
Virulence Factors of Induce Both the Unfolded Protein and Integrated Stress Responses in Airway Epithelial Cells
Pseudomonas aeruginosa causes a devastating infection when it affects patients with cystic fibrosis or other chronic lung diseases. It often causes chronic infection due to its resistance to antibiotic treatment and its ability to form biofilms in these patients. The toxic effects of P. aeruginosa are largely mediated by secreted virulence factors. Efficient functioning of the endoplasmic reticulum is crucial for cell survival and appropriate immune responses, while its dysfunction causes stress and activation of the unfolded protein response. In this study, we found that virulence factors secreted by P. aeruginosa trigger the unfolded protein response in human cells by causing endoplasmic reticulum stress. In addition, secreted virulence factors activate the integrated stress response via a parallel independent pathway. Both stress pathways lead to the induction of the protein GADD34, which appears to provide protection against the toxic effects of the secreted virulence factors.
Published in the journal:
. PLoS Pathog 11(6): e32767. doi:10.1371/journal.ppat.1004946
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004946
Summary
Pseudomonas aeruginosa causes a devastating infection when it affects patients with cystic fibrosis or other chronic lung diseases. It often causes chronic infection due to its resistance to antibiotic treatment and its ability to form biofilms in these patients. The toxic effects of P. aeruginosa are largely mediated by secreted virulence factors. Efficient functioning of the endoplasmic reticulum is crucial for cell survival and appropriate immune responses, while its dysfunction causes stress and activation of the unfolded protein response. In this study, we found that virulence factors secreted by P. aeruginosa trigger the unfolded protein response in human cells by causing endoplasmic reticulum stress. In addition, secreted virulence factors activate the integrated stress response via a parallel independent pathway. Both stress pathways lead to the induction of the protein GADD34, which appears to provide protection against the toxic effects of the secreted virulence factors.
Introduction
The Gram-negative bacterium Pseudomonas aeruginosa is an opportunistic pathogen that increases morbidity and mortality in chronic lung diseases, such as cystic fibrosis (CF) and chronic obstructive pulmonary disease (COPD; GOLD stages III-IV)) [1–3]. P. aeruginosa often causes chronic infection due to its ease of developing antibiotic resistance and its ability to form biofilms in these patients. Furthermore, its survival in the host in the early stages of infection is supported by the secretion of toxins and virulence factors, including pyocyanin and its proteases elastase and alkaline protease (AprA) (reviewed in [4, 5]). Interestingly, their production appears to be lower in the later stages of infection [6, 7]. Therefore, the specific role of these virulence factors in chronic infections is incompletely understood. Pyocyanin is a redox-active toxin that causes cellular senescence [8], ciliary dyskinesia [9], increased expression of IL-8 [10] and disruption of calcium homeostasis [11] in human lung epithelial cells. Pyocyanin inactivates α1-antitrypsin, thereby contributing to the protease-antiprotease imbalance found in CF lungs [12], while P. aeruginosa elastase additionally cleaves many proteins of the extra-cellular matrix, including collagen, fibrinogen and elastin, and opsonin receptors, thus contributing to the invasion of bacteria into the lung parenchyma [13]. AprA is thought to modulate the host response and prevent bacterial clearance by degrading proteins of the host immune system, including TNFα and complement factors [14–16].
P. aeruginosa requires iron both for its respiration and for biofilm formation [17, 18]. Competition with the host is fierce and so P. aeruginosa has evolved specific strategies to obtain iron [19]. It produces redox-active phenazine compounds to turn insoluble Fe3+ to the more soluble Fe2+, siderophores to scavenge iron and receptors for the uptake of iron-siderophore complexes, proteases to degrade host iron-binding proteins, and bacteriocins to eliminate competitors (reviewed in [19]). Moreover, iron availability regulates the production of virulence factors such as pyocyanin, AprA and exotoxin A [20].
The endoplasmic reticulum (ER) functions to fold secretory and membrane proteins and its quality control systems ensure that only properly folded proteins exit the organelle. Accumulation of incompletely folded proteins can impair ER homeostasis and induces “ER stress”, which activates intracellular signal transduction pathways collectively called the “unfolded protein response” (UPR; Fig 1). This response restores ER homeostasis by reducing the influx of new proteins into the lumen of the ER and by enhancing the organelle’s capacity to fold proteins; however, if the stress cannot be resolved then apoptotic cell death pathways are invoked (reviewed in [21]).

Three distinct sensors detect ER stress: protein kinase RNA (PKR)-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1) and activating transcription factor 6 (ATF6) [21]. Early during ER stress, the kinase PERK phosphorylates eukaryotic translation initiation factor 2 on its alpha subunit (eIF2α) causing the inhibition of protein synthesis and thus preventing the load on the ER from increasing further [22–24]. In addition, this promotes the translation of specific mRNAs, for example that encoding the transcription factor ATF4 [25]. One important target of ATF4 is the transcription factor called C/EBP homologous protein (CHOP), and both individually can trans-activate the GADD34 gene [26]. GADD34 is a phosphatase that selectively dephosphorylates eIF2α, completing a negative feedback loop and enabling the translation of other targets of the UPR [27]. In parallel, IRE1 initiates the unconventional splicing of the mRNA encoding X-box binding protein-1 (XBP-1) [28]. Spliced XBP-1 mRNA encodes an active transcription factor that, in concert with ATF6, induces expression of UPR genes, such as the chaperones GRP78 (also known as BiP) and GRP94 [28–30].
The phosphorylation of eIF2α is a point at which the responses to several forms of stress are integrated [31]. During ER stress, PERK phosphorylates eIF2α, but eIF2a can also be phosphorylated by PKR responding to double-stranded RNA during viral infection [32, 33], by GCN2 during amino acid starvation [25, 34, 35], and by HRI during iron deficiency (reviewed in [31]). For this reason, the events initiated by eIF2α phosphorylation have been termed the “integrated stress response” (ISR; Fig 1 and [36]).
Abnormal function of the ER has been implicated in the pathogenesis of many diseases, including diabetes mellitus, atherosclerosis, Alzheimer’s disease and cancer [21, 37]. Remarkably, the ER also plays an important role during immune responses to infection and malignancy. For example, during bacterial infection, Toll-like receptor (TLR) activation triggers splicing of XBP1 mRNA, possibly in response to the increased biosynthesis of secreted inflammatory mediators, increasing the capacity for protein secretion and thus contributing to an augmented inflammatory response [38–40]. In addition, induction of GADD34 is required for cytokine expression during viral infection; however, in contrast to ER stress, pathogen-induced induction of GADD34 appears to be independent of CHOP [41, 42]. Nevertheless, sustained activation of the UPR can impair the immune response by triggering cell death [26, 43].
Previously, it has been shown that infection of airway epithelia or Caenorhabditis elegans with P. aeruginosa can elicit an UPR [39, 44, 45]. In worms, activation of the IRE1-XBP-1 branch of the UPR was dependent on p38 MAPK-signalling [39], but it is unknown if this signalling response is conserved in humans. Moreover, it is unclear whether living bacteria are required for the induction of ER stress or if unidentified secreted factors are sufficient.
In the present study, we set out to test the hypothesis that virulence factors secreted by P. aeruginosa trigger the UPR in human cells via the p38 MAPK pathway. We found that p38 MAPK signalling was required for the response of human epithelial cultures to bacterial conditioned medium and that the secreted factors pyocyanin and AprA contribute to the induction of ER stress. Furthermore, we showed that induction of the ISR target GADD34 is mediated by the iron-regulated kinase HRI and this induction protects the host against the toxic effects of P. aeruginosa.
Results
Conditioned medium of P. aeruginosa strain PAO1 causes ER stress in primary bronchial epithelial cells
Infection with live P. aeruginosa has previously been shown to induce the UPR in mouse macrophages and human immortalized bronchial epithelial cells [40, 45]. To identify whether P. aeruginosa could induce the UPR in primary bronchial epithelial cells (PBEC) and whether living bacteria were necessary for this, we stimulated PBEC with filter-sterilised conditioned medium (CM) from P. aeruginosa strain PAO1 (CM-PAO1), containing secreted virulence factors without living bacteria. Treatment with CM-PAO1 induced ER stress in a time - and dose-dependent manner, as evidenced by a 9.9-fold increase of splicing of XBP1 mRNA (p<0.01), a 12.8-fold increase of CHOP mRNA (p = 0.02) and a 16.2-fold increase of GADD34 mRNA (p<0.05) after 8–12 hours (Fig 2A and 2B). This was accompanied by an increase in phosphorylation of eIF2α and protein expression of GADD34 and GRP78 (Fig 2C). This increase in phosphorylated eIF2α was accompanied by a decrease in global protein translation as assessed by puromycin incorporation in nascent proteins (Fig 2D) [46]. In line with previous reports [47–49], CM-PAO1 gradually impaired epithelial integrity until the monolayer was completely disrupted after 24 hours. Although the epithelial layer was disrupted by CM-PAO1 (as reported by trans-epithelial resistance; S1A Fig and visualised by light microscopy; S1B Fig), the cell membranes themselves remained intact as reported by exclusion of trypan blue stain (S1B Fig).

Induction of ER stress in human bronchial epithelial cells by P. aeruginosa is dependent on p38 MAPK
Infection of C. elegans with P. aeruginosa has been reported to cause splicing of XBP1 mRNA in a p38 MAPK-dependent manner [39]. To exclude the effects of donor variation and complex nutrient/growth factor requirement of primary cells, we tested whether exposure of 16HBE cells, a SV-40 transformed bronchial epithelial cell line, to P. aeruginosa conditioned medium would trigger phosphorylation of p38 MAPK and activate the UPR. We observed that CM-PAO1 caused prolonged phosphorylation of p38 MAPK in 16HBE cells up to 6 hours (Fig 3A). We reasoned that the activation of p38 MAPK after 15 minutes might represent the activation of TLR signalling, since stimulation of HEK-TLR2 or HEK-TLR4 cells [50] with CM-PAO1 demonstrated robust TLR2 and TLR4 activation. The sustained activation was similar to that observed in C. elegans infected with Pseudomonas [39], which suggests the importance of p38 MAPK in the induction of the UPR. To examine if p38 MAPK signalling was required for the ER stress response, we pre-treated 16HBE cells with an inhibitor of p38 MAPK (SB203580) or an inhibitor of TAK1 (5Z-7-oxozeanol, better known as LL-Z1640-2), a kinase upstream of p38 MAPK. We then exposed cells to CM-PAO1 and observed that both compounds markedly reduced activation of p38 by CM-PAO1 (Fig 3B). In addition, both compounds reduced secretion of IL-8 in response to CM-PAO1 treatment (Fig 3C). Of note, these compounds strongly inhibited splicing of XBP1 mRNA and abrogated the induction of CHOP and GRP78 mRNA (Fig 3D). However, the induction of GADD34 was insensitive to the inhibitors (Fig 3D) suggesting the involvement of an additional pathway independent of CHOP.

Pyocyanin is able to induce ER stress
To prepare P. aeruginosa conditioned medium, cultures were grown for 5 days (see Experimental procedures and [47]) to a high optical density, at which quorum-sensing is activated in this strain, thus triggering the production of a variety of virulence factors among which the cytotoxic exoproduct pyocyanin. When pyocyanin levels in CM-PAO1 were measured, values up to 5.5 μg/ml (26 μM) were detected (Fig 4A), which were similar to values observed in sputum of CF patients colonised with P. aeruginosa [51]. We first wished to determine if pyocyanin was an important mediator of the observed ER stress response by CM-PAO1. To this end, P. aeruginosa bacterial cultures were supplemented with iron to suppress pyocyanin production together with other iron-regulated factors (Fig 4A). The conditioned medium prepared in this manner was significantly less efficient at triggering the splicing of XBP1 mRNA and at increasing expression of GRP78 mRNA (Fig 4B). Surprisingly, CHOP mRNA was not significantly affected (Fig 4B), whereas GADD34 mRNA induction was completely abrogated.

These experiments provided only indirect support for the involvement of pyocyanin, since iron supplementation also affects production of other P. aeruginosa virulence factors and may also affect host cells. We therefore tested whether purified pyocyanin could induce ER stress in 16HBE cells. Treatment with purified pyocyanin caused dose-dependent splicing of XBP1 mRNA, induction of CHOP and GRP78 mRNAs and expression of GRP78 and GRP94 protein (Fig 4C and 4D), maximal at 10 μM (2.1 μg/ml). In contrast, GADD34 mRNA continued to rise up to a maximum at ≥ 30 μM (6.3 μg/ml) of pyocyanin (Fig 4D). Once again, this suggested that induction of GADD34 in this system might not simply reflect activation by ER stress. As expected, pyocyanin potently induced secretion of IL-8 by 16HBE cells (Fig 4E) [10].
Since pyocyanin is a redox active toxin, we tested the effect of co-administration of the anti-oxidants N-acetylcysteine (10 mM) and glutathione reduced ethyl-ester (10 mM) for 24 hours. Both failed to ameliorate the ER stress response suggesting that pyocyanin caused ER dysfunction independent of causing oxidative stress [52, 53] (see online repository).
Taken together, these observations suggested that conditioned medium of P. aeruginosa caused ER stress via multiple virulence factors, including pyocyanin. Furthermore, the induction of GADD34 appeared to involve an additional pathway independent of CHOP.
Identifying other factors mediating ER stress
Having found evidence for the involvement of multiple virulence mechanisms in the induction of ER stress, we next attempted to determine their identities. The P. aeruginosa AB toxin exotoxin A is known to cause translational attenuation by catalysing the ADP-ribosylation of elongation factor 2 (EF2) [54]. We investigated whether purified exotoxin A could also induce ER stress, but detected no increase in spliced XBP1, CHOP, GADD34 or GRP78 mRNA (S2A Fig) nor the phosphorylation of eIF2α (S2B Fig). Next, to more broadly explore the involvement of other potential virulence factors, we made use of strains of P. aeruginosa that lacked specific toxic products: PAN8, a lasB aprE double mutant, which is deficient in the production of elastase [55] and the secretion of AprA; PAN11, an xcpR lasB mutant, which is deficient in the production of elastase and the secretion of all other substrates of the type II protein secretion system but still produces AprA; and PAO25, a leu arg double mutant derivative of PAO1 and the direct parental strain of both mutants (Table 1). CM-PAO25 did not differ from CM-PAO1 in the content of all toxins measured (S3A and S3B Fig) and in inducing spliced XBP1, CHOP, GADD34 and GRP78 mRNA (S3C Fig). In spite of the aprE mutation, still traces of AprA were detected in the culture supernatant of the PAN8 strain (Fig 5A), presumably due to cell lysis during the 5-days growth period.

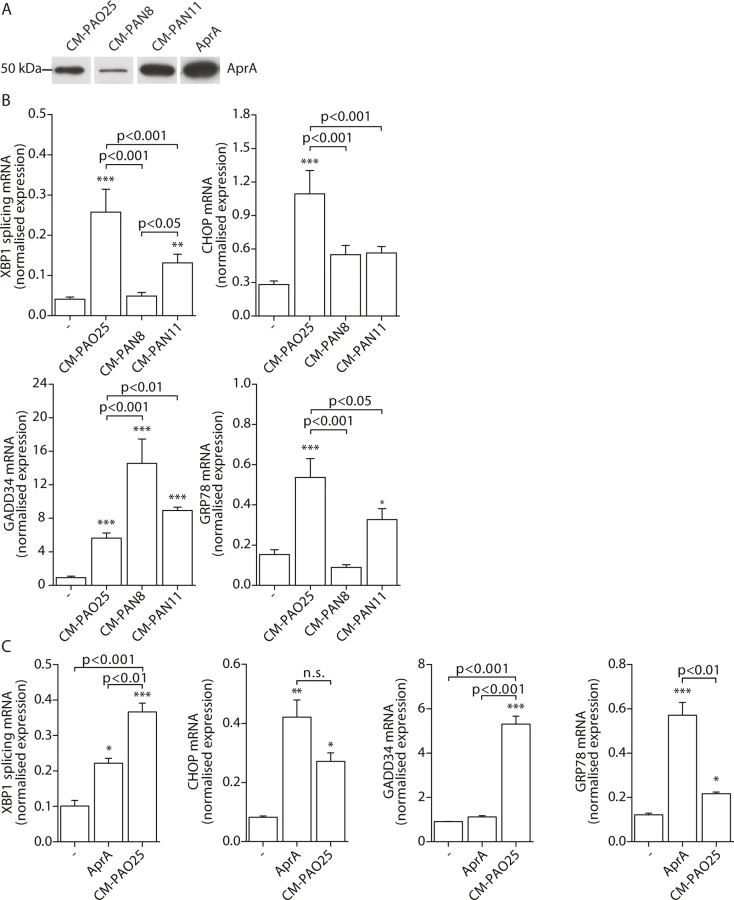
When 16HBE cells were incubated with CM-PAN8 (lacking elastase and AprA), XBP1 mRNA splicing and induction of GRP78 mRNA were completely abolished, and only low induction of CHOP mRNA remained (Fig 5B). In contrast, the response of 16HBE cells to CM-PAN11 (containing AprA, but no elastase or other substrates of the type 2 secretion system) was much less affected relative to CM-PAO1 treatment (Fig 5B), indicating that the reduced response to CM-PAN8 is primarily due to the absence of AprA in this CM rather than to the absence of elastase. Indeed, stimulating 16HBE cells with purified elastase did not elicit an ER stress response within 24 hours (see online repository). On the other hand, incubation with 10 nM purified AprA induced the splicing of XBP1 mRNA, and up-regulated CHOP and GRP78 mRNA (Fig 5C). These experiments suggested that, in addition to pyocyanin, AprA also contributed to the induction of ER stress in 16HBE cells. We therefore next generated conditioned medium of a series of specific AprA and pyocyanin mutant strains to demonstrate the relative contribution of AprA and pyocyanin to the induction of ER stress. However, these experiments were inconclusive because the corresponding wild type strains did not induce sufficient ER stress (see online repository).
Remarkably, once again the induction of GADD34 mRNA followed a distinct trend from the other markers of ER stress. Particularly a lack of AprA (in CM-PAN8) was correlated with an increased expression of GADD34 (Fig 5B), whilst purified AprA did not induce GADD34 mRNA (Fig 5C). This suggested that an unrelated mechanism regulated GADD34 induction by CM-PAO1 and that this might be independent of ER stress.
GADD34 is regulated via the integrated stress response (ISR) independent of PERK
To examine the involvement of ER stress-dependent and-independent responses to CM-PAO1, we next made use of the specific inhibitor of IRE1, 4μ8C, which blocks splicing of XBP1 mRNA during ER stress ([56] and Fig 6). Of note, this compound not only attenuated the splicing of XBP1 mRNA elicited by CM-PAO1, but interestingly, it also attenuated the secretion of IL-8 by 16HBE in response to CM-PAO1 (S4A Fig).

During ER stress, the kinase PERK phosphorylates eIF2α, thereby activating the ISR. When Perk-/- mouse embryonic fibroblasts (MEFs) were exposed to CM-PAO1, the induction of Gadd34 mRNA was unaffected, while the response to the ER stress-inducing agent tunicamycin (Tm) was abrogated (Fig 7A). However, phosphorylation of eIF2α was required for the induction of Gadd34 mRNA in response to CM-PAO1 as demonstrated by the failure of the conditioned medium to induce Gadd34 mRNA in fibroblasts homozygous for the eIF2αAA mutation, which renders them insensitive to all eIF2α kinases (Fig 7B). Moreover, ATF4, a transcription factor translationally up-regulated upon phosphorylation of eIF2α, was essential for the induction Gadd34 mRNA by CM-PAO1 (Fig 7C). As we have shown previously [26], CHOP was only partially required for tunicamycin (ER stress)-induced expression of Gadd34 mRNA (S4B Fig). The same was observed for CM-PAO1, although it did not reach statistical significance (S4B Fig). Interestingly, murine fibroblasts stimulated with CM-PAO1 failed to splice Xbp1 mRNA (S4C Fig), suggesting that activation of IRE1 by CM-PAO1 may be less important in this cell type than in human epithelial cells. However, reassuringly, ISR-dependent signalling in response to pseudomonal toxins was preserved in these cells and, once again, expression of Chop mRNA was regulated via eIF2α and ATF4. As had been observed for Gadd34, Chop induction was independent of PERK, suggesting that in MEFs treated with CM-PAO1, Chop was induced by a stimulus other than ER stress (S4D–S4F Fig).

We next examined which eIF2α kinase was responsible for activation of the ISR by CM-PAO1. To this end, we made use of Pkr-/-, Gcn2-/- and Hri-/- MEFs [25, 57, 58] and observed a significant deficit of CM-PAO1 induction of Chop and Gadd34 mRNA in Hri-/- cells, suggesting the involvement of the iron-sensing kinase HRI (Fig 7D–7F and S4G–S4I Fig). In contrast, although it has been suggested previously that GCN2 is involved in the stress response induced by P. aeruginosa in gut epithelial cells [59], we observed no significant effect on the induction of Gadd34 mRNA in Gcn2-/- cells (Fig 7E). We therefore went on to deplete either GCN2 or HRI in HeLa cells using two separate siRNA oligonucleotides for each gene and obtained similar results: whereas both siRNAs directed against HRI decreased induction of Gadd34 mRNA, one siRNA directed against GCN2 had no effect whereas the other even increased Gadd34 mRNA expression (Fig 7H and S4J Fig). Whereas we cannot exclude the possibility that this increasing effect of one siRNA directed against GCN2 may result from putative off-target effects, we conclude that these data support a role for HRI rather than GCN2.
Since RPMI is an iron-poor medium, we reasoned that the CM-PAO1 would limit iron availability to epithelial cells, e.g. by the presence of siderophores [60], which might activate HRI through depletion of iron from the culture medium. We therefore first evaluated the effect of iron depletion of the epithelial cell culture medium using deferoxamine (DFO). DFO treatment resulted in a marked increase in the expression of the ISR and UPR related genes CHOP and GADD34, whereas GRP78 and spliced XBP1 were not affected (Fig 7H). This is line with selective activation of the ISR by iron depletion. We next confirmed the presence of the iron-chelating siderophore pyoverdine in the CM-PAO1 by the bright fluorescence of the medium upon exposure to UV light (see online repository). To test the possible involvement of iron depletion in CM-PAO1-mediated Gadd34 induction, we supplemented the epithelial cell culture medium with iron, which indeed completely suppressed the induction of Gadd34 mRNA (Fig 7I and S4K Fig).
Taken together, these data demonstrate that CM-PAO1 induces splicing of XBP1 mRNA (ER stress) in human bronchial epithelial cells, while induction of GADD34 predominantly reflects an iron-dependent ISR mediated by the eIF2α kinase HRI.
The role of Gadd34 induction in cell survival
During chronic ER stress in cell and animal models of disease, the induction of GADD34 appears to mediate cellular toxicity [26, 43]. In contrast, during the acute stress of SERCA pump inhibition by thapsigargin, GADD34 has been shown to be protective [61]. To test the role of ER stress-independent induction of GADD34 by exposure to CM-PAO1, we made use of Gadd34ΔC/ΔC MEFs [61], which lack GADD34 phosphatase activity. Cells expressing wild-type GADD34 were more resistant to the cytotoxic effects of CM-PAO1 compared with Gadd34ΔC/ΔC fibroblasts, as reported by the release of lactate dehydrogenase (LDH) (Fig 8A). To confirm these findings, we repeated these experiments in HeLa cells expressing GADD34 from a tetracycline-responsive promoter. The induction of GADD34 with doxycycline significantly increased cell viability upon exposure to CM-PAO1 (Fig 8B). When the cell culture medium of wild-type cells was supplemented with iron, the release of LDH was prevented (Fig 8C, left panel). Iron supplementation was also observed to rescue cell viability reported by MTT assay (Fig 8C, right panel).

Taken together, these data suggest that the toxicity of CM-PAO1 is sensitive to iron and that HRI-mediated induction of GADD34 is protective in this context. Supplementation with iron relieves both the cytotoxicity and the requirement for induction of GADD34.
Discussion
It is known that a normal response to ER stress is required for an efficient innate immune response to bacterial infection [39], but whether live bacteria are required for this has been unclear. In this study, we have shown that secreted virulence factors of P. aeruginosa cause ER stress in primary bronchial epithelial cells and in a cell line, and that this is mediated by TAK1 and phosphorylated p38 MAPK. In addition, we have identified GADD34 induction via an ER-stress independent ISR. We have demonstrated pyocyanin to be one of the factors eliciting these responses, while AprA contributes to the activation of the UPR. We were however unable to establish the relative contribution of pyocyanin and AprA to the activation of the UPR. In contrast, activation of the ISR with induction of GADD34 mRNA is most likely a response to reduced iron availability and may serve a cytoprotective role during exposure to conditioned medium of P. aeruginosa.
In line with these observations, phosphorylation of p38 MAPK has previously been shown to be involved in the splicing of XBP1 upon infection with P. aeruginosa [39, 45], although the involvement of TAK1 upstream of p38 MAPK and its essential involvement in the activation of CHOP and GRP78 are novel findings. Interestingly, GADD34, classically a downstream target of CHOP, was regulated independently of the TAK1-p38 MAPK pathway. The induction of GADD34 is only partially dependent on CHOP (S4B Fig and [26]), but it is absolutely reliant on phosphorylation of eIF2α and ATF4 [26]. This is concordant with the recent description of a virus-induced “microbial stress response” mediated via the PKR/eIF2α/ATF4 pathway, which fails to induce CHOP, but potently induces GADD34 [41, 42].
In contrast to the response of human airway epithelial cells, P. aeruginosa conditioned medium failed to cause splicing of Xbp1 mRNA in murine fibroblasts, suggesting that ER stress may not be a conserved feature of the cellular response to this insult. This is unsurprising, as induction of ER stress is known to be highly cell-type dependent [40]. In the absence of ER stress in the murine fibroblasts, the induction of Chop and Gadd34 suggests that activation of the ISR by the secreted virulence factors may be a more conserved response. Of note, in human bronchial epithelial cells, the induction of CHOP seems primarily subordinate to an ER stress-induced ISR, rather than the microbial stress response (S7 Fig). Consequently, induction of CHOP was dependent on the TAK1-p38 MAPK pathway in those cells (Fig 3D) and its induction was only partially inhibited when bacterial cultures were supplemental with iron (Fig 4B), in contrast to MEFs where Chop induction was dependent on HRI (S4I Fig).
Recent evidence suggests that bacterial components may function as triggers for the UPR. Flagellin has been shown to induce an atypical ER stress response in CF bronchial epithelial cells during live infection [45], while N-(3-oxo-dodecanoyl) homoserine lactone (C12) has been observed to phosphorylate eIF2α and activate p38 MAPK [62]. We have now shown that at least two secreted virulence factors, pyocyanin and AprA, also contribute to this ER stress response to Pseudomonas. More research has to be done to assess the involvement of (other) individual virulence factors.
High concentrations of pyocyanin also mediated an ER stress-independent, ISR-dependent induction of GADD34 (Fig 4E). We were able to identify a crucial role for iron availability and for the iron-sensing kinase HRI in this response, although we cannot fully exclude a role for the kinase GCN2 that has been previously implicated in responses to Pseudomonas spp [59]. Of note, it is possible that the protective effect of GADD34 is unrelated to its ability to dephosphorylate p-eIF2alpha. Interestingly, AprA was not involved in the induction of the ISR response but rather appeared to dampen it, since considerably higher GADD34 expression was observed when conditioned medium of the aprE mutant PAN8 was used to stimulate the cells (Fig 5B). Among other possibilities, an explanation for this observation could be that AprA present in the conditioned medium of the wild-type strain partially degrades HRI, a possibility that warrants further investigations. The discovery of this ER-independent ISR may plausibly offer novel potential therapeutic targets.
It has been shown recently that spliced XBP1 is required for C12-mediated apoptosis [62]. Remarkably, exposure of cells to C12 does not itself trigger the splicing of XBP1 mRNA suggesting that basal levels of XBP1 splicing are both necessary and sufficient for this response. Moreover, the transcriptional activity of spliced XBP1 does not appear to be required for this cell death, indicating that the spliced XBP1 protein may have additional, as yet unidentified, activities. C12 appears able to trigger the ISR in an ER stress-independent matter, although the mechanism for this remains to be determined. It would be interesting to determine if C12 can activate HRI.
Chronic elevation of GADD34 in ER stress can mediate cellular toxicity [26], but GADD34 has been shown to be protective during the acute stress of SERCA pump inhibition with thapsigargin, which depletes the ER of calcium [61]. As with thapsigargin, P. aeruginosa has been associated with altered ER calcium signalling [38, 44]. It is therefore of interest that expression of GADD34 reduced cell toxicity and increased cell survival upon iron deficiency caused by treatment with conditioned medium from P. aeruginosa. It has been shown that lungs of cystic fibrosis patients lack the ability to induce GADD34 [45], which might plausibly lead to increased cytotoxicity or altered innate immunity due to Pseudomonas infection of the lungs of CF patients. However, future in vivo studies are required to confirm the observed cytoprotective effect of GADD34 induction during Pseudomonas infections.
In summary, secreted virulence factors of the PAO1 strain of P. aeruginosa, including pyocyanin and AprA, are sufficient to elicit an ER stress response but the relative contribution of these virulence factors remains to be investigated. In contrast to these virulence factors, our findings strongly suggest that iron depletion mediated by the secretion of siderophores causes an ER stress-independent ISR. The induction of GADD34 by this may serve to ameliorate the toxic effects of P. aeruginosa conditioned medium.
Materials and Methods
Bacterial strains and preparation of conditioned medium of P. aeruginosa
All strains used in this study are listed in Table 1. CM was prepared as described previously with slight modifications [47]. Briefly, overnight bacterial cultures in Luria Broth were inoculated 1 : 50 into RPMI 1640 (Gibco, Life Technologies, Breda, the Netherlands) and incubated at 37°C shaking at 200 rpm. After 5 days, the cultures were centrifuged and supernatants were filter-sterilized through 0.22 μm pore-size filter (Whatman, Dassel, Germany) to obtain CM. Pyocyanin and AprA levels in CM were measured as described previously [63, 64].
Cell culture
PBEC were obtained from tumour-free resected lung tissue by enzymatic digestion as described previously [65]. 16HBE cells (passage 4–15; kindly provided by Dr. D.C. Gruenert, University of California, San Francisco, CA, USA) were cultured in MEM (Invitrogen) supplemented with 1 mM HEPES (Invitrogen), 10% (v/v) heat-inactivated FCS (Bodinco, Alkmaar, the Netherlands), 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (all from BioWhittaker). All MEFs were maintained as described previously [23, 26, 36, 66, 67]. HEK-TLR2 and HEK-TLR4 [50] were a kind gift from Prof. Dr. M. Yazdanbakhsh (Leiden University Medical Center, the Netherlands). HeLa cells were transfected for 6 hours with two different ON-TARGETplus Human EIF2AK1 siRNA (GCACAAACUUCACGUUACU and GAUUAAGGGUGCAACUAAA) and knockdown was assessed 48 hours after transfection (S5 Fig).
GADD34-N1-eGFP (kind gift form S. Shenolikar, Duke-NUS Graduate Medical School Singapore, Singapore) was excised with BglII and NotI and ligated into pTRE2-hyg plasmid (Clontech Laboratories, Mountain View, CA, USA) digested with BamHI and NotI. HeLa Tet-On advanced cells (Clontech Laboratories) were transfected with the pTRE2-hyg_GADD34-eGFP plasmid and selected with 600 μM hygromycin to generate a stable cell line conditionally expressing GADD34-GFP (S6 Fig). Positive cell clones were visualised by GFP expression in response to 1 μg/ml of doxycycline. Once identified, expanded and characterized, these clones were maintained in DMEM (Sigma) supplemented with 10% FBS and antibiotics (100 U/ml penicillin G, 100 μg/ml streptomycin, 200 μg/ml G418 and 200 μM hygromycin). Expression of GADD34 was typically induced using 1 μg/ml doxycycline (Sigma) for 24 hours.
Cells were exposed at 80–90% confluence for 24 hours (unless stated otherwise) to CM-PAO1 (1 in 5 dilution, unless stated otherwise), pyocyanin (1–30 μM), ammonium iron (III) citrate (100 μM; Fe3+), exotoxin A (1–10 ng/ml), AprA (10 nM), elastase (16–64 μg/ml) and/or DFO (1–100 nM) as indicated (all from Sigma). Puromycin (10 μg/ml; Sigma) was added 30 minutes before harvesting. Thapsigargin (100 nM; Sigma), TNFα and IL-1β (both 20 ng/ml; Peprotech, Rocky Hill, NJ) were used as positive controls. The compounds SB203580 (10 nM; Sigma) and 5Z-7-oxozeanol (also called LL-Z1640-2; 100 nM; TebuBio, Heerhugowaard, the Netherlands) were added 30 minutes before stimulation for the inhibition of p38 MAPK and TAK1, respectively. The specific IRE1-inhibitor 4μ8C (30 μM) [56] was a kind gift from Prof. Dr. D. Ron, University of Cambridge.
Western blot
Cells were lysed in buffer H (10 mM HEPES, pH 7.9, 50 mM NaCl, 500 mM sucrose, 0.1 mM EDTA, 0.5% (v/v) Triton X-100, 1 mM PMSF, 1X Complete protease inhibitor cocktail (Roche Applied Science, Mannheim, Germany)) supplemented with phosphatase inhibitors (10 mM tetrasodium pyrophosphate, 17.5 mM β-glycerophosphate, and 100 mM NaF [25, 27]) for detection by antibodies directed against phospho-eIF2α (Cell Signaling Technology, Danvers, MA, USA), eIF2α (gift from Prof. Dr. D. Ron), KDEL (Enzo Life Sciences), GADD34 (ProteinTech, Chicago, IL, USA), puromycin (Millipore, Billerica, MA, USA), β-actin and GAPDH (CellSignalling), or in sample buffer (0.2 M Tris-HCl pH 6.8, 16% [v/v] glycerol, 4% [w/v] SDS, 4% [v/v] 2-mercaptoethanol, 0.003% [w/v] bromophenol blue) for detection by antibodies directed against (phospho-) p38 MAPK (both CellSignalling). The proteins in the samples were separated using a 10% SDS-PAGE gel and transferred onto a nitrocellulose membrane. After blocking with PBS containing 0.05% Tween-20 (v/v) and 5% skimmed-milk (w/v), the membrane was incubated overnight with the primary antibody (1 : 1000) in TBS with 0.05% Tween-20 (v/v) and 5% BSA (w/v) at 4°C. Next, the membrane was incubated with HRP-labelled anti-mouse or anti-rabbit antibody (Sigma) in blocking buffer for 1 hour and developed using ECL (ThermoScientific).
Quantitative reverse-transcriptase polymerase chain reaction (qPCR)
Total RNA was isolated using Qiagen RNeasy mini kit (Qiagen/Westburg, Leusden, the Netherlands). Quantitative reverse-transcriptase polymerase chain reaction (qPCR) was performed as described previously [68] using the primer pairs as defined in Table 2. Relative mRNA concentrations of RPL13A and ATP5B (GeNorm, PrimerDesign Ltd., Southampton, UK) were used as housekeeping genes for human genes and Actb (β-actin) and Sdha for mouse genes to calculate normalized expression.
ELISA
IL-8 was measured using commercially available ELISA kit according to manufacturer’s instructions (Sanquin, Amsterdam, the Netherlands).
Cytotoxicity assays
LDH release was measured with a LDH-cytotoxicity colorimetric assay kit following manufacturer’s instructions (Biovision, Milpitas, CA, USA). Thiazolyl blue tetrazolium bromide (MTT; Sigma) was dissolved in a 5 mg/ml stock concentration in sterile water and cells were incubated with a 1 : 10 dilution for 2 hours at 37°C. Next, the water-insoluble formazan formed from MTT in viable cells was dissolved in isopropanol for 10 min before the absorbance was read at 570 nm wavelength.
Electric cell-sensing impedance sensing
Epithelial barrier function was measured using ECIS (Applied Biophysics, Troy, NY, USA) as described previously [69]. Resistance was measured at 1000 Hz and cells were stimulated with CM-PAO1 when the resistance was stable.
Statistical analysis
Results are expressed as mean ± SEM. Data were analysed using one - or two-way analysis of variance (ANOVA) and corrected with the Bonferroni post-hoc test. Differences with P-values <0.05 were considered to be statistically significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Gomez MI, Prince A. Opportunistic infections in lung disease: Pseudomonas infections in cystic fibrosis. Current opinion in pharmacology. 2007;7(3):244–51. 17418640
2. Soler N, Torres A, Ewig S, Gonzalez J, Celis R, El-Ebiary M, et al. Bronchial microbial patterns in severe exacerbations of chronic obstructive pulmonary disease (COPD) requiring mechanical ventilation. American journal of respiratory and critical care medicine. 1998;157(5 Pt 1):1498–505. 9603129
3. Sethi S, Evans N, Grant BJ, Murphy TF. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. The New England journal of medicine. 2002;347(7):465–71. 12181400
4. Rada B, Leto TL. Pyocyanin effects on respiratory epithelium: relevance in Pseudomonas aeruginosa airway infections. Trends in microbiology. 2013;21(2):73–81. doi: 10.1016/j.tim.2012.10.004 23140890
5. Bleves S, Viarre V, Salacha R, Michel GP, Filloux A, Voulhoux R. Protein secretion systems in Pseudomonas aeruginosa: A wealth of pathogenic weapons. International journal of medical microbiology: IJMM. 2010;300(8):534–43. doi: 10.1016/j.ijmm.2010.08.005 20947426
6. Hunter RC, Klepac-Ceraj V, Lorenzi MM, Grotzinger H, Martin TR, Newman DK. Phenazine content in the cystic fibrosis respiratory tract negatively correlates with lung function and microbial complexity. American journal of respiratory cell and molecular biology. 2012;47(6):738–45. doi: 10.1165/rcmb.2012-0088OC 22865623
7. Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, D'Argenio DA, et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(22):8487–92. 16687478
8. Muller M. Premature cellular senescence induced by pyocyanin, a redox-active Pseudomonas aeruginosa toxin. Free radical biology & medicine. 2006;41(11):1670–7.
9. Wilson R, Pitt T, Taylor G, Watson D, MacDermot J, Sykes D, et al. Pyocyanin and 1-hydroxyphenazine produced by Pseudomonas aeruginosa inhibit the beating of human respiratory cilia in vitro. The Journal of clinical investigation. 1987;79(1):221–9. 3098783
10. Denning GM, Wollenweber LA, Railsback MA, Cox CD, Stoll LL, Britigan BE. Pseudomonas pyocyanin increases interleukin-8 expression by human airway epithelial cells. Infection and immunity. 1998;66(12):5777–84. 9826354
11. Denning GM, Railsback MA, Rasmussen GT, Cox CD, Britigan BE. Pseudomonas pyocyanine alters calcium signaling in human airway epithelial cells. The American journal of physiology. 1998;274(6 Pt 1):L893–900. 9609727
12. Britigan BE, Railsback MA, Cox CD. The Pseudomonas aeruginosa secretory product pyocyanin inactivates alpha1 protease inhibitor: implications for the pathogenesis of cystic fibrosis lung disease. Infection and immunity. 1999;67(3):1207–12. 10024562
13. Tamura Y, Suzuki S, Kijima M, Takahashi T, Nakamura M. Effect of proteolytic enzyme on experimental infection of mice with Pseudomonas aeruginosa. The Journal of veterinary medical science / the Japanese Society of Veterinary Science. 1992;54(3):597–9. 1643183
14. Bardoel BW, van der Ent S, Pel MJ, Tommassen J, Pieterse CM, van Kessel KP, et al. Pseudomonas evades immune recognition of flagellin in both mammals and plants. PLoS pathogens. 2011;7(8):e1002206. doi: 10.1371/journal.ppat.1002206 21901099
15. Parmely M, Gale A, Clabaugh M, Horvat R, Zhou WW. Proteolytic inactivation of cytokines by Pseudomonas aeruginosa. Infection and immunity. 1990;58(9):3009–14. 2117578
16. Hong YQ, Ghebrehiwet B. Effect of Pseudomonas aeruginosa elastase and alkaline protease on serum complement and isolated components C1q and C3. Clinical immunology and immunopathology. 1992;62(2):133–8. 1730152
17. Singh PK, Parsek MR, Greenberg EP, Welsh MJ. A component of innate immunity prevents bacterial biofilm development. Nature. 2002;417(6888):552–5. 12037568
18. O'May CY, Sanderson K, Roddam LF, Kirov SM, Reid DW. Iron-binding compounds impair Pseudomonas aeruginosa biofilm formation, especially under anaerobic conditions. Journal of medical microbiology. 2009;58(Pt 6):765–73. doi: 10.1099/jmm.0.004416-0 19429753
19. Vasil ML, Ochsner UA. The response of Pseudomonas aeruginosa to iron: genetics, biochemistry and virulence. Molecular microbiology. 1999;34(3):399–413. 10564483
20. Kim EJ, Wang W, Deckwer WD, Zeng AP. Expression of the quorum-sensing regulatory protein LasR is strongly affected by iron and oxygen concentrations in cultures of Pseudomonas aeruginosa irrespective of cell density. Microbiology (Reading, England). 2005;151(Pt 4):1127–38. 15817780
21. Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiological reviews. 2006;86(4):1133–49. 17015486
22. Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397(6716):271–4. 9930704
23. Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Molecular cell. 2000;5(5):897–904. 10882126
24. Sood R, Porter AC, Ma K, Quilliam LA, Wek RC. Pancreatic eukaryotic initiation factor-2alpha kinase (PEK) homologues in humans, Drosophila melanogaster and Caenorhabditis elegans that mediate translational control in response to endoplasmic reticulum stress. The Biochemical journal. 2000;346 Pt 2 : 281–93. 10677345
25. Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Molecular cell. 2000;6(5):1099–108. 11106749
26. Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes & development. 2004;18(24):3066–77.
27. Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. The Journal of cell biology. 2001;153(5):1011–22. 11381086
28. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–91. 11779464
29. Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Molecular biology of the cell. 1999;10(11):3787–99. 10564271
30. Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, et al. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Molecular and cellular biology. 2000;20(18):6755–67. 10958673
31. Dalton LE, Healey E, Irving J, Marciniak SJ. Phosphoproteins in stress-induced disease. Progress in molecular biology and translational science. 2012;106 : 189–221. doi: 10.1016/B978-0-12-396456-4.00003-1 22340719
32. Clemens MJ, Elia A. The double-stranded RNA-dependent protein kinase PKR: structure and function. Journal of interferon & cytokine research: the official journal of the International Society for Interferon and Cytokine Research. 1997;17(9):503–24.
33. Samuel CE, Kuhen KL, George CX, Ortega LG, Rende-Fournier R, Tanaka H. The PKR protein kinase--an interferon-inducible regulator of cell growth and differentiation. International journal of hematology. 1997;65(3):227–37. 9114594
34. Sood R, Porter AC, Olsen DA, Cavener DR, Wek RC. A mammalian homologue of GCN2 protein kinase important for translational control by phosphorylation of eukaryotic initiation factor-2alpha. Genetics. 2000;154(2):787–801. 10655230
35. Berlanga JJ, Santoyo J, De Haro C. Characterization of a mammalian homolog of the GCN2 eukaryotic initiation factor 2alpha kinase. European journal of biochemistry / FEBS. 1999;265(2):754–62. 10504407
36. Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Molecular cell. 2003;11(3):619–33. 12667446
37. Roussel BD, Kruppa AJ, Miranda E, Crowther DC, Lomas DA, Marciniak SJ. Endoplasmic reticulum dysfunction in neurological disease. Lancet neurology. 2013;12(1):105–18. doi: 10.1016/S1474-4422(12)70238-7 23237905
38. Ribeiro CM, Paradiso AM, Schwab U, Perez-Vilar J, Jones L, O'Neal W, et al. Chronic airway infection/inflammation induces a Ca2+i-dependent hyperinflammatory response in human cystic fibrosis airway epithelia. The Journal of biological chemistry. 2005;280(18):17798–806. 15746099
39. Richardson CE, Kooistra T, Kim DH. An essential role for XBP-1 in host protection against immune activation in C. elegans. Nature. 2010;463(7284):1092–5. doi: 10.1038/nature08762 20182512
40. Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nature immunology. 2010;11(5):411–8. doi: 10.1038/ni.1857 20351694
41. Clavarino G, Claudio N, Couderc T, Dalet A, Judith D, Camosseto V, et al. Induction of GADD34 is necessary for dsRNA-dependent interferon-beta production and participates in the control of Chikungunya virus infection. PLoS pathogens. 2012;8(5):e1002708. doi: 10.1371/journal.ppat.1002708 22615568
42. Clavarino G, Claudio N, Dalet A, Terawaki S, Couderc T, Chasson L, et al. Protein phosphatase 1 subunit Ppp1r15a/GADD34 regulates cytokine production in polyinosinic:polycytidylic acid-stimulated dendritic cells. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(8):3006–11. doi: 10.1073/pnas.1104491109 22315398
43. Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nature cell biology. 2013;15(5):481–90. doi: 10.1038/ncb2738 23624402
44. Martino ME, Olsen JC, Fulcher NB, Wolfgang MC, O'Neal WK, Ribeiro CM. Airway epithelial inflammation-induced endoplasmic reticulum Ca2+ store expansion is mediated by X-box binding protein-1. The Journal of biological chemistry. 2009;284(22):14904–13. doi: 10.1074/jbc.M809180200 19321437
45. Blohmke CJ, Mayer ML, Tang AC, Hirschfeld AF, Fjell CD, Sze MA, et al. Atypical Activation of the Unfolded Protein Response in Cystic Fibrosis Airway Cells Contributes to p38 MAPK-Mediated Innate Immune Responses. Journal of immunology (Baltimore, Md: 1950). 2012;189(11):5467–75. doi: 10.4049/jimmunol.1103661 23105139
46. Liu J, Xu Y, Stoleru D, Salic A. Imaging protein synthesis in cells and tissues with an alkyne analog of puromycin. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(2):413–8. doi: 10.1073/pnas.1111561108 22160674
47. Halldorsson S, Gudjonsson T, Gottfredsson M, Singh PK, Gudmundsson GH, Baldursson O. Azithromycin maintains airway epithelial integrity during Pseudomonas aeruginosa infection. American journal of respiratory cell and molecular biology. 2010;42(1):62–8. doi: 10.1165/rcmb.2008-0357OC 19372247
48. Soong G, Parker D, Magargee M, Prince AS. The type III toxins of Pseudomonas aeruginosa disrupt epithelial barrier function. Journal of bacteriology. 2008;190(8):2814–21. doi: 10.1128/JB.01567-07 18165298
49. Vikstrom E, Tafazoli F, Magnusson KE. Pseudomonas aeruginosa quorum sensing molecule N-(3 oxododecanoyl)-l-homoserine lactone disrupts epithelial barrier integrity of Caco-2 cells. FEBS letters. 2006;580(30):6921–8. 17157842
50. van der Kleij D, van den Biggelaar AH, Kruize YC, Retra K, Fillie Y, Schmitz M, et al. Responses to Toll-like receptor ligands in children living in areas where schistosome infections are endemic. The Journal of infectious diseases. 2004;189(6):1044–51. 14999608
51. Wilson R, Sykes DA, Watson D, Rutman A, Taylor GW, Cole PJ. Measurement of Pseudomonas aeruginosa phenazine pigments in sputum and assessment of their contribution to sputum sol toxicity for respiratory epithelium. Infection and immunity. 1988;56(9):2515–7. 3137173
52. O'Malley YQ, Reszka KJ, Rasmussen GT, Abdalla MY, Denning GM, Britigan BE. The Pseudomonas secretory product pyocyanin inhibits catalase activity in human lung epithelial cells. American journal of physiology Lung cellular and molecular physiology. 2003;285(5):L1077–86. 12871859
53. Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxidants & redox signaling. 2007;9(12):2277–93.
54. Yates SP, Jorgensen R, Andersen GR, Merrill AR. Stealth and mimicry by deadly bacterial toxins. Trends in biochemical sciences. 2006;31(2):123–33. 16406634
55. Braun P, de Groot A, Bitter W, Tommassen J. Secretion of elastinolytic enzymes and their propeptides by Pseudomonas aeruginosa. Journal of bacteriology. 1998;180(13):3467–9. 9642203
56. Cross BC, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM, et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(15):E869–78. doi: 10.1073/pnas.1115623109 22315414
57. Yang YL, Reis LF, Pavlovic J, Aguzzi A, Schafer R, Kumar A, et al. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. The EMBO journal. 1995;14(24):6095–106. 8557029
58. Han AP, Yu C, Lu L, Fujiwara Y, Browne C, Chin G, et al. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. The EMBO journal. 2001;20(23):6909–18. 11726526
59. Chakrabarti S, Liehl P, Buchon N, Lemaitre B. Infection-induced host translational blockage inhibits immune responses and epithelial renewal in the Drosophila gut. Cell host & microbe. 2012;12(1):60–70.
60. Visca P, Imperi F, Lamont IL. Pyoverdine siderophores: from biogenesis to biosignificance. Trends in microbiology. 2007;15(1):22–30. 17118662
61. Novoa I, Zhang Y, Zeng H, Jungreis R, Harding HP, Ron D. Stress-induced gene expression requires programmed recovery from translational repression. The EMBO journal. 2003;22(5):1180–7. 12606582
62. Valentine CD, Anderson MO, Papa FR, Haggie PM. X-Box Binding Protein 1 (XBP1s) Is a Critical Determinant of Pseudomonas aeruginosa Homoserine Lactone-Mediated Apoptosis. PLoS pathogens. 2013;9(8):e1003576. doi: 10.1371/journal.ppat.1003576 23990788
63. Essar DW, Eberly L, Hadero A, Crawford IP. Identification and characterization of genes for a second anthranilate synthase in Pseudomonas aeruginosa: interchangeability of the two anthranilate synthases and evolutionary implications. Journal of bacteriology. 1990;172(2):884–900. 2153661
64. Laarman AJ, Bardoel BW, Ruyken M, Fernie J, Milder FJ, van Strijp JA, et al. Pseudomonas aeruginosa alkaline protease blocks complement activation via the classical and lectin pathways. Journal of immunology (Baltimore, Md: 1950). 2012;188(1):386–93. doi: 10.4049/jimmunol.1102162 22131330
65. van Wetering S, van der Linden AC, van Sterkenburg MA, Rabe KF, Schalkwijk J, Hiemstra PS. Regulation of secretory leukocyte proteinase inhibitor (SLPI) production by human bronchial epithelial cells: increase of cell-associated SLPI by neutrophil elastase. Journal of investigative medicine: the official publication of the American Federation for Clinical Research. 2000;48(5):359–66. 10979241
66. Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Molecular cell. 2001;7(6):1165–76. 11430820
67. Kumar A, Yang YL, Flati V, Der S, Kadereit S, Deb A, et al. Deficient cytokine signaling in mouse embryo fibroblasts with a targeted deletion in the PKR gene: role of IRF-1 and NF-kappaB. The EMBO journal. 1997;16(2):406–16. 9029159
68. van Schadewijk A, van't Wout EF, Stolk J, Hiemstra PS. A quantitative method for detection of spliced X-box binding protein-1 (XBP1) mRNA as a measure of endoplasmic reticulum (ER) stress. Cell stress & chaperones. 2012;17(2):275–9.
69. Wegener J, Keese CR, Giaever I. Electric cell-substrate impedance sensing (ECIS) as a noninvasive means to monitor the kinetics of cell spreading to artificial surfaces. Experimental cell research. 2000;259(1):158–66. 10942588
70. Haas D, Holloway BW. R factor variants with enhanced sex factor activity in Pseudomonas aeruginosa. Molecular & general genetics: MGG. 1976;144(3):243–51.
71. Scott DW, Mutamba S, Hopkins RG, Loo G. Increased GADD gene expression in human colon epithelial cells exposed to deoxycholate. Journal of cellular physiology. 2005;202(1):295–303. 15316935
72. Hirota M, Kitagaki M, Itagaki H, Aiba S. Quantitative measurement of spliced XBP1 mRNA as an indicator of endoplasmic reticulum stress. The Journal of toxicological sciences. 2006;31(2):149–56. 16772704
73. Fitch SR, Kimber GM, Wilson NK, Parker A, Mirshekar-Syahkal B, Gottgens B, et al. Signaling from the sympathetic nervous system regulates hematopoietic stem cell emergence during embryogenesis. Cell stem cell. 2012;11(4):554–66. doi: 10.1016/j.stem.2012.07.002 23040481
74. Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. The Journal of clinical investigation. 2008;118(10):3378–89. doi: 10.1172/JCI34587 18776938
75. Lipson KL, Ghosh R, Urano F. The role of IRE1alpha in the degradation of insulin mRNA in pancreatic beta-cells. PloS one. 2008;3(2):e1648. doi: 10.1371/journal.pone.0001648 18286202
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 6
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- HIV Latency Is Established Directly and Early in Both Resting and Activated Primary CD4 T Cells
- Battling Phages: How Bacteria Defend against Viral Attack
- A 21st Century Perspective of Poliovirus Replication
- Adenovirus Tales: From the Cell Surface to the Nuclear Pore Complex
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy