
Carneyho komplex
Carney complex
Carney complex is a clinically and genetically heterogeneous disease, with at least two genetic loci including the PRKAR1A gene located on chromosome 17 and the CNC2 locus mapped to chromosome 2. Clinically this syndrome is characterized by multiple myxomas occurring in different anatomic sites, mucocutaneous pigmentary lesions, and a variety of non-endocrine and endocrine tumors, often causing endocrine abnormalities, involving various organs.
Knowledge of morphological findings in CNC patients with their typical locations is necessary to raise suspicion of this syndrome by pathologists. Confirmation of the diagnosis allows regular clinical check-ups and early treatment of these patients.
Keywords:
Carney complex – cutaneous myxoma – cardiac myxoma – primary pigmented nodular adrenocortical disease – large-cell calcifying Sertoli cell tumor – psammomatous melanotic schwannoma – PRKAR1A gene – CNC2 locus
Autoři:
D. Kacerovská 1,2; M. Michal 1,2; R. Šíma 1,2; P. Grossmann 1,2; D. V. Kazakov 1,2
Působiště autorů:
Šiklův patologický ústav, FN a LF UK, Plzeň
1; Bioptická laboratoř s. r. o., Plzeň
2
Vyšlo v časopise:
Čes.-slov. Patol., 47, 2011, No. 4, p. 192-197
Kategorie:
Přehledový článek
Souhrn
Carneyho komplex (CNC) představuje klinicky i geneticky heterogenní onemocnění s nejméně dvěma známými genetickými lokusy zahrnujícími gen PRKAR1A lokalizovaný na chromozomu 17 a lokus CNC2 ležící na chromozomu 2. Klinicky je tento syndrom charakterizován mnohočetnými myxomy, kožními a slizničními pigmentovými lézemi a tumory, které mohou postihovat různé orgány a v některých případech vést k endokrinologickým abnormalitám.
Znalost morfologických nálezů CNC a jejich typické lokalizace umožňuje patologům vznést podezření na tento závažný syndrom. Odhalení diagnózy CNC vede k dispenzarizaci těchto pacientů příslušnými specialisty a k časné léčbě.
Klíčová slova:
Carneyho komplex – kožní myxom – srdeční myxom – primární pigmentovaná nodulární adrenokortikální choroba – velkobuněčný kalcifikující nádor ze Sertoliho buněk – psamomatózní melanotický schwannom – gen PRKAR1A – lokus CNC2
Carneyho komplex (CNC, zkratka z angl. Carney complex) je autozomálně dominantní dědičné onemocnění charakterizované mnohočetnými myxomy, kožními a slizničními pigmentovými lézemi a tumory, které mohou postihovat různé orgány a v některých případech vést k endokrinologickým abnormalitám (tab. 1)(1). Tento syndrom byl popsán v roce 1985 J. Aidanem Carneym (2) (pozn. nezaměnit za Carneyho triádu projevující se kombinací gastrointestinálního stromálního tumoru (GIST), paragangliomu a pulmonálního chondromu). Před rozpoznáním CNC jako samostatného syndromu byly tyto případy publikovány pod názvy „NAME“ syndrom (název pochází z počátečních písmen angl. nevi, atrial myxoma, myxoid neurofibromas, ephelides) (3) nebo „LAMB“ syndrom (z angl. lentigines, atrial myxomas, blue nevi) (4). CNC představuje klinicky i geneticky heterogenní onemocnění s nejméně dvěma známými genetickými lokusy zahrnujícími gen PRKAR1A (také známý pod názvem gen CNC1) lokalizovaný na chromozomu 17 a lokus CNC2 ležící na chromozomu 2 (5).
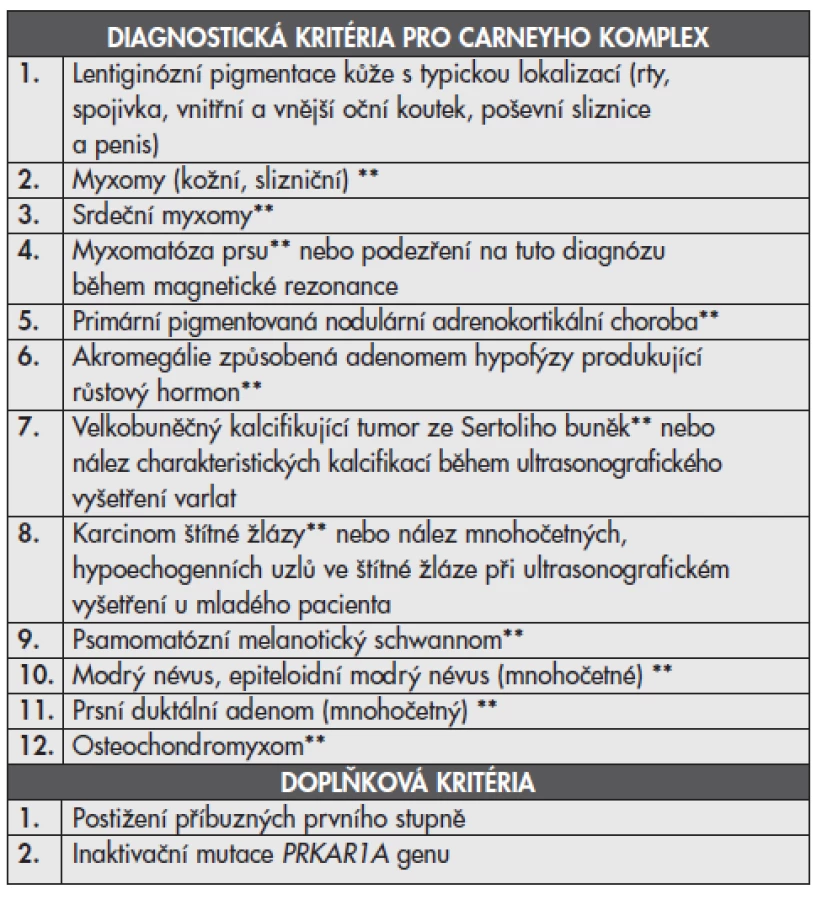
Pro tento syndrom je typická variabilní exprese a vysoká penetrance (téměř 100%). Věkový průměr stanovení diagnózy je 20. rok života. CNC je obvykle diagnostikován na podkladě srdečních myxomů, mnohočetných kožních myxomů nebo Cushingovy nemoci. Více než dvě třetiny případů je familiárních s poměrně nízkým počtem postižených členů rodiny (6). Sporadické případy bývají vzácné. Výskyt u mužů a žen je rovnoměrný. Mezi nejčastěji postižené orgány patří kůže, srdce, varlata, hypofýza a nadledviny (1,6,7).
KOŽNÍ A SLIZNIČNÍ PROJEVY
Kožní projevy vyskytující se u více než 75 % pacientů zahrnují myxomy, lentiginózní pigmentace, modré névy a epiteloidní modré névy (8,9).
Myxomy mohou vznikat kdekoliv na kůži, avšak typickou oblastí jejich výskytu je oční koutek, víčko, zvukovod a prsní bradavka. Tyto benigní tumory bývají mnohočetné a mívají vzhled poměrně dobře ohraničených papul a nodulů, hladkého povrchu, barvy narůžovělé, namodralé nebo zbarvení kůže, velikosti od 0,2 do 5 cm. Větší léze bývají obvykle polypovitého tvaru, vznikající často ve zvukovodu (8). Mikroskopicky jsou myxomy dobře ohraničené, neopouzdřené, na buňky chudé léze složené z roztroušených polygonálních, cípatých nebo vřetenitých buněk na hojném mucinózním pozadí s malými, lehce dilatovanými nebo štěrbinovitými cévami (obr. 1). V některých případech je možné v nádorových buňkách zastihnout jaderné pseudoinkluze. Častým nálezem jsou i změny epidermis ležící nad myxomem charakterizované nápadnou přítomností navzájem propojených pruhů epitelových buněk s periferním palisádováním (obr. 2). Vzácně může být myxoidní oblast vázaná na vlasový folikl. Existují i případy s nápadnou epitelovou proliferací v myxomatózní oblasti, s folikulární a/nebo sebaceózní diferenciací, včetně infundibulocystických struktur, lézí napodobujících trichofolikulom a proliferací napodobujících struktury pláště (mantlu) (8,10).


Lentigo se na kůži klinicky projevuje přítomností malých, hnědých makul, zejména kolem očí, rtů, na prstech, na dlaních a na genitální sliznici. Morfologicky je lentigo na rozdíl od pih, u kterých je počet melanocytů v epidermis normální a je pouze zvýšené množství pigmentu melaninu, charakterizováno hyperplazií melanocytů v bazální vrstvě epidermis, obvykle s prodloužením dermoepidermálních čepů (11,12).
Modré a epiteloidní modré névy nemají na rozdíl od lentiga typickou lokalizaci a mohou se vyskytovat kdekoliv na kůži jako malé pigmentované noduly. Mikroskopicky se v případě epiteloidního modrého névu jedná o intradermální proliferaci silně pigmentovaných polygonálních a/nebo dendritických buněk, které jsou od sebe odděleny kolagenními snopci (obr. 3) (13). Naopak modré névy jsou morfologicky charakterizovány přítomností intradermálních pigmentových dendritických buněk a melanofágů s hrubými melaninovými granuly, často uloženými zejména v okolí adnexálních struktur.

EXTRAKUTÁNNÍ PROJEVY
Kromě zmíněných kožních myxomů jsou pacienti s CNC ohroženi vznikem myxomů uložených v měkkých tkáních, prsu a srdci. Jak už bylo uvedeno výše, biologickým chováním se jedná o benigní tumory, avšak při lokalizaci nádoru v srdci bývá pacient ohrožen na životě. Mortalita u těchto případů je více než 50 % (1). Srdeční myxomy mohou vznikat v jakémkoliv srdečním oddíle, a to jak v síních, tak v komorách. Ve více než v polovině případů bývají myxomy mnohočetné (obvykle 2–3 ale i více). V důsledku srdeční obstrukce způsobené nádorem dochází k náhlým synkopám, srdeční nedostatečnosti, poruchám srdečního rytmu, které mohou vést až k náhlé smrti. Stejně tak odtržení nádorové hmoty s následnou embolizací je častou příčinou úmrtí. Jedinou účinnou léčbou je v tomto případě chirurgické odstranění nádoru, po kterém se však mohou objevovat recidivy. Právě to je důvodem, proč pacienti musí být pravidelně dispenzarizováni kardiologem, a to i po úspěšné kardiochirurgické operaci. Průměrný věk vzniku prvního srdečního myxomu je okolo 50. roku života, ale věkové rozpětí se pohybuje od 3 do 67 let (14). Mikroskopicky jsou srdeční myxomy tvořeny kulatými, polygonálními a cípatými buňkami, které jsou hojně obklopeny stromatem bohatým na mukopolysacharidy, rostoucími kolem cévních struktur (obr. 4A). Vzácně může být součástí tumoru glandulární komponenta (obr. 4B,C) (15).

Myxomy měkkých tkání jsou identické s jejich kožními analogy, avšak chybí zde epitelová komponenta. Myxoidní změny (tzv. „myxomatóza“) mohou být nálezem i u jiných tumorů, jako např. myxoidní neurofibrom, prsní myxoidní fibroadenom nebo mamární duktální adenom (16). Myxoidní fibroadenom může být u CNC solitární nebo mnohočetný, velikosti od několika milimetrů do 3 cm v průměru. Mikrokopicky tumor vykazuje typický obraz fibroadenomu doprovázený ložisky hypocelulárního myxoidního stromatu (obr. 5).

Dalším typickým nálezem u těchto nemocných je postižení nadledvin, tzv. primární pigmentovaná nodulární adrenokortikální choroba (PPNAD; zkratka z angl. primary pigmented nodular adrenocortical disease). Toto postižení vede ve většině případů k ACTH (adrenokortikotropní hormon) independentní formě Cushingova syndromu, který se začíná projevovat mezi 3. a 4. dekádou života. Jelikož se jedná o ACTH nezávislou formu, klinický obraz zahrnuje spíše astenismus nežli obezitu, těžkou osteoporózu, malý vzrůst a úbytek svalové hmoty (11). Ženy jsou postiženy častěji než muži. Do věku 40-ti let se u více než 70 % žen nesoucích mutaci genu PRKAR1A manifestuje PPNAD. Ačkoliv klinicky je odhaleno toto onemocnění pouze u 30–60 % případů, při pitvě je nález PPNAD přítomen u téměř všech pacientů s CNC (1). Postižení nadledvin je obvykle bilaterální. Makroskopický obraz je tvořen mnohočetnými, různě velikými uzly, barvy černé, hnědé a/nebo žluté. Mikroskopicky jsou tyto dobře ohraničené noduly tvořeny z velkých buněk s mírně pleomorfním jádrem a nápadným jadérkem, s hojnou, tmavě eozinofilní cytoplazmou obsahující granulky hnědého pigmentu lipofuscinu (obr. 6) (17).

Testikulární léze vyskytující se cca u 40 % pacientů zahrnují mnohočetné kalcifikace a velkobuněčný kalcifikující nádor ze Sertoliho buněk (LCCSCT). Vzácně se může vyskytovat tumor z Leydigových buněk a ložiska ektopické kůry nadledvin. Léze varlat bývají detekovány při palpačním nebo ultrasonografickém vyšetření. LCCSCT bývá často multicentrický, bilaterální a může vést k produkci estrogenů a tak způsobovat gynekomastii, předčasnou pubertu, event. snížení fertility (1,18,19). Mikroskopicky je LCCSCT obvykle solidní, trabekulární či nádorové buňky vytvářejí pruhy nebo solidní protáhlé struktury připomínající tubuly. Buňky mají objemnou, světle eozinofilní cytoplazmu, střídající se s pruhy buněk, které jsou menší a mají tmavě eozinofilní cytoplazmu. Charakteristickým mikroskopickým nálezem je i přítomnost ložisek koncentrických a lamelárních kalcifikací (obr. 7). Ve stromatu bývá často přítomna zánětlivá celulizace. Vzácně mohou být přítomny světlobuněčná diferenciace a změny napodobující deciduu. Nádorové buňky reagují pozitivně s inhibinem, S-100 proteinem a vimentinem (20). Většina těchto nádorů je jak histologicky tak klinicky benigních (21), avšak byly popsány případy histologicky maligních LCCSCT asociovaných s CNC (22).

Dalším nálezem u CNC je tzv. psamomatózní melanotický schwannom (PMS), který bývá obvykle multicentrický, s častým postižením zažívacího traktu (zejména jícnu a žaludku) a paraspinálních sympatických vláken (23). Tyto tumory jsou obvykle benigní, ale až 10 % z nich může metastazovat, a to zejména do plic. Léze postihující kost mohou vyústit v její destrukci. Velikost PMS se pohybuje v rozpětí od 0,5 do 25 cm v průměru. Morfologicky se jedná o solidní nebo fascikulární struktury složené z vřetenitých a/nebo epiteloidních buněk s jemnými nebo hrubými melaninovými granuly v cytoplazmě. Typickým mikroskopickým nálezem je i přítomnost kalcifikovaných psamomatózních tělísek (obr. 8). Až u poloviny případů může být zachycena lipomatózní metaplazie. Vzácně může být také přítomno vírovité uspořádání nebo palisádování buněk (23).

Až u 75 % postižených se nacházejí mnohočetné nefunkční uzly ve štítné žláze. Většina z nich představuje folikulární adenomy, ze kterých mohou vzácně vznikat papilární nebo folikulární karcinomy (1,24).
Pravidlem u těchto nemocných je postižení hypofýzy, ale pouze u menšiny pacientů (10–12 %) dochází k akromegálii vzniklé na podkladě adenomu produkujícího růstový hormon. Byly také popsány adenomy produkující prolaktin (1).
Součástí tohoto syndromu je i osteochondromyxom, který může být vrozený, anebo vzniká v neonatálním či dětském období. Prezentuje se jako nebolestivá masa postihující diafýzu distálních dlouhých kostí, nazální kosti a sinusy (25). Mikroskopicky je charakterizován nálezem polymorfních buněk nezhoubného vzhledu uspořádaných do pruhů a lobulů na myxoidním, chrupavčitém, kostním a fibrózně hyalinním pozadí.
MOLEKULÁRNÍ BIOLOGIE
CNC je geneticky heterogenní onemocnění. Doposud byly zmapovány dvě chromozomální oblasti související s tímto syndromem. V oblasti 17q24 byl nalezen gen PRKAR1A (někdy označován jako gen CNC1), který kóduje regulační podjednotku cAMP-dependentní protein kinázy A, významnou efektorovou molekulu v mnohých endokrinních signálních drahách, a v oblasti 2p16 byl detekován lokus CNC2 (5,26). Inaktivační mutace PRKAR1A genu bývá zjištěna cca u 70 % všech případů CNC (obr. 9) (14,27). Doposud bylo popsáno více než 80 mutací zahrnujících bodové mutace, krátké delece, inzerce nebo komplexní změny. Rovněž byly detekovány rozsáhlé delece tohoto genu postihující až 4165 bp (28). Mutace může být zastižena v jakékoliv části PRKAR1A genu, avšak nejvíce se vyskytuje v exonech 3, 4, 6, 8 a 9 (číslování exonů podle NCBI referenční sekvence NG_007093.2). Dvě mutace vyskytující se nezávisle v několika nepříbuzných rodinách různého etnického původu byly navrženy jako tzv. „hot spots“ mutace genu PRKAR1A, a to c.709-7del6 v intronu 8 a c.491-492delTG v exonu 6 (14).

V naší laboratoři rutinně provádíme vyšetření deseti kódujících exonů genu PRKAR1A, včetně exon-intronových spojů, z nádorové tkáně nebo periferní krve za použití PCR amplifikace a metody přímého sekvenování.
KORELACE GENOTYP – FENOTYP
Korelace mezi genotypem a fenotypem u CNC nemohla být po dlouhou dobu stanovena až do doby, než byla publikována sestava 353 pacientů s CNC s 80 odlišnými fenotypy (14). Tato studie umožnila rozdělit CNC minimálně do 3 skupin. První skupina (70 %) zahrnuje pacienty, kteří jsou nositeli mutace v genu PRKAR1A. Ve druhé skupině jsou postižení, u kterých nebyla prokázána žádná alterace genu PRKAR1A. Poslední skupina je tvořena malým počtem nemocných s izolovaným nálezem PPNAD. Pacienti patřící do první skupiny (nositelé mutace genu PRKAR1A) představovali většinou familiární případy s typickým klinickým obrazem (mnohočetné myxomy, schwannomy, tumory varlat a štítné žlázy) vznikajícím v mladém věku. U těchto nemocných byla hot spot mutace c.709-7del6 asociována hlavně s PPNAD, zatímco hot spot mutace c.491-492delTG byla častěji spojena s výskytem srdečních myxomů, kožních lentiginózních projevů a tumorů štítné žlázy. Druhá skupina, která splňovala diagnostická kritéria CNC, ale u které nebyly nalezeny žádné alterace v genu PRKAR1A, byla tvořena většinou sporadickými a méně familiárními případy asociovanými s abnormalitami v oblasti lokusu CNC2. Manifestace onemocnění u těchto nemocných byla v pozdějším věku charakterizována vznikem myxomů, tumorů štítné žlázy, LCCSCT a PMS (14). Nemocní s izolovaným nálezem PPNAD vykazovali mírný fenotyp (téměř výhradně Cushingův syndrom vzhledem k přítomné PPNAD), s nenápadnými specifickými dermatologickými nálezy. U některých z nich byla prokázaná zárodečná mutace v intronu 8 [exon 7 IVS del (-7→-2), dle současné nomenklatury je tato mutace zapisována c.709-7del6] genu PRKAR1A (29). Další pacienti v této skupině byly zejména děti s ACTH independentní mikronodulární adrenokortikální chorobou bez dalších klinických manifestací CNC a bez nálezu alterace ve dvou zmíněných lokusech (tj. gen PRKAR1A a lokus CNC2) (14).
ZÁVĚR
Znalost morfologických nálezů a jejich typické lokalizace u CNC umožňuje patologům vznést podezření na tento závažný syndrom. Dalším krokem jsou pak příslušná klinická vyšetření (tj. vyšetření kardiologem, dermatologem, endokrinologem, urologem atd.) a molekulárně biologická analýza z periferní krve nebo nádorové tkáně k průkazu zárodečné mutace v příslušném genu. Včasné odhalení diagnózy CNC umožňuje těmto pacientům jejich dispenzarizaci příslušnými specialisty a včasnou léčbu. Důležité jsou především pravidelné kardiologické kontroly vedoucí k detekci srdečních myxomů. Právě tyto nádory pacienty ohrožují na životě a mohou vést k náhlému úmrtí bez ohledu na věk.
Adresa pro korespondenci:
MUDr. Denisa Kacerovská, Ph.D.
Šiklův patologicko-anatomický ústav
Alej Svobody 80, 304 60 Plzeň
tel: +420-377104645, fax: +420-377104650
e-mail: kacerovska@medima.cz
Zdroje
1. Stratakis CA, Kirschner LS, Carney JA. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab 2001; 86 : 4041–4046.
2. Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VL. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine (Baltimore) 1985; 64 : 270–283.
3. Atherton DJ, Pitcher DW, Wells RS, MacDonald DM. A syndrome of various cutaneous pigmented lesions, myxoid neurofibromata and atrial myxoma: the NAME syndrome. Br J Dermatol 1980; 103 : 421–429.
4. Rhodes AR, Silverman RA, Harrist TJ, Perez-Atayde AR. Mucocutaneous lentigines, cardiomucocutaneous myxomas, and multiple blue nevi: the “LAMB” syndrome. J Am Acad Dermatol 1984; 10 : 72–82.
5. Casey M, Mah C, Merliss AD, et al. Identification of a novel genetic locus for familial cardiac myxomas and Carney complex. Circulation 1998; 98 : 2560–2566.
6. DeLellis RA, Lloyd RV, Heitz PU, Eng CE. World Health Organization of Tumours. Pathology and Genetics of Tumours of Endocrine organs. Lyon: IARCPress; 2004.
7. LeBoit PE, Günter B, Weedon D, Sarasin A. Pathology & Genetics Skin Tumours. World Health Organization Classification of Tumours. Lyon: IARCPress; 2006.
8. Carney JA, Headington JT, Su WP. Cutaneous myxomas. A major component of the complex of myxomas, spotty pigmentation, and endocrine overactivity. Arch Dermatol 1986; 122 : 790–798.
9. Carney JA. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Arch Intern Med 1987; 147 : 418–419.
10. Kacerovska D, Sima R, Michal M, et al. Carney complex: a clinicopathologic and molecular biological study of a sporadiccase, including extracutaneous and cutaneous lesions and a novel mutation of the PRKAR1A gene. J Am Acad Dermatol 2009; 61 : 80–87.
11. Stratakis CA. Genetics of Peutz-Jeghers syndrome, Carney complex and other familial lentiginoses. Horm Res 2000; 54 : 334–343.
12. Cook CA, Lund BA, Carney JA. Mucocutaneous pigmented spots and oral myxomas: the oral manifestations of the complex of myxomas, spotty pigmentation, and endocrine overactivity. Oral Surg Oral Med Oral Pathol 1987; 63 : 175–183.
13. Carney JA, Stratakis CA. Epithelioid blue nevus and psammomatous melanotic schwannoma: the unusual pigmented skin tumors of the Carney complex. Semin Diagn Pathol 1998; 15 : 216–224.
14. Bertherat J, Horvath A, Groussin L, et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5’-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab 2009; 94 : 2085–2091.
15. Pucci A, Bartoloni G, Tessitore E, Carney JA, Papotti M. Cytokeratin profile and neuroendocrine cells in the glandular component of cardiac myxoma. Virchows Arch 2003; 443 : 618–624.
16. Carney JA, Stratakis CA. Ductal adenoma of the breast and the Carney complex. Am J Surg Pathol 1996; 20 : 1154–1155.
17. Lloyd RV, Douglas BR, Young WF. Endocrine diseases. Atlas of nontumor pathology. First series. Fascicle 1. AFIP, Washington, DC; 2002.
18. Premkumar A, Stratakis CA, Shawker TH, Papanicolaou DA, Chrousos GP. Testicular ultrasound in Carney complex: report of three cases. J Clin Ultrasound 1997; 25 : 211–214.
19. Washecka R, Dresner MI, Honda SA. Testicular tumors in Carney’s complex. J Urol 2002; 167 : 1299–1302.
20. Hes O, Michal M, Mukenšnabl P, Veličkinová H, Hora M, Boudová L. Nádory varlat. EUROVERLAG: Plzeň; 2007.
21. Jayasena SN, Ariyasinghe JT, Gunawardena DM, Gunawardena SA, de Silva MV. Large-cell calcifying sertoli cell tumour of the testis detected at screening of a family with Carney syndrome. Urol Int 2005; 75 : 365–367.
22. Kratzer SS, Ulbright TM, Talerman A, et al. Large cell calcifying Sertoli cell tumor of the testis: contrasting features of six malignant and six benign tumors and a review of the literature. Am J Surg Pathol 1997; 21 : 1271–1280.
23. Carney JA. Psammomatous melanotic schwannoma. A distinctive, heritable tumor with special associations, including cardiac myxoma and the Cushing syndrome. Am J Surg Pathol 1990; 14 : 206–222.
24. Stratakis CA, Courcoutsakis NA, Abati A, et al. Thyroid gland abnormalities in patients with the syndrome of spotty skin pigmentation, myxomas, endocrine overactivity, and schwannomas (Carney complex). J Clin Endocrinol Metab 1997; 82 : 2037–2043.
25. Carney JA, Boccon-Gibod L, Jarka DE, et al. Osteochondromyxoma of bone: a congenital tumor associated with lentigines and other unusual disorders. Am J Surg Pathol 2001; 25 : 164–176.
26. Stratakis CA, Carney JA, Lin JP, et al. Carney complex, a familial multiple neoplasia and lentiginosis syndrome. Analysis of 11 kindreds and linkage to the short arm of chromosome 2. J Clin Invest 1996; 97 : 699–705.
27. Kirschner LS, Sandrini F, Monbo J, Lin JP, Carney JA, Stratakis CA. Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with the carney complex. Hum Mol Genet 2000; 9 : 3037–3046.
28. Horvath A, Bossis I, Giatzakis C, et al. Large deletions of the PRKAR1A gene in Carney complex. Clin Cancer Res 2008; 14 : 388–395.
29. Groussin L, Horvath A, Jullian E, et al. A PRKAR1A mutation associated with primary pigmented nodular adrenocortical disease in 12 kindreds. J Clin Endocrinol Metab 2006; 91 : 1943–1949.
Štítky
Patologie Soudní lékařství ToxikologieČlánek vyšel v časopise
Česko-slovenská patologie

2011 Číslo 4
Nejčtenější v tomto čísle
- Carneyho komplex
- Prediktivní diagnostika HER2 v adenokarcinomu žaludku
- Prediktivní diagnostika u karcinomu prsu
- „Gigantický“ bazocelulárny karcinóm kože hlavy s intrakraniálnou propagáciou – kazuistika
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy