
Histopatologická diagnostika mitochondriálních myopatií – indikace a přínos svalové biopsie
Histopathological Diagnosis of Mitochondrial Myopathies – Indications and the Utility of Muscle Biopsy
Mitochondrial myopathies make up a complex heterogenous group of diseases characterized by mitochondrial dysfunction. Mitochondrial disorders are caused biochemically, by impairment of the oxidative phosphorylation system formed by five multi-subunit polypeptide complexes (I-V) located within the inner mitochondrial membrane, along with two electron carriers (Q10 and cytochrome c). The electron carriers and complex II are nuclear DNA-encoded, while the remaining complexes are encoded both by nuclear and mitochondrial DNA. Classification of mitochondrial disorders is rendered complicated by clinical and genetic heterogeneity. Morphological examination is based on evaluation of the general pattern of histopathological changes in a muscle biopsy, of quantitative and qualitative alteration of the mitochondria and, last but not least, on assessment of the presence/absence of ragged red fibres in relation to the cytochrome c oxidase and succinyldehydrogenase reactivity. Combined with immunohistochemistry and in situ hybridisation, this may allow assessment of the type of heredity and/or point to a disorder in a certain complex of the oxidative phosphorylation chain.
Key words:
mitochondrial myopathy – muscle biopsy – diagnosis – classification
Autoři:
O. Souček 1
![]() ; P. Ješina 2; J. Zeman 2; M. Elleder 3; H. Hůlková 3; Z. Lukáš 1
; P. Ješina 2; J. Zeman 2; M. Elleder 3; H. Hůlková 3; Z. Lukáš 1
Působiště autorů:
Ústav patologie LF MU a FN Brno
1; Klinika dětského a dorostového lékařství 1. LF UK a VFN v Praze
2; Ústav dědičných a metabolických poruch 1. LF UK a VFN v Praze
3
Vyšlo v časopise:
Cesk Slov Neurol N 2011; 74/107(4): 428-436
Kategorie:
Přehledný referát
Souhrn
Mitochondriální myopatie jsou komplexní heterogenní skupina onemocnění charakterizovaná funkční poruchou mitochondrií, která může být spojena s jejich morfologickými abnormitami. Klasifikace mitochondriálních myopatií je komplikovaná. Stejný typ mutace se může projevovat variabilním klinickým obrazem, a naopak velmi podobný klinický obraz může být způsoben různými mutacemi. Bioptická diagnostika spočívá v posouzení základního vzorce histopatologických změn v biopsii, zhodnocení morfologických odchylek mitochondrií a v neposlední řadě ve zjištění přítomnosti či absence tzv. ragged red fibres ve vztahu k reaktivitě na cytochrom-c-oxidázu (COX) a sukcinyldehydrogenázu (SDH). Spolu s imunohistochemickým vyšetřením a in situ hybridizací může posoudit i typ dědičnosti či příslušnost onemocnění některému z komplexů oxidativně-fosforylačního řetězce.
Klíčová slova:
mitochondriální myopatie – svalová biopsie – diagnóza – klasifikace
Úvod
Mitochondriální poruchy energetického metabolizmu představují velkou a heterogenní skupinu klinicky závažných onemocnění projevujících se izolovaným nebo kombinovaným postižením různých tkání a orgánových systémů. Nejčastěji jsou postiženy tkáně s vysokými energetickými nároky na funkci, především centrální nervová soustava, svaly a srdce. Mitochondriální onemocnění vznikají na podkladě metabolické poruchy v systému oxidativní fosforylace [1–3], tedy poruchy způsobené funkčním defektem v mitochondriálním respiračním řetězci. V tomto sdělení úvodem nastíníme stavbu tohoto řetězce, strukturu a vlastnosti mitochondriální DNA (mtDNA) a základy mitochondriální dědičnosti. V další části shrneme základní klinické a biochemické znaky mitochondriálních onemocnění, které by indikovaly svalovou biopsii, a nakonec možný přínos svalové biopsie k jejich diagnostice.
Genetická klasifikace
Mitochondriální DNA (mtDNA) je nositelka mimojaderné dědičnosti. V buňce je molekula mtDNA přítomna v mnoha kopiích, jedna mitochondrie obsahuje cca 10–20 kopií, každá buňka má přibližně 1 000 mitochondrií (polyplazmie); mutací postižená mtDNA se obvykle nachází pouze v části mitochondrií v buňce (heteroplazmie) a k porušení oxidativní fosforylace je nutné určité kritické procento kopií mutované mtDNA (prahový efekt). Vzhledem k tomu, že mtDNA pochází z mateřské zárodečné buňky, onemocnění způsobená patogenními mutacemi mtDNA se dědí po mateřské linii. V následných buněčných generacích se podíl mutované mtDNA či stupeň heteroplazmie může měnit (replikativní segregace) a podle toho může konkrétní onemocnění v různé míře postihovat jednotlivé orgány (tkáňová heterogenita). Ojedinělé bývají spontánní rozsáhlé delece mtDNA, které se vyskytují sporadicky. Těmito základními rysy se mitochondriální dědičnost liší od dědičnosti mendelovské.
Mitochondriální DNA kóduje 13 z cca 90 proteinů respiračního řetězce, všechny ostatní jsou kódovány jadernou DNA (nDNA). Transkript nDNA přechází z jádra do cytoplazmy a odtud do mitochondrie, kde se translací syntetizovaný protein začlení do vnitřní mitochondriální membrány, v níž se spojuje s proteiny kódovanými mtDNA. Ta kromě strukturálních proteinů kóduje dva ribozomální rRNA a 22 transferových tRNA využívané při transkripci a translaci v mitochondriích.
Elektronový transportní řetězec spřažuje chemické reakce mezi donory elektronů a akceptorem elektronů (O2). NADH a sukcinát generované v cyklu kyseliny citronové jsou začleněny do transportního řetězce, kde jsou oxidovány, čímž je získávána energie pro ATP-syntázu. Struktura respiračního řetězce, řazení jednotlivých komplexů a toku protonů a elektronů jsou schematicky znázorněny na obr. 1.


V komplexu I (podjednotky NADH-dehydrogenázy) jsou z NADH odebrány dva elektrony a přeneseny na nosič ubichinon (Q) rozpustný v lipidech. Redukovaný ubichinol (QH2) difunduje uvnitř membrány. Komplex I přemístí čtyři protony přes membránu, čímž vytvoří protonový gradient.
V komplexu II (čtyři podjednotky sukcinyldehydrogenázy, SDH) jsou další elektrony ze sukcinátu předány na ubichinon (Q).
V komplexu III (cytochrom bc) jsou odebrány dva elektrony z QH2 a přeneseny na dvě molekuly cytochromu c, ve vodě rozpustného nosiče umístěného v mezimembránovém prostoru. Protonový gradient se tvoří oxidací dvou molekul ubichinolu (4QH2 + 4e–) s tvorbou chinolu (2QH + 2e).
Komplex IV (cytochrom c oxidáza) je komplex 10 podjednotek kódovaných nDNA a tří kódovaných mtDNA. V komplexu jsou čtyři elektrony odebrány ze čtyř molekul cytochromu c a přeneseny na molekulární kyslík za vzniku dvou molekul vody. Kyslík se dostane do mitochondrie difuzí. Současně jsou čtyři protony přemístěny přes membránu a přispívají k protonovému gradientu. Aktivita cytochromu c je inhibována kyanidem.
Spřažení s oxidativní fosforylací. Elektronový transportní řetězec a oxidativní fosforylace jsou spřaženy protonovým gradientem přes vnitřní mitochondriální membránu. Tok elektronů z mitochondriální matrix vytváří elektrochemický (protonový) gradient. Tento gradient využívá komplex ATP-syntázy k tvorbě ATP oxidativní fosforylací. ATP-syntáza se někdy nazývá komplex V. Jedna složka ATP-syntázy funguje jako iontový kanál, který zprostředkuje tok protonů zpět do mitochondriální matrix. Reflux uvolňuje volnou energii produkovanou při generaci oxidovaných forem elektronových nosičů NADH a Q. Volné energie je použito k uskutečnění syntézy ATP.
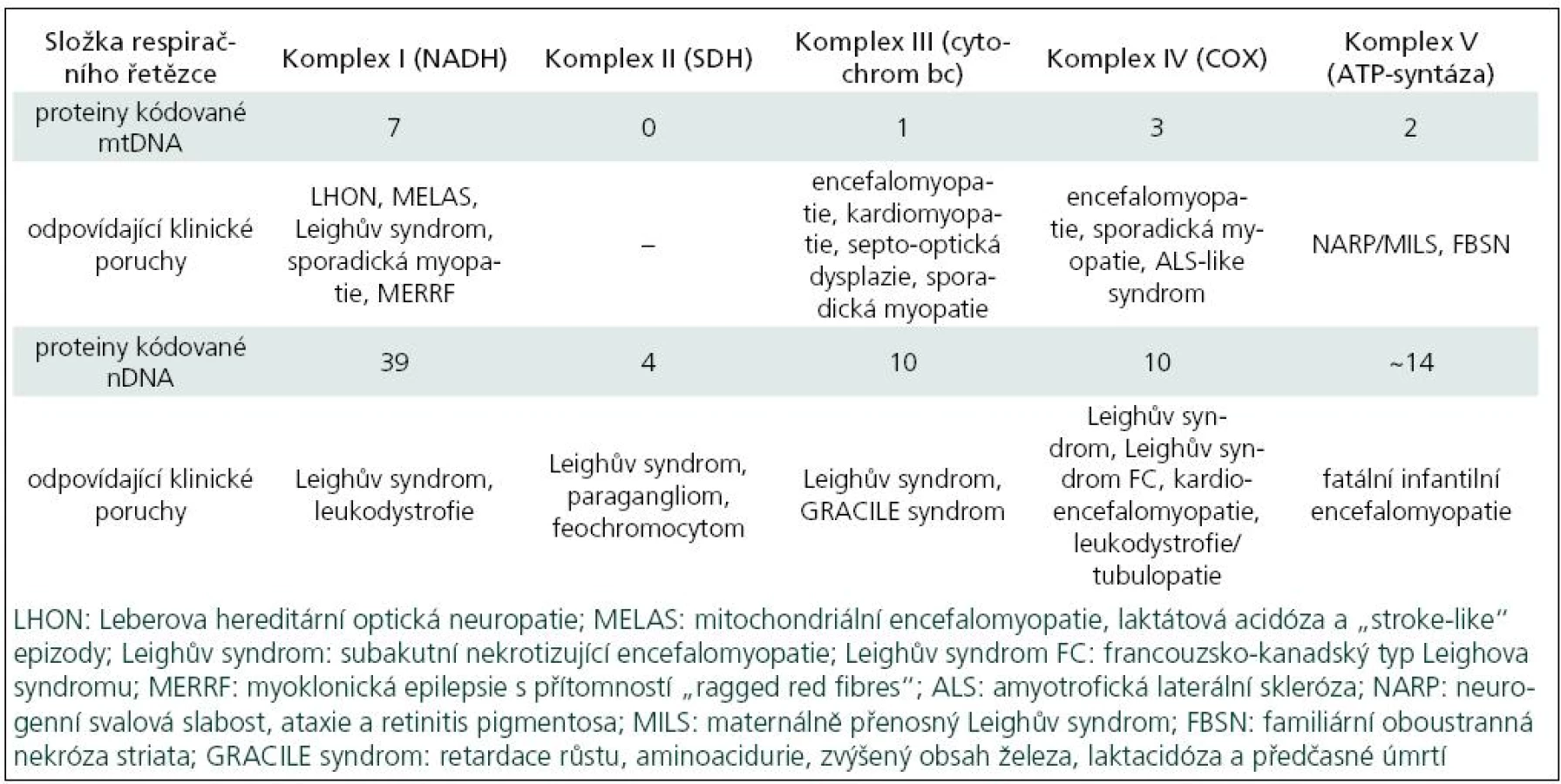
Poruchy oxidativní fosforylace je možno klasifikovat podle klinických, biochemických a molekulárně-genetických znaků. Z biochemického pohledu může jít o izolované nebo mnohočetné enzymové deficity. Tab. 1 ukazuje vztah genetických poruch k některým klinickým syndromům. Lze z ní vyčíst, že některé syndromy (Leighův, sporadická myopatie aj.) se mohou vztahovat k několika různým genetickým a biochemickým poruchám.
Klinická a biochemická klasifikace
Klinická klasifikace mitochondriálních onemocnění je komplikovaná: z předchozího textu vyplývá, že stejný typ mutace se může projevit různými klinickými příznaky, a naopak velmi podobný klinický obraz může být způsoben mutacemi v různých genech [2,3].
Výskyt mitochondriálních onemocnění v populaci se odhaduje 1 : 4 000, ale díky kvalitnější a dostupnější diagnostice se nadále zvyšuje. První klinické projevy mitochondriálního onemocnění se mohou objevit ve všech věkových skupinách od novorozeneckého věku až po stáří, časnější projevy jsou obvykle spojeny s těžším průběhem onemocnění a méně příznivou prognózou. Příznaky mitochondriálního onemocnění mohou postihnout kteroukoliv tkáň, a to jak izolovaně, například zrak u Leberovy hereditární optické neuropatie (syndrom LHON), tak i v rozmanitých kombinacích. Nejčastější klinické projevy mitochondriálních onemocnění jsou uvedeny v tab. 2 [4].

Diagnostika mitochondriálních onemocnění vychází z podrobného klinického vyšetření a rodinné i osobní anamnézy, ale pro diagnostiku jsou obvykle potřebné i zobrazovací metody a biochemické a molekulární analýzy. Například zvýšená hladina laktátu a alaninu v krvi a mozkomíšním moku u předškolního dítěte s podezřením na neurodegenerativní onemocnění a symetrická ložiska v bazálních gangliích a/nebo v mozkovém kmeni viditelná při vyšetření MR svědčí pro Leighův syndrom, ale až enzymatické a molekulárněbiologické vyšetření ve svalové biopsii nebo v kultivovaných kožních fibroblastech určí konkrétní etiologii onemocnění a typ dědičnosti. Nejčastější laboratorní nálezy u pacientů s mitochondriální poruchou jsou uvedeny v tab. 3.

Spektrofotometrická a radioizotopová měření aktivit jednotlivých mitochondriálních komplexů, elektroforetické/imunochemické analýzy stanovující množství a složení jednotlivých enzymů a molekulárně genetické vyšetření mitochondriálně nebo jaderně kódovaných genů jsou finančně i časově velice náročná [5]. U některých klinických jednotek, mezi které patří syndromy NARP (neuronální ataxie, retinitis pigmentosa), MELAS (mitochondriální encefalopatie, laktátová acidóza a iktu podobné příhody), MERRF (myoklonická epilepsie a ragged red fibres) i syndrom LHON, je dostupná přímá molekulárně genetická diagnostika v DNA izolované ze vzorku krve, vlasových foliklů či bukálního stěru, ale negativní nález z důvodu tkáňové heterogenity neznamená vyloučení nemoci.
Proto i v dnešní době má nezastupitelnou roli při diagnostice mitochondriálních onemocnění i histologické a histochemické vyšetření svalové biopsie. V tomto příspěvku chceme ukázat, jaké jsou základní histopatologické rysy mitochondriálních myopatií (MM), stejně jako znaky, dovolující vymezit některé skupiny těchto onemocnění.
Metodika zpracování svalové biopsie
Přestože poruchy oxidativně-fosforylačního systému mají multiorgánový rozsah (hepatopatie, kardiomyopatie, encefalopatie s leukodystrofií, retinopatie), zůstává nejčastější bioptickou metodou k histopatologické diagnostice svalová biopsie. Protože je biopsie vysoce invazivní metoda, je třeba k její indikaci přistoupit na základě důkladného zhodnocení klinických a laboratorních dat, jak je uvedeno v předchozím oddíle.
Nativní tkáň musí být hluboce zmrazena a zpracována pomocí přehledných barvení (kromě barvení hematoxylinem a eozinem je velmi přínosný tzv. Gomoriho trichrom) a konvenčních histochemických a imunohistochemických reakcí běžně používaných v diagnostické myopatologii [6,7]. Tyto reakce slouží k typové klasifikaci svalových vláken (myozinová ATP-áza), znázornění lyzosomálních hydroláz (kyselá fosfatáza), mitochondriálních dehydrogenáz (cytochromoxidáza (COX)), sukcinyldehydrogenáza (SDH) a NADH--tetrazolium reduktáza), glycidů (PAS) a lipidů (olejová červeň, černý sudan). V poslední době se v diagnostice MM uplatňují i imunohistochemické metody ke znázornění jednotlivých polypeptidových komplexů i metody flourescenční in situ hybridizace. Část vzorku ve formě svazečku vláken bývá po odběru vložena do glutaraldehydu k eletronmikroskopickému vyšetření.
Histopatologické nálezy u mitochondriáních myopatií
Základním histopatologickým vzorcem u MM je tzv. myogenní léze s kolísajícím rozsahem strukturálních změn od minimálních až po velmi zřetelné, jako atrofie či nekróza. Barvení Gomoriho trichromem je u svalových biopsií metodou volby. V normálním svalu nacházíme zeleno-modré zbarvení sarkoplazmy, jádra jsou červená, mitochondrie jsou patrné ve formě drobných červených teček. U MM barvení Gomoriho trichromem umožní identifikovat tzv. ragged red fibres (RRF) – jde o svalová vlákna „rozedraného“ vzhledu, s hrubými subsarkolemálními a intermyofibrilárními depozity červené barvy (obr. 2). Vzhled RRF je podmíněn proliferací a akumulací mitochondrií [8], kterou lze prokázat i histoenzymatickou detekcí zvýšené aktivity mitochondriálních dehydrogenáz, a to zejména SDH, neboť sukcinyldehydrogenáza je lokalizována výlučně v mitochondriích. V normálním svalu je rozložení reakčního produktu mitochondriálních dehydrogenáz odstupňováno v závislosti na typu svalového vlákna – vlákna typu 1 (převážně oxidativní) jsou tmavší, vlákna typu 2 (převážně glykolytická) světlejší. U encefalomyopatie MELAS dále nacházíme intenzivní reakci na SDH ve stěně cév [9]. V případě poruchy komplexu II bylo popsáno vymizení SDH reaktivity [10,11]. Další metodou průkazu mitochondriálních abnormit je hodnocení reaktivity cytochromoxidázy (COX), jejíž podjednotky tvoří komplex IV. Oslabená nebo vymizelá reakce COX je výrazem poruchy jejích podjednotek ve vnitřní mitochondriální membráně. V normě je intenzita reakce svalových vláken závislá na typu svalového vlákna obdobně jako u SDH – vlákna typu 1 jsou tmavší, vlákna typu 2 světlejší. Při některých metabolických poruchách nebo MM bývá pozorována nadměrná akumulace hydrofobních lipidů v sarkoplazmě (viz níže).

Ultrastrukturální změny se týkají odchylek v počtu, distribuci nebo velikosti mitochondrií, jejich případných strukturálních změn a přítomnosti inkluzí [12,13]. V normálním svalu jsou mitochondrie lokalizovány intermyofibrilárně v blízkosti I-proužku a subsarkolematicky ve formě menších shluků. MM jsou elektronmikroskopicky charakterizovány nálezem velkých agregátů strukturálně abnormálních mitochondrií (obr. 3). Změny struktury mitochondrií se týkají abnormit mitochondriálních krist. Může se jednat o jejich ztluštění, koncentrické nebo podélně řazení, případně angulární nebo tubulární transformaci. Inkluze u mitochondriálních myopatií jsou dvojího typu – kulovité osmiofilní nebo podélné krystaloidní (obr. 4, 5). Světelně mikroskopickým korelátem mitochondriálních agregátů jsou RRF a zvýšená intenzita reakce na SDH popsané výše.



Obecně lze říci, že histopatologický obraz myogenní léze je u MM zcela necharakteristický; i ultrastrukturální změny mitochondrií jsou samy o sobě rovněž málo specifické, neboť obdobné změny lze prokázat i u myozitid a svalových dystrofií [14] a jejich nález zhodnocuje jen přítomnost histochemických a dalších znaků spojených s mitochondriálními abnormitami.
Přítomnost RRF rovněž není zcela patognomická pro mitochondriální poruchy, neboť podobné struktury se vyskytují sekundárně i u některých jiných nervosvalových onemocnění, např. myozitid typu „inclusion body myositis“ nebo dermatomyozitidy, a případně i v průběhu stárnutí [15–18]. Deficit COX bývá někdy omezen na určité úseky svalových vláken (několik µm až několik milimetrů), takže negativní nález ve tkáňových řezech neznamená jejich absenci [19].
Skupinové zařazení MM na základě histopatologického nálezu
Histoenzymatický profil mitochondriální reaktivity je třeba interpretovat v kontextu klinického obrazu i biochemického nálezu. Zjištění přítomnosti či absence RRF ve vztahu k reaktivitě COX a reaktivita SDH ve svalových vláknech dovoluje v rámci MM rozlišit několik skupin, uvedených v tab. 4 [20–33].

Imunohistochemie přinesla řadu poznatků o vlastnostech MM, avšak teprve recentně připravené protilátky proti jednotlivým podjednotkám oxidativně-fosforylačního komplexu slibují pokrok k jejich rozlišení: normální svalová vlákna vykazují charakteristickou mozaiku reaktivity na komplexy I–V podle obsahu mitochondrií. U pacientů s defektem tRNA byla zjištěna vyšší heterogenita pro komplexy I a IV. RRF u pacientů se syndromem MERRF byla v průkazu komplexů I a IV negativní, zatímco u pacientů se syndromem MELAS byl komplex I negativní a komplex IV pozitivní. Znázornění komplexu I odhalilo dva pacienty s defektem tRNA, kteří unikli běžnému histochemickému vyšetření [34].
Z našeho archivu
Na závěr demonstrujeme přínos popsaného postupu na konkrétních případech.
Případ 1
Žena 34 let, s inzulin-dependentním diabetem, těžkou myopií a hypakuzí. V rodinné anamnéze zjištěny kalcifikace CNS u otce, u bratra blíže nespecifikovaná porucha chůze (nebyl vyšetřován). Pacientka byla poprvé hospitalizována v roce 2007 po epileptickém záchvatu, v dalším průběhu následovaly záchvaty se sekundární generalizací s ložiskovou abnormitou parieto-okcipitálně a temporo-okcipitálně vpravo. Na CT mozku popsány ischemické změny temporo-parietálně vpravo a dorzo-parietálně vlevo, kalcifikace v centrálních oblastech mozečkových hemisfér a v ncl. lentiformis, caput ncl. caudati a thalamu bilaterálně, laboratorně intermitentně zvýšená hladina laktátu. Klinický i biochemický nález tedy budil podezření na encefalomyopatii MELAS.
Histologicky ze svalové biopsie zjištěny nevýrazné velikostní a strukturální změny svalových vláken v HE, avšak přítomnost RRF v Gomoriho trichromu i v SDH. Intenzita reakce COX kolísala podle typu vláken, ojediněle byla přítomna i vlákna negativní. RRF vykazovala silně pozitivní reakci na COX (obr. 6). Nález tedy odpovídal výše popsanému zařazení do skupiny 1 (RRF přítomna/COX+) kompatibilnímu s MELAS. Vyšetření ultrastruktury přineslo nález různých mitochondriálních abnormit (velikostních, strukturálních i distribučních, včetně krystaloidních inkluzí). Neobvyklým nálezem byla v tomto případě přítomnost vláknitých inkluzí v sarkoplazmě (obr. 7, 8), s největší pravděpodobností jde o svazky aktinových filament.



Mutační analýza přinesla nález potvrzující mutace A3243G (bodová mutace tRNA), která je zodpovědná za cca 80 % případů MELAS.
Případ 2
1,5letá dívka s poruchou motorického vývoje od 10. měsíce života progredující do těžkého myopatického syndromu, od 12 měsíců imobilní, v 15 měsících závislá na umělé plicní ventilaci, objevuje se rovněž porucha polykání s nutností gastrostomie a rozvíjí se hypertrofické kardiomyopatie. Mentální projev po celou dobu bez známek patologie. U pacientky bylo provedeno MR vyšetření mozku s nálezem difuzní signálových změn v bílé hmotě obou mozkových hemisfér, subkortikálně a při IV. komoře. Biochemická analýza kosterního svalu odhalila sníženou aktivitu komplexu IV. mitochondriálního respiračního řetězce (COX) a mírně sníženou aktivitu komplexu I. Vzhledem k výsledkům vyšetření bylo vysloveno podezření na mitochondriální etiologii onemocnění na úrovni cytochromoxidázy.
Histologickým vyšetřením byl zjištěn myopatický vzorec s velikostním kolísáním průměrů svalových vláken, regresivními změnami typu flokulace a nekróz a fibrolipomatózním rozšířením intersticia. Průkaz mitochondriálních dehydrogenáz odhalil kolísající reaktivitu od slabší až po velmi intenzivní, přítomnost RRF nebyla jednoznačná. Vyšetření COX ukázalo prakticky totální absenci reaktivity v extrafuzálních svalových vláknech, v intrafuzálních vláknech byla reaktivita nicméně zachována (obr. 9). Nález tedy odpovídal skupinovému zařazení 5.

Molekulárně-geneticky byla vyšetřena mtDNA na delece a bodové mutace A8344G, A3243G, T3271C a T8993G, analýza však v tomto případě nepřinesla potvrzující nález poruchy mtDNA. Vyšetřena ale byla pouze oblast nejčastějších delecí mtDNA (8150–14276), což může být zdrojem falešně negativního výsledku. Dále použitá metodika vyšetření nezachytí mutace genu pro mitochondriálně kódovanou podjednotku 6 ATP-syntázy a některých genů pro mitochondriální tRNA (MTTK, MTTL1).
Pacientka za příznaků progrese neurologické symptomatologie zemřela v necelých dvou letech věku na perakutně probíhající infekci. Materiál k histologickému vyšetření byl získán postmortálně.
Závěr
Zhodnocení svalové biopsie slouží ke zjištění histopatologických změn typických pro mitochondriální onemocnění a v některých případech (reaktivita na COX versus přítomnost RRF) může posoudit i typ dědičnosti či příslušnost k některému z komplexů oxidativně-fosforylačního řetězce. K bližšímu zařazení do jednotlivých komplexů bezpochyby přispěje i imunohistochemická detekce polypeptidů jednotlivých komplexů nebo in situ hybridizace. Negativní bioptický nález, tedy nepřítomnost RRF nebo mitochondriálních abnormit, nemůže MM jednoznačně vyloučit, biopsie však může naznačit jiný směr pro další diagnostickou rozvahu. Je třeba zdůraznit, že žádný z uvedených histopatologických znaků není sám o sobě pro diagnózu MM specifický, a proto je diagnosticky významnější jejich seskupení, zejména v návaznosti na biochemickou analýzu oxidativní kapacity a stanovení aktivity enzymů v čerstvé tkáni. Konečné diagnostické zhodnocení má přinést mutační analýza.
Seznam použitých zkratek
- ALS - amyotrofická laterální skleróza
- ATP - adenosintrifosfát
- CK - kreatinkináza
- CNS - centrální nervový systém
- COX - cytochromoxidáza
- CT - počítačová tomografie
- DNA - deoxyribonukleová kyselina
- EM - elektronová mikroskopie
- FBSN - familiární oboustranná nekróza striata
- GRACILE - syndrom retardace růstu, aminoacidurie, zvýšený obsah železa, laktacidóza a předčasné úmrtí
- HE - hematoxylin-eozin, základní přehledné barvení v histologii
- LHON - Leberova hereditární optická neuropatie
- MELAS - mitochondriální encefalomyopatie, laktátová acidóza a „stroke-like“ episody
- MERRF - myoklonická epilepsie s přítomností „ragged red fibres“
- MILS - maternálně přenosný Leighův syndrom
- MM - mitochondriální myopatie
- MRI - magnetic resonance imaging
- mtDNA - mitochondriální DNA
- MTTL1 - mitochondriální tRNA pro leucin
- MTTK - mitochondriální tRNA pro lysin
- NADH - redukovaná forma nikotinamid-adenin-dinukleotidu
- NARP - neurogenní svalová slabost, ataxie a retinitis pigmentosa
- nDNA - jaderná DNA
- OGGT - orální glukózový toleranční test
- PAS - period acid Schiff, histologické barvení k průkazu glykogenu a glykoproteinů
- PEO - progresivní externí oftalmoplegie
- Q - ubichinon
- QH2 - ubichinol
- RRF - ragged red fibres
- rRNA - ribozomální RNA
- SDH - sukcinyldehydrogenáza
- STH - somatotropin
- tRNA - transferová RNA
Podpořeno grantem IGA MZ ČR NS 9782-4/2008.
MUDr. Ondřej Souček
Ústav
patologie LF MU a FN Brno
Jihlavská
20
625
00 Brno
e-mail:
osoucek@fnbrno.cz
Přijato
k recenzi: 22. 3. 2010
Přijato
do tisku: 6. 1. 2011
Zdroje
1. DiMauro S. Mitochondrial diseases. Biochim Biophys Acta 2004, 1658(2–3): 80–88.
2. DiMauro S, Hirano M. Mitochondrial encephalomyopathies: an update. Neuromuscul Disord 2005, 15(4): 276–286.
3. Wallace DC. Mitochondrial diseases in man and mouse. Science 1999; 283(1): 1482–1488.
4. Zeviani M. Mitochondrial disorders. Brain 2004; 127 (10): 2153–2172.
5. Munnich A, Rustin P. Clinical spectrum and diagnosis of mitochondrial disorders. Am J Med Genet 2001; 106(1): 4–17.
6. Dubowitz V, Sewry CA. Muscle biopsy. A Practical Approach. London: Saunders Elsevier 2007.
7. Bednařík J et al. Nemoci kosterního svalstva. Praha: Triton 2001.
8. Taylor RW, Schaefer AM, Barron MJ, McFarland R, Turnbull DM. The diagnosis of mitochondrial muscle disease. Neuromuscul Disord 2004, 14(4): 237–245.
9. Hasegawa H, Matsuoka T, Goto Y, Nonaka I. Strongly succinate dehydrogenase-reactive blood vessels in muscles from patients with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes. Ann Neurol 1991; 29(6): 610–615.
10. Haller RG, Henriksson KG, Jorfeldt L, Hultman E, Wibom R, Sahlin K et al. Deficiency of skeletal muscle succinate dehydrogenase and aconitase: pathofysiology of exercise in a novel human muscle oxidative defect. J Clin Invest 1991; 88(4): 1197–1206.
11. Hall RE, Henriksson KG, Lewis SF, Haller RG, Kennaway NG. Mitochondrial myopathy with succinate dehydrogenase and aconitase deficiency: abnormalities of several iron-sulfur proteins. J Clin Invest 1993; 92(6): 2660–2666.
12. Lindal S, Lund I, Torbergsen T, Aasly J, Mellgren SI, Borud O et al. Mitochondrial diseases and myopathies: a series of muscle biopsy specimens with ultrasctructural changes in the mitochondria. Ultrastruct Pathol 1992; 16(3): 263–275.
13. Kyriacou K, Hadjisavvas A, Zenios A, Papacharalambous R, Kyriakides T. Morphological methods in the diagnosis of mitochondrial encephalomyopathies: the role of electron microscopy. Ultrastruct Pathol 2005; 29(3–4): 169–174.
14. Carpenter S, Karpáti G. Pathology of skeletal muscle. 2nd ed. New York: Oxford University Press 2001.
15. Bourgeois JM, Tarnopolsky MA. Pathology of skeletal muscle in mitochondrial disorders. Mitochondrion 2004; 4(5–6): 441–452.
16. Rifai Z, Welle S, Kamp C, Thornton CAl. Ragged red fibers in normal aging and inflammatory myopathy. Ann Neurol 1995; 37(1): 24–29.
17. Nelson RL, Prayson RA. Mitochondrial abnormalities and inclusion body myositis: a histopathologic and ultrastructural study of 36 patients. J Surg Pathol 1997; 2 : 63–68.
18. Askanas V, Engel WK. Inclusion-body myositis: muscle-fiber molecular pathology and possible pathogenic significance of its similarity to Alzheimer’s and Parkinson’s disease brains. Acta Neuropathol 2008; 116(6): 583–595.
19. Yerroum M, Pham-Dang C, Authier FJ, Monnet I, Gherardi R, Chariot P. Cytochrome c oxidase deficiency in the muscle of patients with zidovudine myopathy is segmental and affects both mitochondrial DNA - and nuclear DNA-encoded subunits. Acta Neuropathol 2000; 100(1): 82–86.
20. Pavlakis SG, Phillips PC, DiMauro S, De Vivo DC, Rowland LP. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes: a distinctive clinical syndrome. Ann Neurol 1984; 16(4): 481–488.
21. Andreu AL, Hanna MG, Reichmann H, Bruno C, Penn AS, Tanji K et al. Exercise intolerance due to mutations in the cytochrome b gene of mitochondrial DNA. N Engl J Med 1999; 341(14): 1037–1044.
22. Andreu AL, Tanji K, Bruno C, Hadjigeorgiou GM, Sue CM, Jay C et al. Exercise intolerance due to a nonsense mutation in the mtDNA ND4 gene. Ann Neurol 1999; 45(6): 820–823.
23. Horváth R, Schoser BGH, Müller-Höcker J, Völpel M, Jaksch M, Lochmüller Hl. Mutations in mt-DNA encoded cytochrome c oxidase subunit genes causing isolated myopathy or severe encephalomyopathy. Neuromuscul Disord 2005; 15(12): 851–857.
24. Gempel K, Topaloglu H, Talim B, Schneiderat P, Schoser BG, Hans VH et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007; 130(8): 2037–2044.
25. Carta A, Carelli V, D‘Adda T, Ross-Cisneros FN, Sadun AA. Human extraocular muscles in mitochondrial diseases: comparing chronic progressive external ophthalmoplegia with Leber‘s hereditary optic neuropathy. Br J Ophthalmol 2005; 89(7): 825–827.
26. Zhu Z, Yao J, Johns T, Fu K, De Bie I, Macmillan C et al. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat Genet 1998; 20(4): 337–343.
27. Tiranti V, Hoertnagel K, Carrozzo R, Galimberti C, Munaro M, Granatiero M et al. Mutations of SURF-1 in Leigh disease associated with cytochrome c oxidase deficiency. Am J Hum Genet 1998; 63(6): 1609–1621.
28. McFarland R, Clark KM, Morris AA, Taylor RW, Macphail S, Lightowlers RN et al. Multiple neonatal deaths due to a homoplasmic mitochondrial DNA mutation. Nat Genet 2002, 30(2): 145–146.
29. Taylor RW, Giordano C, Davidson MM, d‘Amati G, Bain H, Hayes CM et al. A homoplasmic mitochondrial transfer ribonucleic acid mutation as a cause of maternally inherited hypetrophic cardiomyopathy. J Am Coll Cardiol 2003; 41(10): 1786–1796.
30. Bresolin N, Zeviani M, Bonilla E, Miller RH, Leech RW, Shanske S et al. Fatal infantile cytochrome c oxidase deficiency: decrease of immunologically detectable enzyme in muscle. Neurology 1985; 35(6): 802–812.
31. Tritschler HJ, Bonilla E, Lombes A, Andreetta F, Servidei S, Schneyder B et al. Differential diagnosis of fatal and benign cytochrome c oxidase-deficient myopathies of infancy: an immunohistochemical approach. Neurology 1991; 41(1): 300–305.
32. DiMauro S, Lombes A, Nakase H, Mita S, Fabrizi GM, Tritschler HJ et al. Cytochrome c oxidase deficiency. Pediatr Res 1990; 28(5): 536–541.
33. Cacić M, Wilichowski E, Mejaski-Bosnjak V, Fumić K, Lujić L, Marusić Della Marina B et al. Cytochrome c oxidase partial deficiency-associated Leigh disease presenting as an extrapyramidal syndrome. J Child Neurol 2001; 16(8): 616–619.
34. De Paepe B, Smet J, Lammens M, Seneca S, Martin JJ, De Bleecker J et al. Immunohistochemical analysis of the oxidative phosphorylation complexes in skeletal muscle from patients with mitochondrial DNA encoded tRNA gene defects. J Clin Pathol 2009; 62(2): 172–176.
Štítky
Dětská neurologie Neurochirurgie NeurologieČlánek vyšel v časopise
Česká a slovenská neurologie a neurochirurgie
2011 Číslo 4
- Přibývající důkazy o přínosu léčebného konopí u pacientů s chronickou bolestí
- Nízká hladina vitamínu D v prenatálním období může zvýšit riziko roztroušené sklerózy
- Pacientům s roztroušenou sklerózou pomáhá intermitentní pohybová aktivita
- Zahájení léčby intramuskulárním interferonem beta-1a během první epizody demyelinizace u RS
- Časné klinické prediktory a progrese ireverzibilní invalidity u RS
Nejčtenější v tomto čísle
- Porucha pozornosti s hyperaktivitou (attention deficit/hyperactivity disorder – ADHD)
- Opožděný akutní subdurální hematom
- Neurologické komplikace při onemocnění herpes zoster – kazuistika
- Vrozená myotonie na podkladě mutací v genu pro chloridový kanál ClC-1
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy