
Neurodegenerativní onemocnění: přehled současné klasifikace a diagnostických neuropatologických kritérií
Neurodegenerative Disorders: Review of Current Classification and Diagnostic Neuropathological Criteria
Neurodegenerative disorders are progressive diseases characterized by loss of specific neuronal populations followed by a clinical picture of a different neurodegenerative entity. Current classification of these diseases respects the names of the main pathophysiological processes involved in the groups of disorders. This is the reason why key proteins which represent neuropathological and biochemical hallmarks of diseases are found in their names. Neuropathological diagnosis is a synthesis of neurohistological changes in the brain and spinal cord and identification of pathological proteinaceous aggregates in neurons and/or glial cells. These inclusions are predominant diagnostic micromorphological and biochemical markers of disease. In the text, there is a brief summary of current knowledge about pathophysiology of neurodegenerations and diagnostic criteria for the most frequent entities.
Keywords:
neurodegeneration – Alzheimer disease – classification – neuropathology
Autoři:
R. Matěj 1; R. Rusina 2
Působiště autorů:
Centrum pro diagnostiku a studium neurodegenerativních onemocnění
; Oddělení patologie a molekulární medicíny FTNsP, Praha
1; Neurologická klinika IPVZ a FTNsP, Praha
2
Vyšlo v časopise:
Čes.-slov. Patol., 48, 2012, No. 2, p. 83-90
Kategorie:
Přehledové články – Neuropatologie
Souhrn
Neurodegenerativní onemocnění jsou progredující onemocnění charakterizována úbytkem specifických skupin neuronů, což následně podmiňuje klinický obraz daného onemocnění. Současně platná klasifikace těchto onemocnění respektuje v názvech jednotlivých skupin chorob uplatnění nejvýznamnějších patofyziologických dějů, proto v názvu skupin onemocnění bývá zastoupen klíčový změněný protein. Neuropatologická diagnóza zohledňuje soubor neurohistologických změn v různých oblastech mozku a míchy a průkaz specifických neuronálních a/nebo gliových proteinových inkluzí, které jsou pro jednotlivá onemocnění určující. V textu je uveden přehled nejvýznamnějších neurodegenerací spolu s patofyziologickými aspekty i základními diagnostickými kritérii.
Klíčová slova:
neurodegenerace – Alzheimerova nemoc – neuropatologie – klasifikace
Neurodegenerativní onemocnění jsou charakterizována úbytkem specifických skupin neuronů, což následně podmiňuje klinický obraz daného onemocnění. Podstatou je kombinace různých patogenetických vlivů, z nichž jsou čtyři hlavní.
Apoptóza – interakce proapoptotických a anti-apoptotických faktorů spustí nezadržitelnou kaskádovitou reakci s výsledným zánikem postižené buňky za spoluúčasti volných kyslíkových radikálů. Abnormální patologické proteinové agregáty jsou specifické pro jednotlivé nosologické jednotky (tauopatie, synukleinopatie, Alzheimerova nemoc atd). Zásadní je i genetické pozadí, tedy vliv různých genových polymorfismů a postižení genomu patogenními mutacemi.
Obecně se tedy jedná o specifický děj (agregace určitého, pro dané onemocnění typického, proteinu v CNS), v kombinaci s obecnými apoptotickými mechanismy, které jsou společné pro celou skupinu neurodegenerativních onemocnění (1,2).
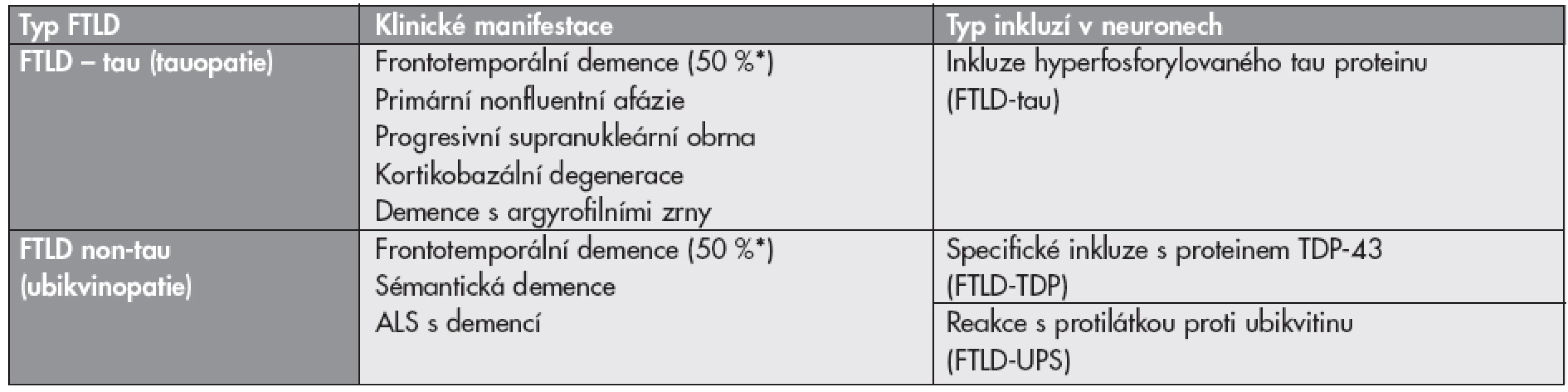
Klasifikace neurodegenerativních onemocnění respektuje v názvech jednotlivých skupin těchto chorob uplatnění nejvýznamnější patofyziologické dějů, proto v názvu skupin onemocnění bývá zastoupen změněný protein (tab. 1).

ALZHEIMEROVA NEMOC
U nejčastějšího neurodegenerativního onemocnění – Alzheimerovy nemoci (AN) – se specifický neuroanatomický nález překrývá se změnami doprovázejícími stárnutí. Ukazuje se, že úbytek neuronů vlivem stárnutí je menší, než vyplývalo z výsledků klasických prací (1–3). V rozmezí 24—100 let dosahuje ve frontální kůře asi 15 %, v kůře temporální asi 10 %, zatímco podkorové neuronální populace jsou stárnutím ovlivněny jen minimálně (3).
Klasickými diagnostickými znaky doprovázejícími Alzheimerovu nemoc jsou senilní (amyloidové) plaky s depozity ß-amyloidu a neuronální klubka (tangles) (4,5) (obr. 1).

Nejčastěji se depozita ß-amyloidu objevují v neokortikálních oblastech. Neuritické (senilní) plaky jsou nepravidelně okrouhlé 10–200 Ķm velké útvary s obsahem amyloidu, peptidových fragmentů tvořených 40 a 42 aminokyselinami. V klasických (neuritických) plakách tvoří ložisková A-ß 40 depozita husté jádro a periferie obsahuje A-ß 42. Součástí plak jsou heterogenní neurity s filamenty a dystrofickými organelami. V primitivních (nezralých) plakách jsou sférické uloženiny A-ß 42 v neuropilu, husté jádro není přítomno, běžně se nalézají v mozku starších nedementních lidí často ve striatu. Kompaktní („vyhořené“) plaky tvořené jen hustým amyloidovým jádrem z A-ß 42 a proměnlivým počtem neuritů, typicky jsou ve vrstvě Purkyňových buněk a v globus palidus. Difúzní plaky, časté u starých nedementních lidí, v molekulární vrstvě mozečku a u demence s Lewyho tělísky, se podobají primitivním, špatně se barví histochemicky na amyloid, nebývají přítomné dystrofické neurity.
V plakách se dá prokázat aktivovaná mikroglie, tvarově odlišná od klidové mikroglie, která se může podílet na patogenezi AN v rámci zánětlivých mechanismů společně s oxidativním stresem.
Součástí AN je dále cerebrální amyloidová angiopatie (CAA), která se vyskytuje i v několika familiárních variantách bez klinických příznaků a anatomických známek AN (1).
Neuronální klubka (tangles, Alzheimerovy změny neurofibril) vypadají při impregnaci solemi stříbra jako chumáče hrubších vláken v cytoplazmě neuronů, případně jako plamének nebo tenisová raketa. Pro rozpadu perikarya jsou volně v neuropilu. Základní složkou párových spirálních vláken je protein s relativně nízkou molekulární hmotností pojmenovaný tau.
Klubka se nejdříve objevují v amygdale a parahipokampálním závitu, poté v hipokampu a temporálním kortexu. Distribuce klubek je laminární, postiženy bývají jen vybrané vrstvy mozkové kůry, například v entorhinální kůře vrstvy II a IV, v asociačních korových oblastech vrstvy II, III a V, nebo ve zrakové asociační kůře vrstvy II, III a V.
Imunohistochemický průkaz monoklonální protilátkou proti hyperfosforylované formě tau proteinu (klon AT8) by měl být podle nejnovějších doporučení BrainNetEurope (BNE) standardní vyšetřovací metodou pro správnou diagnostiku definitivní Alzheimerovy nemoci.
Významnou roli hrají genetické vlivy. AN má v 5 % prokazatelný genetický původ. První patogenní mutace byly popsány v genu APP na chromosomu 21, v současnosti je jich známo 25. V genu pro presenilin 1 (PSEN1) na chromosomu 14 je známo 155 mutací, onemocnění začíná ve středním věku 44–46 let, popsáno je celosvětově přes 300 rodin. Gen presenilinu 2 (PSEN2) na chromosomu 1 obsahuje známých 10 mutací, vyvolávající chorobu ve věku 58–59 let. Rizikovým (nikoliv kauzálním!) faktorem pro pozdní podobu AN (po 65. roce věku) u bělochů je polymorfismus apoliproproteinu E (homozygoti pro APOE 4 mají až 8x vyšší četnost AN než v populaci nejčastější homozygoti APOE 3; heterozygotů s jednou alelou APOE 4 je v populaci asi 16 %, ale u pacientů s AN nacházíme APOE 4 homozygoty v nadpoloviční většině). Mechanismus, jímž APOE 4 zvyšuje riziko choroby, není však přesně znám (6).
Definitivní diagnóza Alzheimerovy nemoci se opírá o doporučení Reaganova ústavu (NIA-Reagan Working Group), která navazují na starší kriteria skupiny CERAD a kriteria vypracovaná Braakovými (4,7).
Neuropatologická diagnóza Alzheimerovy nemoci má tři kroky: 1. semikvantitativně se určí počet plak a klubek v místech, kde je jejich denzita nejvyšší; 2. určí se počet plak ve vztahu k věku pacienta v době úmrtí (kombinací pacientova věku a semikvantitativní míry počtu plak v místě jejich největší denzity); 3. zjištěné skóre se porovná s klinickými informacemi (míra demence).
Braakovi stanovili vývoj Alzheimerovy nemoci měřený denzitou neuronálních klubek v šesti stádiích: I a II – transentorhinální stadia, III a IV – limbická stadia, V a VI – neokortikální stádia (4) (tab. 2). Určení stádia vyžaduje imunohistochemický průkaz reakce s monoklonální protilátkou proti hyperfosforylované formě tau proteinu (BNE doporučuje klon AT8) (8).

TAUOPATIE
(Mikromorfologie tauopatií je dokumentována na obrázku S1 v elektronickém suplementu – viz www.CSpatologie.cz)
Tato skupina onemocnění odvozuje svůj název od proteinu tau (tubulin associated unit), který se váže na tubulin mikrotubulů, startuje jejich polymerizaci a účastní se intracelulárního transportu. Jakmile je defosforylován, podílí se na jejich stabilizaci (9).
V normálním mozku se tau protein vyskytuje v šesti isoformách o molekulové hmotnosti 50–65 kDa, obsahujících 352–441 aminokyselin a kódovaných genem MAPT (microtubulin associated unit) na 17. chromozomu. Za patologických okolností dochází k hyperfosforylaci na specifických vazebných místech. Vzniknou tak obtížně odbouratelné proteinové agregáty následně spouštějící apoptózu (1).
Tauopatie se dělí do 3 základních skupin (tab. 3) dle počtu opakovaní vazebného místa pro molekuly mikrotubulů (direct-repeats – DR), které mohou být převážně tři (DR3) nebo čtyři (DR4) a rovněž se zohledňuje haplotypizace genu MAPT (H1 a H2).

Tauopatie jsou podskupinou onemocnění zvaných fronto-temporální lobární degenerace (FTLD) a označují se jako FTLD-tau. Roztříštěnost poznatků o této skupině onemocnění vedlo k různým klasifikacím, které se často prolínaly a zaměňovaly (10).
Revidovaná klinická kritéria používají termín frontotemporální demence (FTD), v jejímž rámci vymezují tři klinicko-patologické jednotky: „vlastní“ frontotemporální demenci (FTD – někdy se uvádí termín behaviorální varianta – bvFTD), progresivní neplynulá (nonfluentní) afázie (PNFA) a sémantická demence (SD) (10,11).
Obraz behaviorální varianty frontotemporální demence je charakterizován těžkými poruchami chování a časnými změnami osobnosti s postupným rozvojem demence frontálního typu. Progresivní afázie se projevují jako zpočátku izolovaná porucha řeči expresivního typu (připomínající Brocovu afázii) u PNFA nebo porucha sémantického obsahu řeči (připomínající Wernickeovu afázii) u SD s postupnou progresí do těžké frontální demence (12,13).
Atrofie frontálních a temporálních laloků bývá asymetrická, pravá strana je více postižená u FTD, levá strana u progresivních afázií. Mimořádně atrofické gyry mohou makroskopicky připomínat „ostří nože“ či listy („listovité gyry“). Postiženy bývají i hipokampy a amygdala, v menší míře rovněž bazální ganglia. Makroskopickému nálezu odpovídá těžká numerická atrofie (úbytek počtu) neuronů, astrocytóza a spongióza, zpočátku zejména v korových vrstvách II. a III (1,2).
FTD je v polovině případů tauopatie (tab. 4), progresivní afázie mohou mít různý strukturální podklad (tab 4). Pickova nemoc je paradoxně (přesto, že byla dlouho chápána jako synonymum frontotemporální demence) oproti původním představám vzácné onemocnění, je podkladem jen necelých 5 % případů FTD. Pickovy buňky jsou zduřelé balónovité neurony s oxyfilním plazmatem, vyplněné kulovitými inkluzemi, tzv. Pickovými tělísky (argyrofilní intraneuronální inkluze, nejčastěji ve fascia dentata a pyramidových neuronech hipokampu). Většinu familiárních případů Pickovy nemoci doprovázejí mutace genu MAPT (9,10).
Kortikobazální degenerace byla popsána Rebeizovou skupinou v roce 1968 jako kortikodentatonigrální degenerace a neuronální achromázií s imunohistochemickou pozitivitou v barvení tau proteinu, typicky s predominancí DR4 isoformy. Numerická atrofie neuronů a glióza s basofilními inkluzemi se najdou v substantia nigra a v palidu. Ve zbývajících pigmentových neuronech substantia nigra jsou tzv. bledé inkluze. Nejsou přítomna Pickova ani Lewyho tělíska, neuritické plaky ani neuronální klubka. Charakteristické je rovněž postižení gliálních elementů s průkazem tau-pozitivních lézí zejména patognomonických astrocytárních plak a „threads“ v bílé i šedé hmotě kortikálních struktur, bazálních ganglií a substantia nigra (1,9).
Progresivní supranukleární obrnu (Steelův-Richardsonův-Olszewského syndrom – PSO) provází globoidní neuronální klubka s hyperfosforylovaným tau proteinem s predominancí DR4 isoformy hlavně v subkortikálních lokalizacích. Míra numerické atrofie neuronů a gliózy kolísá. Imunohistochemicky se rovněž prokazují změněné astrocyty a oligodendroglie jakož i patologická vlákna v bazálních gangliích, mezimozku a mozkovém kmeni. Charakteristické je těžké postižení substantia nigra, locus coeruleus, globus pallidus, nc. subthalamicus, periakveduktální šedi mezencefala, jádra hlavových nervů v pontu a mezencefalu a nc. dentatus mozečku. Časté postižení se týká striata, hippokampu a prefrontalního kortexu. Prakticky nikdy nebývá postižen cerebellární kortex (1,9,14).
Diagnostická kritéria PSO vyžadují splnění inkluzních kriterií pro typickou PSO (vysoká denzita neurofibrilárních klubek – tangles a „neuropil threads“ a klinický obraz kompatibilní s PSO) nebo pro kombinovanou PSO (přítomné vaskulární změny v pontu a bazálních gangliích) (14).
Nemoc s argyrofilními zrny (argyrophilic grain disease – AGD) vykazuje drobná vřetenitá, argyrofilní, v imunohistochemickém barvení tau protein positivní “zrna”, která se dají prokázat i v oligodendroglii, kde se prokazují “svinutá” (coiled) tau positivní tělíska s predominancí DR4 isoformy. AGD se často vyskytuje společně s Alzheimerovou nemocí nebo jinými tauopatiemi. Hlavní složkou argyrofilních zrn je hyperfosforylovaný tau protein s predominancí DR4 isoformy (15).
Problémem je klinická definice tohoto onemocnění, protože z několika srovnávacích klinicko-neuropatologických studií především japonských autorů vyplývá, že až 50 % jednoznačně neuropatologicky definovaných případů AGD se neprojevovalo demencí (16).
FRONTOTEMPORÁLNÍ LOBÁRNÍ DEGENERACE
Z neuropatologického a etiologického hlediska jde o heterogenní skupinu onemocnění, u nichž abnormálně transformovaný protein interaguje s metabolickými drahami neuronů a vede k apoptóze. Podle názvu patologicky změněné bílkoviny v inkluzích mluvíme zejména o tauopatiích a ubikvitinopatiích, nově i proteinu TDP-43 (TAR DNA-binding protein 43), který se ukázal být zásadním u non-tau FTLD (tab. 4). TDP-43 hraje klíčovou roli i u FTLD spojené s onemocněním motorického neuronu (amyotrofická laterální skleróza) (FTLD-MND) (17–19).
Makroskopický nález u FTLD charakterizuje obvykle lehká symetrická atrofie závitů čelní a přední temporální kůry, histologicky patrné změny jsou v kůře frontální konvexity, v orbitofrontální kůře, v přední třetině spánkového laloku a gyrus cinguli. Mikroskopicky se najde mikrovakuolizace a mírná až střední astrocytární glióza zejména I.–III. korové vrstvy. Numerická atrofie neuronů je patrná zejména ve vrstvě II a III. Typické je chybění specifických alzheimerovských změn (senilních plak a neurofibrilárních klubek) a Pickových či Lewyho tělísek (20).
Podle imunohistochemického obrazu a charakteru intracelulárních inkluzí dělíme FTLD na tauopatie a non-tau FTLD s neuronálními a/nebo gliálními inkluzemi pozitivními v imunohistochemickém průkazu s protilátkou proti ubikvitinu (ubikvitinopatie – FTLD-UPS), FTLD s inkluzemi proteinu TDP-43 – (FTLD-TDP) různých typů a tvarů a další méně časté jednotky podle reakce s dalšími protilátkami (tab. 4) (21). (Mikromorfologie proteinových depozit u FTLD/MND je dokumentována na obrázku S2 v elektronickém suplementu - viz www.CSpatologie.cz)
V této oblasti molekulární neuropatologie probíhá v současnosti velmi dynamický vývoj, který akceptuje teorii, že neurodegenerativní onemocnění podmíněná patologií proteinu TDP-43 jsou obdobnými systémovými onemocněními jako synukleinopatie nebo tauopatie (19).
FTLD mají poměrně častý familiární výskyt (30–50 % postižených mezi příbuznými I. stupně). Byly popsány mutace genu pro tau protein (MAPT), genu pro protein TDP-43 (TARDBP), genu pro progranulin (PGRN), genu pro valosin (VCP) a jiné. Klinický obraz je pestrý s behaviorálními poruchami, často i amyotrofickou laterální sklerózou. Stejná mutace se v rámci jedné postižené rodiny může projevovat různými klinickými symptomy. Přehledné schéma současného chápání frontotemporálních lobárních degenerací je patrné na schématu č. 1 (6,21).

SYNUKLEINOPATIE
Typickým zástupcem této skupiny je Parkinsonova nemoc s degeneraci neuronů ventrolaterální oblasti pars compacta substantia nigra (obr. 2) a dalších kmenových jader doprovázenou výskytem alfa synuklein pozitivních cytoplasmatických inkluzí – Lewyho tělísek – v cytoplazmě blízko jádra zachovaných neuronů. Ve standardním barvení můžeme rozeznat eozinofilní centrum a periferní projasnění (někdy bývá přirovnáváno k podšálku), obklopené zbytky melaninového pigmentu. Lewyho tělíska jsou složena z různých proteinů, pro diagnostiku je nejdůležitější přítomnost ubikvitinu, proteinu p62 a alfa synukleinu (22).

Neuropatologická diagnóza Parkinsonovy nemoci může být zatížena rozpaky. Při systematickém vyšetřování mozků starších lidí se totiž nachází:
- několik procent případů klinicky němých, u nichž jsou Lewyho tělíska v neuronech substantia nigra (“incidentální choroba s Lewyho tělísky”, “subklinická Parkinsonova nemoc”),
- klinicky a neuropatologicky prokázaná „čistá“ Parkinsonova nemoc s převažujícím postižním s. nigra, k tomu žádné nebo podprahové “azheimerovské” změny (plaky a neuronální klubka)
- případy klinicky rovněž charakterizované Parkinsonovým syndromem s rostoucí mírou alzheimerovských změn a rostoucím počtem Lewyho tělísek v korových oblastech (nález se někdy interpretuje jako současný výskyt Alzheimerovy a Parkinsonovy nemoci).
- případy s klinicky a neuropatogicky zřejmou Alzheimerovou nemocí, nicméně se najdou Lewyho tělíska v substantia nigra i jinde.
Definitivní diagnóza demence s Lewyho tělísky (DLB) je také neuropatologická a opírá se o průkaz numerické atrofie neuronů zejména pigmentových neuronů substantia nigra, locus coeruleus a neuronů nc. basalis Meynerti a nález Lewyho tělísek a Lewyho neuritů (23).
Lewyho tělíska jsou kmenová (“klasická”) a korová (“neklasická”). Klasická Lewyho tělíska jsou sférické (někdy i vřetenovité) eozinofilní převážně intraplasmatické inkluze, s typickým “halo” kolem hyalinního jádra. Korová tělíska jsou méně zřetelné, víceméně sférické útvary v plasmatu korových neuronů. “Bledá tělíska” (pale bodies) se objevují v kmenových neuronech obvykle společně s Lewyho tělísky, mohou být jejich prekurzory.
Lewyho tělíska se hledají v pigmentových neuronech s. nigra a locus coeruleus, jsou průkazná ve standardním barvení, korová Lewyho tělíska lze detekovat pomocí protilátek proti ubiquitinu, proteinu p62 nebo lépe alfa synukleinu. Lewyho neurity se prokazují imunohistochemicky stejně jako korová Lewyho tělíska, objevují se v CA2/3 sektoru hippokampu, v amygdale, nc. basalis Meynerti, nc. dorsalis n. X a v dalších kmenových jádrech hlavových nervů.
Ve vývoji DLB existuje dynamika: Lewyho tělíska a neurity se objevují nejdříve v korových vrstvách V–VI, poté ve vrstvě III, nakonec ve vrstvě II. Topograficky viděno postihují tyto změny nejprve amygdalu, posléze limbickou kůru, nakonec neokortex. V konečném výsledku pak dojde ke stratifikaci na stádium prevážně kmenové (I), stádium limbické (II) a difúzní neokortikální stádium (III) (23,24).
Součásti DLB jsou alzheimerovské změny: plaky a v malém množství neuronální klubka. Vývoj alzheimerovských změn se diagnostikuje dle kritérií uvedených v části o Alzheimerově nemoci a ke standardní interpretaci neuropatologického nálezu by mělo patřit i stanovení, jaká je pravděpodobnost, že klinické symptomy u pacienta byly v souvislosti s postižením mozku alfa synuklein pozitivními depozity (tab. 5) (1,2,23).

Vzácnější synukleinopatií je soubor onemocnění zvané mnohotná systémová atrofie – MSA. V klinickém obraze se objevují projevy mozečkové, parkinsonské, pyramidové a dysautonomie, a to v různé kombinaci. Onemocnění má dvě základní formy, mozečkovou (MSA-C – s výraznou atrofií mozečku a jeho středního pedunklu) a parkinsonskou (MSA-P, kde bývá prostým okem viditelné šedavě nazelenalé zabarvení velmi atrofického putamen v čelním řezu na úrovni corpora mammillaria, někdy je zřejmá atrofie pigmentované části substantia nigra a locus coeruleus).
MSA charakterizuje zejména apoptotický zánik oligodendroglí, neuronální apoptóza se při tomto onemocnění nápadněji nepozoruje (25). Mikroskopicky převládá numerická atrofie neuronů, reaktivní astroglióza a demyelinizace. Nejpostiženější oblastí jsou dorzální části kaudálního putamen, laterální oblasti s. nigra, locus coeruleus, nc. olivae inferioris et accessorius, nc. pontis, Purkyňovy buňky jsou postiženy ve vermis cerebelli více než v hemisférách, nc. dorsalis n. X, nc. intermediolateralis a Onufovo jádro v sakrální míše. Kromě toho se morfometricky prokazuje numerická atrofie neuronů v primární a asociační motorické kůře, při běžném prohlížení však obvykle unikne.
Za poruchy autonomních funkcí odpovídá zejména postižení katecholaminergních neuronů rostrální ventromediální prodloužené míchy (skupina C1), a noradrenergních neuronů v kaudální ventrolaterální prodloužené míše (skupina A1).
Charakteristickým diagnostickým příznakem jsou argyrofilní cytoplasmatické inkluze v oligodendroglii, nazývané plaménkové či Pappovy-Lantosovy. Tvoří je 10–15 nm silná filamenta, imunohistochemicky pozitivní v reakci s protilátkami proti alfa synukleinu, proteinu p62 a ubikvitinu (1,2,25).
(Mikromorfologie synukleinopatií je dokumentována na obrázku S3 v elektronickém suplementu - viz www.CSpatologie.cz)
ONEMOCNĚNÍ S OPAKOVÁNÍM TRIPLETŮ
Prototypem této skupiny onemocnění je Huntingtonova nemoc, způsobená mutací genu, který kóduje protein huntingtin, na 4. chromozomu. V prvním exonu je kolem 20 opakování glutaminových zbytků kodonem CAG. Chorobný fenotyp se projevuje při počtu opakování nad 35. Čím je počet repetic vyšší, tím dříve choroba nastupuje a její průběh je těžší.
Kaskáda, kterou mutovaný huntingtin poškozuje neurony, je mimořádně složitá a v podrobnostech ne zcela jasná. Makroskopické změny mozku odpovídají v 80 % případů atrofii čelního laloku s často přidružením postižením i bílé hmoty, v 95 % případů je makroskopicky patrná atrofie striata. Nejpostiženější oblastí je ocas nc. caudatus, následuje tělo, po něm caput. Podobně je postižena kaudální část putamen víc než část rostrální.
Numerickou atrofii neuronů doprovází astroglióza. Vonsattel vytvořil pětistupňový grading (0–4) postižení striata (26). Mezi postižením striata a dalších mozkových oblastí existuje korelace. Při vyšších stupních postižení atrofují palidum, čelní kůra, talamus, nc. subtalamicus a mozeček. V dystrofických neuronech lze nalézt různé typy převážně intranukleárních inkluzí, s imunohistochemickou pozitivitou ubikvitinu a huntingtinu.
ONEMOCNĚNÍ MOTORICKÉHO NEURONU
V histopatologickém vyšetření nejčastějšího onemocnění motorického neuronu amyotrofické laterální sklerózy (ALS) převažuje numerická atrofie velkých motorických neuronů předních rohů míšních a motorických neuronů jader hlavových nervů v mozkovém kmeni. Postiženy bývají i Betzovy pyramidy v primární motorické kůře. Příčně pruhované svaly jeví známky chronické denervační neurogenní atrofie s tukovou pseudohypertrofií. Degenerace kortikospinálního traktu je kromě míchy nejvíce nápadná v mozkovém kmeni a diencefalu.
Klíčovým diagnostickým znakem je výrazná numerická atrofie velkých motorických neuronů předních rohů míšních, především v cervikální a lumbální intumescenci. Zbylé neuronální struktury jeví známky regresivních změn s depozity lipofuscinu v cytoplazmě, v jádrech může být patrná centrální chromatolýza. Může se objevit neuronofagie a perivaskulární lymfocytární infiltráty (1,2).
V cytoplazmě postižených neuronů lze najít různé inkluze, největší specificitu mají Buninova tělíska – malé eozinofilní granulární inkluze (velikost 2–4 Ķm) s pozitivitou v reakci s protilátkou proti cystatinu C.
Pomocí protilátky proti ubiquitinu či proteinu P62 a proteinu TDP-43 lze ozřejmit tzv. „skein-like“ inkluze protaženého či vláknitého tvaru, které tvoří až bizarní sférické struktury v perikariu neuronů (obr. 3A), jednak světle eozinofilní kulaté hyalinní inkluze. Impregnací solemi stříbra dle Bielschowského lze prokázat cytoplasmatické inkluze zvané axonální sferoidy a zejména v případech juvenilních forem ALS je možné za pomoci metylové zeleni pyroninu najít basofilní inkluze, které obsahují kondenzovanou RNA (1,2).

U familiárních forem ALS nacházíme vysoce argyrofilní cytoplazmatické Lewyho tělískům podobné hyalinní inkluze, a u mutací genu pro superoxid dismutázu 1 (SOD1) silnou reaktivitu s protilátkou proti SOD1 (6).
Protein TDP-43, zásadní u non-tau FTLD (tab. 4), hraje klíčovou roli i u ALS s demencí (FTLD-MND: frontotemporální lobární degenerace spojená s onemocněním motoneuronu). Překvapivě však TDP-43 pozitivní inkluze je možné najít i u ALS bez postižení kognice (27).
(Amyotrofická laterální skleróza je dokumentována na obrázku S4 v elektronickém suplementu - viz www.CSpatologie.cz)
PRIONOVÁ ONEMOCNĚNÍ (PŘENOSNÉ SPONGIFORMNÍ ENCEFALOPATIE)
Nejčastější lidské prionové onemocnění, Creutzfeldtova-Jakobova nemoc (CJN) je vzácné, obvykle rychle progredující neurodegenerace s infaustní prognózou. V různé míře postihuje mozkovou kůru a oblasti podkorové šedi. Projevuje se rychle progredující demencí a řadou neurologických příznaků (tab. 6).

Rozlišují se 3 základní typy CJN: sporadická, genetická (familiární) a náhodně přenesená v souvislosti s lékařským výkonem (iatrogenní). Nejvíce případů je sporadických. Jejich příčina je neznámá a doposud nebyl zjištěn žádný vztah k prionovému onemocnění zvířat. Asi 10–15 % je genetických, na podkladě mutací v genu pro prionový protein (PRNP), na 20. chromozomu. Vzácně (v méně než 5 %) byl popsán iatrogenní přenos (výtažky z lidských hypofýz, meningeální štěpy, transplantace rohovky) (28).
Priony (zkratka ze slov „proteinaceous infectious particles“) (prionový protein – PrP) jsou malé proteinové částice, jediné známé patogeny, které nemají nukleové kyseliny. Dostane-li se patogenní PrPSc do kontaktu s tkáňovým PrPc, stimuluje jeho přeměnu na PrPSc - hraje úlohu „šablony“, podle které mění existující PrPc svoji prostorovou strukturu. Přeměna PrPc na PrPSc může být spontánní (sporadická forma) nebo je usnadněna patogenní mutací prionového proteinového genu PRNP (genetická forma) (29).
Klasickou neurohistologickou trojicí změn při prionových nemocech je spongiformní dystrofie, numerická atrofie neuronů a glióza (obr. 3A,B). Spongiformní dystrofii charakterisují vakuoly v neuropilu jejichž průměr je 2–20 Ķm. Objevují se ložiskově, je nutné odlišit je od artefaktů a od vakuol, které se objevují v horních korových vrstvách při různých příčinách korové atrofie. Někdy jsou patrná depozita prionového proteinu ve formě plak (obr. 3C) s charakteristickými vlastnostmi amyloidu (obr. 3D). Imunohistochemické vyšetření užívá různé typy protilátek ozřejmujících výskyt prionů ve tkáni (obr. 3E,F). Vysoce charakteristické je, že priony ve tkáni nevyvolávají zánětlivou odpověď, což byl jeden z důvodů dlouhého tápání při objasňování povahy této nemoci (1,2,29).
Genetickou predispozicí pro vznik CJN (sporadické a získané) je polymorfizmus PRNP v oblasti kodonu 129, kde je kódován buď metionin, nebo valin. Většina chorob postihuje homozygoty metionin/metionin (30).
Původce nové varianty CJN (vCJN) je neodlišitelný od prionu bovinní spongiformní encefalopatie (BSE), jde tedy zřejmě o lidskou formu BSE. Vzniká pravděpodobně alimentární nákazou člověka masem hovězího dobytka postiženého BSE. Poprvé byla popsána v 90. letech ve Velké Británii. V České republice dosud nebyl zaznamenán žádný případ vCJN.
Na rozdíl od klasické sporadické CJN postihuje vCJN většinou mladší jedince (20–40 let), průměrný věk postiženého v době úmrtí je 29 let. Její průběh je o něco pomalejší. Na počátku jsou v popředí psychiatrické příznaky, přidávají se poruchy hybnosti a bolestivé parestézie, demence bývá až velmi pozdním příznakem (28,29).
U vCJN se priony nacházejí, na rozdíl od jiných forem CJN, i v periferních lymforetikulárních tkáních (slezina, lymfatické uzliny, tonzily, apendix), proto může být definitivní diagnóza stanovena již za života nemocného biopsií krční tonsily. Novou variantu CJN charakterizuje velký počet “floridních” plak v kůře mozku i mozečku, shluky malých plak v imunohistochemickém obraze, amorfní pericelulární a perivaskulární kupení PrP (28,29).
PODĚKOVÁNÍ
Podpořeno granty 309/09/P204 Grantové agentury ČR a IGA NT 12094-5 Interní grantové agentury Ministerstva zdravotnictví ČR.
Seznam použitých zkratek
AGD: nemoc s argyrofilními zrny; ALS: amyotrofická laterální skleróza; AN: Alzheimerova nemoc; bvFTD: behaviorální forma frontotemporální demence; BSE: bovinní spongiformní encefalopatie; CJN: Creutzfeldtova-Jakobova nemoc; DLB: demence s Lewyho tělísky; DR: opakovaní vazebného místa pro molekuly mikrotubulů (direct-repeats); FTD: frontotemporální demence; FTLD: frontotemporální lobární degenerace; FTLD-MND: frontotemporální lobární degenerace spojená s onemocněním motoneuronu; FTLD-TDP: FTLD s inkluzemi proteinu TDP-43; FTLD-UPS: FTLD s ubiquitin pozitivními inkluzemi; MAPT: gen pro tau protein, MSA: mnohotná systémová atrofie; PGRN gen pro progranulin; PNFA primární progresivní nonfluentní (neplynulá) afázie; PRNP: gen pro prionový protein; PrP: prionový protein; PSO: progresivní supranukleární obrna; SOD: superoxid dismutáza; SD: sémantická demence; TARDBP: gen pro protein TDP-43; TDP-43:TAR DNA-binding protein 43; vCJN: nová varianta Creutzfeldtovy-Jakobovy nemoci;
Adresa pro korespondenci:
MUDr. Radoslav Matěj, Ph.D.
Centrum pro diagnostiku a studium neurodegenerativních onemocnění
Oddělení patologie a molekulární medicíny FTNsP
Vídeňská 800, 14059 Praha 4 – Krč
tel: 261083741
e-mail: radoslav.matej@ftn.cz
Zdroje
1. Dickson DW ed. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. Basel: ISN Neuropath Press, 2003.
2. Love S, Louis DN, Ellison DW. (eds). Greenfieldęs Neuropatology 8th edition. Hodder Arnold, 2008 : 889–1264.
3. Quertfurth HW, LaFerla FM. Alzheimeręs disease. New Engl J Med 2010; 362 : 329–344.
4. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica 1991; 82 : 239–259.
5. Jack CR Jr, Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7(3): 257–62.
6. OMIM On-line Mendelian Inheritance in Man. http://www.ncbi.nlm.nih.gov/omim
7. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7 : 263–269.
8. Alafuzoff I, Arzberger T, Al-Sarraj S, et al. Staging of neurofibrillary patology in Alzheimeręs disease: a study of the BrainNet Europe Consortium. Brain Pathol 2008; 18 : 484–496.
9. Dickson DW, Kouri N, Murray ME, et al. Neuropathology of Frontotemporal Lobar Degeneration-Tau (FTLD-Tau). J Mol Neurosci 2011: Jul 1. [Epub ahead of print]
10. Bigio EH. Update on recent molecular and genetic advances in frontotemporal lobar degeneration. J Neuropathol Exp Neurol 2008; 67 : 635–648.
11. Sleegers K, Cruts M, Van Broeckhoven C. Molecular pathways of frontotemporal lobar degeneration. Annu Rev Neurosci 2010; 33 : 71–88.
12. Kertesz A, McMonagle P, Blair M, et al. The evolution and pathology of frontotemporal dementia. Brain 2005; 128 : 1996–2005.
13. Hodges JR, Patterson K. Semantic dementia: a unique clinicopathological syndrome. Lancet Neurol 2007; 6 : 1004–1014.
14. Williams DR, Lees AJ. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol 2009; 8 : 270–279.
15. Fujino Y, Wang DS, Thomas N, et al. Increased frequency of argyrophilic grain disease in Alzheimer disease with 4R tau-specific immunohistochemistry. J Neuropathol Exp Neurol 2005; 65 : 209–214.
16. Josephs KA, Whitwell JL, Parisi JE, et al. Argyrophilic grains: a distinct disease or an additive pathology? Neurobiol Aging 2008; 29 : 566–573.
17. Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006; 314 : 130–133.
18. Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 2006; 351 : 602–611.
19. Chen-Plotkin AS, Lee VM, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol 2010; 6 : 211–220.
20. Cairns NJ, Bigio EH, McKenzie IRA. Neuropathologic and nosologic kriteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Denegeration. Acta Neuropathol 2007; 114 : 5–22.
21. Mackenzie IR, Neumann M, Cairns NJ, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendation. Acta Neuropathol 2009; 117 : 15–18.
22. Ferrer I, Martinez A, Blanco R, et al. Neuropathology of sporadic Parkinson disease before the appearance of parkinsonism: preclinical Parkinson disease. J Neural Transm 2011; 118 : 821–839.
23. Beach TG, Adler Ch H, Lue L, et al. Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 2009; 117 : 613–634.
24. McKeith IG, Dickson DW, Lowe DM, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology 2005; 65 : 1–11.
25. Wenning GK, Stefanova N, Jellinger KA, et al. Multiple system atrophy: a primary oligodendrogliopathy. Ann Neurol 2008; 64 : 239-246.
26. Vonsattel J-PG. Huntington disease models and human neuropatology: similarities and differences. Acta Neuropathol 2008; 115 : 55–69.
27. Geser F, Martinez-Lage M, Kwong LK, et al. Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: the TDP-43 diseases. J Neurol 2009; 256 : 1205–1214.
28. WHO manual for surveillance of human transmissible spongiform encephalopaties including variant Creutzfeldt-Jakob disease 2003. [http://whqlibdoc.who.int/publications/2003/9241545887.pdf]
29. Aguzzi A, Calella AM. Prions: protein aggregation and infectious diseases. Physiol Rev 2009; 89 : 1105–1152.
30. Hill AF, Joiner S, Wadsworth JD, et al. Molecular classification of sporadic Creutzfeldt-Jakob disease. Brain 2003; 126 : 1333–1346.
Štítky
Patologie Soudní lékařství ToxikologieČlánek vyšel v časopise
Česko-slovenská patologie

2012 Číslo 2
Nejčtenější v tomto čísle
- Neurodegenerativní onemocnění: přehled současné klasifikace a diagnostických neuropatologických kritérií
- Neuropatologie farmakorezistentní epilepsie - strukturální podklad a mechanismy epileptogeneze
- Vybrané biomarkery primárnych nádorov centrálneho nervového systému: krátky prehľad
- NÁDORY ASOCIOVANÉ S EPILEPSIÍ
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy