
Kombinovaná léčba recidivujícího maligního schwannomu
Multimodal Therapy of Recurrent Malignant Schwannoma
Background:
Malignant peripheral nerve sheath tumor schwannoma (MPNST), also known as malignant schwannoma, is a very rare tumor accounting for only 2% of all sarcomas. The prognosis is relatively poor, with a 5-year survival rate of 46–69%. The treatment of MPNST has not been standardized yet. Mainstay treatment is radical resection. Oncological adjuvant or neoadjuvant treatment has equivocal indications with unclear effects.
Case:
The case report presents a 55-year-old patient who showed resistance in the medial-ventral area of the left lower limb. An MRI scan showed a tumor adjacent to the femoral nerve. Tumor extirpation was performed. Histology revealed malignant schwannoma (MPNST) and the resection was assessed as R0. Postoperative whole-body PET/CT revealed no viable tumor tissue. The patient was regularly followed-up. On a follow-up MRI scan, performed 53 months after initial surgery, tumor recurrence was detected in the left thigh. Extirpation of the recurrent tumor was performed. Histology confirmed MPNST and the resection radicality was assessed as R2. Postoperative PET/CT revealed tumor residues. Therefore, 58 months after the initial surgery, another operation of the residual tumor was performed with R0 resection. Three applicators for interstitial brachytherapy were placed in the resection cavity. Following the operation, radiotherapy with an interstitial brachytherapy boost of 18 Gy followed by external fractionated radiotherapy of 50 Gy were administered. The latest MRI scan, performed 66 months after the diagnosis of MPNST, showed no tumor tissue. The patient had no neurological deficit.
Conclusion:
The mainstay of treatment for MPNST is radical en bloc resection. The use of subsequent oncological therapy depends on the radicality of the resection. In our case, because of the good radicality of the initial surgery, adjuvant oncological therapy was postponed. As part of recurrence management, we again attempted to achieve the most radical resection possible and then apply adjuvant radiotherapy. In MPNST, as in all soft tissue sarcomas, high doses are chosen because of potential radioresistance. Given the confined nature of the disease, we chose this locally intensified therapeutic strategy, which resulted in this case in disease remission. Due to the low incidence of MPNST, it is not possible to test the efficacies of individual oncologic therapeutic procedures in larger patient cohorts.
Key words:
malignant schwannoma – soft tissue sarcoma – multimodal therapy
The authors declare they have no potential confl icts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted:
13. 3. 2016
Accepted:
25. 4. 2016
Autoři:
O. Kalita 1; K. Cwiertka 2; D. Vrána 2; M. Vaverka 1; L. Tučková 3; M. Megová 4
Působiště autorů:
Neurochirurgická klinika LF UP a FN Olomouc
1; Onkologická klinika LF UP a FN Olomouc
2; Laboratoř molekulární patologie, Oddělení patologie, Ústav molekulární a translační medicíny, LF UP a FN Olomouc
3; Ústav molekulární a translační medicíny, LF UP v Olomouci
4
Vyšlo v časopise:
Klin Onkol 2016; 29(5): 364-368
Kategorie:
Kazuistiky
doi:
https://doi.org/10.14735/amko2016364
Souhrn
Východiska:
Maligní nádor pochvy periferního nervu (malignant peripheral nerve sheath tumour – MPNST), zvaný též maligní schwannom, je velmi vzácný nádor, který tvoří pouze 2 % všech sarkomů. Nádor má poměrně špatnou prognózu, kdy 5leté celkové přežití dosahuje 46–69 %. Léčba MPNST není nadále standardizována. Základem je radikální resekce. Onkologická adjuvantní či neoadjuvantní léčba má nejasné indikace s nejasným efektem.
Případ:
Naše kazuistika prezentuje 55letého pacienta vyšetřovaného pro rezistenci na medio-ventrální ploše levé dolní končetiny. MRI ukázala nádor levého stehna nasedajícího na n. femoralis. Byla provedena radikální exstirpace nádoru. Histologický nález odhalil maligní schwannom (MPNST) a resekce byla hodnocena jako stupeň R0. Pooperační celotělové PET/CT neodhalilo viabilní nádorovou tkáň. Nemocný byl dále dispenzarizován. Na kontrolním MRI, 53 měsíců od úvodní operace, se objevil nález recidivy nádoru. Provedena exstirpace recidivy nádoru na stehně levé dolní končetiny. Histologické vyšetření potvrdilo MPNST, radikalita resekce byla hodnocena jako R2. Pooperační PET/CT objevilo ve stěně resekční dutiny rezidua nádoru. Proto proběhla 58 měsíců od první operace další operace zbývajícího tumoru, a to do stupně R0. Do resekční dutiny byly umístěny tři aplikátory intersticiální brachyterapie. Po operaci proběhla radioterapie – intersticiální brachyterapie (18 Gy) a následně zevní frakcionované radioterapie (50 Gy). Poslední MRI 66 měsíců od diagnózy MPNST neukázala žádnou nádorovou tkáň. Pacient je nadále bez neurologického deficitu.
Závěr:
Základem terapie MPNST je radikální en bloc resekce. Použití následné onkologické léčby závisí právě na radikalitě resekce. V našem případě vzhledem k dobré radikalitě první operace byla adjuvantní onkologická terapie postponována. V rámci řešení recidiv jsme se opět snažili o dosažení co nejradikálnější resekce s následnou adjuvantní radioterapií. U MPNST jako u všech sarkomů měkkých tkání se z důvodu potenciální radiorezistence volí vysoké dávky radioterapie. Vzhledem k ohraničenosti onemocnění jsme zvolili tento lokálně intenzifikovaný strategický postup, který vedl v této chvíli k remisi onemocnění. Vzhledem k nízké incidenci MPNST není možné ověřit efektivitu jednotlivých onkologických léčebných postupů na větším souboru pacientů.
Klíčová slova:
maligní schwannom – sarkomy měkkých tkání – multimodální léčba
Úvod
Maligní nádor pochvy periferního nervu (malignant peripheral nerve sheath tumour – MPNST) je současný doporučený název pro všechny zhoubné nádory vyrůstající z periferní nervové tkáně, popř. z obalů nervů a zahrnuje nádory předtím známé jako maligní neurom, maligní neurilemom, neurogenní sarkom, neurofibrosarkom a maligní schwannom [1]. MPNST je velmi vzácný nádor, který tvoří pouze 2 % všech sarkomů, s incidencí 0,2 případu na 100 000 obyvatel. Nádor má poměrně špatnou prognózu, kdy 5leté celkové přežití (overal survival – OS) dosahuje 46−69 % [1–4].
Dle literatury jsou MPNST děleny do dvou, někdy do tří rozdílných forem [5–7]. První je sporadická forma tvořící cca polovinu případů. Druhá forma MPNST, tvořící 22−50 %, se váže na neurofibromatózu I. typu (NF1), což je nejčastější autozomálně dominantní onemocnění. MPNST zde často vzniká maligní transformací plexiformního neurofibromu. Mimoto pacienti mají i jiné charakteristické klinické projevy, jako jsou mnohočetné benigní neurofibromy, Lischovy noduly a café-au-lait kožní skvrny. Poslední formou jsou radioterapií indukované MPNST, které tvoří cca 10 %. MPNST se typicky manifestuje mezi 25. a 50. rokem, tedy v nižším věku než většina ostatních sarkomů měkkých tkání [7−10]. Pokud hodnotíme věkovou distribuci v rámci skupiny MPNST, nemocní s NF1 jsou mladší než nemocní se sporadickou formou tohoto onemocnění. Navzdory skutečnosti, že jednotlivé tři subtypy tohoto nádoru vyrůstají v rozdílných kontextech a pravděpodobně mají i rozdílné biologické charakteristiky, jsou nadále léčeny jako jedna entita [11].
Léčba MPNST není nadále standardizována. Základem je co možná nejradikálnější resekce, nejlépe na úrovni R0, a to i za cenu opakovaných operací. Onkologická adjuvantní či neoadjuvantní léčba má nejasné indikace s nejasným efektem [12].
Popis případu
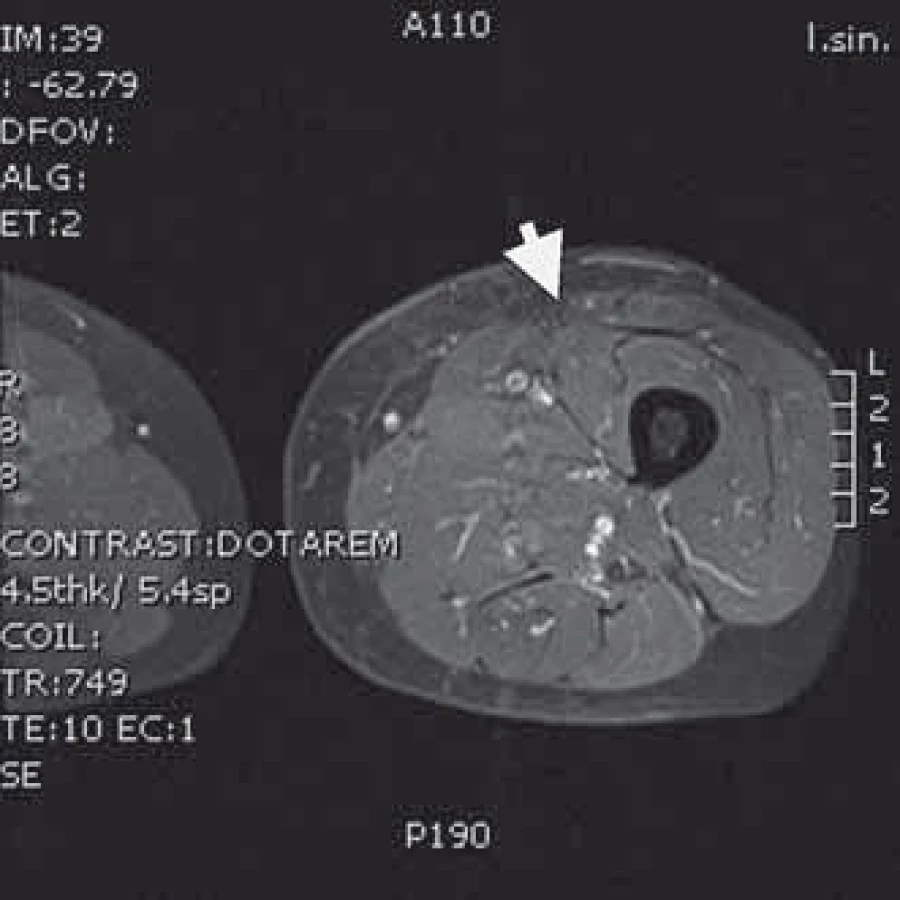
Naše kazuistika prezentuje 55letého pacienta vyšetřovaného v 9/2010 pro palpačně citlivou rezistenci na medio-ventrální ploše levého stehna. Byla doplněna MRI, která ukázala ovoidní tumor velikosti 36 × 24 × 30 mm (obr. 1) nasedající na levý n. femoralis, několik centimetrů po jeho výstupu zpod tříselného vazu. V 10/2010 byla provedena radikální exstirpace nádoru vyrůstajícího ze svalové větve femorálního nervu na stehně. Histologický nález odhalil maligní schwannom (MPNST) a resekce byla hodnocena jako R0. Imunohistochemicky nádorové buňky exprimovaly vimentin, CD99 a asi 50 % populace S-100. Negativní bylo vyšetření na Melan A, HMB45, SMA. Proliferační aktivita (Ki 67 %) dosáhla 10 %. Pooperační celotělové PET/CT neodhalilo viabilní nádorovou tkáň, která by vychytávala radiofarmakum. Nemocný byl dispenzarizován pravidelnými MRI a klinickými kontrolami, které byly opakovaně negativní. Až při kontrolním MRI 53 měsíců od operace se objevil nález recidivy tumoru na levém stehně, velikosti 70 × 40 × 40 mm. Sám pacient uvedl, že již cca 3 měsíce sleduje opětovný vznik hmatné, palpačně velmi bolestivé rezistence v místě jizvy. Proběhla exstirpace recidivy, která vzhledem k těsnému vztahu k femorálnímu nervu a nejasným okrajům nádoru byla stran radikality nedostatečná. Histologické vyšetření opětovně potvrdilo MPNST, radikalita resekce byla, dle očekávání, hodnocena jako R2. Pooperační celotělové PET/CT objevilo ve stěně resekční dutiny tři menší rezidua nádoru (obr. 2), a to především v kraniální části resekční dutiny směrem k hlavnímu nervovému kmeni. Pacient následně souhlasil s opakovanou operací a akceptoval možnost pooperačního neurologického deficitu. Proto proběhla 58 měsíců od první operace radikální exstirpace zbývajícího tumoru. Během výkonu byly zároveň do resekční dutiny umístěny tři aplikátory pro intersticiální brachyterapii (obr. 3), v dostatečné vzdálenosti od femorálního nervově-cévního svazku. Histologické vyšetření opětovně potvrdilo diagnózu MPNST. Imunohistochemické vyšetření odhalilo pozitivní expresi CD99 v 45 % buněk populace, S-100 v 45 % buněk, NSE v 50 % buněk a synaptophysinu v 60 % buněk. Negativní byla v nádoru exprese Melan A, HMB45, chromograninu, p53 DO 7 a SMA. Ki 67 % dosáhlo 20 %. Po operaci proběhla radioterapie, v rámci které byl cestou intersticiální brachyterapie podán boost 18 Gy s následnou zevní frakcionovanou radioterapií 50 Gy. Poslední MRI 66 měsíců od diagnózy MPNST neukázala žádnou reziduální či recidivující nádorovou tkáň (obr. 4). Jak po opakovaných operacích, tak po onkologické léčbě je pacient bez senzitivního či motorického neurologického deficitu. Přítomen je jen okrsek hypestezie v okolí jizvy na stehně a lehká lokální pigmentace.



Diskuze
Za posledních 100 let od popsání neurofibromatózy I. typu a MPNST došlo k výraznému vzestupu znalostí o biologii tohoto nádoru, stejně tak se výrazně proměnily chirurgické či onkologické techniky [13]. Nejlépe to dokumentuje zlepšení 5letého OS, které se z 23−34 % v 80. letech minulého století [3,14] posunulo na současných 46−69 % [4,6,7]. Z důvodu chybění standardizace léčba MPNST však nadále představuje velkou výzvu. Jak již bylo napsáno, základem je radikální resekce s cílem dosáhnout mikroskopicky čistých okrajů resekátu, tedy stupně R0 [4,11]. Chemoterapie (doxorubicin, někdy v kombinaci s ifosfamidem), ať už v neoadjuvantním, či v adjuvantním režimu, je využívána u pacientů s neresekovatelnými nebo metastatickými nádory. Radioterapie v konkomitantním nebo v sekvenčním režimu je využívána ve větší frekvenci [4,11]. Z důvodu malého počtu případů není možné provést randomizovanou studii, která by zhodnotila efekt adjuvantní léčby [15−19].
Stále probíhá diskuze týkající se rozdílu přežívání mezi sporadickými a na NF1 vázanými MPNST. Dle posledních metaanalýz [4,11] jednoznačně signifikantní rozdíl nebyl nalezen, ačkoliv pacienti s NF1 mají tendenci jak ke kratšímu OS, tak ke kratšímu přežívání bez progrese (progression-free survival – PFS). Jako důvod se předpokládá častější výskyt hluboko uložených a lokálně pokročilých nádorů, vč. vyššího výskytu resekce stupně R2 [4,7,8]. Tendence ke kratšímu OS a PFS se u MPNST spojených s NF1 zvýrazňuje u metastatických a neresekovatelných forem tohoto nádoru [3]. Dle jiných autorů [20−25] existují nejméně čtyři vysvětlení, proč NF1 pacienti mají horší výsledky v léčbě MPNST: 1. MPNST u NF1 pacientů jsou biologicky odlišné a vrozeně více agresivní; 2. přirozený obranný systém u NF1 je kompromitován, což umožňuje rychlejší růst maligního nádoru; 3. MPNST diagnóza je u NF1 opožděná, což rezultuje ve vznik více pokročilých nádorů; 4. léčba MPNST u NF1 pacientů je jiná než u non-NF1 pacientů. První dvě alternativy se týkají biologických rozdílů, druhé dvě alternativy se týkají klinických otázek. U biologických rozdílů lze očekávat molekulární diference na úrovni DNA, RNA nebo proteinů, ale zatím žádné významnější rozdíly nebyly nalezeny. Stejná mutace NF1 genu se nachází jak u sporadických, tak u s neurofibromatózou spojených nádorů, takže samotný výskyt této mutace nemůže vysvětlit rozdílné výsledky [26]. Nadto u neurofibromatózy stejný gen zapříčiňuje vznik jak benigního plexiformního neurofibromu, tak MPNST [27,28]. Rozdíly existují pouze v klinických parametrech, kdy pacienti s MPNST vázanými na NF1 jsou mladší než pacienti se sporadickou formou MPNST. Populační studie NF1 pacientů ukázala, že mortalita nepojící se k MPNST je obecně vyšší, především z důvodu výskytu nádorů mozku a onemocnění plic, což by mohlo vysvětlovat i nižší OS v této skupině [29−32]. Asociace mezi NF1 statutem a dalšími klinickými parametry je méně jasná a nebyla potvrzena nezávislými studiemi. Jako klinicky negativní prognostický faktor se ukázala lokalizace MPNST, kdy pacienti s hluboko uloženými nádory, s MPNST v oblasti hlavy, krku a trupu nebo lokálně pokročilými tumory mají signifikantně kratší OS oproti povrchově či v končetinách lokalizovanými, resp. lokálně ohraničenými nádory [4,18,19]. Při metaanalýze sama velikost nádoru neměla významnější vliv na prognózu nemocných [4]. Kratší OS měli pacienti s nádorem hodnoceným dle škály FNCLCC stupněm velikosti III oproti stupni I a II. Dle očekávání byl odhalen jako negativní prognostický faktor stupeň resekce R2 oproti resekci R1 či R0 [4,11,33].
Typ a strategie onkologické léčby MPNST nejsou přesně určeny. Dle poslední větší studie kompletní či částečná odpověď na chemoterapii byla u 24 % a kontrola nemoci byla dosažena u 62 % pacientů [4]. Chemoterapie však významněji neměnila OS těchto pacientů. Naopak pacienti s radioterapií měli signifikantně delší OS.
Použití této multimodální terapie s kombinací radikální resekce s adjuvantní intersticiální brachyterapií a zevní radioterapie u recidivy tohoto typu sarkomu měkkých tkání v dostupné literatuře nebylo popsáno.
Závěr
MPNST patří mezi vzácná maligní onemocnění periferního nervového systému s poměrně špatnou prognózou. Základem terapie je radikální en bloc resekce, která se odvíjí především od lokalizace a charakteru nádoru. Použití následné onkologické léčby závisí právě na radikalitě resekce. V našem případě sporadické formy MPNST, vzhledem k dobré radikalitě první operace, byla adjuvantní onkologická terapie postponována, což se vzhledem k dlouhé době bezpříznakového období ukázalo být správnou strategií. V rámci řešení recidiv jsme se opět snažili i za cenu opakované operace o dosažení co nejradikálnější resekce, ale již s plánem použití adjuvantní radioterapie. Vzhledem k negativnímu nálezu jiných ložisek na opakovaném celotělovém PET/CT nebyla indikována chemoterapie.
Bylo pomýšleno i na MPNST vzniklý v rámci neurofibromatózy I. typu, proti čemuž stojí nepřítomnost jakýchkoliv dalších známek této nemoci a věk manifestace maligního schwannomu. Taktéž dospělé děti nemocného nevykazují žádné známky tohoto geneticky přenosného onemocnění.
U MPNST jako u všech sarkomů měkkých tkání se vzhledem k potenciální radiorezistenci volí vysoké dávky. U našeho pacienta bylo cestou intersticiální brachyterapie aplikováno 18 Gy a dalších 50 Gy pomocí zevní frakcionované radioterapie. Vzhledem k ohraničenosti onemocnění jsme zvolili tuto lokálně intenzifikovanou radioterapii. Tento postup vedl v této chvíli k remisi onemocnění, i když vzhledem ke krátkému času od operace nelze jednoznačné závěry postulovat. Cílem práce bylo především ukázat možnosti řešení u vzácného onemocnění, které nemá jasně stanovený terapeutický postup. Jak již bylo napsáno, vzhledem k nízké incidenci MPNST není možné ověřit efektivitu jednotlivých léčebných postupů na větších souborech pacientů.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Ondřej Kalita, Ph.D., MBA
Neurochirurgická klinika LF UP a FN Olomouc
I. P. Pavlova 6
779 00 Olomouc
e-mail: ondrej.kalita@fnol.cz
Obdrženo: 13. 3. 2016
Přijato: 25. 4. 2016
Zdroje
1. Ng VY, Scharschmidt TJ, Mayerson JL et al. Incidence and survival in sarcoma in the United States: a focus on musculoskeletal lesions. Anticancer Res 2013; 33 (6): 2597–2604.
2. Žaloudík J, Taláč R, Vagunda V et al. Sarkomy měkkých tkání – přehled novějších diagnostických a léčebných postupů. Klin Onkol 2000; 13 (5): 141–150.
3. Ducatman BS, Scheithauer BW, Piepgras DG et al. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer 1986; 57 (10): 2006–2021.
4. Valentin T, Le Cesne A, Ray-Coquard I et al. Management and prognosis of malignant peripheral nerve sheath tumors: the experience of the French Sarcoma Group (GSF-GETO). Eur J Cancer 2016; 56 (3): 77–84. doi: 10.1016/j.ejca.2015.12.015.
5. Zou C, Smith KD, Liu J et al. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg 2009; 249 (6): 1014–1022. doi: 10.1097/SLA.0b013e3181a77 e9a.
6. LaFemina J, Qin LX, Moraco NH et al. Oncologic outcomes of sporadic, neurofibromatosis-associated, and radiation-induced malignant peripheral nerve sheath tumors. Ann Surg Oncol 2013; 20 (1): 66–72. doi: 10.1245/s10434-012-2573-2.
7. Stucky CC, Johnson KN, Gray RJ et al. Malignant peripheral nerve sheath tumors (MPNST): the Mayo clinic experience. Ann Surg Oncol 2012; 19 (3): 878–885. doi: 10.1245/s10434-011-1978-7.
8. Anghileri M, Miceli R, Fiore M et al. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer 2006; 107 (5): 1065–1074.
9. Longhi A, Errani C, Magagnoli G et al. High grade malignant peripheral nerve sheath tumors: outcome of 62 patients with localized disease and review of the literature. J Chemother 2010; 22 (6): 413–418.
10. Porter DE, Prasad V, Foster L et al. Survival in malignant peripheral nerve sheath tumors: a comparison between sporadic and neurofibromatosis type 1-associated tumors. Sarcoma 2009; 2009: ID 756395. doi: 10.1155/2009/756395.
11. Kolberg M, Høland M, Agesen TH et al. Survival meta-analyses for > 1800 malignant peripheral nerve sheath tumor patients with and without neurofibromatosis type 1. Neuro Oncol 2013; 15 (2): 135–147. doi: 10.1093/ neuonc/nos287.
12. Soft tissue and visceral sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2014; 25 (Suppl 3): iii102–iii112. doi: 10.1093/annonc/mdu254.
13. Harbitz F. Multiple neurofibromatosis (von Recklinghausen’s disease). Arch Int Med 1909; 3 (1): 32–65. doi: 10.1001/archinte.1909.00050120047003.
14. Sordillo PP, Helson L, Hajdu SI et al. Malignant schwannomaeclinical characteristics, survival, and response to therapy. Cancer 1981; 47 (10): 2503–2509.
15. Grobmyer SR, Reith JD, Shahlaee A et al. Malignant peripheral nerve sheath tumor: molecular pathogenesis and current management considerations. J Surg Oncol 2008; 97 (4): 340–349. doi: 10.1002/jso.20971.
16. Gupta G, Mammis A, Maniker A. Malignant peripheral nerve sheath tumors. Neurosurg Clin N Am 2008; 19 (4): 533–543. doi: 10.1016/j.nec.2008.07.004.
17. Widemann BC. Current status of sporadic and neurofibromatosis type 1 – associated malignant peripheral nerve sheath tumors. Curr Oncol Rep 2009; 11 (4): 322–328.
18. Beneš V III, Kramář F, Hrabal P et al. Maligní tumor z pochvy periferního nervu – dvě kazuistiky. Cesk Slov Neurol N 2009; 72/105 (2): 163–167.
19. Kadaňka Z Jr, Hanák J, Gál B. Maligní tumor z pochvy periferního nervu v oblasti cervikálního plexu – kazuistika. Cesk Slov Neurol N 2013; 76/109 (6): 751–755.
20. Watson MA, Perry A, Tihan T et al. Gene expression profiling reveals unique molecular subtypes of neurofibromatosis type I – associated and sporadic malignant peripheral nerve sheath tumors. Brain Pathol 2004; 14 (3): 297–303.
21. Henderson SR, Guiliano D, Presneau N et al. A molecular map of mesenchymal tumors. Genome Biol 2005; 6 (9): R76.
22. Karube K, Nabeshima K, Ishiguro M et al. cDNA microarray analysis of cancer associated gene expression profiles in malignant peripheral nerve sheath tumours. J Clin Pathol 2006; 59 (2): 160–165.
23. Miller SJ, Rangwala F, Williams J et al. Large-scale molecular comparison of human schwann cells to malignant peripheral nerve sheath tumor cell lines and tissues. Cancer Res 2006; 66 (5): 2584–2591.
24. Francis P, Namløs HM, Muller C et al. Diagnostic and prognostic gene expression signatures in 177 soft tissue sarcomas: hypoxia-induced transcription profile signifies metastatic potential. BMC Genomics 2007; 8 : 73.
25. Lévy P, Ripoche H, Laurendeau I et al. Microarray-based identification of tenascin C and tenascin XB, genes possibly involved in tumorigenesis associated with neurofibromatosis type 1. Clin Cancer Res 2007; 13 (2 Pt 1): 398–407.
26. Agesen TH, Flørenes VA, Molenaar WM et al. Expression patterns of cell cycle components in sporadic and neurofibromatosis type 1 – related malignant peripheral nerve sheath tumors. J Neuropathol Exp Neurol 2005; 64 (1): 74–81.
27. Cichowski K, Shih TS, Schmitt E et al. Mouse models of tumor development in neurofibromatosis type 1. Science 1999; 286 (5447): 2172–2176.
28. Vogel KS, Klesse LJ, Velasco-Miguel S et al. Mouse tumor model for neurofibromatosis type 1. Science 1999; 286 (5447): 2176–2179.
29. Zöller M, Rembeck B, Akesson HO et al. Life expectancy, mortality and prognostic factors in neurofibromatosis type 1. A twelve-year follow-up of an epidemiological study in Goteborg, Sweden. Acta Derm Venereol 1995; 75 (2): 136–140.
30. Rasmussen SA, Yang Q, Friedman JM. Mortality in neurofibromatosis 1: an analysis using U. S. death certificates. Am J Hum Genet 2001; 68 (5): 1110–1118.
31. Evans DG, O’Hara C, Wilding A et al. Mortality in neurofibromatosis 1: in NorthWest England: an assessment of actuarial survival in a region of the UK since 1989. Eur J Hum Genet 2011; 19 (11): 1187–1191. doi: 10.1038/ejhg.2011.113.
32. Masocco M, Kodra Y, Vichi M et al. Mortality associated with neurofibromatosis type 1: a study based on Italian death certificates (1995–2006). Orphanet J Rare Dis 2011; 6 : 11. doi: 10.1186/1750-1172-6-11.
33. Coindre JM. Grading of soft tissue sarcomas: review and update. Arch Pathol Lab Med 2006; 130 (10): 1448–1453.
Štítky
Dětská onkologie Chirurgie všeobecná OnkologieČlánek vyšel v časopise
Klinická onkologie

2016 Číslo 5
- Srovnání vlivu tamoxifenu a exemestanu na tloušťku endometria
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
- Incidence mozkových metastáz a intrakraniální aktivita sotorasibu u pacientů s NSCLC s mutací G12C onkogenu KRAS
- Přibývající důkazy o přínosu léčebného konopí u pacientů s chronickou bolestí
Nejčtenější v tomto čísle
- Vliv nutriční podpory s vysokým obsahem bílkovin na výsledky léčby a náklady u pacientů s kolorektálním karcinomem
-
Průvodce mladého onkologa infuzní terapií a výživou
Díl 5 – Hyperkalemie. Indikace umělé výživy. Kazuistika 5 - Melanom plosky nohy
- Léčba relabovaného a refrakterního Hodgkinova lymfomu – doporučení české studijní skupiny Hodgkinův lymfom
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy