
Esenciální trombocytemie a jiné myeloproliferace s trombocytemií léčené Thromboreductinem. Výstupy z databáze Registru k 1. čtvrtletí roku 2010
Essential thrombocythaemia and other myeloproliferative disorders with thrombocythaemia treated with Thromboreductin. A report from the database of Register for the 1st quarter of 2010
In the Czech Republic, anagrelid is used according to the recommendations of the Czech Working Group on Myeloproliferative Disorders for treatment of thrombocythaemias associated with chronic myeloproliferative disorders – mainly essential thrombocythaemia and, regularly, reports are being presented from the Register of Patients Treated with Thromboreductin®, most recently last year (Vnitř Lék 2009; 55: I – XII). The Register commenced in 2005 and from then it aims to determine detailed clinical and laboratory profiles of the patients. The structure of the Register has changed significantly in the course of its existence, reflecting the reports from each of the analyses conducted so far. Also, the data entry in the database improves every year and it reaches 97% on some of the items. The longest evaluation period in some of the patients is 108 months. By April 2010, the Register database contained data on 717 patients. Of these, 672 patients with the diagnosis of a Ph ‑ negative chronic myeloproliferative disorder were evaluated. This year’s analysis included the patients with essential thrombocythaemia, polycythaemia vera and primary myelofibrosis only. The analysis included 418 women and 254 men with median age of 50 years. Unlike the first years, 2/ 3 of the current sample are non pretreated patients, meaning that the patients reach the specialized centres early in their treatment. Also, patients, and the older patients in particular, are more frequently treated with combined regimens including Thromboreductin®. We increasingly observe hypertension as one of the monitored risk factors preceding the disease and laboratory parameters show JAK2 mutation in more than a half of patients while some form of thrombotic diathesis is found in the anamnesis of 7 – 10% of patients. Some bleeding is observed in 1 – 5% of the registered patients. In comparison to the previous years, this is a decrease in the prevalence of clinical symptoms prior to the disease onset; this is very likely associated with an earlier patient diagnosis within the asymptomatic phase of the disease. Therapeutically, we achieve a fast treatment response but there still are 16.3% of sufficient after one year of treatment. Thromboreductin® dose is increasing but even in this group it does not exceeds the mean of 2.38 mg per 24 hours. Complications are observed in 6.2% of patients in the first year of therapy, and of these, thrombotic events in about 2.5% and (small) bleeding complications in 4% of patients. The data suggest that we still do not reach treatment response in a certain proportion of patients after a year of their therapy. Even though the care results from the analysed data improve every year, the Register helps to uncover some issues that still remain, such as treatment intensification and other treatment modifications.
Key words:
essential thrombocytopenia – myeloproliferative disorders – anagrelid (Thromboreductin®) – Register – JAK2 mutation – thrombophilia
Autoři:
M. Penka 1; J. Schwarz 2; P. Ovesná 3; A. Hluší 4; Z. Kořístek 5; M. Doubek 5; P. Ďulíček 6; D. Pospíšilová 7; J. Kissová 1; A. Buliková, (ČHS) 1; T. Pavlík 3; Kolektiv České Pracovní Skupiny Pro Myeloproliferativní Choroby (czemp)
Působiště autorů:
Oddělení klinické hematologie FN Brno, pracoviště Bohunice, přednosta prof. MU Dr. Miroslav Penka, CSc. 2Ústav hematologie a krevní transfuze Praha, ředitel prof. MU Dr. Marek Trněný, CSc. 3Institut biostatiky a analýz Lékařské a Přírodovědecké fakulty M
1
Vyšlo v časopise:
Vnitř Lék 2010; 56(6): 503-512
Kategorie:
Původní práce
Souhrn
Anagrelid je v ČR používán dle doporučení České pracovní skupiny pro myeloproliferativní choroby k léčbě trombocytemie provázející chronické myeloproliferace – především esenciální trombocytemii – a opakovaně byly již také prezentovány výstupy z Registru nemocných léčených Thromboreductinem, naposledy vloni (Vnitř Lék 2009; 55: I – XII). Zmíněný registr je veden od roku 2005 a od počátku je jeho cílem stanovení podrobného klinického a laboratorního profilu nemocných. Během svého užívání doznává struktura registru významných změn, které reagují na výstupy získávané z každé dosavadní analýzy dat. Také vyplněnost databáze se rok od roku zvyšuje a v některých položkách dosahuje až 97 %. Doba sledování u nejdéle sledovaných nemocných činí 108 měsíců. V databázi registru jsou k dubnu roku 2010 údaje o 717 nemocných. Z toho bylo hodnoceno 672 nemocných s diagnózou chronické Ph negativní myeloproliferativní choroby. Letošní analýze byli podrobeni pouze nemocní s esenciální trombocytemií, polycytemia vera a primární myelofibrózou. Jedná se o 418 žen a 254 mužů s věkovým mediánem 50 let. Na rozdíl od prvních let v loňském roce představovali nepředléčení pacienti již 2/ 3 nově registrovaných nemocných, což znamená, že se nemocní dostávají na specializovaná pracoviště brzy. Častěji se také setkáváme s kombinovanou léčbou Thromboreductinem, a to zejména u starších nemocných. Ze sledovaných rizikových parametrů předchorobí narůstá výskyt hypertenze, z laboratorních ukazatelů se u sledovaných zjišťuje u více než poloviny případů JAK2 mutace, zatímco nějakou formu trombotické diatézy zjišťujeme v anamnéze u 7 – 10 % nemocných. S rozličným krvácením v anamnéze se setkáváme u 1 – 5 % zaregistrovaných pacientů. Oproti předchozím letům se jedná o pokles výskytu klinických symptomů v předchorobí, což je velmi pravděpodobně dáno rychlejším záchytem nemocných v bezpříznakovém období. Z hlediska léčby dosahujeme u sledovaných nemocných rychlé odpovědi, přesto do roka dosahuje nedostatečné odpovědi 16,3 % pacientů. Průměrná dávka Thromboreductinu se sice zvyšuje, ale ani v této skupině nepřesahuje v průměru 2,38 mg na 24 hod. Ke komplikacím dochází v prvním roce léčby u 6,2 % nemocných, z toho k trombotickým příhodám v asi 2,5 %, zatímco k (malým) krvácivým projevům ve 4 % případů. Z údajů vyplývá, že stále nedosahujeme v ročním horizontu u určitého procenta nemocných ani částečné léčebné odpovědi. I přesto, že výsledky péče v analyzovaných údajích vyznívají rok od roku příznivěji, přetrvávají rezervy v intenzifikaci či jiných úpravách léčby, které Registr pomáhá odhalit.
Klíčová slova:
esenciální trombocytemie – myeloproliferace – anagrelid (Thromboreductin®) – Registr – JAK2 mutace – trombofilie
Úvod
WHO klasifikace z roku 2008 zařazuje nejen jednotlivá onemocnění celé skupiny myeloproliferativních neoplazií (MPN), jak byla tato skupina přejmenována, ale upravuje i diagnostické požadavky na jednotlivé klinické jednotky. Sami se skupinou Ph negativních myeloproliferací soustředěně zabýváme od roku 2004 a od roku 2005 existuje i Registr, který střádá data u nemocných s esenciální trombocytemií léčených Thromboreductinem. Postupně docházelo při důslednějším uplatňování WHO kritérií k překlasifikovávání esenciální trombocytemie, nejčastěji na primární myelofibrózu, k čemuž vedl jak vývoj choroby, který již náznak primární myelofibrózy původně nesl, nebo byl zařazen podle klinického nálezu a periferních hodnot krevního obrazu nebo skutečně do myelofibrózy přešel, tak se ve skupině sledovaných objevují i jiné diagnózy myeloproliferativní skupiny chorob. V některých případech zase musela být léčba Thromboreductinem ukončena nebo doplněna dalším cytoredukčním lékem, a tak sem byli zařazováni i nemocní na jiné léčbě než (jen) Thromboreductinem. Ze zmíněných důvodů se soubor rozrůstal i do stran původně neplánovaných. Když byl k dispozici již velmi sofistikovaný systém sbírání a hodnocení dat, bylo možné využívat Registr šířeji. Proto má dnes povahu trošku odlišnou od původní představy.
Esenciální trombocytemie (ET) se vyskytuje v asi 0,1 – 2,5 případů na 100 000 osob ročně [2]. Má klonální povahu [8,31] a diagnostiku usměrňuje řada klasifikací – PVSG [19], WHO z roku 2001 [9], ECMP z roku 2007 [18], WHO z roku 2008 [31]. Některé klasifikace neumožňují odlišení hraničních nálezů (např. časné fáze primární myelofibrózy – PMF) či prepolycytemického stadia polycythaemia vera (PV) jako např. PVSG, některé jsou přímo k určení zmíněných rozdílů detailně propracovány (WHO z roku 2001, ECMP z roku 2007). Rozdíl klasifikací je vyjádřen zejména v odlišnosti histologického nálezu v kostní dřeni. Poslední klasifikační schémata využívají navíc komplexní hodnocení, které zahrnuje výsledky molekulárně biologického vyšetření, především stanovení JAK2 mutace V617F. Jelikož jednotlivé typy MPN odpovídají na léčbu odlišně [17,32], je nutné upřednostnit klasifikaci, která vychází z vyšetření histologie kostní dřeně [30] a vyšetření klonality onemocnění. Za klinicky důležité se považuje co nejpřesnější určení diferenciální diagnózy hned od počátku.
Jak již bylo řečeno, v současné době se stále více do popředí pozornosti dostává vyšetření mutace JAK2V617F, která může být nalézána až u 98 % případů polycythaemia vera, u 34 – 67 % případů primární myelofibrózy a u 23 – 57 % esenciální trombocytemie [12].
JAK2 mutace zvyšuje na hematopoetických buňkách citlivost k růstovým faktorům, a tím přispívá ke zvýšení proliferační aktivity a přežívání buněk [12,13]. Pacienti mají agresivnější fenotyp – mutovaná JAK2 má spontánní aktivitu [11], vyšší výskyt trombotických komplikací [30], a je nutno u JAK2 pozitivních případů dříve zahajovat terapii [4]. Průkaz JAK2 mutace V617F se stal součástí klasifikačních kritérií. V současné době se doporučuje vyšetření dalších genetických změn – exon 12, mutace trombopoetinového receptoru MPLW515L apod. [9].
ET není onemocněním dramatickým, pokud není provázeno závažnými krvácivými či trombotickými projevy [7]. K nim disponují nemocní z řady důvodů – v závislosti na počtu krevních destiček a také v závislosti na výskytu souběžných predispozičních faktorů. Těmi může být trombofilní či krvácivá zátěž a také, jak se ukazuje a bylo již zmíněno, i přítomnost JAK2 mutace [20,25,27]. Méně častou komplikací, o to závažnější, je však progrese choroby až její přechod – transformace do akutní leukemie. Právě z důvodu účinné profylaxe vzniku zmíněné klinické symptomatologie indikujeme léčbu nemocných [15].
Tam, kde je přítomna v souvislosti s ET či MPN trombocytemie přesahující 1 000 × 109/ l nebo dochází k nárůstu počtu trombocytů do 2 měsíců o více než 200 × 109/ l, a zvláště za výskytu dalšího rizika (viz výše), doporučujeme léčbu vedoucí ke snížení, resp. normalizaci počtu destiček [20,25]. Především u mladších nemocných (< 60 let) je lékem volby anagrelid. Průměrná dávka léku při zavedené léčbě může činit asi 2,0 – 2,5 mg denně [22], přičemž se nedoporučuje překračovat denní dávku 5 mg [1,20].
Léčbu anagrelidem lze také kombinovat – s interferonem [5] či hydroxyureou [3], čímž se dosáhne možnosti redukce dávky obou léků proti dávce léku při monoterapii každým z nich. V některých případech je vhodné kombinovat léčbu cyto - , resp. tromboreduktivní, s antiagregační léčbou – nejčastěji s kyselinou acetylsalicylovou (ASA) [14]. Jedná se o případy s počtem destiček od 400 do 1 500 × 109/ l, kdy je vyšší riziko trombózy, a/ nebo o případy zvýšeného kardiovaskulárního rizika – tedy hrozby tepenné (kardiaci, pacienti s cévním onemocněním mozku apod.) a mikrovaskulární trombózy. V daných případech je však nutno vyloučit možnou současně se vyskytující prokrvácivou dispozici – ať již z důvodu choroby samotné, nebo náchylnosti spojené s výskytem jiné choroby či patologického stavu (např. získaný von Willebrandův syndrom nebo von Willebrandova choroba). Aspekt opatrnosti současného podávání ASA či jiných antitrombocytárních léků (tienopyridiny, nesteroidní antiflogistika apod.) je umocněn i přítomností funkční poruchy krevních destiček u nemocných s trombocytemií, které se klinicky manifestují především při počtu destiček nad 1 200 × 109/ l. Tento fenomén popisuje velmi trefně známý Michielsův model „doutníku a klínu“ [17].
Je známo, že z nežádoucích účinků léčby anagrelidem jsou v našem souboru nejčastější bolesti hlavy – 29 pří-padů a palpitace – 25 případů. Dále se však mohou objevovat také slabost, otoky, nauzea a bolest břicha, flatulence, zvracení, horečka, rash, závrať, dušnost, bolest na hrudi, anorexie, tachykardie, faryngitida, malátnost, kašel, parestezie, bolesti zad, svědění, dyspepsie, chřipkové obtíže a dehydratace. Většina obtíží přichází v úvodu terapie a většinou docela rychle a spontánně ustupuje [4,22].
Při podávání anagrelidu je doporučována zvýšená opatrnost u kardiaků (NYHA IV, resp. III), u nemocných s jaterním (jaterní transaminázy zvýšeny 5krát proti normálním hodnotám) a ledvinným selháním (clearance endogenního kreatininu pod 30 ml/ min). Vedle počtu trombocytů je nutné sledovat jaterní a renální parametry a v rámci objektivního vyšetření kardiologický stav nemocného.
V tomto roce jsme se zaměřili vedle shrnutí obvyklých popisných údajů opět na hodnocení léčebné odpovědi a dále na komplikace onemocnění a nežádoucí účinky léčby. Součástí analýzy byl také rozbor hodnot krevních destiček v době výskytu komplikací a rozbor příčin ukončení léčby a úmrtí nemocných.
Soubor nemocných
V současné době jsou k dispozici data od 717 nemocných, hodnoceno je však 672 záznamů u pacientů s esenciální trombocytemií (76,3 %), primární myelofibrózou (12,9 %) a pravou polycystemií (10,7 %) léčených Thromboreductinem. V souboru je 418 žen a 254 mužů s věkovým mediánem 50 let (graf 1). Do databáze jsou shromažďovány údaje o základních laboratorních ukazatelích, rizikových dispozicích žilního tromboembolizmu a kardiovaskulárních komplikací včetně rodinné anamnézy i získaných přitěžujících okolností. Data od jednotlivých pacientů jsou zaznamenávána ve čtvrtletních intervalech, v 1. pololetí dokonce jednou za měsíc. Celková doba sledování činí již 108 měsíců s mediánem 30 měsíců a vyplněnost jednotlivých záznamů kolísá mezi 63 a 96 %.

Takřka 3/ 4 nemocných celého sou-boru byly již před započetím soustavného sledování předléčeny jakoukoliv jinou trombocytoreduktivní léčbou, procento se však snižuje a trend svědčí pro to, že se nemocní dostávají časně do specializovaných ambulancí ještě před dokončením stanovení diagnózy, započetím léčby a zařazením do registru (graf 2).

Statistické hodnocení
Popis souboru pacientů a sledovaných charakteristik byl proveden pomocí frekvenčních tabulek a standardních popisných statistik: průměru, mediánu,minima, maxima a kvantilů. Pro vizualizaci byly použity koláčové a krabicové grafy, v případě kategoriálních dat pak kontingenční tabulky. Hodnocení vztahu dvou kategoriálních proměnných binárního charakteru bylo provedeno pomocí Fisherova exaktního testu. Pro stanovení statistické významnosti byla použita standardní hladina a = 0,05. Data byla zpracována v programu Statistica 9 [28].
Výsledky a diskuze
Ukazuje se, že odpovědi na léčbu je dosahováno velmi rychle – při zhodnocení nálezů v prvním mezičase systematického sledování, tedy v době 3 měsíců od započetí léčby Thromboreductinem, je jí již dosaženo (graf 3). Při neměnné průměrné denní dávce však zůstává odezva přesahující limit počtu destiček 400 × 109/ l až do 27. měsíce (tab. 1). Od 27. měsíce je zaznamenávána průměrná hodnota destiček odpovídající kritériím kompletní odpovědi (počet destiček nepřesahující hodnotu 400 × 109/ l). Průměrná dávka léku přitom činí 2,1 mg na den a ta se od po celou dobu sledování výrazně nemění (tab. 2). V této souvislosti není bez zajímavosti, že se zmíněný profil oproti předchozím létům nemění a neposouvá se k časnější kompletní odpovědi vyjádřené počtem krevních destiček nepřesahujícím 400 × 109/ l. Ke zmírnění tohoto méně příznivého hodnocení je třeba dodat, že z této skupiny přirozeně odpadávají ti nemocní, u nichž je třeba přejít na jinou nebo kombinovanou terapii, kdy se Thromboreductin® buď zcela vysazuje, nebo se snižuje jeho dávka. U nemocných s kombinovanou terapií vede snížení dávky Thromboreductinu ke snížení jeho celkové průměrné dávky.
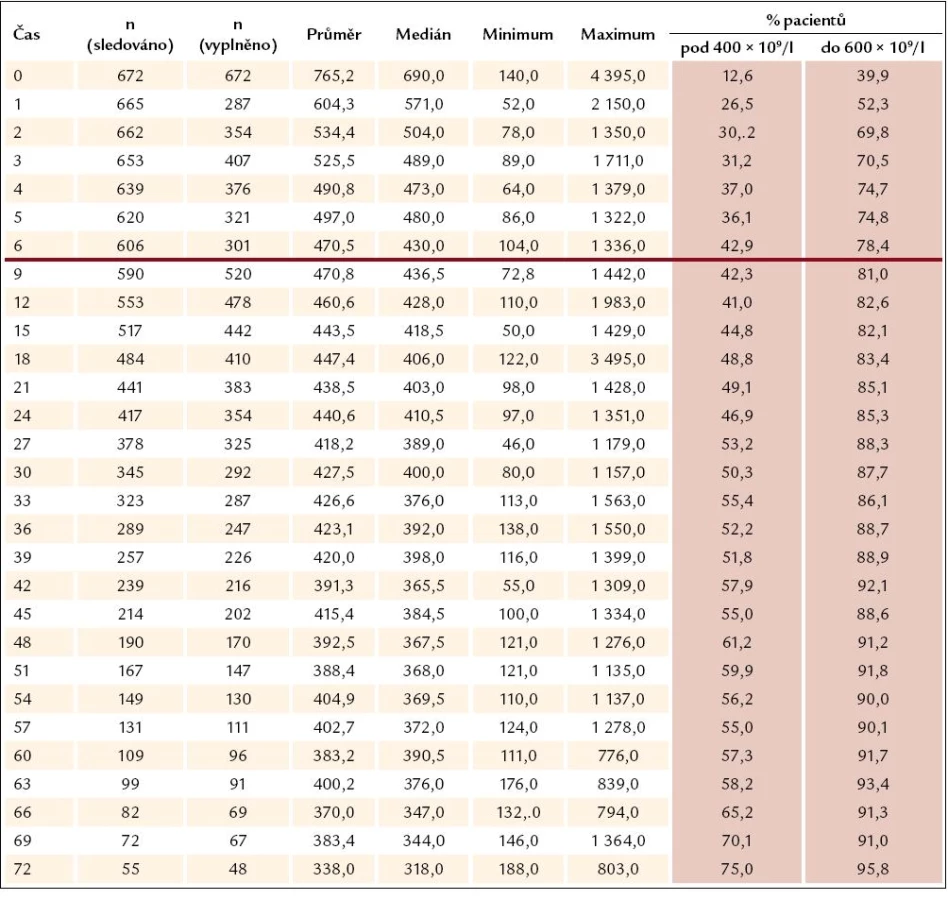


K podrobnějšímu hodnocení jsme podobně jako v předchozích analýzách soubor rozdělili na 3 skupiny podle dosahovaného snížení počtu trombocytů – a to na skupinu s počtem destiček pod 400 × 109/ l (kompletní léčebná odpověď), s počtem kolísajícím mezi 400 a 600 × 109/ l (částečná léčebná odpověď) a počtem přesahujícím 600 × 109/ l (nedostatečná odpověd na léčbu). V jednotlivých skupinách nemocných je přitom hodnoceno kolísání dávky Thromboreductinu v periodách 3 měsíců pravidelného sledování a z něho vyplývá, že k navyšování dávky dochází především ve skupině pacientů s přetrvávajícím počtem destiček převažujícím 600 × 109/ l. Ani zde ale medián denní dávky léku nepřesahuje 2,0 mg, a i když počet nemocných v této skupině od 3. měsíce klesá (182 ... 71 ... 54 ... 45), medián dávky léku stoupá od 1 přes 1,5 na 2 mg denně (graf 4). K dalšímu navyšování dávky pak nedochází. Jak vyplývá z dalšího, není tomu tak jen z důvodu nežádoucích účinků léčby (graf 5). Téměř polovina, tj. 9/ 19 nemocných ze skupiny bez dostatečné odpovědi na léčbu po celou dobu sledování (v čase 0, 3, 6 a 12 měsíců) je léčeno pouze Thromboreductinem. U 2 z nich byly zjištěny 3, resp. 4 rizikové faktory a dávka anagrelidu 2, resp. 0,5 mg na 24 hod. U ostatních případů lze říci, že s ohledem na nízký výskyt rizikových faktorů nebývá pozorována snaha dosáhnout normalizace počtu krevních destiček (tedy kritérií kompletní odpovědi), rozhodně bychom se však měli pokusit dosáhnout alespoň odpovědi částečné. Tam, kde je kombinovaná léčba, je nutno brát průměrnou dávku anagrelidu s rezervou, neboť ta tvoří pouze část komplexní kombinované léčby, a tudíž je její dávka nižší, než by byla užita v monoterapii. Kombinovaná léčba zůstává pouze u jedné čtvrtiny z celkového počtu nemocných (n = 672) a zde jako doplňující léčba k Thromboreductinu převažuje hydroxyurea (80 %) (graf 6).



Po 6 měsících terapie 43 % pacientů dosáhlo kompletní odpovědi na léčbu a 78 % dostatečné (kompletní nebo částečné) odpovědi.
Na léčbě zůstává po 2 letech 86,7 % nemocných, po 4 letech 79,8 % a po 6 letech od jejího nasazení 71,9 % pacientů sledovaného souboru (graf 7). To je výsledek zcela jistě uspokojivý.

Předléčených nemocných jsou v celémsouboru necelé 3/ 4 (n = 478) a většinou se jedná o nemocné s předchozím zavedením léčby hydroxyureou, méně interferonem nebo řadou různých kombinací. Dochází však ke zvyšování podílu nepředléčených nemocných (z 59 na 68 % v meziročním srovnání za poslední 2 roky) [21,22], což svědčí o tom, že se nemocní dostávají na specializovaná pracoviště časně, jak již bylo konstatováno výše.
U sledovaných pacientů jsme hodnotili komplikace onemocnění. Ty bylyrozděleny na velké tepenné a žilní trombózy, malé tepenné a žilní trombózy, mikrotrombózy, velká a malá krvácení a dále progresi a transformaci choroby. V souboru bylo zjištěno celkem 107 zmíněných komplikací u 82 pacientů, což činí 12,2 % z celkového počtu pacientů. Problematika klinické symptomatologie byla rozebírána v předchozích analýzách [21,22] a ukázalo se, že podíl na vzniku trombotické diatézy, jako dominantního příznaky choroby, má především přítomnost JAK2 mutace a může k ní přispívat i FVL a deficit proteinu S w[21,26]. Tyto závěry potvrdily i letošní výsledky. Fakt, že u nemocných s nižší hodnotou trombocytů nacházíme i krvácivé projevy, může být dán právě současným podáváním ASA u těchto nemocných.
Parametrem, jenž se dnes ukazuje významným i z hlediska výskytu tromboembolických komplikací, je leukocytóza. Průměr počtu leukocytů po celou dobu sledování nepřesahuje hodnotu 13,3 × 109/ l. Při rozboru jednotlivých případů pozorujeme i kolísání k vysokým hodnotám, ale ani v těchto případech není výskyt trombotických projevů vyšší než u pacientů, u nichž k vysoké leukocytóze nedochází.
Podobně ani hemoglobin nezasahuje v průměrných hodnotách a jejich rozptylu do naopak nízkých hodnot – a to ani u žen, ani u mužů (jsou sem však zavzaty i hodnoty pacientů s polycytemia vera, které mohou být zkreslením zmíněného kolísání).
Nežádoucí účinky léčby se objevily u 103 nemocných, z toho ve 3 případech jsou hodnoceny jako závažné. Šedesát nemocných trpělo jedním a 23 nemocných dvěma nežádoucími účinky, 9 třemi a více než 4 nežádou-cími účinky 8 nemocných. Ve 29 případech (4,3 %) se jednalo o bolesti hlavy, ve 25 případech (3,7 %) o palpitace, v 11 případech o symptomatickou anémii a v 10 případech o otoky kotníků (graf 5).
K ukončení léčby došlo u 115 (17,1%) nemocných, přičemž v 16 případech z důvodu úmrtí (graf 8). Příčinu smrti lze klást do souvislosti s onemocněním v 6 případech, v 6 naopak souvislost chybí a ve 4 případech nejsou okolnosti smrti dostatečně známy.

Dalším důvodem ukončení léčby byly nežádoucí účinky (18 případů), absence odpovědi na léčbu (12 případů) a komplikace léčby byly příčinou jejího přerušení u 15 případů. Na žádost pacienta byla léčba přerušena 14krát, 6krát byla léčba přerušena pro graviditu a z důvodu normalizace nálezu v 7 případech. U 16 nebyla příčina ukončení léčby zjištěna a u 11 případů došlo k jejich ztrátě z evidence.
K progresi choroby došlo u 53 pacientů – u 7 k přechodu onemocnění do akutní leukemie, 39 jeví známky myeloidní metaplazie – 27 nemocných s ET, 9 s PMF a 3 nemocní s PV. U 7 byl zaznamenán přechod do polycytemia vera. Hodnocení známek myeloidní metaplazie je z hlediska průběhu onemocnění problematické, neboť u všech pacientů nebylo na začátku provedeno histologické vyšetření kostní dřeně.
Závěr
Je možno konstatovat, že se stále zvyšuje objem dat databáze registru v jejich jednotlivých položkách a její výpovědní hodnota. Je potvrzována rychlá odpověď na Thromboreductin®, stále však ne zcela uspokojivé dávkování.
I letošní analýza registru potvrzuje, že Thromboreductin® je účinný lék, s nímž je dosahováno rychlé léčebné odpovědi při výskytu buď srovnatelného, nebo ještě nižšího počtu nežádoucích účinků, které jsou referovány v literatuře. Lék je velmi dobře tolerován. Ze všech těchto důvodů je Thromboreductin® dle doporučených postupů vypracovaných Českou pracovní skupinou pro Ph negativní myeloproliferace určen jako lék první volby pro nemocné do 60 let, přesahuje‑li počet trombocytů hranici 1 000 × 109/ l nebo jsou‑li přítomny rizikové faktory.
Z věcného hlediska vyplývá přetrvávající rezerva v možnosti navyšování dávky anagrelidu k zajištění vyšší úrovně odpovědi na léčbu a detailní rozbor jednotlivých případů za účelem zjištění, proč k navýšení léčby, a tudíž i dosahování vyšší úrovně odpovědi, nedochází, zejména u pacientů s více rizikovými faktory.
Do dalšího období je třeba zajistit podrobnější rozbor nemocných na kombinované léčbě a zrevidovat histologické nálezy s event. překlasifikováním nemocných k určení, zda k selhávání léčebné odpovědi nedochází právě v případech, u nichž dochází ke změně původní diagnózy. V tomto směru je také stále více kladen důraz na možnost zajištění rutinního druhého čtení histologických preparátů.
Registr má zcela konkrétní požadavky na zpracování i v následujícím roce. Fakt, že došlo ke zvýšení podílu nemocných odesílaných časně na specializovaná pracoviště, lze považovat za jeden z velkých úspěchů, na němž má registr pacientů podíl.
prof. MU Dr. Miroslav Penka, CSc.
www.fnbrno.cz
e-mail: m.penka@fnbrno.cz
Doručeno do redakce: 19. 5. 2010
Zdroje
1. Barbui T, Barosi G, Grossi A et al. Practice guidelines for the therapy of essential thrombocythemia. A statement from the Italian Society of Hematology, the Italian Society of Experimental Hematology and the Italian Group for Bone Marrow Transplantation. Haematologica 2004; 89 : 215 – 232.
2. Brière JB. Essential thrombocythemia. Orphanet J Rare Dis 2007; 2 – 3 : 1 – 17.
3. Cortelazzo S, Finazzi G, Ruggeri M et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 1995; 332 : 1132 – 1136.
4. Costello R, O’Callaghan TO, Sébahoun G. Traitement de la thrombocythémie essentielle. Rev Med Interne 2005; 26 : 947 – 955.
5. Elliott MA, Tefferi A. Interferon‑alpha therapy in polycythemia vera and essential thrombocythemia. Semin Thromb Hemost 1997; 23 : 463 – 472.
6. Fruchtman SM, Petitt RM, Gilbert HS et al. Anagrelid: analysis of long‑term efficacy, safety and leukemogenic potential in myeloproliferative disorders. Leuk Res 2005; 29 : 481 – 491.
7. Gisslinger H, Kralovics R, Gotic M et al. Non ‑ inferiority of anagrelide compared to hydroxyurea in newly diagnose patients with essential thrombocythemia. The ANAHYDRET ‑ Study. Blood 2007; 110 : 1038A.
8. Green A, Campbell P, Buck G et al. The Medical Research Council PT1 trial in essentials thrombocythemia. Blood 2004; 104 : 5a – 6a.
9. Hoffman R, Prchal JT, Samuelson S et al. Philadelphia chromosome ‑ negative myeloproliferative disorders: biology and treatment. Biol Blood Marrow Transplant 2007; 13 (Suppl 1): 64 – 72.
10. Jaffe ES, Harris NL, Stein H et al. World Health Organization classification of tumours. Pathology and genetics of tumours of haematopoietic and lymphoid tissues. Lyon: IARC Press 2001.
11. Kaushansky K. The chronic myeloproliferative disorders and mutation of JAK2: Dameshek’s 54 year old speculation comes of age. Best Pract Res Clin Haematol 2007; 20 : 5 – 12.
12. Kralovics R, Passamonti F, Buser AS et al. A gain‑of ‑ function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 352 : 1779 – 1790.
13. Kralovics R, Skoda RC. Molecular pathogenesis of Philadelphia chromosome negative myeloproliferative disorders. Blood Rev 2005; 19 : 1 – 13.
14. Landolfi R, Rocca B, Patrono C. Bleeding and thrombosis in myeloproliferative disorders: mechanism and treatment. Crit Rev Oncol Hematol 1995; 20 : 203 – 222.
15. Michiels JJ, Kutti J, Stark P et al. Diagnosis, pathogenesis and treatment of the myeloproliferative disorders essential thrombocythemia, polycythemia vera and essential megakaryocytic granulocytic metaplasia and myelofibrosis. Neth J Med 1999; 54 : 46 – 62.
16. Michiels JJ, Barbui T, Finazzi G et al. Diagnosis and treatment of polycythemia vera and possible future study designs of the PVSG. Leuk Lymphoma 2000; 36 : 239 – 253.
17. Michiels JJ, Thiele J. Clinical and pathological criteria for the diagnosis of essential thrombocythemia, polycythemia vera and idiopathic myelofibrosis (agnogenic myeloid metaplasia). Int J Hematol 2002; 76 : 133 – 145.
18. Michiels JJ, De Raeve H, Hebeda K et al. WHO bone marrow features and European clinical, molecular, and pathological (ECMP) criteria for the diagnosis of myeloproliferative disorders. Leuk Res 2007; 31 : 1031 – 1038.
19. Murphy S, Peterson P, Iland H et al. Experience of the Polycythemia Vera Study Group with essential thrombocythemia: a final report on diagnostic criteria, survival, and leukemic transition by treatment. Semin Hematol 1997; 34 : 29 – 39.
20. Penka M, Schwarz J, Pytlík R et al. Doporučený postup diagnostiky a terapie esenciální trombocytémie a trombocytémie provázející myeloproliferativní onemocnění. Vnitř Lék 2005; 51 : 741 – 751.
21. Penka M, Schwarz J, Pavlík T et al. Esenciální trombocytemie a další myeloproliferace s trombocytemií v údajích registru pacientů léčených Thromboreductinem® do konce roku 2007. Vnitř Lék, 2008; 54 : 775 – 782.
22. Penka M, Schwarz J, Pavlík T et al. Výsledky léčby nemocných s esenciální trombocytemií a dalšími myeloproliferacemi provázenými trombocytemií – zpráva z registru pacientů léčených Thromboreductinem. Vnitř Lék 2009; 55: I – XII.
23. Petrides PE. Anagrelid: A decade of clinical experiences with its use for the treatment of primary thrombocythaemia. Expert Opin Pharmacother 2004; 5 : 1781 – 1798.
24. Puigdecanet E, Spinet B, Villa O et al. Detection of abnormalities of PRV-1, TPO, and c‑MPL genes detected by fluorescence in situ hybridization in essential thrombocythemia. Cancer Genet Cytogenet 2006; 167 : 39 – 42.
25. Schwarz J, Hrachovinova I, Vorlova Z et al. Thromboembolism in thrombocythemia patients with an additional thrombophilic state. Hematol J 2004; 5 (Suppl 2): S321.
26. Schwarz J, Penka M, Doubek M et al. JAK2 mutation and an additional thrombophilic state are major prothrombotic risk factors in myeloproliferations with thrombocythemia – data from a registry of anagrelide‑treated patients. Haematologica 2008; 1 : 93.
27. StatSoft, Inc. Statistica (Data Analysis Software System) version 9.0. 2009. www.statsoft.com.
28. Silverstein MN, Tefferi A. Treatment of essential thrombocythemia with anagrelid. Semin Hematol 1999; 36 (1 Suppl 2): 23 – 25.
29. Steurer M, Gastl G, Jedrzejczak W et al. Anagrelide for thrombocytosis in myeloproliferative disorders: a prospective study to assess efficacy and adverse event profile. Cancer 2004; 101 : 2239 – 2246.
30. Thiele J, Kvasnicka HM, Vardiman J. Bone marrow histopathology in the diagnosis of chronic myeloproliferative disorders: a forgotten pearl. Best Pract Res Clin Haematol 2006; 19 : 413 – 437.
31. Tefferi A, Thiele J, Orazi A et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycytemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood 2007; 110 : 1092 – 1097.
32. Tsimberidou MA, Colburn DE, Welch MA et al. Anagrelid and imatinib mesylate combination therapy in patients with chronic myeloproliferative disorders. Cancer Chemother Pharmacol 2003; 52 : 229 – 234.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2010 Číslo 6
- Plicní hypertenze – syndrom mnoha tváří – vyžaduje přesnou diagnostiku a specializovanou léčbu
- Proces hojení ran krok za krokem a co ho může zkomplikovat
- Limity glykovaného hemoglobinu a význam dalších glykovaných proteinů
- Korelace dávky levothyroxinu s titrem autoimunitních protilátek u primární hypotyreózy
- Zeolit-jodový komplex pomáhá v péči o infikované rány
Nejčtenější v tomto čísle
- Sepse a septický šok u onkologických a imunokompromitovaných pacientů – diagnostika, terapie
-
Hyperlipoproteinemie a dyslipoproteinemie I.
Klasifikace, diagnostika, kardiovaskulární, kardiometabolické a reziduální riziko - Esenciální trombocytemie a jiné myeloproliferace s trombocytemií léčené Thromboreductinem. Výstupy z databáze Registru k 1. čtvrtletí roku 2010
- Odlišné průběhy recidivující anebo multisystémové formy histiocytózy z Langerhansových buněk u dospělých osob – popis 22 případů z jednoho pracoviště
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy