
Cílený rozklad obtížně inhibovatelných proteinů způsobujících maligní hematologická onemocnění pomocí dvoufunkčních molekul využívajících polyubikvitinaci a rozklad v proteazomech
Targeted degradation of undruggable, malignant haematological disorder-causing proteins using bifunctional molecules, exploiting polyubiquitination and degradation in proteasomes
Inhibition of proteins with small-molecule inhibitors is restricted to receptors and enzymes which make up approximately 25% of the human proteome. The majority of proteins such as transcription factors, scaffolding proteins, regulatory proteins and non-enzymatic proteins are difficult (“undruggable“) targets for small-molecule inhibitors. In the past 20 years, new procedures have been developed based on the mechanism of inducible protein degradation that exploits polyubiquitination and degradation in proteasomes using proteolysis targeting chimeras (PROTACs). Bifunctional molecules (PROTACs) are composed of specific ligand for the protein of interest connected via a linker to a specific ligand for E3 ubiquitin ligase used in protein of interest polyubiquitination and its subsequent degradation in proteasomes. The presented bifunctional molecules have been successfully used experimentally in degradation of many proteins of interest that have important functions in the pathogenesis of haematological malignancies and are the subject of this review.
Keywords:
protein degradation – proteolysis-targeting chimera (PROTAC) – ubiquitin – proteasome – E3 ubiquitin ligase
Autoři:
O. Fuchs; R. Bokorová
Působiště autorů:
Ústav hematologie a krevní transfuze, Praha
Vyšlo v časopise:
Transfuze Hematol. dnes,26, 2020, No. 4, p. 292-301.
Kategorie:
Souhrnné/edukační práce
Souhrn
Inhibice proteinů nízkomolekulárními inhibitory je omezena na enzymy a receptory, které tvoří přibližně 25 % lidského proteomu. Většina proteinů, jako jsou transkripční faktory, strukturální proteiny, regulační proteiny a proteiny bez funkce enzymu, je nedostupná (undruggable) pro inhibiční molekuly. Během posledních 20 let byl vyvinut postup založený na mechanismu indukované degradace proteinů, využívající polyubikvitinaci a rozklad v proteazomech pomocí PROTACs (proteolysis-targeting chimeras). Dvoufunkční molekuly PROTACs jsou složeny ze specifického ligandu pro protein určený k degradaci, spojky (linker) a ze specifického ligandu pro E3 ubikvitinligázu využívanou v polyubikvitinaci cílového proteinu pro jeho následnou degradaci v proteazomech. Uvedené dvoufunkční molekuly byly experimentálně úspěšně použity k degradaci řady proteinů, které hrají důležité funkce v patogenezi hematologických malignit a jsou předmětem tohoto souhrnného článku.
Klíčová slova:
degradace proteinů – chimera pro cílenou proteolýzu (PROTAC) – ubikvitín – proteazom – E3 ubikvitinligáza
ÚVOD
RNA interference a nízkomolekulární inhibitory nebo protilátky lze použít k inhibici biologické funkce proteinů způsobujících různá onemocnění. Při RNA interferenci dochází k zablokování translace nebo rozkladu mediátorové RNA (mRNA) pro cílený protein [1, 2]. Vybrané a optimalizované nízkomolekulární inhibitory nebo protilátky se váží specificky do dobře definovaného aktivního místa cílového proteinu (enzymu nebo receptoru) a blokují jeho biologickou aktivitu. Jak jsme již uvedli v souhrnu, přibližně 75 % proteinů nemá aktivní místo a nelze jejich biologickou funkci zablokovat pomocí nízkomolekulárních inhibitorů a navíc často dochází při dlouhodobé terapii k rezistenci. Protilátky jsou obvykle o velké molekulové hmotnosti a jsou schopné blokovat jen proteiny na vnější straně buněčné membrány a nepronikají do buňky [1].
Tyto obtíže vedly k nové strategii využívající E3 ubikvitinligázy v ubikvitin-proteazomovém systému (UPS). Uvedené E3 ubikvitinligázy jsou součástí dvoufunkčních molekul PROTACs (proteolysis-targeting chimeras). Kromě obou funkcí, vázat cílový protein určený k degradaci a E3 ubikvitinligázu pomocí specifických ligand, je zde ještě linker (spojka), který oba ligandy spojuje a na jehož optimální délce též závisí výsledná polyubikvitinace a degradace cílového proteinu v 26S proteazomech [1–4].
Za objev ubikvitinu, evolučně konzervativního malého proteinu obsahujícího 76 aminokyselinových zbytků, a jeho funkci v nelysosomální cestě degradace proteinu obdrželi doktoři Avram Hershko, Aaron Ciechanover a Irwin Rose v roce 2004 Nobelovu cenu za chemii [5–7]. Schematický průběh polyubikvitinace proteinů v UPS je ukázán na obrázku 1. Ubikvitinace je posttranslační kovalentní modifikace proteinu. Ubikvitin je aktivován pomocí enzymu E1 v závislosti na ATP. Aktivovaný glycin v poloze 76 na C-konci molekuly ubikvitinu vytvoří thioesterovou vazbu s cysteinem enzymu E1. Následně je přenesen na ubikvitin – přenášející, nebo konjugující enzym E2. Přenos ubikvitinu na cílový protein, který má být degradován, je uskutečněn pomocí E3 ubikvitinligázy, která rozpozná právě tento protein a obvykle i několik dalších proteinů. Je tedy ze všech tří enzymů vybavena nejvyšší specificitou. Ubikvitinace probíhá na primární amino skupině lysinu jako nukleofilní útok na vazbu E2-Ub za vzniku stabilní izopeptidové vazby. Po navázání prvého ubikvitinu (Ub) následuje navázání dalších ubikvitinů. Nejběžnější způsob vazby dvou ubikvitinů v polyubikvitinovém řetězci je přes lysin 48 (K48). Nejméně čtyřčlenný řetězec ubikvitinových zbytků je vyžadován pro následné rozpoznání cílového proteinu 26S proteazomem a jeho degradaci v uvedeném proteazomu. Lidské buňky obsahují dva E1 enzymy, přibližně 40 E2 enzymů a více než 600 E3 ubikvitinligáz [8]. Existují dvě hlavní skupiny E3 ubikvitinligáz. Byly pojmenovány podle charakteristických oblastí RING (really interesting new gene) a HECT (homologous to E6-associated protein carboxyl terminus) [9–12]. HECT druh E3 ubikvitinligáz funguje jako kovalentní meziprodukt v ubikvitinaci. Ubikvitin je nejprve dopraven do aktivního místa cysteinu E3 enzymu transthiolací před konjugací k substrátu. RING druh E3 ubikvitinligáz zprostředkuje přímo vazbu z E2-Ub k substrátu. Několik E3 ubikvitinligáz tohoto typu RING bylo zatím využito pro rozklad cílového proteinu pomocí PROTACs. Jedná se především o VHL (von Hippel Lindau disease tumor suppressor protein E3 ubikvitinligáza /CRL2VHL/), CRL4ACRBN (cullin-4 really interesting new gene /RING/ E3 ubikvitinligáza s cereblonem /CRBN/ vázajícím deriváty thalidomidu a cIAP (cellular inhibitor of apoptosis protein 1 E3 ubikvitin ligáza).

PRVNÍ GENERACE MOLEKUL PROTACs VYUŽÍVAJÍCÍ PEPTIDY A VHL
Schematické znázornění molekuly PROTAC je prezentováno na obrázku 2. Molekula PROTAC je složena ze tří částí. První součástí je ligand pro cílový protein určený k polyubikvitinaci a následné degradaci v proteazomu. Druhou částí je linker. Třetí složkou je ligand pro E3 ubikvitinligázu vybranou pro proces ubikvitinace.

Molekuly PROTACs první generace využívají jako ligand pro E3 ubikvitinligázu CRL2VHL malé peptidové řetězce, které napodobují interakci protein-protein mezi VHL a jejím endogenním substrátem – hypoxií indukovatelným faktorem 1α (HIF-1α) [13, 14]. V roce 2019 byla Nobelova cena za fyziologii nebo lékařství udělena americkým onkologům (William G. Kaelin a Gregg L. Semenza) a britskému nefrologu (Peter J. Ratcliffe) za objev mechanismu, jak buňky vnímají a přizpůsobují se dostupnosti kyslíku, kde hydroxylace prolinových zbytků 402 a 564 v molekule HIF-1α, polyubikvitinace za pomoci E3 ubikvitinligázy VHL a následná degradace HIF-1α v proteazomu při normální koncentraci kyslíku hraje klíčovou úlohu [14]. Uvedený malý peptidový řetězec obsahuje sekvenci ALA-Hyp-YIP, podobnou sekvenci zahrnující hydroxylovaný prolinový zbytek Hyp 564 v molekule HIF-1α. Tento peptid byl prodloužen pro lepší prostupnost molekuly PROTAC buněčnou membránou o osm argininových zbytků (poly-D-Arg) [15–17]. Výsledný peptid byl konjugován s dihydrotestosteronem pro polyubikvitinaci androgenního receptoru (AR) a jeho rozklad v buňkách HEK 293. Tento PROTAC v koncentraci 25μM způsobil za 1 hodinu působení degradaci AR. V komplexu E3 ubikvitinligázy VHL jsou dále obsaženy elonginy B a C, kulin 2 a Rbx1 (RING-box protein 1) [16]. Pokud je použit jako ligand pro cílový protein určený k polyubikvitinaci a následné degradaci estradiol místo dihydrotestosteronu, slouží PROTAC k polyubikvitinaci a degradaci estrogenního receptoru [18].
První generace PROTACs měla jako hlavní nedostatek příliš velkou molekulární hmotnost. Dalšími nevýhodami byly labilní peptidové vazby, špatná prostupnost přes buněčnou membránu a nízká účinnost [19].
DRUHÁ GENERACE MOLEKUL PROTACs VYUŽÍVAJÍCÍ NÍZKOMOLEKULÁRNÍ LÁTKY JAKO LIGANDY PRO CÍLOVÉ PROTEINY
E3 ubikvitinligáza CRL4ACRBN je nejvíce využívána u této generace molekul PROTACs. Cereblon je součástí komplexu této E3 ubikvitinligázy vedle DDB1 (damaged DNA binding protein 1), kulin 4A a ROC 1 (regulator of cullins 1). CRL4A se za normálních podmínek podílí na autoubikvitinaci cereblonu [20] a polyubikvitinaci proteinu MEIS 2 (transkripční faktor obsahující homeobox) [21], glutaminsyntetázy [22] a amyloidového prekurzorového proteinu [23]. Thalidomid a jeho deriváty (imunomodulační látky, IMiDs) se váží k hydrofobní kapse na C-konci cereblonu tvořené třemi tryptofanovými zbytky a jedním fenylalaninovým zbytkem. Po působení IMiDs mění CRL4ACRBN specificitu a místo uvedených substrátů za normálních podmínek se podílí na polyubikvitinaci transkripčních faktorů IKZF1 (Ikaros) a IKZF3 (Aiolos) v buňkách mnohočetného myelomu [24–27]. Působení IMiDs je schematicky ukázáno na obrázku 3. IMiDs jsou používány jako ligand pro CRL4ACRBN při konstrukci PROTACs [28].

DVOUFUNKČNÍ MOLEKULY PROTACs PRO POLYUBIKVITINACI A DEGRADACI PROTEINŮ S BROMO - A EXTRATERMINÁLNÍ DOMÉNOU
Rodina proteinů BET byla pojmenována podle dvou tandemových bromodomén a extraterminálního motivu [29–31]. Obsahuje proteiny BRD2, BRD3, BRD4 a BRDT. Bromodoména je evolučně konzervativní motiv a přednostně se váže na acetylované lysinové zbytky histonů. Acetylované histony jsou asociovány výhradně s transkripčně aktivní, méně kondenzovanou strukturou chromatinu a zvýšenou přístupností transkripčních faktorů k DNA. Nejlépe prostudovaným členem proteinů BET je BRD4, který se často nachází v oblasti superenhancerů důležitých onkogenů c-myc, bcl-xL a bcl-6 a zvyšují jejich expresi [32, 33].
Celá řada nízkomolekulárních inhibitorů BET proteinů (JQ1/TEN-010, BI 894999, GSK525762/Molibresib, I-BET151/GSK1210151A, AZD5153, CPI-0610, INCB057643, ZEN-3694, Birabresib/OTX015/MK-8628, HYD276, I-BET762, RX-37, HJ897, NHWD-870) byla a stále je zkoušena u hematologických malignit i solidních nádorů v klinických studiích fáze I a II [34–36]. Často však dochází k rezistenci na tyto nízkomolekulární inhibitory, a proto byly vyvinuty molekuly PROTACs, které překonávají uvedenou rezistenci (obr. 4) [37]. Degradace cílového proteinu je tedy výhodnější než jeho pouhá inhibice specifickým nízkomolekulárním inhibitorem, která je často spojena s rezistencí na použité inhibitory [37].

Účinnější PROTACs používají k vazbě proteinů BET určených k degradaci právě vazebné motivy z uvedených nízkomolekulárních inhibitorů proteinů BET, spojené linkerem s ligandem pro CRL4ACRBN, obvykle thalidomidem nebo lenalidomidem. První PROTAC dBET1 pro polyubikvitinaci a následnou degradaci proteinu BRD4 obsahoval jako ligand pro BRD4 BET inhibitor JQ1, který byl přes N-butyl-2-hydroxy-aceta-mid spojen s thalidomidem jako ligandem pro CRL4CRBN. Zvýšená apoptóza primárních buněk pacientů s akutní myeloidní leukemií (AML) byla pozorována po působení dBET1 [38].
Dašími molekulami PROTACs k cílené polyubikvitinaci a následné degradaci proteinů BET jsou ARV-825 a ARV-771 (Arvinas Inc.). Zde byl použit thiazolo-diazepin acetamid odvozený z vazebné části inhibitoru proteinů BET OTX015 jako ligand pro cílové T proteiny BET a lenalidomid nebo peptid odvozený z molekuly HIF-1α pro E3 ubikvitinligázy CRL4CRBN a CRL2VHL. Saenz a spolupracovníci porovnali účinnost ARV-825 a ARV-771 s nízkomolekulárním inhibitorem OTX015 [35]. Při koncentraci 100–500 nM byla účinnost uvedených PROTACs 10–100× vyšší než nízkomolekulárního inhibitoru OTX015. Bylo také zjištěno větší snížení hladin c-myc, JAK2, STAT3, STAT5 a fosforylovaných forem obou proteinů STAT, PIM1 a bcl-xL. PIM1 je protoonkogen, který stabilizuje c-myc. Bcl-xL je antiapoptotický protein, který brání uvolnění cytochromu c z mitochondrií a inhibuje kaspázy a apoptózu [39]. Porucha regulace signální dráhy STAT, zejména silně zvýšená exprese STAT5, je spojená se zvýšenou proliferací a sníženou diferenciací a zablokováním apoptózy u buněk AML.
PROTAC ARV-825 silně snížil membránové CXCR4 (chemokinový receptor obsahující C-X-C motiv, také pojmenovaný fusin nebo CD184) a CD44, který hraje úlohu v mezibuněčné interakci. Snížení exprese těchto glykoproteinů v buňkách AML vede k poruše adheze a migrace buněk a ke zvýšenému oxidačnímu stresu [40].
ARV-825 a ARV-771 byly použity k polyubikvitinaci a následné degradaci proteinů BET též v buňkách lymfomu z plášťových buněk. Indukovaly zde apoptózu účinněji než nízkomolekulární inhibitory proteinů BET a snížily hladinu c-myc, cdk4, cyklinu D1 a několika cílových proteinů transkripčního faktoru NF-κB (bcl-xL, induktor apoptózy spojený s chromozomem X /XIAP/ a Brutonova tyrozinkináza /BTK/) [41].
ARV-825 snížil hladinu c-myc také v buňkách Burkittova lymfomu (buněčné linie Namalwa a Ramos) [33].
PROTAC ARV-825 byl také zkoumán u buněk mnohočetného myelomu (MM) in vitro a in vivo. ARV-825 inhiboval proliferaci buněk MM u 13 lidských linií a 3 čerstvých vzorků z pacientů s MM. Způsobil zastavení buněčného cyklu a apoptózu. Proteiny BRD2 a BRD4 byly účinně degradovány. Citlivost buněk MM k ARV-825 korelovala s hladinou cereblonu. Byla snížena hladina myc, myb, chemokinového receptoru s motivem C-C typu 1 (CCR1) a regulátoru signální dráhy G-proteinu (RGS) [42, 43].
PROTAC PRO POLYUBIKVITINACI A NÁSLEDNOU DEGRADACI ONKOPROTEINU-TYROZINKINÁZY BCR-ABL1
Inhibitory tyrozinových kináz (TKI) potlačují aktivitu onkoproteinu bcr-abl1. Významným mechanismem rezistence buněk chronické myeloidní leukemie (CML) vůči TKI jsou mutace v kinázové doméně bcr-abl1. Rozklad fúzního onkoproteinu bcr-abl1 by překonal uvedenou rezistenci. Burslem et al. připravili řadu PROTACs na základě alosterického inhibitoru GNF-5, spojky a ligandu pro CRL2VHL. Nejúčinnějším byl PROTAC GMB-475, který způsobil rychlou degradaci bcr-abl1 v buňkách K562 a myších buňkách Ba/F3 a snížil též aktivitu STAT5 [44].
SELEKTIVNÍ DEGRADACE SERIN/THREONIN PROTEIN KINÁZY 6 ZÁVISLÉ NA CYKLINECH (CDK6) POMOCÍ PROTAC ZALOŽENÉM NA PALBOCIKLIBU
Cyklin dependentní kinázy hrají zásadní úlohu v signalizační kaskádě, která vede k progresi buněčného cyklu a buněčné proliferace. Palbociklib je selektivní inhibitor cyklin dependentních kináz CDK4 a CDK6. Palbociklib proto blokuje buněčný cyklus a redukuje tak proliferaci nádorových buněk. Působení molekuly PROTAC připravené za použití palbociklibu jako ligandu pro CDK6, která je polyubikvitována a následně degradována v proteazomech, je schematicky ukázáno na obrázku 5. Účinek PROTAC je proti účinku nízkomolekulárního inhibitoru – palbociklibu – selektivní a specificky předurčí k degradaci pouze CDK6 a nemá vliv na CDK4. PROTAC navíc ve srovnání s nízkomolekulárními inhibitory zasáhne i na kinázové aktivitě nezávislé vlivy CDK6 [45–47].

Exprese CDK6 je na rozdíl od exprese CDK4 nezbytná pro růst buněk akutní lymfoblastické leukemie s filadelfským chromozomem (Ph+ALL). PROTAC YX-2-107 vedle specifické inhibice CDK6 v buňkách Ph+ALL snížil fosforylaci retinoblastoma proteinu (Rb) a tím přechod z kontrolního bodu fáze G1 do fáze S buněčného cyklu, který je řízen komplexem cyklinu D s CDK4 a CDK6 [47]. PROTAC YX-2-107 snížil i expresi transkripčního faktoru FOXM1 (Forkhead box protein M1) zahrnutého v regulaci buněčného cyklu a nezbytného pro DNA replikaci a mitózu. Jeho nadměrná exprese je spojena s maligní transformací buněk. Tyto vlivy přidaného PROTAC YX-2-07 nebyly pozorovány v normálních hematopoetických CD34+ progenitorových buňkách, i když CDK6 byla účinně degradována. Leukemie byla potlačena i v myších injikovaných s de novo nebo na inhibitor tyrozinkinázy rezistentními buňkami Ph+ALL a YX-2-107. Tento účinek PROTAC YX-2-107 byl srovnatelný nebo lepší než účinek palbociklibu. Je zde i menší nebezpečí vzniku neutropenie při působení PROTAC YX-2-107 než při použití nízkomolekulárního inhibitoru – palbociklibu [47].
DEGRADACE BRUTON TYROZINKINÁZY (BTK) POMOCÍ PROTAC
Aktivita BTK je důležitá pro přežití a proliferaci leukemických B buněk a pro jejich interakci s buňkami mikroprostředí. BTK se kromě buněčné signalizace přes BCR (B cell receptor) účastní i signalizace přes TLR (toll-like receptor) a Fc receptor. Nízkomolekulární inhibitory BTK (ibrutinib a řada dalších experimentálních látek) vykázaly silný protinádorový účinek zejména u chronické lymfocytární leukemie (CLL) a lymfomu z plášťových buněk (MCL).
Konstrukce PROTAC za účelem polyubikvitinace a následné degradace BTK využily nízkomolekulární inhibitory BTK, např. ibrutinib, CGI1746 nebo spebrutinib (CC-292, AVL-292) jako ligandy pro BTK a lenalidomid [48], pomalidomid [49–53] jako ligand pro CLR4CRBN nebo RG-7112 jako ligand pro E3 ubikvitinligázu MDM2 (murine double minute 2) [48]. Tyto konstrukty byly účinné in vitro v modelech B buněčných leukemií a lymfomů a v myším xenograftovém modelu ustaveném z buněk pacientů s těmito B buněčnými malignitami. Tyto PROTACs způsobily polyubikvitinaci a následnou degradaci BTK vedle transkripčních faktorů Ikaros a Aiolos [48–51]. PROTACs s CLR4CRBN byly účinnější než PROTACs s E3 ubikvitinligázou MDM2 [52].
DEGRADACE S RECEPTOREM PRO INTERLEUKIN-1 ASOCIOVANÉ KINÁZY 4 (IRAK4)
Firma Kymera (Cambridge, Massachusetts, USA) vyvinula PROTAC KYM-001 cílený na degradaci serin/threonin kinázy IRAK4. Tento protein fosforyluje TLR a IL-1R (receptor pro interleukin-1) a je zahrnut v signálních drahách přes uvedené receptory. Funguje i jako lešení pro uspořádání a aktivaci myddozomu s funkcí v NF-кB a MAPK signálních drahách a produkci zánětlivých cytokinů. IRAK4 hraje klíčovou úlohu u difuzních velkobuněčných B-lymfomů s mutací v genu MYD88 (myeloid differentiation primary response 88). Podání KYM-001 způsobilo 75% redukci v hladině IRAK4 a regresi nádoru v myším modelu lymfomu s mutací v genu MYD88 [54, 55].
DEGRADACE TRANSKRIPČNÍHO FAKTORU STAT3 POMOCÍ PROTAC SD-36
Transkripční faktor STAT3 (signal transducer and activator of transcription 3) je aktivován různými cytokiny, růstovými factory a onkogeny v různých lidských maligních onemocněních, včetně leukemií a lymfomů. Degradace STAT3 se zdá proto jako slibný terapeutický přístup. Pro syntézu molekuly PROTAC byly použity dva nízkomolekulární inhibitory STAT3 (SI-109 nebo SI-108) jako ligandy pro STAT3 a dále ligandy pro CRBN nebo VHL v komplexu E3 ubikvitinligáz (CRL4CRBN nebo CRL2VHL). PROTAC SD-36 obsahuje ligandy SI-109 a lenalidomid spojené linkerem. SD-36 degradoval STAT3 v závislosti na čase i koncentraci v buňkách AML linie MOLM-16 a v buňkách anaplastického velkobuněčného lymfomu linií SU-DHL-1, DEL a KI-JK [56, 57].
DEGRADACE LIDSKÉHO HOMOLOGU E3 UBIKVITINLIGÁZY MDM2
MDM2 (murine double minute 2) je E3 ubikvitinligáza, která se účastní na polyubikvitinaci a následné degradaci nádorového supresoru – proteinu p53. Působí také jako inhibitor transkripční aktivity p53 a jeho translokace z jádra do cytoplazmy. Existuje několik skupin nízkomolekulárních inhibitorů MDM2 (nutliny, imidazoly, benzodiazepiny, spirooxindoly, indoly, isochinoliny, piperidony, pyrrolidony a pyrrolidiny). Uvedené nízkomolekulární inhibitory, hlavně nutliny, byly použity při syntéze PROTAC jako ligandy pro cílový protein HDM2. Tento ligand MI-1061 byl přes spojku konjugován s lenalidomidem jako ligandem pro CRL4CRBN při přípravě PROTAC MD-224 [58].
Zvýšená exprese MDM2 a její aktivační mutace byly nalezeny u AML a CLL a právě v těchto případech byly použity inhibitory MDM2 a PROTAC MD-224. Tento PROTAC indukoval rychlý rozklad MDM2 v koncentracích < 1 nM u lidských leukemických buněk. MD-224 inhiboval růst buněk AML linie RS4;11 s přestavbou chromozomů 4 a 11 a také růst nádoru v xenograftovém myším modelu RS4;11 [58].
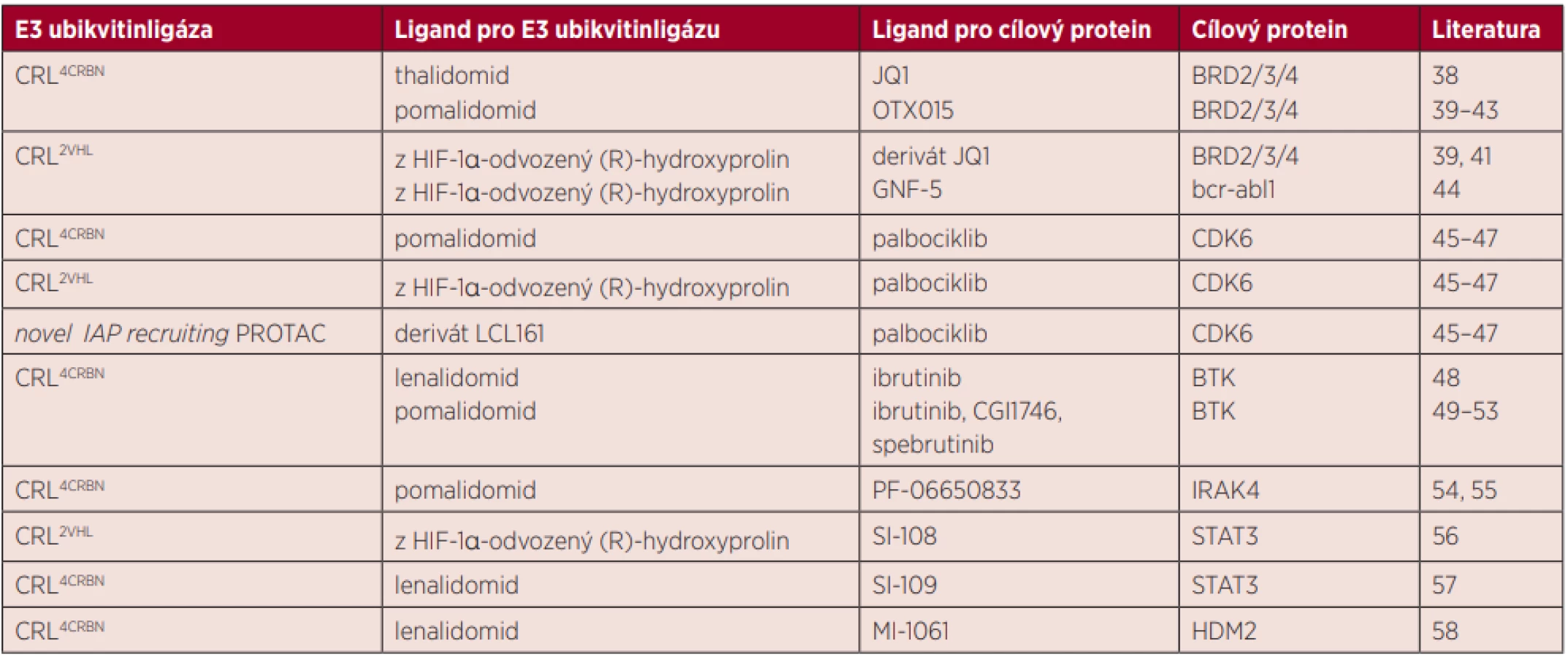
Přehled ligandů pro E3 ubikvitinligázy a cílové proteiny, které byly použity při syntéze molekul PROTACs pro popsané cílové proteiny je ukázán v tabulce 1.
DEGRADACE E3 UBIKVITINLIGÁZ POMOCÍ HOMO-PROTACs NEBO HETERO-PROTACs
Homo-PROTACs jsou bivalentní molekuly, které obsahují dva identické ligandy pro cílovou E3 ubikvitinligázu spojené linkerem [59–62]. Homo-PROTACs degradují tedy jako cílový protein stejnou E3 ubikvitinligázu, kterou využívají jako prostředek ke svému účinku. Před rozkladem je tato E3 ubikvitinligáza vlastně dimerizována a následně dochází k jejímu sebezničení uvnitř buňky chemickým odbouráním prostřednictvím ubikvitin proteazomového systému. Degradace CRBN v komplexu CRL4CRBN pomocí homo-PROTAC využívající CRL4CRBN neovlivnila proliferaci buněk MM, znemožnila degradaci transkripčních faktorů Ikaros a Aiolos a způsobila rezistenci na pomalidomid a lenalidomid u různých linií buněk MM.
Obdobně dochází k rozkladu VHL pomocí homo-PROTAC dimerizujícího dvě molekuly CRL2VHL [60].
Dvouvalentní molekuly PROTAC mohou také přivést do interakce dvě molekuly různých E3 ubikvitinligáz, například CRL4CRBN a CRL2VHL [63]. Zde dochází k heterodimerizaci obou E3 ubikvitinligáz a jejich vzájemnému odbourání prostřednictvím ubikvitin proteazomového systému.
ZÁVĚR
Nízkomolekulární inhibitory hrají důležitou úlohu při syntéze molekul PROTACs, kde jsou využívány jako ligandy pro cílový protein k polyubikvitinaci a následné degradaci v proteazomech. Uvedené molekuly PROTACs překonávají rezistenci na nízkomolekulární inhibitory a jejich mechanismus působení se úplně liší od nízkomolekulárních inhibitorů. Mechanismus působení PROTACs je založen na polyubikvitinaci cílového proteinu a jeho degradaci v proteazomu. Připravené PROTACs jsou zatím testovány u hematologických malignit v preklinických studiích jako potenciální léčiva in vitro a in vivo. Popsaný PROTAC KYM-001 pro polyubikvitinaci a následnou degradaci IRAK4 má nejblíže ke klinickým zkouškám. Pouze dva PROTACs jsou již v klinických zkouškách u karcinomu prsu a karcinomu prostaty. PROTAC ARV-110 vykazoval vysokou účinnost jak u divokého typu cílového proteinu androgenního receptoru, tak u jeho mutované formy vyhovující bezpečnost a snášenlivost. PROTAC ARV-110 je nyní v klinickém testování (NCT03888612) u metastazující formy rakoviny prostaty, kde není možná kastrace a kde vybraní pacienti již absolvovali nejméně dva druhy terapie. Oba tyto orálně aplikované PROTACs byly vyvinuty firmou Arvinas, Inc., založenou předním badatelem v této oblasti – Craigem Crewsem z Yaleovy univerzity v New Haven v americkém státě Connecticut. Jedná se o již zmíněný PROTAC ARV-110 pro degradaci androgenního receptoru a PROTAC ARV-471 pro degradaci estrogenního receptoru u žen s pokročilým lokálním nebo metastazujícím na estrogenní receptor pozitivním nádorem prsu a naopak na HER2 (člen receptorů pro epidermální růstový faktor s tyrozinkinázovou aktivitou) negativním nálezem.
Četné farmaceutické společnosti (Arvinas, C4 Therapeutics, Kymera Therapeutics, Captor Therapeutics, AstraZeneca, Bayer, Novartis, Amgen, Pfizer, Glaxo SmithKline, Merck a Boehringer Ingelheim) vyvíjejí PROTACs pro cílenou degradaci proteinů. Prověřením knihoven nízkomolekulárních látek na vazbu k cílovým proteinům bez známých vazebných míst pro nízkomolekulární inhibitory lze rozšířit množství proteinů, které by bylo možno tímto způsobem polyubikvitinovat a rozložit v proteazomech. PROTACs mají výhodu orální aplikace a schopnosti polyubikvitinovat a degradovat jinými způsoby neinhibovatelné (např. transkripční faktor STAT3) nebo přechodně inhibovatelné ale po rezistenci na nízkomolekulární inhibitory již neovlivnitelné cílové proteiny (např. AR, ER, BTK, BET, BCR-ABL a další proteiny). Nevýhodou PROTACs může být mutace v komplexu E3 ubikvitinligáz v ubikvitin proteazomovém systému a s ní související rezistence na PROTACs.
Použité zkratky
AR – androgenni receptor
Bcl2 – B-cell lymphoma 2
Bcl-xL – B-cell lymphoma-extra large
BET – bromodomain and extraterminal domain
BRD – bromodomain and extraterminal domain
BRT – Brutonova tyrozinkináza
CCR1 – receptor pro chemokin s motivem C-C typu 1
CDK – cyclin-dependent kinase
cIAP1 – cellular inhibitor of apoptosis protein 1
CRBN – cereblon
CRL – cullin-RING E3 ubiquitin ligase
ER – estrogenní receptor
FLT3 – FMS–like tyrosine kinase 3
FOXM1 – Forkhead box protein 1
HECT – homologous to E6 associated protein carboxyl terminus
HIF1α – hypoxií indukovatelný faktor 1α
IKZF – Ikaros family zinc finger protein
IL-1R – receptor pro interleukin 1
IMiDs – immunomodulatory drugs
IRAK – interleukin-1 receptor-associated kinase
MAPK – proteinkináza aktivovaná mitogeny
MCL – mantle cell lymphoma
MDM2 – murine double minute 2
MYD88 – myeloid differentiation primary response
NPM1 – nukleofosmin
PROTAC – proteolysis targeting chimera
RB – retinoblastoma protein
RING – really interesting new gene
Rbx1 – RING box protein 1
RGS – regulátor signální dráhy G-proteinu
STAT – signal transducer and activator of transcription
T-ALL – T-cell acute lymmphoblastic leukemia
TKI – inhibitor tyrozinkinázy
TLR – toll-like receptor
VHL – von Hippel-Lindau
XIAP – X-linked inhibitor of apoptosis protein
Podíl autorů na přípravě rukopisu
OF, RB – příprava, revize, korekce a schválení konečné verze rukopisu
Poděkování
Práce vznikla s podporou grantu MZČR pro koncepční rozvoj vědecké organizace číslo 00023736 (ÚHKT) a grantové agentury UK, projekt č. 924616.
Čestné prohlášení
Autoři práce prohlašují, že v souvislosti s tématem, vznikem a publikací tohoto článku nejsou ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou.
Do redakce doručeno dne 19. 5. 2020.
Přijato po recenzi dne 18. 6. 2020.
Rozklad proteinů pomocí dvoufunkčních molekul
Ing. Ota Fuchs, CSc.
Ústav hematologie a krevní transfuze
U Nemocnice 1
128 20 Praha 2
e-mail: Ota.Fuchs@uhkt.cz
Zdroje
- Neklesa TK, Winkler JD, Crews CM. Targeted protein degradation by PROTACs. Pharmacol Ther. 2017;174 : 138–144.
- Burslem GM, Crews CM. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell. 2020;181(1):102–114.
- Gao H, Sun X, Rao Y. PROTAC technology: opportunities and chal-lenges. ACS Med Chem Lett. 2020;11(3):237–240.
- Li X, Song Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J Hematol Oncol. 2020;13(1):50.
- Hershko A. Review: Nobel Lecture. The ubiquitin system for protein degradation and some of its roles in the control of the cell division cycle. Cell Death Differ. 2005;12(9):1191–1197.
- Ciechanover A. Intracellular protein degradation from a vague idea through the lysosome and the ubiquitin-proteasome system and on to human diseases and drug targeting: Nobel Lecture, December 8, 2004. Ann NY Acad Sci. 2007;1116 : 1–28.
- Rose I. Review: Nobel Lecture. Ubiquitin at fox chase. Cell Death Differ. 2005;12(9):1198–1201.
- Nguyen LK. Dynamics of ubiquitin – mediated signalling: insights from mathematical modelling and experimental studies. Brief Bioinform. 2016;17(3):479–493.
- Matějíková J, Kubiczková L, Sedlaříková L, et al. Degradace proteinů ubikvitin-proteazomovou drahou. Klin Onkol. 2013;26(4):251–256.
- Metzger MB, Pruneda JN, Klevit RE, Weissman AM. RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim Biophys Acta. 2014;1843(1):47–60.
- Rusnac DV, Zheng N. Structural biology of CRL ubiquitin ligases. Adv Exp Med Biol. 2020;1217 : 9–31.
- Wang Y, Argiles-Castillo D, Kane IE, et al. HECT E3 ubiquitin ligases-emerging insights into their biological roles and disease relevance. J Cell Sci. 2020;133(7):jcs228072.
- Sýkora V, Nečas E. Buněčná a tkáňová hypoxie-úloha von Hippel Lindau genu a hypoxií indukovatelného faktoru 1. Čas Lék Čes. 2003;142(10):595–598.
- Zhang Q, Yan Q, Yang H, Wei W. Oxygen sensing and adaptibility won the 2019 Nobel Prize in Physiology and Medicine. Genes Dis. 2019;6(4):328–332.
- Ohh M, Park CW, Ivan M, et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2(7):423–427.
- Hon WC, Wilson MJ, Harlos K, et al. Structural basis for the recognition of hydroxyproline in HIF-1α by pVHL. Nature. 2002;417(6892):975–978.
- Schneekloth JS, Fonseca FN, Koldobskly M, et al. Chemical genetic control of protein levels selective in vivo targeted degradation. J Am Chem Soc. 2004;126(12):3748–3754.
- Rodriguez-Gonzalez A, Cyrus K, Salcius M, et al. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene. 2008;27(57):7201–7211.
- Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017;16(2):101–114.
- Lopez-Girona A, Mendy D, Ito T, et al. Cereblon is a distinct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. 2012;26(11):2326–2335.
- Fischer ES, Böhm K, Lydeard JR, et al. (2014) Structure of the DDB1--CRBN E3 ubiquitin ligase in complex with thalidomide. Nature. 2014;512(7512):49–53.
- Nguyen TV, Lee JE, Sweredoski MJ, et al. Glutamine triggers acetylation-dependent degradation of glutamine synthetase via the thalidomide receptor cereblon. Mol Cell. 2016;61(6):809–820.
- Del Prete D, Rice RC, Rajadhyaksha AM, D‘Adamio L. Amyloid precursor protein (APP) may act as a substrate and a recognition unit for CRL4CRBN and Stub1 E3 ligases facilitating ubiquitination of proteins involved in presynaptic functions and neurodegeneration. J Biol Chem. 2016;291(33):17209–17227.
- Krönke J, Udeshi ND, Narla A, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343(6168):301–305.
- Gandhi AK, Kang J, Havens CG, et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN). Br J Haematol. 2014;164(6):811–821.
- Lu G, Middleton RE, Sun H, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2014;343(6168):305–309.
- Stanková M, Bešše L, Sedlaříková L, et al. Cereblon - nový terapeutický ciel´ v liečbe mnohopočetného myelómu. Klin Onkol. 2014;27(5):326–330.
- Fuchs O, Bokorova R. Rationale of targeting protein cereblon as a potential strategy for cancer treatment. Drugs Fut. 2020;45(5):305–317.
- Haynes SR, Dollard C, Winston F, et al. The bromodomain: a conserved sequence found in human, Drosophila and yeast proteins. Nucleic Acids Res. 1992;20(10):2603.
- Doroshow DB, Eder JP, LoRusso PM. BET inhibitors: a novel epigenetic approach. Ann Oncol. 2017;28(8):1776–1787.
- Stathis A, Bertoni F. BET proteins as targets for anticancer treat-ment. Cancer Discov. 2018;8(1):24–36.
- Chapuy B, McKeown MR, Lin CY, et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B-cell lymphoma. Cancer Cell. 2013;24(6):777–790.
- Lu J, Qian Y, Altieri M, et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem Biol. 2015;22(6):755–763.
- Braun T, Gardin C. Investigational BET bromodomain protein inhibitors in early stage clinical trials for acute myelogenous leukemia (AML). Expert Opin Investig Drugs. 2017;26(7):803–811.
- Massé A, Roulin L, Pasani J, et al. BET inhibitors impair leukemic stem cell function only in defined oncogenic subgroups of acute myeloid leukaemias. Leuk Res. 2019;87 : 106269.
- Pan D, Rampal R, Mascarenhas J. Clinical developments in epigenetic-directed therapies in acute myeloid leukemia. Blood Adv. 2020;4(5):970–982.
- Sun X, Rao Y. PROTACs as potential therapeutic agents for cancer drug resistance. Biochemistry. 2020;59(3):240–249.
- Winter GE, Buckley DL, Paulk J, et al. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348(6241):1376–1381.
- Saenz DT, Fiskus W, Qian Y, et al. Novel BET protein proteolysis targeting chiméra exerts superior lethal actity than bromodomain inhibitor (BETi) against post-myeloproliferative neoplasm secondary (s) AML cells. Leukemia. 2017;31(9):1951–1961.
- Piya S, Mu H, Bhattacharya S, et al. Myelogenous leukemia stem cells and the microenvironment. J Clin Invest. 2019;129(5):1878–1894.
- Sun B, Fiskus W, Qian Y, et al. BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia. 2018;32(2):343–352.
- Lim SL, Damnernsawad A, Shyamsunder P, et al. Proteolysis targeting chimeric molecules as therapy for multiple myeloma: efficacy, biomarker and drug combinations. Haematologica. 2019;104(6):1209–1220.
- Zhang X, Lee HC, Shirazi F, et al. Protein targeting chimeric molecules specific for bromodomain and extra-terminal motif family proteins are active against pre-clinical models of multiple myeloma. Leukemia. 2018;32(10):2224–2239.
- Burslem GM, Schultz AR, Bondeson DP, et al. Targeting BCR-ABL1 in chronic myeloid leukemia by PROTAC-mediated targeted protein degradation. Cancer Res. 2019;79(18):4744–4753.
- Rana S, Bendjennat M, Kour S, et al. Selective degradation of CDK6 by a palbociclib based PROTAC. Bioorg Med Chem Lett. 2019;29(11):1375–1379.
- Anderson NA, Cryan J, Ahmed A, et al. Selective CDK6 degradation mediated by cereblon, VHL, and novel IAP-recruiting PROTACs. Bioorg Med Chem Lett. 2020;30(9):127106.
- De Dominici M, Porazzi P, Xiao Y, et al. Selective inhibition of Ph-positive ALL cell growth through kinase-dependent and -independent effects by CDK6-specific PROTACs. Blood. 2020;135(18):1560–1573.
- Dobrovolsky D, Wang ES, Morrow S, et al. Bruton tyrosine kinase degradation as a therapeutic strategy for cancer. Blood. 2019;133(9):952–961.
- Zorba A, Nguyen C, Xu Y, et al. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc Natl Acad Sci USA. 2018;115(31):E7285–E7292.
- Buhimschi AD, Armstrong HA, Toure M, et al. Targeting the C481S ibrutinib-resistance mutation in Bruton´s tyrosine kinase using PROTAC-mediated degradation. Biochemistry. 2018;57(26):3564–3575.
- Jaime-Figueroa S, Buhimschi AD, Toure M, et al. Design, synthesis and biological evaluation of proteolysis targeting chimeras (PROTACs) as a BTK degraders with improved pharmacokinetic properties. Bioorg Med Chem Lett. 2020;30(3):126877.
- Sun Y, Zhao X, Ding N, et al. PROTAC-induced BTK degradation as novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 2018;28(7):779–781.
- Sun Y, Ding N, Song Y, et al. Degradation of Bruton´s tyrosine kinase mutants by PROTACs for potential treatment of ibrutinib-resistant non-Hodgkin lymphomas. Leukemia. 2019;33(8):2105–2110.
- Nunes J, McGonagle GA, Eden J, et al. Targeting IRAK4 for degradation with PROTACs. ACS Med Chem Lett. 2019;10(7):1081–1085.
- Kargbo RB. PROTAC degradation of IRAK4 for the treatment of cancer. ACS Med Chem Lett. 2019;10(10):1370–1371.
- Bai L, Zhou H, Xu R, et al. A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell. 2019;36(5):498–511.
- Zhou H, Bai L, Xu R, et al. Structure-based discovery of SD-36 as a potent, selective, and efficacious PROTAC degrader of STAT3 protein. J Med Chem. 2019;62(24):11280–11300.
- Li Y, Yang J, Aguilar A, et al. Discovery of MD-224 a first-in-class highly potent, and efficacious proteolysis targeting chimera murine double minute 2 degrader capable of achieving complete and durable regression. J Med Chem. 2019;62(2):448–466.
- Steinbach C, Lindner S, Udeshi ND, et al. Homo-PROTACs for the chemical knockdown of cereblon. ACS Chem Biol. 2018;13(9):2771–2782.
- Steinbach C, Kehm H, Lindner S, et al. PROTAC-mediated crosstalk between E3 ligases. Chem Commun (Camb). 2019;55(12):1821–1824.
- Lindner S, Steinbach C, Kehm H, et al. Chemical inactivation of the E3 ubiquitin ligase cereblon by pomalidomomide-based homo-PROTACs. J Vis Exp. 2019;147:e59472.
- Maniaci C, Hughes SJ, Testa A, et al. Homo-PROTACs: bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat Commun. 2017;8(1):830.
- Giardini M, Maniaci C, Hughes SJ, et al. Cereblon versus VHL: Hijacking E3 ligases against each other using PROTACs. Bioorg Med Chem. 2019;27(12):2466–2479.
Štítky
Hematologie a transfuzní lékařství Interní lékařství OnkologieČlánek vyšel v časopise
Transfuze a hematologie dnes

2020 Číslo 4
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Evaluace skóre m7-FLIPI u pacientů s folikulárním lymfomem odhalila slibný prediktivní biomarker k určení vhodné chemoterapie
- Neuropatie výrazně zpomaluje proces hojení u povrchových ran
- Diagnostika von Willebrandovy choroby krok za krokem
- Vybíráme z Červené knihy ČHS: Co nového v letošních doporučeních pro diagnostiku a léčbu AML?
Nejčtenější v tomto čísle
- Stillova nemoc – vzácné autoinflamatorní onemocnění dospělých: popis případu a přehled léčebných možností
- Odporúčania pre skríning, diagnostiku, profylaxiu a liečbu hepatitíd u hematoonkologických pacientov – odporúčania CELL
- Současná role alogenní transplantace krvetvorby v léčbě akutní myeloidní leukemie
- Léčba Erdheimovy-Chesterovy choroby
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy