’Connell RM, Chaudhuri AA et al. MicroRNA-34a perturbs B lymphocyte development by repressing the forkhead box transcription factor Foxp1. Immunity 2010; 33 (1): 48–59. doi: 10.1016/j.immuni.2010.06.013.
34. Mraz M, Chen L, Rassenti LZ et al. miR-150 influences B-cell receptor signaling in chronic lymphocytic leukemia by regulating expression of GAB1 and FOXP1. Blood 2014; 124 (1): 84–95. doi: 10.1182/blood-2013-09-527234.
35. O’Donnell KA, Wentzel EA, Zeller KI et al. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005; 435 (7043): 839–843.
36. Li Y, Choi PS, Casey SC et al. MYC through miR-17-92 Suppresses Specific Target Genes to Maintain Survival, Autonomous Proliferation and a Neoplastic State. Cancer Cell 2014; 26 (2): 262–272. doi: 10.1016/j.ccr.2014.06.014.
37. Šmardová J, Moulis M, Lišková K et al. Double-hit lymphomas – review of the literature and case report. Klin Onkol 2014; 27 (1): 24–32. doi: 10.14735/amko201424.
38. Maniati E, Marzec J, Okosun J et al. Investigating the role of MLL2 (Mll4) in B cell development. Blood 2013; 122 (21): 343–343.
39. Berg T, Thoene S, Yap D et al. A transgenic mouse model demonstrating the oncogenic role of mutations in the polycomb-group gene EZH2 in lymphomagenesis. Blood 2014; 123 (25): 3914–3924. doi: 10.1182/blood-2012-12-473439.
40. Guo S, Chan JK, Iqbal J et al. EZH2 mutations in follicular lymphoma from different ethnic groups and associated gene expression alterations. Clin Cancer Res 2014; 20 (12): 3078–3086. doi: 10.1158/1078-0432.CCR-13-1597.
41. Lu C, Han HD, Mangala LS et al. Regulation of tumor angiogenesis by EZH2. Cancer Cell 2010; 18 (2): 185–197. doi: 10.1016/j.ccr.2010.06.016.
42. Knutson SK, Wigle TJ, Warholic NM et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol 2012; 8 (11): 890–896. doi: 10.1038/nchembio.1084.
43. McCabe MT, Ott HM, Ganji G et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012; 492 (7427): 108–112. doi: 10.1038/nature11606.
44. Pasqualucci L, Dominguez-Sola D, Chiarenza A et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011; 471 (7337): 189–195. doi: 10.1038/nature09730.
45. Fontes JD, Kanazawa S, Jean D et al. Interactions between the class II transactivator and CREB binding protein increase transcription of major histocompatibility complex class II genes. Mol Cell Biol 1999; 19 (1): 941–947.
46. Kretsovali A, Agalioti T, Spilianakis C et al. Involvement of CREB binding protein in expression of major histocompatibility complex class II genes via interaction with the class II transactivator. Mol Cell Biol 1998; 18 (11): 6777–6783.
47. Green MR, Kihira S, Liu CL et al. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc Natl Acad Sci U S A 2015; 112 (10): E1116–E1125. doi: 10.1073/pnas.1501199112.
48. Pasini D, Malatesta M, Jung HR et al. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res 2010; 38 (15): 4958–4969. doi: 10.1093/nar/gkq 244.
49. Han A, He J, Wu Y et al. Mechanism of recruitment of class II histone deacetylases by myocyte enhancer factor-2. J Mol Biol 2005; 345 (1): 91–102.
50. Morin RD, Mendez-Lago M, Mungall AJ et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011; 476 (7360): 298–303. doi: 10.1038/nature10351.
51. Ying CY, Dominguez-Sola D, Fabi M et al. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat Immunol 2013; 14 (10): 1084–1092. doi: 10.1038/ni.2688.
52. Li H, Kaminski MS, Li Y et al. Mutations in linker histone genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A underlying the pathogenesis of follicular lymphoma. Blood 2014; 123 (10): 1487–1498. doi: 10.1182/blood-2013-05-500264.
53. Morschhauser F, Terriou L, Coiffier B et al. Phase 1 study of the oral histone deacetylase inhibitor abexinostat in patients with Hodgkin lymphoma, non-Hodgkin lymphoma, or chronic lymphocytic leukaemia. Invest New Drugs 2015; 33 (2): 423–431. doi: 10.1007/s10637-015-0206-x.
54. Chen R, Frankel P, Popplewell L et al. A phase II study of vorinostat and rituximab for treatment of newly diagnosed and relapsed/refractory indolent non-Hodgkin lymphoma. Haematologica 2015; 100 (3): 357–362. doi: 10.3324/haematol.2014.117473.
55. Westin JR, Chu F, Zhang M et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: a single group, open-label, phase 2 trial. Lancet Oncol 2014; 15 (1): 69–77. doi: 10.1016/S1470-2045 (13) 70551-5.
56. Leonard JP, Jung SH, Johnson J et al. Randomized Trial of Lenalidomide Alone Versus Lenalidomide Plus Rituximab in Patients With Recurrent Follicular Lymphoma: CALGB 50401 (Alliance). J Clin Oncol 2015; 33 (31): 3635–3640. doi: 10.1200/JCO.2014.59.9258.
57. Musilova K, Mraz M. MicroRNAs in B-cell lymphomas: how a complex biology gets more complex. Leukemia 2015; 29 (5): 1004–1017. doi: 10.1038/leu.2014.351.
58. Calin GA, Dumitru CD, Shimizu M et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 2002; 99 (24): 15524–15529.
59. Ventura A, Young AG, Winslow MM et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008; 132 (5): 875–886. doi: 10.1016/j.cell.2008.02.019.
60. Medina PP, Nolde M, Slack FJ. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature 2010; 467 (7311): 86–90. doi: 10.1038/nature09284.
61. Thompson RC, Vardinogiannis I, Gilmore TD. Identification of an NF-κB p50/p65-responsive site in the human MIR155HG promoter. BMC Mol Biol 2013; 14 : 24. doi: 10.1186/1471-2199-14-24.
62. Rai D, Kim SW, McKeller MR et al. Targeting of SMAD5 links microRNA-155 to the TGF-beta pathway and lymphomagenesis. Proc Natl Acad Sci U S A 2010; 107 (7): 3111–3116. doi: 10.1073/pnas.0910667107.
63. Costinean S, Zanesi N, Pekarsky Y et al. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E (mu) -miR155 transgenic mice. Proc Natl Acad Sci U S A 2006; 103 (18): 7024–7029.
64. Cheng CJ, Bahal R, Babar IA et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature 2015; 518 (7537): 107–110. doi: 10.1038/nature13905.
Štítky
Dětská onkologie Chirurgie všeobecná OnkologieČlánek vyšel v časopise
Klinická onkologie

2017 Číslo 4
- Profesionální expozice azbestu a riziko vzniku karcinomu plic
- Plazmatický fibrinogen jako možný prognostický marker u nemetastatického renálního karcinomu
- Aktuální možnosti léčby mnohočetného myelomu
- Proces hojení ran krok za krokem a co ho může zkomplikovat
- Účinnost ceftazidimu/avibaktamu v léčbě infekcí způsobených enterobakteriemi produkujícími karbapenemázy
Nejčtenější v tomto čísle
- Metastatické postižení hypofýzy
- Reaktivní lymfoidní hyperplazie jater
- Radioterapie nádorů s plicní lokalizací u idiopatické plicní fibrózy
- Radionekróza horní krční míchy po protonovém ozáření u nemocné po radikální resekci ependymomu IV. komory mozkové
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy