
IRF-5-Mediated Inflammation Limits CD8 T Cell Expansion by Inducing HIF-1α and Impairing Dendritic Cell Functions during Infection
Inflammation is essential for inducing, sustaining, and regulating CD8+ T cell responses. The transcription factor IRF-5 is mainly responsible for initiating the inflammatory response following experimental Leishmani donovani infection. IRF-5 activates several genes encoding key pro-inflammatory cytokines, such as IL-6 and TNF. In this study, we investigate the role of IRF-5-mediated inflammation in regulating antigen-specific CD8+ T cell responses during L. donovani infection. Our data demonstrate that the inflammatory response induced by IRF-5 limits the expansion CD8+ T cell. This negative effect is mediated by the induction of HIF-1α in dendritic cells. Indeed, we observed a significant increase in CD8+ T cell expansion in mice lacking HIF-1α expression in dendritic cells. Moreover, these mice had a significantly lower parasite burden in the spleen, suggesting that induction of HIF-1α may represent an immune evasive mechanism adopted by Leishmania parasites to establish persistent infections.
Published in the journal:
. PLoS Pathog 11(6): e32767. doi:10.1371/journal.ppat.1004938
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004938
Summary
Inflammation is essential for inducing, sustaining, and regulating CD8+ T cell responses. The transcription factor IRF-5 is mainly responsible for initiating the inflammatory response following experimental Leishmani donovani infection. IRF-5 activates several genes encoding key pro-inflammatory cytokines, such as IL-6 and TNF. In this study, we investigate the role of IRF-5-mediated inflammation in regulating antigen-specific CD8+ T cell responses during L. donovani infection. Our data demonstrate that the inflammatory response induced by IRF-5 limits the expansion CD8+ T cell. This negative effect is mediated by the induction of HIF-1α in dendritic cells. Indeed, we observed a significant increase in CD8+ T cell expansion in mice lacking HIF-1α expression in dendritic cells. Moreover, these mice had a significantly lower parasite burden in the spleen, suggesting that induction of HIF-1α may represent an immune evasive mechanism adopted by Leishmania parasites to establish persistent infections.
Introduction
Maintenance of a proper balance between inflammatory and anti-inflammatory responses is essential for achieving effective immunity against infectious pathogens while limiting collateral inflammatory damage to the tissue. However, immunosuppressive responses are sometimes generated in excess. This event often results in the strong inhibition of protective pro-inflammatory responses and leads to susceptibility to infectious pathogens, such as Plasmodium [1,2], Leishmania [3,4], lymphocytic choriomeningitis virus [5,6], and Mycobacteria spp. [7].
Visceral leishmaniasis (VL) is a good example of dysregulated balance between inflammatory and anti-inflammatory responses. VL is a potentially lethal disease caused by Leishmania donovani and L. infantum/chagasi. Leishmania are protozoan parasites, existing as flagellated promastigotes within sandflies and as intracellular amastigotes in infected mammals. In the host, Leishmania preferentially infects macrophages; however, it can also be found in other cells, such as DCs, neutrophils, and fibroblasts [8–12]. VL is characterized by persistent infection of the spleen and by immunodeficiency during the chronic stage [13]. Experimental infection with L. donovani results in pathogen-induced disruption of the splenic microarchitecture, which involves both the disruption of the marginal zone and the B-cell follicles, and the progressive loss of stromal cells [14,15]. This disruption is mediated by TNF [16], a cytokine that is overexpressed during VL [17,18]. Interestingly, TNF deficient mice infected with L. donovani have a lower IL-10 mRNA accumulation in the spleen than do their wild type counterparts [14], suggesting that TNF may be involved as a positive regulator of IL-10 production.
We have recently demonstrated that the inflammatory response following L. donovani infection is largely mediated by the transcription factor IRF-5 [19]. IRF-5 can be activated by TLR7 and TLR9 via the MyD88 signaling pathway and/or directly by viral infections and Type I interferon [20]. This transcription factor is responsible for the activation of genes encoding for various key inflammatory cytokines [21–24]. Interestingly, L. donovani infected Irf5-/- mice do not show the hallmark symptoms of VL, which are hepato-and splenomegaly, due to the lack of inflammatory cell infiltration. Furthermore, these mice generate profoundly defective Th1 responses during chronic disease [19]. The role of IRF-5-mediated inflammation on the development of CD8+ T cell responses during VL has not yet been explored.
We have previously shown that L. donovani induces defective CD8+ T cell responses with limited clonal expansion [25]. Moreover, the majority of CD8+ T cells that survive clonal contraction are central memory-like cells, suggesting that perhaps effector responses are not sustained.
Pro-inflammatory cytokines are known to tune CD8+ T cell responses and provide the critical third signal necessary for the development of effector CD8+ T cells [26–30]. For instance, IL-12 seems to regulate T-bet and eomesodermin (Eomes) expression [30,31], the differentiation of short-lived effector cells (SLEC) [30], and the cytolytic activity of CTLs [32]. Type I IFN, IFNγ, and IL-4 also appear to be required for efficient CD8+ T cells priming and memory differentiation [33–38]. The inflammatory milieu was also shown to control antigen sensitivity by enhancing T cell receptor signaling [39]. In contrast, Stelekati et al. recently reported that a bystander chronic inflammatory milieu impairs the development of CD8+ T cell memory following immunization [40]. This implies that inflammation does not always play a positive role in supporting the development of CD8+ T cell responses and that the role of inflammation might depend on the specific inflammatory milieu induced by each pathogen.
In this study, we investigated the role of IRF-5 mediated inflammation in regulating CD8+ T cell expansion following L. donovani infection. Our data shows that IRF-5 participates in limiting antigen-specific CD8+ T cell expansion at the very early stages of infection by indirectly inducing HIF-1α expression in DCs. Upregulation of HIF-1α in DCs resulted in decreased IL-12 and increased IL-10 expression. Ablation of HIF-1α in CD11c+ cells led to a higher frequency of short-lived effector CD8+ T cells (SLEC), enhanced CD8+ T cell expansion, and significantly reduced parasite burden.
Results
IRF-5-mediated inflammation limits CD8+ T cell expansion during acute infection
We have previously demonstrated that IRF-5 is essential for initiating the inflammatory response following experimental L. donovani infection. Indeed, Irf5-/- mice fail to generate mature granulomas in the liver and exhibit a severely reduced inflammatory infiltration in the liver and spleen [19]. Hence, to investigate the role of inflammation in the development of antigen-specific CD8+ T cells, we monitored CD8+ T cell responses in L. donovani infected IRF-5-deficient mice. To this end, we adoptively transferred CD45.1-OT-I CD8+ T cells into Irf5flox/flox-CMV-Cre- and Irf5flox/flox-CMV-Cre+ mice. Mice were subsequently infected with ovalbumin-transgenic L. donovani amastigotes. We have previously reported that OT-I CD8+ T cells undergo clonal expansion between day 3 and 7 after parasite inoculation; by day 14, about 80% of the cells display a central memory-like phenotype [25]. To our surprise, we observed a 3–4 fold increase in the number (Fig 1A) and frequency (Fig 1B) of OT-I CD8+ T cells present in the spleen of IRF-5-deficient mice at d7p.i.; no difference was detected at d14p.i. We next analysed the phenotype of OT-I CD8+ T cells found in the spleen at d7 and 14p.i. Despite the fact that about 35% of OT-I CD8+ T cells had downregulated CD127 (Fig 1C and S1 Fig), only 7% of the OT-I cells were CD127-KLRG1+ in the Cre- group (Fig 1D and S1 Fig), suggesting that only a small percentage of the cells were short-lived effector cells (SLEC). In contrast, a significant increase in frequency of CD127- (Fig 1C and S1 Fig) cells and SLEC (Fig 1D and S1 Fig) was noticed in IRF-5-deficient mice at d7 p.i.

Because Leishmania is known to induce strong IL-6 and TNF responses [17,18] and IRF-5 is known to govern the expression of both cytokines [21–24], we next assessed the mRNA levels for IL-6 and TNF in the spleen of L. donovani infected mice. As expected, IRF-5-deficient mice had a lower expression of IL-6 during the first week of infection (Fig 1E). By d14 p.i., however, both experimental groups expressed similar mRNA levels. In contrast, TNF mRNA levels were higher in Cre- mice compared to IRF-5 deficient mice during the first 2 weeks of infection (Fig 1F).
Taken together, our data suggest that IRF-5 participates in limiting CD8+ T cell expansion and hampering the development of SLEC.
Upregulation of HIF-1α in the spleen restricts CD8+ T cell expansion
Because inflammation is known to induce HIF-1α, we next proceeded to assess HIF-1α expression in splenocytes from L. donovani infected mice. The hypoxia-inducible transcription factor (HIF-1α) is the key regulator in the cellular response to hypoxia [41,42]. Under hypoxic conditions HIF-1α accumulates and translocates into the nucleus, where it binds the constitutively expressed HIF-1β [43]. The resultant heterodimer binds and triggers transcription of the hypoxia response element-containing genes in all cells [44]. Nevertheless, accumulation and transcriptional activity of HIF-1α can also be induced at normoxic conditions by pro-inflammatory cytokines such as TNF and IL-1β [45,46], or by TLR ligation [47–49]. Furthermore, Leishmania promastigotes were reported to induce HIF-1α in macrophages [50–52]. Hence, we first investigated whether HIF-1α expression was at all upregulated in splenocytes following the inoculation of L. donovani amastigotes. As expected, HIF-1α mRNA expression was upregulated in the spleen of L. donovani infected mice (Fig 2A). HIF-1α upregulation was confirmed on the protein level by western blot (Fig 2B). We next determined if the upregulation of HIF-1α in splenocytes had a negative effect on CD8+ T cell expansion and function. Thus, we generated hemizygous Hif1a+/- mice by crossing Hif1aflox/flox with CMV-Cre mice. We then monitored adoptively transferred CD45.1-OT-I CD8+ T cells at day 7 and 14 p.i. in L. donovani infected Hif1a+/- and Hif1aflox/flox mice. Interestingly, we observed a significant increase in the number (Fig 2C) as well as frequency (Fig 2D) of OT-I CD8+ T cells at day 7 p.i. in Hif1a+/- compared to the control group; at d14 p.i., however, both groups had similar number and frequency of OT-I CD8+ T cells in the spleen. As previously seen in IRF-5-deficient mice, increased expansion was paralleled by a higher frequency of effector cells (Fig 2E and S2 Fig). We also notice a significant decrease in the frequency of CD44hiCD62LhiCD127+ cells in Hif1a+/- mice compared to the control group (Fig 2F and S2 Fig). Collectively, our results suggest that HIF-1α might be involved in limiting CD8+ T cell expansion and effector cell differentiation.

L. donovani infection induces HIF-1α expression in CD11chi splenic DCs in an IRF-5-dependent manner
Enhanced CD8+ T cell responses were only observed at peak expansion in Hif1a+/- mice. Hence, it is possible that this transcription factor exerts different functions among various cell types. Since dendritic cells play a crucial role during T cell priming, we next assessed the effect of HIF-1α expression in these cells. First, we wanted to determine if HIF-1α was at all upregulated in dendritic cells during L. donovani infection. As shown in Fig 3A, HIF-1α was progressively expressed in DCs during the first week of infection. Increased expression was also confirmed on the protein level by western blot (Fig 3B). To determine whether IRF-5-mediated inflammation was at all involved in HIF-1α upregulation in DCs, we next assessed HIF-1α mRNA levels in DCs from IRF-5 deficient mice following L. donovani infection. Interestingly, HIF-1α expression was only upregulated in DCs purified from infected IRF-5-sufficent mice, but not in those purified from IRF-5 deficient mice (Fig 3C and 3D), suggesting that IRF-5 is involved in the induction of HIF-1α in DCs. Similar results were obtained when HIF-1α expression was assessed in CD11c- splenocytes (Fig 3E). HIF-1α was not directly induced by IRF-5 in DCs, since we could observe an increase in mRNA levels for HIF-1α in IRF-5 deficient bone marrow derived DCs infected with L. donovani in vitro (Fig 3F). This implies that IRF-5 is not required for the induction of HIF-1α when DCs are directly infected by the parasite. Because only a very small percentage of splenic DCs bury parasites during acute infection (S3 Fig), accumulation of HIF-1α mRNA in DCs is most likely due to the inflammatory milieu induced by IRF5.

HIF-1α expression in CD11c+ cells limits expansion of CD8+ T cells and favours the induction of MPEC
HIF-1α upregulation in DCs was shown to reduce their stimulatory capacity for T cell functions in vitro [53]. To investigate the role of HIF-1 α expression in DCs during experimental VL, we generated HIF-1α conditional knock-out mice by crossing Hif1aflox/flox with Cd11c-Cre mice. Firstly, we assessed whether HIF-1α expression was abolished in our conditional knock-outs. As shown in S4A Fig, HIF-1α mRNA levels did not increase in L. donovani infected BMDC from Hif1aflox/flox—Cd11c-Cre+ mice compared to the Cre- group as measured by real time PCR. Similar results were obtained with CD11c+ cells purified from the spleen following in vivo infection with L. donovani (S4B Fig).
It has been reported that Leishmania promastigotes require HIF-1α for their survival inside macrophages and DCs [50,51]. Thus, we next evaluated whether this transcription factor was also needed for survival inside DCs by the intracellular and clinically relevant form of the parasite, namely the amastigote. In contrast to promastigotes, amastigotes seemed to survive equally well in BMDC from Hif1aflox/flox—Cd11c-Cre+ mice compared to those derived from the Cre- group (Fig 4A). We also assessed amastigote survival in purified splenic CD11chi DCs and found no differences at any time point between the two groups of mice (Fig 4B). This suggests that amastigotes were able to survive inside DCs from Hif1aflox/flox—Cd11c-Cre+ mice.

We next proceeded to assess CD8+ T cell responses in our conditional knock-out mice. CD45.1-OT-I CD8+ T cells were adoptively transferred into Hif1aflox/flox—Cd11c-Cre+ mice and their Cre- littermates a day prior to infection with L. donovani amastigotes. CD8+ T cell responses were then monitored at d7 (peak expansion) and 14 p.i. (end of contraction). Remarkably, OT-I CD8+ T cells underwent a greater expansion in Hif1aflox/flox—Cd11c-Cre+ mice compared to their Cre- littermates as indicated by a larger number and higher frequency of cells present in the spleen at d7 p.i. (Fig 4C and 4D, left panels). Unlike in IRF-5 deficient and Hif+/- mice, this difference was still observed at d14 p.i., when OT-I CD8+ T cell numbers were 3–4 fold higher in the conditional knock-out group (Fig 4C and 4D, right panels). When we analyzed the phenotype of the adoptively transferred cells, we noticed a significant decrease in CD44hiCD62LhiCD127+ OT-I CD8+ T cells in Hif1aflox/flox—Cd11c-Cre+ mice compared to littermate controls at both time points analyzed (Fig 5A and S5 Fig), suggesting that less memory precursor effector cells (MPEC) were generated in those mice. These results were paralleled by an increase in effector cell frequency at d7 and 14 p.i. (Fig 5B and S5 Fig), which was reflected in a higher percentage of CD127-KLRG1+ cells (Fig 5C and S5 Fig) in Hif1aflox/flox—Cd11c-Cre+.
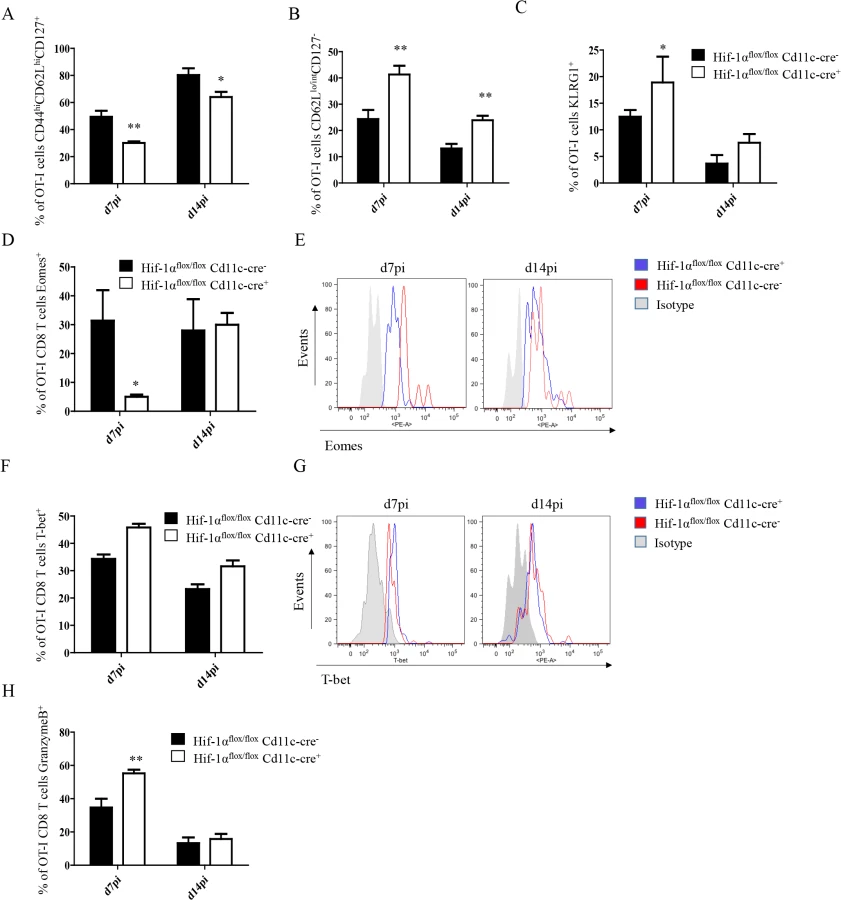
The T-box transcription factors Eomes and T-bet appear to regulate effector/memory differentiation program of CD8+ T cells. Because we observed an increase in frequency of SLEC, we were wondering whether Eomes and T-bet were expressed at different level in OT-I CD8+ T cells adoptively transferred into Hif1aflox/flox—Cd11c-Cre+ mice compared to their Cre- counterpart. As expected, we noticed a significant decrease in Eomes in Hif1aflox/flox—Cd11c-Cre+ mice at d7 p.i. compared to the HIF-1α sufficient group (Fig 5D and 5E). No differences, however, were observed at d14. A light upregulation of T-bet was also observed in OT-I CD8+ T cells from the Cre+ group compared to the Cre- littermates, but this was not statistically significant (Fig 5F and 5G).
Interestingly, OT-I CD8+ T cells also showed a higher cytotoxic capacity in Hif1aflox/flox—Cd11c-Cre+ mice compared to the control group (Fig 5H and S5C Fig), while maintaining similar levels of IFNγ, TNF, and IL-2 production upon restimulation in vitro. Taken together, our data suggest that HIF-1α expression in CD11c+ cells favours the development of MPEC and limits CD8+ T cell expansion following L. donovani infection.
HIF-1α hampers IL-12 expression by splenic CD11chi DCs
IL-12 plays an important role in promoting CD8+ T cell responses. Particularly, this cytokine has been shown to induce the development of SLEC by regulating T-bet expression [30,31]. In VL, IL-12 is mainly produced by DCs, however this production is not sustained after 24h [17]. Because more SLEC with a greater cytotoxic capacity were observed in Hif1aflox/flox—Cd11c-Cre+ mice, we were wondering whether HIF-1α deficient DCs were producing more IL-12. Hence, we purified CD11chi splenic DCs from L. donovani infected mice at various time points after infection and assessed the expression of IL-12p35 by real time PCR. As shown in Fig 6A, IL-12p35 expression was sustained in Hif1aflox/flox—Cd11c-Cre+ mice compared to the Cre- control group. Similar results were obtained when we measured IL-12p40 mRNA levels (Fig 6B).

We also assessed the expression of IL-10, a cytokine that is known to be associated with susceptibility to experimental VL. IL-10 mRNA accumulation is readily detected in DCs few days after L. donovani inoculation and is continuously produced until the chronic phase of the disease [54]. In agreement with the literature, we noticed an increase in IL-10 mRNA levels in the Cre- group during the first 2 weeks of infection (Fig 6C). In contrast, DCs from Hif1aflox/flox—Cd11c-Cre+ mice failed to upregulate IL-10 expression during the first week of infection. At d14 p.i., however, IL-10 mRNA levels in the Cre- group increased but were still significantly lower than the control group. No differences were observed between both groups in the IL-10 mRNA levels of the CD11c negative fraction. Interestingly, CD11chi splenic DCs lacking HIF-1α also expressed lower TNF mRNA levels compared to their HIF-1α-sufficient counterparts (Fig 6D); similarly to IL-10, TNF mRNA levels of the CD11c negative fraction were comparable in both groups between d5 and 14 p.i. (S6 Fig).
HIF-1α expression in CD11c+ cells exacerbates disease
Finally, we wanted to determine whether HIF-1α ablation in DCs had any biological effect on the course of L. donovani infection. Hence, we assessed the splenic parasite burden at various time points after infection (Fig 7A). We noticed a significant reduction in the splenic parasite burden in Hif1aflox/flox—Cd11c-Cre+ mice compared to the Cre- littermates already at d14p.i. Reduction in the parasite number in Hif1aflox/flox—Cd11c-Cre+ mice at d14p.i was also confirmed by limiting dilutions (Fig 7B). Interestingly, the hepatic parasite burden in Hif1aflox/flox—Cd11c-Cre+ mice was not different than their Cre- counterpart (Fig 7C). Finally, we proceeded to determine whether CD8+ T cells participate at all to the reduction in parasite numbers observed in the HIF-1α conditional knock-out group. Hence, we depleted CD8+ T cells in L. donovani infected Hif1aflox/flox—Cd11c-Cre+ and Cre- mice, and assessed the parasite burden at d14 p.i. As shown in Fig 7D, CD8+ T cell depletion only caused a mild and not significant increase in the parasite burden in Cre - mice; in contrast, CD8+ T cells seem to play an essential role in controlling parasite growth in HIF-1α conditional knockout mice, suggesting that HIF-1α induction in DCs is responsible for limiting protective CD8+ T cell responses.
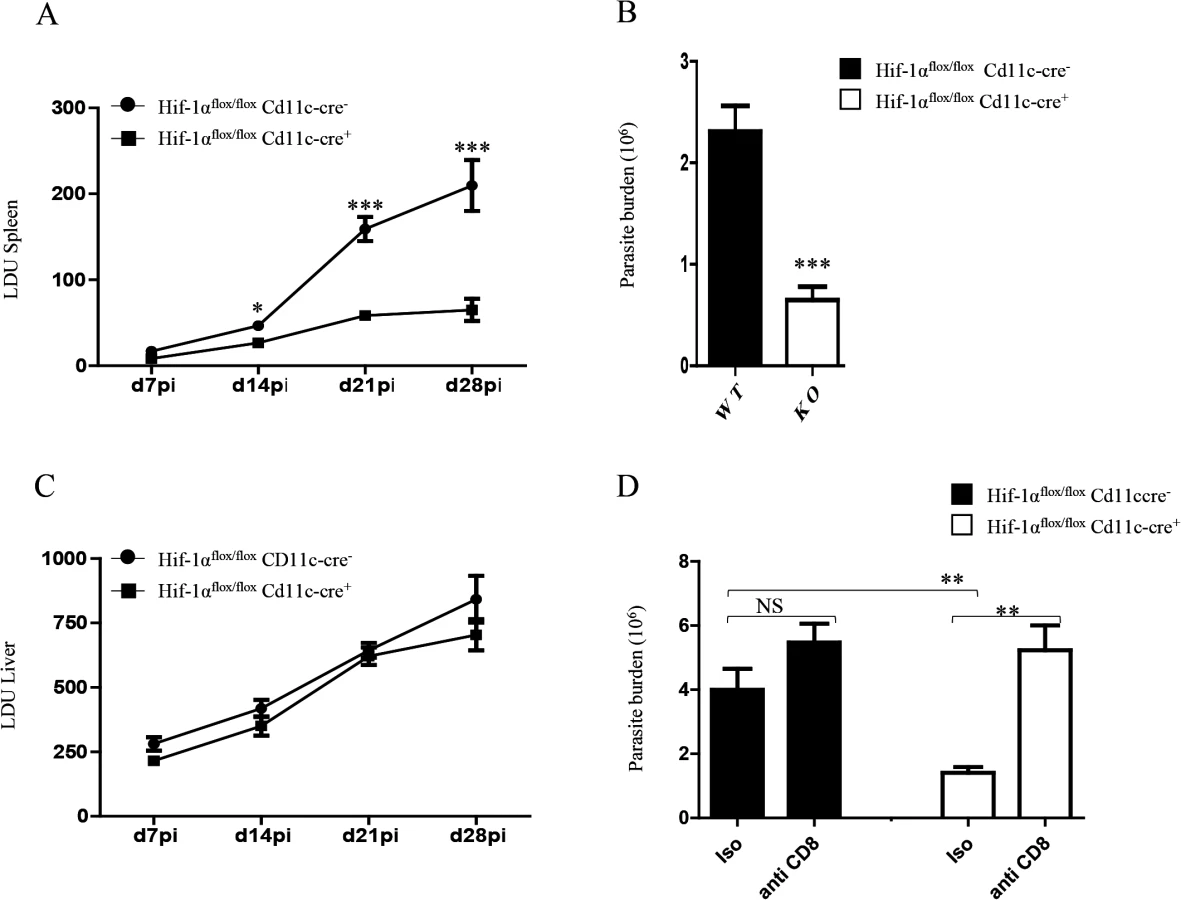
In conclusion, HIF-1α induction in DCs represents an immune-evasive mechanism adopted by Leishmania to establish chronic infection.
Discussion
The natural reaction of an organism to a foreign pathogen is to initiate an inflammatory response. This response is essential for effective immunity against the pathogen. In the present study though, we show that the IRF-5-dependent inflammatory milieu induced by Leishmania during the first week of infection inhibits CD8+ T cell expansion and the development of SLECs by inducing HIF-1α in dendritic cells and consequently altering DC functions. Ablation of HIF-1α in CD11c+ cells resulted in a lower parasite burden.
Following infection, antigen-specific CD8+ T cells expand and acquire effector function necessary for protective immunity. This process is initiated and sustained by inflammatory cytokines which play a major role during the development of CD8+ T cell responses by providing “signal 3”. Indeed, inflammatory cytokines were reported to tune effector/memory differentiation, expansion, and survival of antigen-specific CD8+ T cells [26–30,55]. IL-12, for instance, is crucial for effector function acquisition following infection with Listeria monocytogenes [55], vaccinia virus [56], and Toxoplasma gondii [57]. IL-12 was also shown to participate in the induction of T-bet while repressing Eomes [30,31,58]. In contrast, IL-12 deficiency does not seem to interfere with CD8+ T cell development following LCMV infection, where type I IFN play the most critical role [36,37]. Hence, it appears that inflammatory cytokines promote and maintain CD8+ T cell differentiation to effectors while preventing the development of memory cells. It is important to note that most of the work on the role of inflammation in promoting CD8+ T cells was done in models of acute infection. A recent work showed that a chronic inflammatory environment negatively impacted the development of CD8+ T cell responses [40], suggesting that perhaps the composition of the inflammatory milieu rather than inflammation itself determines the outcome of CD8+ T cell responses. It is thus crucial to identify the pathogen-specific signature of proinflammatory cytokines to determine the effect of inflammation on CD8+ T cell responses. Our data suggest that IRF-5-mediate inflammation induced by Leishmania at early stages of infection plays a detrimental role in the development of protective CD8+ T cell responses.
IRF-5 is involved in the transcriptional activation of both Type I IFN genes and genes encoding for key pro-inflammatory cytokines such as IL-12, TNF, IL-23 and IL-6 [21,22,24,59]. Our recent study using Irf5-/- mice indicates that IRF-5 is essential for the development of protective Th1 responses. In fact, L. donovani infected Irf5-/- mice generate profoundly defective Th1 cells during chronic disease. However, we observed stronger Th1 responses in Irf5-/- mice during the first two weeks of infection, suggesting that priming of Th1 cells was more effective in the absence of IRF-5 and that IRF-5 is mainly needed for sustaining Th1 responses [19]. These observations prompted us to think that the strong TNF-dominated inflammatory response induced by Leishmania at the onset of infection [18] [60] plays a dual role and may help the establishment of chronic infection by inhibiting T cell responses.
To control the inflammatory response launched by foreign pathogens upon encounter with the immune system, the host has evolved several defense mechanisms aimed at tissue protection. One of these mechanisms is the hypoxia-driven, adenosine receptor-mediated immune suppressive pathway. The hypoxia-adenosinergic tissue-protecting pathway is a physiologic response to inflammatory damage, low oxygen tension, and hypoxia-driven accumulation of extracellular adenosine. HIF-1α is the key regulator in the cellular response to hypoxia [41]. This pathway is designed to allow the cells to survive in an environment of low oxygen tension, but it also leads to suppression of pro-inflammatory responses [61–64]. Indeed, HIF-1α suppresses cytokine production in T-cells [64–66], induces the potency and the number of CD25+CD4+ regulatory T-cells [67], induces IL-10 production [68,69], and inhibits DC maturation [53]. Moreover, HIF-1α expression in tumor-associated macrophages enhances the T cell suppressive capacity of these cells [70,71]; HIF-1α also causes upregulation of PD-L1 on MDSC, exacerbating their suppressive capacity [72,73].
Under normoxic conditions, HIF-1α can be upregulated by inflammatory cytokines such as TNF and IL-1β, by TLR agonists, and by several viruses [74,75], bacteria [75,76], and parasites [51,75,77,78]. Leishmania promastigotes are known to stabilize HIF-1α and induce HIF-1α mRNA in host cells [51,52]. Our results indicate that L. donovani amastigotes are also able to induce HIF-1α mRNA in BMDC; however, unlike for promastigotes [51], HIF-1α was not required for amastigote survival inside DCs. Interestingly, HIF-1α mRNA was induced in dendritic cells following L. donovani infection in an IRF-5-dependent manner. Because HIF-1α mRNA levels in IRF-5 deficient BMDC infected with L. donovani amastigotes were comparable to those observed in infected IRF-5 sufficient BMDC, we assume that HIF-1α in DCs is mainly induced by the inflammatory milieu generated by IRF-5 rather than the parasite itself. This suggests that IRF-5 is not involved in the induction of HIF-1α in DCs upon direct infection with L. donovani. The factor(s) responsible for the induction of HIF-1α in DCs following infection with L. donovani amastigotes are yet unknown. We tried to block IL-6R and TNF, but this blockade did not affect HIF-1α expression or CD8+ T cell expansion, suggesting that the upregulation of HIF-1α in DCs does not rely on the effect of a single cytokine. Further investigations are warranted in order to identify the mechanism(s) of HIF-1α induction in DCs during experimental VL.
HIF-1α appears to play a dual role in myeloid cells. An important body of literature has demonstrated that HIF-1α is involved in myeloid cell-mediated inflammation and is critical in the defence against various pathogens [76,79,80]. For instance, macrophages phagocytose and kill bacteria better under hypoxic conditions rather than under normoxic conditions [76,80]. Thus it is not surprising that mice with a targeted deletion of HIF-1α in myeloid cells are more susceptible to many bacterial and viral pathogens [76,79,81]. In contrast, some pathogens such as Toxoplasma gondii and Leishmania (promastigotes) require HIF-1α for their survival inside the cell [50–52,78]. This transcription factor was also shown to enhance replication of some viruses [82–84]. In the tumor microenvironment, HIF-1α expression in tumor-associated macrophages suppresses T cell responses [70] and induces expression of arginase 1 [71]; HIF-1α also enhances the inhibitory effect of MDSC [72].
Divergent effects of HIF-1α upregulation have also been observed in DCs. HIF-1α appears to play an important role in DC differentiation and migration in a hypoxic environment [53,85]. Nevertheless, this transcription factor impairs the upregulation of CCR7, a chemokine involved in homing of mature DCs to secondary lymphoid organs [53,85]. Moreover, HIF-1α acts as a negative regulator of plasmacytoid dendritic cell development [86]. Our results reveal a detrimental role for HIF-1α in DC function during experimental VL. Indeed, the absence of HIF-1α in DCs resulted in increased CD8 T cell expansion and a higher frequency of SLECs. Increased expansion directly correlated with a lower splenic parasite burden in HIF-1α conditional knock out mice. In contrast, the parasite load in the liver remained unchanged. This is possibly due to the differences in the cellular environment between liver and spleen, and also to the type of cell that hosts the parasite at the onset of infection. The two organs generate very diverse immune responses that lead to parasite clearance in the liver and chronic infection in the spleen [13].
Interestingly, OT-I CD8+ T cells were still detectable at a higher frequency at d14 p.i. in Hifflox/flox - Cd11c-cre+ mice; in contrast in Hif+/-mice, the frequency at d14 p.i. was similar to the control group. This difference could possibly be caused by the general reduction in the level of IL-10 expression in infected Hif+/- mice compared to Hifflox/flox - Cd11c-cre+ mice. We have previously reported that IL-10 blockade induces increased expansion of CD8+ T cells [87]. However, in the absence of IL-10, CD8+ T cells display a more dramatic contraction, at the end of which CD8+ T cell numbers are similar to those observed in infected wild type mice. This suggests that HIF-1α expression in some cells is necessary to counterbalance the strong inflammatory responses induced by L. donovani and slow down clonal contraction.
L. donovani typically induces a swift IL-12 production by DCs at very early stages of infection; this production, however, ceases after 24h of infection [17]. We observed a sustained IL-12p35 and IL12p40 expression in HIF-1α deficient DCs. This expression was paralleled by a decrease in IL-10 and TNF mRNA levels. This suggests that HIF-1α directly or indirectly regulates the expression of IL-12, TNF, and IL-10 in dendritic cells. Increased expression of IL-12p35 and p40 may explain the higher frequency of SLECs detected in mice with a targeted deletion of HIF-1α in CD11c+ cells and therefore the enhanced expansion. Identifying the mechanism of HIF-1α induction in DCs will help to understand the physiological role of this transcription factor. It is possible that the pathway of activation as well as the duration of the signal determine the target gene selection and the final effect on DC functions.
In conclusion, we demonstrate that IRF-5-mediated inflammation induced by L. donovani at the onset of the infection participates in limiting CD8+ T cell expansion by upregulating HIF-1α in DCs and subsequently impairing DC functions. Several pathogens that cause chronic infections induce upregulation of HIF-1α, which is then directly required for their survival. Our data additionally shows that HIF-1α induction in DCs that are not the main target of Leishmania results in disease susceptibility. It is thus tempting to speculate that targeting the HIF-1α pathway may represent a major immune evasive mechanism adopted by some pathogens to establish chronic infection.
Materials and Methods
Mice and parasites
C57BL/6-Tg(OT-I)-RAG1tm1Mom mice and B6-Ly5.1 congenic mice were purchased from The Jackson Laboratory. Mice hemizygous for HIF-1α were generated by crossing once Hif1aflox/flox mice with mice expressing the cre-recombinase under the CMV promoter. Mice with a targeted HIF-1α mutation in CD11c+ cells were generated by crossing C57BL/6 Hif1aflox/flox mice with mice expressing the cre-recombinase under the CD11c promoter (C57BL/6 Cd11c-Cre+/-), both purchased by The Jackson Laboratory. IRF-5-deficient mice were generated by crossing C57BL/6 Irf5flox/flox mice with mice expressing the cre-recombinase under the CMV promoter (The Jackson Laboratory). C57BL/6 Irf5flox/flox mice were a kind gift from Dr. Paula Pitha-Rowe (The Johns Hopkins University). All mice were housed at the INRS animal facility under specific pathogen-free conditions and used at 6–10 weeks of age. Ovalbumin-transgenic parasites were a gift from Drs. P. Kaye and D.F. Smith (University of York, UK). Wild type and ovalbumin transgenic Leishmania donovani (strain LV9) were maintained by serial passage in B6.129S7-Rag1tm1Mom mice, and amastigotes were isolated from the spleens of infected animals. Mice were infected by injecting 2×107 amastigotes intravenously via the lateral tail vein. Splenic parasite burdens were determined either by limiting dilutions or by examining methanol-fixed, Giemsa stained tissue impression smears [87]. Data are presented as number of parasites per spleen or as Leishman Donovan Units (LDU).
Ethics statement
Experiments involving mice were carried out under protocols approved by the Comité Institutionel de Protection des Animaux of the INRS-Institut Armand-Frappier (1110–06, 1110–07). These protocols respect procedures on good animal practice provided by the Canadian Council on animal care.
Adoptive transfer of OT-I cells
CD45.1-OT-I/RAG1 mice, transgenic for a T cell receptor specific for chicken ovalbumin 257–264 presented by the MHC class I molecule H-2 Kb, were used as T cell donors. CD8+ T cells were enriched from splenocytes of naïve CD45.1-OT-I/RAG1 animals as previously described [25]. 2x104 CD45.1 - OT-I CD8+ T cells were injected into the lateral tail vein of Irf5flox/flox CMV-Cre+, Hif1a+/-, Hif1αflox/flox CD11c-Cre+ mice and their respective Cre- littermate controls. Animals were infected the day after with ovalbumin-transgenic Leishmania donovani amastigotes.
PKH67 labelling of parasites
L. donovani parasites were stained with PKH67 (Sigma) following manufacturer’s instructions, as previously described [87]. Mice received 5 x 107 PKH67 labeled parasites. Spleens from naïve and infected mice were harvested 24h later and surface stained for flow cytometric analysis.
Flow cytometry
Adoptively transferred OT-I CD8+ T cells were identified by staining splenocytes with FITC-conjugated anti-CD45.1 antibody and Pacific Blue-conjugated anti-CD8 (BD Biosciences). The following antibodies were used to further characterize the OT-I response: APC-conjugated anti-CD44 and anti-KLRG1, PE-Cy5-conjugated anti-CD62L, and PE-conjugated anti-CD127 (all obtained from eBioscience). For all surface markers, cells were directly stained as previously described [25]. For intracellular staining, splenocytes were stimulated with the SIINFEKL peptide for 4 hours in the presence of Brefeldin A and recombinant IL-2, and then stained with FITC-conjugated anti-CD45.1 and Pacific Blue-conjugated anti-CD8. After fixation, cells were permeabilized and stained with PE-conjugated anti-granzyme B (Invitrogen), PE-conjugated anti-Eomes and anti-IL-2 (BD Biosciences). APC-conjugated anti-INFγ and PE-Cy7-conjugated anti-TNF were also used. Flow cytometric analysis was performed with a BD LSRFortessa cell analyzer (Becton Dickinson). One to two millions cells per sample were acquired and analyzed with the FACSDiva or with the Flowjo software.
For DCs analysis after injection of PKH67-labelled parasites, spleens were removed 24h after parasite injection and digested with collagenase. Splenocytes were then stained with anti-CD11c-APC, anti-MHCII-PE, anti-CD8-PB, and anti-CD4-PE-Cy7. Flow cytometric analysis was performed with a BD LSRFortessa cell analyzer (Becton Dickinson).
Real-time PCR analysis
Real-time PCR (Stratagene mx3005p Real time PCR System) was used to analyze transcripts levels of HPRT, HIF-1α, IL-10, IL-12p35, IL-12p40, IL-6, and TNF [19,54,88,89]. Total RNA was insolated using RNeasy (Qiagen) to perform real-time RT-PCR. cDNA was prepared using 500 ng of total RNA using High capacity cDNA Reverse Transcription kit (Bio Rad). Real time PCR was performed using standard cycle of amplification.
Western blot analysis
Total cell protein extracts of CD11c+ cells purified by MACS and cell sorting from infected and naive mice were pooled and lysed in RIPA buffer (sigma Aldrich, Germany). Equal amounts of protein (15 μg) were fractionated by 10% SDS-PAGE. Monoclonal anti-HIF-1α antibody Hif-1α67 (Novus Biologicals, Littleton, CO, USA) was used for immunoblot assays. Blots were stripped and reprobed with a polyclonal antibody against β-actin to confirm equal protein loading. [90]. Densitometric analysis was performed by spot densitometry using AlphaImager 3400 imaging software (Alpha Innotech Corporation) and normalized to ß-actin control. Values are presented as fold induction compared to the level in naive mice.
Survival of L. donovani following in vitro infection
CD11c+ dendritic cells (DCs) were isolated using anti-CD11c beads (Miltenyi Biotec) [19]. BMDC were generated as previously described [91] and seeded at 106 cells/ml onto 24-well plates. Cells were subsequently infected with PKH26-stained L. donovani amastigotes (Sigma, staining done according to manufacturer’s instructions) at a MOI of 5 : 1. Infected cells were then harvested 24, 48, and 72 h later, fixed with 2% PFA, stained with Hoechst (Invitrogen, staining done according to manufacturer’s instructions) and cytospined on PBS/ 1% BSA-prepared slides. Mounted slides were than analyzed using a fluorescent microscope, to evaluate parasite survival and target infection rate.
CD8 T cell depletion
Hifflox/flox - Cd11c cre+ and cre- mice were treated bi-weekly with 0.2 mg of anti-CD8 antibody (clone 2.43), starting one day before infection. At day 14 p.i., mice were sacrificed and parasite burdens were determined by limiting dilutions.
Statistical analysis
Statistical analysis was performed using a Student’s t-test, with p<0.05 considered significant. All experiments were conducted independently at least three times.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Omer FM, de Souza JB, Riley EM (2003) Differential induction of TGF-beta regulates proinflammatory cytokine production and determines the outcome of lethal and nonlethal Plasmodium yoelii infections. J Immunol 171 : 5430–5436. 14607947
2. Wu Y, Wang QH, Zheng L, Feng H, Liu J, et al. (2007) Plasmodium yoelii: distinct CD4(+)CD25(+) regulatory T cell responses during the early stages of infection in susceptible and resistant mice. Exp Parasitol 115 : 301–304. 17084842
3. Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL (2002) CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature 420 : 502–507. 12466842
4. Anderson CF, Mendez S, Sacks DL (2005) Nonhealing infection despite Th1 polarization produced by a strain of Leishmania major in C57BL/6 mice. J Immunol 174 : 2934–2941. 15728505
5. Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, et al. (2006) Interleukin-10 determines viral clearance or persistence in vivo. Nat Med 12 : 1301–1309. 17041596
6. Ejrnaes M, Filippi CM, Martinic MM, Ling EM, Togher LM, et al. (2006) Resolution of a chronic viral infection after interleukin-10 receptor blockade. J Exp Med 203 : 2461–2472. 17030951
7. Hernandez-Pando R, Aguilar D, Hernandez ML, Orozco H, Rook G (2004) Pulmonary tuberculosis in BALB/c mice with non-functional IL-4 genes: changes in the inflammatory effects of TNF-alpha and in the regulation of fibrosis. Eur J Immunol 34 : 174–183. 14971043
8. Beil WJ, Meinardus-Hager G, Neugebauer DC, Sorg C (1992) Differences in the onset of the inflammatory response to cutaneous leishmaniasis in resistant and susceptible mice. J Leukoc Biol 52 : 135–142. 1506767
9. Bogdan C, Donhauser N, Doring R, Rollinghoff M, Diefenbach A, et al. (2000) Fibroblasts as host cells in latent leishmaniosis. J Exp Med 191 : 2121–2130. 10859337
10. Gorak PM, Engwerda CR, Kaye PM (1998) Dendritic cells, but not macrophages, produce IL-12 immediately following Leishmania donovani infection. Eur J Immunol 28 : 687–695. 9521079
11. Iezzi G, Frohlich A, Ernst B, Ampenberger F, Saeland S, et al. (2006) Lymph node resident rather than skin-derived dendritic cells initiate specific T cell responses after Leishmania major infection. J Immunol 177 : 1250–1256. 16818784
12. Peters NC, Egen JG, Secundino N, Debrabant A, Kimblin N, et al. (2008) In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science 321 : 970–974. doi: 10.1126/science.1159194 18703742
13. Kaye PM, Svensson M, Ato M, Maroof A, Polley R, et al. (2004) The immunopathology of experimental visceral leishmaniasis. Immunol Rev 201 : 239–253. 15361245
14. Ato M, Stager S, Engwerda CR, Kaye PM (2002) Defective CCR7 expression on dendritic cells contributes to the development of visceral leishmaniasis. Nat Immunol 3 : 1185–1191. 12436111
15. Smelt SC, Engwerda CR, McCrossen M, Kaye PM (1997) Destruction of follicular dendritic cells during chronic visceral leishmaniasis. J Immunol 158 : 3813–3821. 9103448
16. Engwerda CR, Ato M, Cotterell SE, Mynott TL, Tschannerl A, et al. (2002) A role for tumor necrosis factor-alpha in remodeling the splenic marginal zone during Leishmania donovani infection. Am J Pathol 161 : 429–437. 12163368
17. Engwerda CR, Murphy ML, Cotterell SE, Smelt SC, Kaye PM (1998) Neutralization of IL-12 demonstrates the existence of discrete organ-specific phases in the control of Leishmania donovani. Eur J Immunol 28 : 669–680. 9521077
18. Engwerda CR, Smelt SC, Kaye PM (1996) An in vivo analysis of cytokine production during Leishmania donovani infection in scid mice. Exp Parasitol 84 : 195–202. 8932769
19. Paun A, Bankoti R, Joshi T, Pitha PM, Stager S (2011) Critical role of IRF-5 in the development of T helper 1 responses to Leishmania donovani infection. PLoS Pathog 7: e1001246. doi: 10.1371/journal.ppat.1001246 21253574
20. Schoenemeyer A, Barnes BJ, Mancl ME, Latz E, Goutagny N, et al. (2005) The interferon regulatory factor, IRF5, is a central mediator of toll-like receptor 7 signaling. J Biol Chem 280 : 17005–17012. 15695821
21. Barnes BJ, Richards J, Mancl M, Hanash S, Beretta L, et al. (2004) Global and distinct targets of IRF-5 and IRF-7 during innate response to viral infection. J Biol Chem 279 : 45194–45207. 15308637
22. Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, et al. (2005) Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature 434 : 243–249. 15665823
23. Honda K, Takaoka A, Taniguchi T (2006) Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25 : 349–360. 16979567
24. Honda K, Taniguchi T (2006) IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol 6 : 644–658. 16932750
25. Joshi T, Rodriguez S, Perovic V, Cockburn IA, Stager S (2009) B7-H1 blockade increases survival of dysfunctional CD8(+) T cells and confers protection against Leishmania donovani infections. PLoS Pathog 5: e1000431. doi: 10.1371/journal.ppat.1000431 19436710
26. Pham NL, Badovinac VP, Harty JT (2011) Differential role of "Signal 3" inflammatory cytokines in regulating CD8 T cell expansion and differentiation in vivo. Front Immunol 2 : 4. doi: 10.3389/fimmu.2011.00004 22566795
27. Curtsinger JM, Gerner MY, Lins DC, Mescher MF (2007) Signal 3 availability limits the CD8 T cell response to a solid tumor. J Immunol 178 : 6752–6760. 17513722
28. Curtsinger JM, Mescher MF (2010) Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol 22 : 333–340. doi: 10.1016/j.coi.2010.02.013 20363604
29. Haring JS, Badovinac VP, Harty JT (2006) Inflaming the CD8+ T cell response. Immunity 25 : 19–29. 16860754
30. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, et al. (2007) Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 27 : 281–295. 17723218
31. Takemoto N, Intlekofer AM, Northrup JT, Wherry EJ, Reiner SL (2006) Cutting Edge: IL-12 inversely regulates T-bet and eomesodermin expression during pathogen-induced CD8+ T cell differentiation. J Immunol 177 : 7515–7519. 17114419
32. Curtsinger JM, Lins DC, Johnson CM, Mescher MF (2005) Signal 3 tolerant CD8 T cells degranulate in response to antigen but lack granzyme B to mediate cytolysis. J Immunol 175 : 4392–4399. 16177080
33. Badovinac VP, Tvinnereim AR, Harty JT (2000) Regulation of antigen-specific CD8+ T cell homeostasis by perforin and interferon-gamma. Science 290 : 1354–1358. 11082062
34. Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF (2005) Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol 174 : 4465–4469. 15814665
35. Stager S, Alexander J, Kirby AC, Botto M, Rooijen NV, et al. (2003) Natural antibodies and complement are endogenous adjuvants for vaccine-induced CD8+ T-cell responses. Nat Med 9 : 1287–1292. 14502281
36. Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K (2005) Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med 202 : 637–650. 16129706
37. Aichele P, Unsoeld H, Koschella M, Schweier O, Kalinke U, et al. (2006) CD8 T cells specific for lymphocytic choriomeningitis virus require type I IFN receptor for clonal expansion. J Immunol 176 : 4525–4529. 16585541
38. Whitmire JK, Tan JT, Whitton JL (2005) Interferon-gamma acts directly on CD8+ T cells to increase their abundance during virus infection. J Exp Med 201 : 1053–1059. 15809350
39. Richer MJ, Nolz JC, Harty JT (2013) Pathogen-specific inflammatory milieux tune the antigen sensitivity of CD8(+) T cells by enhancing T cell receptor signaling. Immunity 38 : 140–152. doi: 10.1016/j.immuni.2012.09.017 23260194
40. Stelekati E, Shin H, Doering TA, Dolfi DV, Ziegler CG, et al. (2014) Bystander chronic infection negatively impacts development of CD8(+) T cell memory. Immunity 40 : 801–813. doi: 10.1016/j.immuni.2014.04.010 24837104
41. Semenza GL (2009) Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology (Bethesda) 24 : 97–106. doi: 10.1152/physiol.00045.2008 19364912
42. Semenza GL (2009) Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin Cancer Biol 19 : 12–16. doi: 10.1016/j.semcancer.2008.11.009 19114105
43. Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, et al. (1996) Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem 271 : 32529–32537. 8955077
44. Semenza GL (2007) Hypoxia and cancer. Cancer Metastasis Rev 26 : 223–224. 17404692
45. Jung Y, Isaacs JS, Lee S, Trepel J, Liu ZG, et al. (2003) Hypoxia-inducible factor induction by tumour necrosis factor in normoxic cells requires receptor-interacting protein-dependent nuclear factor kappa B activation. Biochem J 370 : 1011–1017. 12479793
46. Zhou J, Schmid T, Brune B (2003) Tumor necrosis factor-alpha causes accumulation of a ubiquitinated form of hypoxia inducible factor-1alpha through a nuclear factor-kappaB-dependent pathway. Mol Biol Cell 14 : 2216–2225. 12808024
47. Spirig R, Djafarzadeh S, Regueira T, Shaw SG, von Garnier C, et al. (2010) Effects of TLR agonists on the hypoxia-regulated transcription factor HIF-1alpha and dendritic cell maturation under normoxic conditions. PLoS One 5: e0010983. doi: 10.1371/journal.pone.0010983 20539755
48. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, et al. (2008) NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 453 : 807–811. doi: 10.1038/nature06905 18432192
49. Tacchini L, Gammella E, De Ponti C, Recalcati S, Cairo G (2008) Role of HIF-1 and NF-kappaB transcription factors in the modulation of transferrin receptor by inflammatory and anti-inflammatory signals. J Biol Chem 283 : 20674–20686. doi: 10.1074/jbc.M800365200 18519569
50. Degrossoli A, Arrais-Silva WW, Colhone MC, Gadelha FR, Joazeiro PP, et al. (2011) The influence of low oxygen on macrophage response to Leishmania infection. Scand J Immunol 74 : 165–175. doi: 10.1111/j.1365-3083.2011.02566.x 21517930
51. Degrossoli A, Bosetto MC, Lima CB, Giorgio S (2007) Expression of hypoxia-inducible factor 1alpha in mononuclear phagocytes infected with Leishmania amazonensis. Immunol Lett 114 : 119–125. 17983667
52. Singh AK, Mukhopadhyay C, Biswas S, Singh VK, Mukhopadhyay CK (2012) Intracellular pathogen Leishmania donovani activates hypoxia inducible factor-1 by dual mechanism for survival advantage within macrophage. PLoS One 7: e38489. doi: 10.1371/journal.pone.0038489 22701652
53. Mancino A, Schioppa T, Larghi P, Pasqualini F, Nebuloni M, et al. (2008) Divergent effects of hypoxia on dendritic cell functions. Blood 112 : 3723–3734. doi: 10.1182/blood-2008-02-142091 18694997
54. Maroof A, Beattie L, Zubairi S, Svensson M, Stager S, et al. (2008) Posttranscriptional regulation of II10 gene expression allows natural killer cells to express immunoregulatory function. Immunity 29 : 295–305. doi: 10.1016/j.immuni.2008.06.012 18701085
55. Ohteki T, Fukao T, Suzue K, Maki C, Ito M, et al. (1999) Interleukin 12-dependent interferon gamma production by CD8alpha+ lymphoid dendritic cells. J Exp Med 189 : 1981–1986. 10377194
56. Xiao Z, Casey KA, Jameson SC, Curtsinger JM, Mescher MF (2009) Programming for CD8 T cell memory development requires IL-12 or type I IFN. J Immunol 182 : 2786–2794. doi: 10.4049/jimmunol.0803484 19234173
57. Wilson DC, Grotenbreg GM, Liu K, Zhao Y, Frickel EM, et al. (2010) Differential regulation of effector - and central-memory responses to Toxoplasma gondii Infection by IL-12 revealed by tracking of Tgd057-specific CD8+ T cells. PLoS Pathog 6: e1000815. doi: 10.1371/journal.ppat.1000815 20333242
58. Rao RR, Li Q, Odunsi K, Shrikant PA (2010) The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity 32 : 67–78. doi: 10.1016/j.immuni.2009.10.010 20060330
59. Barnes BJ, Moore PA, Pitha PM (2001) Virus-specific activation of a novel interferon regulatory factor, IRF-5, results in the induction of distinct interferon alpha genes. J Biol Chem 276 : 23382–23390. 11303025
60. Arango Duque G, Fukuda M, Turco SJ, Stager S, Descoteaux A (2014) Leishmania Promastigotes Induce Cytokine Secretion in Macrophages through the Degradation of Synaptotagmin XI. J Immunol 193 : 2363–2372. doi: 10.4049/jimmunol.1303043 25063865
61. Ohta A, Sitkovsky M (2001) Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 414 : 916–920. 11780065
62. Sitkovsky M, Lukashev D (2005) Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat Rev Immunol 5 : 712–721. 16110315
63. Hasko G, Linden J, Cronstein B, Pacher P (2008) Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov 7 : 759–770. doi: 10.1038/nrd2638 18758473
64. Csoka B, Himer L, Selmeczy Z, Vizi ES, Pacher P, et al. (2008) Adenosine A2A receptor activation inhibits T helper 1 and T helper 2 cell development and effector function. Faseb J 22 : 3491–3499. doi: 10.1096/fj.08-107458 18625677
65. Lukashev D, Klebanov B, Kojima H, Grinberg A, Ohta A, et al. (2006) Cutting edge: hypoxia-inducible factor 1alpha and its activation-inducible short isoform I.1 negatively regulate functions of CD4+ and CD8+ T lymphocytes. J Immunol 177 : 4962–4965. 17015677
66. Thiel M, Caldwell CC, Kreth S, Kuboki S, Chen P, et al. (2007) Targeted deletion of HIF-1alpha gene in T cells prevents their inhibition in hypoxic inflamed tissues and improves septic mice survival. PLoS One 2: e853. 17786224
67. Ben-Shoshan J, Maysel-Auslender S, Mor A, Keren G, George J (2008) Hypoxia controls CD4+CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1alpha. Eur J Immunol 38 : 2412–2418. doi: 10.1002/eji.200838318 18792019
68. Panther E, Corinti S, Idzko M, Herouy Y, Napp M, et al. (2003) Adenosine affects expression of membrane molecules, cytokine and chemokine release, and the T-cell stimulatory capacity of human dendritic cells. Blood 101 : 3985–3990. 12446452
69. Brenner S, Prosch S, Schenke-Layland K, Riese U, Gausmann U, et al. (2003) cAMP-induced Interleukin-10 promoter activation depends on CCAAT/enhancer-binding protein expression and monocytic differentiation. J Biol Chem 278 : 5597–5604. 12493739
70. Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, et al. (2010) Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res 70 : 7465–7475. doi: 10.1158/0008-5472.CAN-10-1439 20841473
71. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, et al. (2014) Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513 : 559–563. doi: 10.1038/nature13490 25043024
72. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, et al. (2010) HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med 207 : 2439–2453. doi: 10.1084/jem.20100587 20876310
73. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, et al. (2014) PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med 211 : 781–790. doi: 10.1084/jem.20131916 24778419
74. Kilani MM, Mohammed KA, Nasreen N, Tepper RS, Antony VB (2004) RSV causes HIF-1alpha stabilization via NO release in primary bronchial epithelial cells. Inflammation 28 : 245–251. 16133997
75. Nizet V, Johnson RS (2009) Interdependence of hypoxic and innate immune responses. Nat Rev Immunol 9 : 609–617. doi: 10.1038/nri2607 19704417
76. Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, et al. (2005) HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest 115 : 1806–1815. 16007254
77. Spear W, Chan D, Coppens I, Johnson RS, Giaccia A, et al. (2006) The host cell transcription factor hypoxia-inducible factor 1 is required for Toxoplasma gondii growth and survival at physiological oxygen levels. Cell Microbiol 8 : 339–352. 16441443
78. Brown KM, Suvorova E, Farrell A, McLain A, Dittmar A, et al. (2014) Forward genetic screening identifies a small molecule that blocks Toxoplasma gondii growth by inhibiting both host - and parasite-encoded kinases. PLoS Pathog 10: e1004180. doi: 10.1371/journal.ppat.1004180 24945800
79. Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, et al. (2003) HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112 : 645–657. 12628185
80. Anand RJ, Gribar SC, Li J, Kohler JW, Branca MF, et al. (2007) Hypoxia causes an increase in phagocytosis by macrophages in a HIF-1alpha-dependent manner. J Leukoc Biol 82 : 1257–1265. 17675562
81. Hwang II, Watson IR, Der SD, Ohh M (2006) Loss of VHL confers hypoxia-inducible factor (HIF)-dependent resistance to vesicular stomatitis virus: role of HIF in antiviral response. J Virol 80 : 10712–10723. 16928739
82. Vassilaki N, Kalliampakou KI, Kotta-Loizou I, Befani C, Liakos P, et al. (2013) Low oxygen tension enhances hepatitis C virus replication. J Virol 87 : 2935–2948. doi: 10.1128/JVI.02534-12 23269812
83. Veeranna RP, Haque M, Davis DA, Yang M, Yarchoan R (2012) Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen induction by hypoxia and hypoxia-inducible factors. J Virol 86 : 1097–1108. doi: 10.1128/JVI.05167-11 22090111
84. Tomaskova J, Oveckova I, Labudova M, Lukacikova L, Laposova K, et al. (2011) Hypoxia induces the gene expression and extracellular transmission of persistent lymphocytic choriomeningitis virus. J Virol 85 : 13069–13076. doi: 10.1128/JVI.00829-11 21957293
85. Kohler T, Reizis B, Johnson RS, Weighardt H, Forster I (2012) Influence of hypoxia-inducible factor 1alpha on dendritic cell differentiation and migration. Eur J Immunol 42 : 1226–1236. doi: 10.1002/eji.201142053 22539295
86. Weigert A, Weichand B, Sekar D, Sha W, Hahn C, et al. (2012) HIF-1alpha is a negative regulator of plasmacytoid DC development in vitro and in vivo. Blood 120 : 3001–3006. doi: 10.1182/blood-2012-03-417022 22936665
87. Bankoti R, Gupta K, Levchenko A, Stager S (2012) Marginal zone B cells regulate antigen-specific T cell responses during infection. J Immunol 188 : 3961–3971. doi: 10.4049/jimmunol.1102880 22412197
88. Stager S, Maroof A, Zubairi S, Sanos SL, Kopf M, et al. (2006) Distinct roles for IL-6 and IL-12p40 in mediating protection against Leishmania donovani and the expansion of IL-10+ CD4+ T cells. Eur J Immunol 36 : 1764–1771. 16791879
89. Patel TH, Kimura H, Weiss CR, Semenza GL, Hofmann LV (2005) Constitutively active HIF-1alpha improves perfusion and arterial remodeling in an endovascular model of limb ischemia. Cardiovasc Res 68 : 144–154. 15921668
90. Sarkar K, Cai Z, Gupta R, Parajuli N, Fox-Talbot K, et al. (2012) Hypoxia-inducible factor 1 transcriptional activity in endothelial cells is required for acute phase cardioprotection induced by ischemic preconditioning. Proc Natl Acad Sci U S A 109 : 10504–10509. doi: 10.1073/pnas.1208314109 22699503
91. Ranatunga D, Hedrich CM, Wang F, McVicar DW, Nowak N, et al. (2009) A human IL10 BAC transgene reveals tissue-specific control of IL-10 expression and alters disease outcome. Proc Natl Acad Sci U S A 106 : 17123–17128. doi: 10.1073/pnas.0904955106 19805095
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 6
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- HIV Latency Is Established Directly and Early in Both Resting and Activated Primary CD4 T Cells
- Battling Phages: How Bacteria Defend against Viral Attack
- A 21st Century Perspective of Poliovirus Replication
- Adenovirus Tales: From the Cell Surface to the Nuclear Pore Complex
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy