
Surface-Associated Lipoproteins Link Virulence to Colitogenic Activity in IL-10-Deficient Mice Independent of Their Expression Levels
Enterococcus faecalis is a commensal of the human intestinal core microbiota harboring several putative virulence factors, which highlight its role as opportunistic pathogen. This dualistic character is supported by recent evidence linking Enterococcus spp. to the pathogenesis of inflammatory bowel diseases (IBD). Although several studies suggest a crucial role for opportunistic pathogens in IBD pathogenesis targeting genetically susceptible individuals, the dynamic relationship between disease-relevant host compartments and specific bacterial structures able to trigger intestinal inflammation remain unclear. Here, we report that cell surface-associated lipoproteins and the enterococcal polysaccharide antigen, which are relevant for E. faecalis virulence in invertebrate infection models, but whose expression is minimally affected by the intestinal inflammatory milieu, exhibit colitogenic activity in a mouse model susceptible for chronic colitis. Bacterial lipoproteins trigger innate immune cell activation and are a critical prerequisite for E. faecalis-induced colitis. The enterococcal polysaccharide antigen mediates bacterial mucus penetration and adhesion to mucosal surfaces, promotes the formation of biofilm and contributes to E. faecalis colitogenic activity. Using E. faecalis as a model organism, we demonstrate that colitogenic activity of opportunistic pathogens can be assigned to specific bacterial structures, a finding that may help to identify the most essential steps in IBD-related microbe-host interactions.
Published in the journal:
. PLoS Pathog 11(6): e32767. doi:10.1371/journal.ppat.1004911
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004911
Summary
Enterococcus faecalis is a commensal of the human intestinal core microbiota harboring several putative virulence factors, which highlight its role as opportunistic pathogen. This dualistic character is supported by recent evidence linking Enterococcus spp. to the pathogenesis of inflammatory bowel diseases (IBD). Although several studies suggest a crucial role for opportunistic pathogens in IBD pathogenesis targeting genetically susceptible individuals, the dynamic relationship between disease-relevant host compartments and specific bacterial structures able to trigger intestinal inflammation remain unclear. Here, we report that cell surface-associated lipoproteins and the enterococcal polysaccharide antigen, which are relevant for E. faecalis virulence in invertebrate infection models, but whose expression is minimally affected by the intestinal inflammatory milieu, exhibit colitogenic activity in a mouse model susceptible for chronic colitis. Bacterial lipoproteins trigger innate immune cell activation and are a critical prerequisite for E. faecalis-induced colitis. The enterococcal polysaccharide antigen mediates bacterial mucus penetration and adhesion to mucosal surfaces, promotes the formation of biofilm and contributes to E. faecalis colitogenic activity. Using E. faecalis as a model organism, we demonstrate that colitogenic activity of opportunistic pathogens can be assigned to specific bacterial structures, a finding that may help to identify the most essential steps in IBD-related microbe-host interactions.
Introduction
The Gram-positive commensal Enterococcus faecalis is a member of the human intestinal core microbiota [1], but is also known for harboring several putative virulence genes mediating its pathogenicity [2]. The ability to acquire antibiotic resistance genes [3] and the emerging importance in nosocomial infections [4,5] highlight its role as an opportunistic pathogen. While opportunistic pathogens are important triggers of infectious inflammation, they might also play a role in pathogenesis of inflammatory bowel diseases (IBD) targeting genetically susceptible populations [6].
IBD are a heterogeneous group of chronic relapsing inflammatory conditions of the intestine comprising the two main manifestations Crohn’s disease (CD) and Ulcerative Colitis (UC). Several factors have been suggested to trigger the pathogenesis of IBD including genetic [7] and environmental factors together with a loss of immune tolerance to endogenous commensal microbiota (reviewed by [8,9]). Although changes in composition, diversity and function of the intestinal microbiota were demonstrated in IBD patients (reviewed by [10]), the specific contributions of individual bacteria and their virulence-relevant structures to chronic intestinal inflammation remain mainly unclear. Consequently, known putative virulence factors of commensal bacteria such as E. faecalis need to be reconsidered in the context of IBD pathogenesis. The investigation of colitogenic structure-function relationships in mouse models will help to understand the pathogenesis of this complex human disease.
Fecal samples from CD patients show higher numbers of enterococci [11], especially of Enterococcus faecium [12] and UC patients have increased mucosal growth of E. faecalis correlating with high titers of E. faecalis-specific antibodies [13] and disease severity [14]. Consistently, E. faecalis isolates from IBD patients are more likely to harbor virulence-related genes and activity [15]. In the IL-10-deficient mouse model (IL-10-/-), which is a valuable model to mimic conditions of human chronic colitis [16], monoassociation with E. faecalis induces severe intestinal inflammation [17]. This E. faecalis-driven inflammation is limited to the distal colon [18] and characterized by defective resolution of pro-inflammatory gene expression in the intestinal epithelium of IL-10-/ - mice [19].
We have recently identified that the zinc-dependent metalloprotease, gelatinase E (GelE), a known virulence factor of E. faecalis partially impairs intestinal epithelial barrier integrity in IL-10-/ - mice [20]. Since the loss of GelE did not result in total diminution of intestinal inflammation, we targeted two additional bacterial genes that have been linked to virulence in different infection models (Table 1) and are thought to be critical for E. faecalis interaction with the host during chronic colitis.

First, the enterococcal polysaccharide antigen (Epa) locus consists of 18 genes (epaA-epaR) encoding for enzymes and transporters that are involved in bacterial cell wall polysaccharide metabolism [21] and are important for intestinal colonization capability of E. faecalis in a mouse model of transient colonization [22]. Epa genes are suggested to synthesize a rhamnopolysaccharide, which together with wall teichoic acid forms the secondary wall polysaccharides [23]. The epaB gene encodes a putative glycosyl transferase that mediates transfer of rhamnose to cell wall polysaccharides and seems to be critical for enterococcal shape determination [24]. Disruption of epaB results in reduced virulence in mouse peritonitis [25] and urinary tract infection models [26], attenuated translocation across polarized human enterocyte monolayers [27], impaired resistance to polymorphonuclear leukocyte killing [28] and to phagocytosis in a zebrafish larva infection model [29].
Second, the prolipoprotein diacylglyceryl transferase (Lgt) contributes to maturation of bacterial lipoproteins by mediating the transfer of a diacylglyceryl moiety to conserved cysteine residues in Gram-positive bacteria [30]. Loss of Lgt results in enhanced growth of E. faecalis under oxidative stress or high Mn2+ concentrations in vitro and impaired virulence in an invertebrate infection model [31]. Although some lipoprotein-deficient Gram-positive pathogens show hypervirulent phenotypes [32,33], most of them have attenuated virulence [34] and impair toll-like receptor-2 (TLR2)-mediated activation of immune cells [32,35,36]. Bacterial lipoproteins were described as predominant immunobiologically active agonists of TLR2 [37,38].
In this study we aim at unraveling novel colitogenic activity of E. faecalis virulence factors other than gelatinase E in the context of chronic intestinal inflammation. For this reason we monoassociate germ-free wild type and IL-10-/ - mice with E. faecalis wild type OG1RF or isogenic mutants that lack either epaB or lgt. We are able to establish a novel correlation of intestinal inflammation and the presence of enterococcal polysaccharide antigen or cell surface-associated lipoproteins. Using these isogenic mutant strains we identify bacterial structures with critical colitogenic function and determine novel mechanisms by which opportunistic pathogens direct microbe-host interaction in the context of experimental colitis.
Results
Enterococcal polysaccharide antigen and lipoproteins promote colitogenic activity and virulence of E. faecalis
To screen for expression of virulence traits of E. faecalis under conditions of chronic inflammation, we monoassociated germ-free wild type and IL-10-/ - mice with E. faecalis wild type OG1RF and performed RNA-sequencing of E. faecalis isolated from colon content (Fig 1A). A focused screening for selected genes known to be involved in E. faecalis virulence (see Table 2 for a list of genes and selection criteria in the Materials and Methods section) confirmed our previous findings that GelE partially contributed to E. faecalis-induced inflammation (Table 1). Expression of gelE (OG1RF_11526) and related genes including sprE (OG1RF_11525) as well as the fsr-locus (OG1RF_11527, OG1RF_11528, OG1RF_11529), which controls the expression of these two proteases, were up-regulated in E. faecalis OG1RF under conditions of chronic colitis (Fig 1A). Surprisingly, the expression levels of most known E. faecalis virulence genes did not undergo substantial alterations in the inflamed environment raising questions about additional GelE-independent virulence factors contributing to the colitogenic activity of E. faecalis.
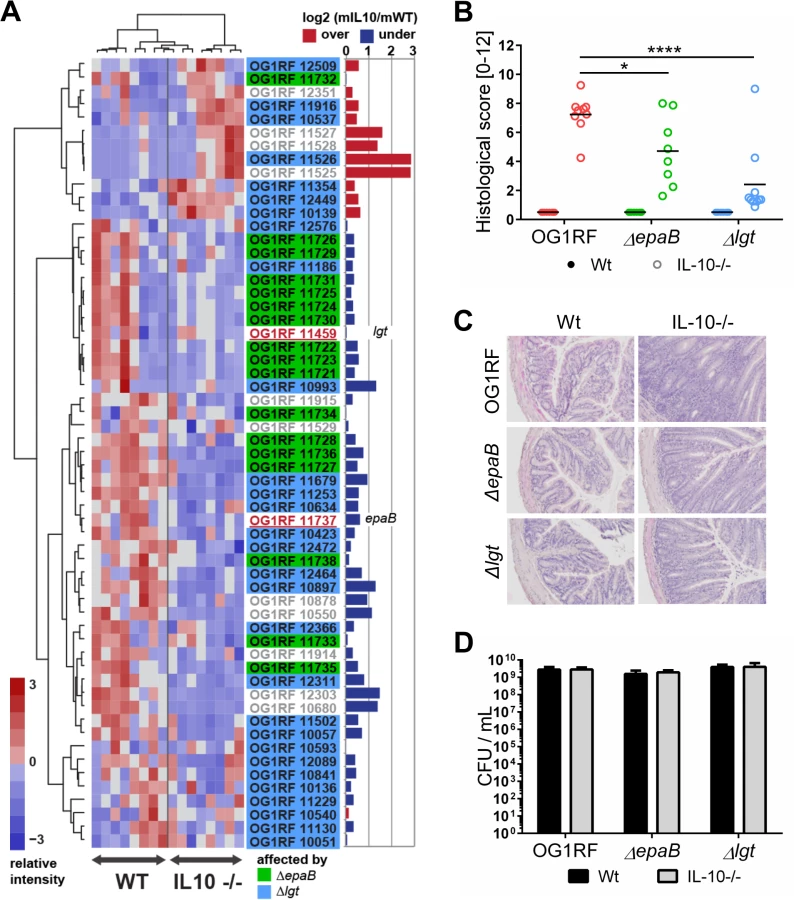

To determine whether E. faecalis virulence factors that are not up-regulated during colitis affect the pathogenesis of colitis, we studied two genes known to substantially impact on E. faecalis virulence by different mechanisms, but whose expression levels were not altered in the inflammatory milieu. These include epaB (OG1RF_11737) from the enterococcal polysaccharide antigen cluster and lgt (OG1RF_11459), a prolipoprotein diacylglyceryl transferase (Fig 1A). The respective E. faecalis mutants were generated and germ-free wild type and IL-10-/ - mice were monoassociated with E. faecalis wild type OG1RF, ΔepaB or Δlgt mutant strains for 16 weeks. While IL-10-/ - mice monoassociated with E. faecalis OG1RF had severe inflammation in the distal colon (OG1RF histopathological score: 7.2±1.2), colitis was partially impaired in E. faecalis ΔepaB (ΔepaB score: 4.7±2.3; *p<0.05) and was almost completely abrogated in E. faecalis Δlgt monoassociated IL-10-/ - mice (Δlgt score: 2.3±2.3; ****p<0.0001) (Fig 1B and 1C). All monoassociated wild type mice remained disease free (Fig 1B and 1C). E. faecalis wild type OG1RF and both mutant strains showed a similar colonization density as detected by countable colony forming units (CFU) in luminal contents from the colon (Fig 1D), ileum and cecum (S1 Fig). Immunofluorescence analysis revealed that infiltration of F4/80-positive (+), Ly6G+ and CD3+ cells into the colonic mucosa of monoassociated IL-10-/ - mice correlated with severity of inflammation in the distal colon (Fig 2A and 2B). However, the number of CD11c+ cells in the colonic mucosa was independent of disease activity or the strain used for monoassociation (Fig 2A and 2B).

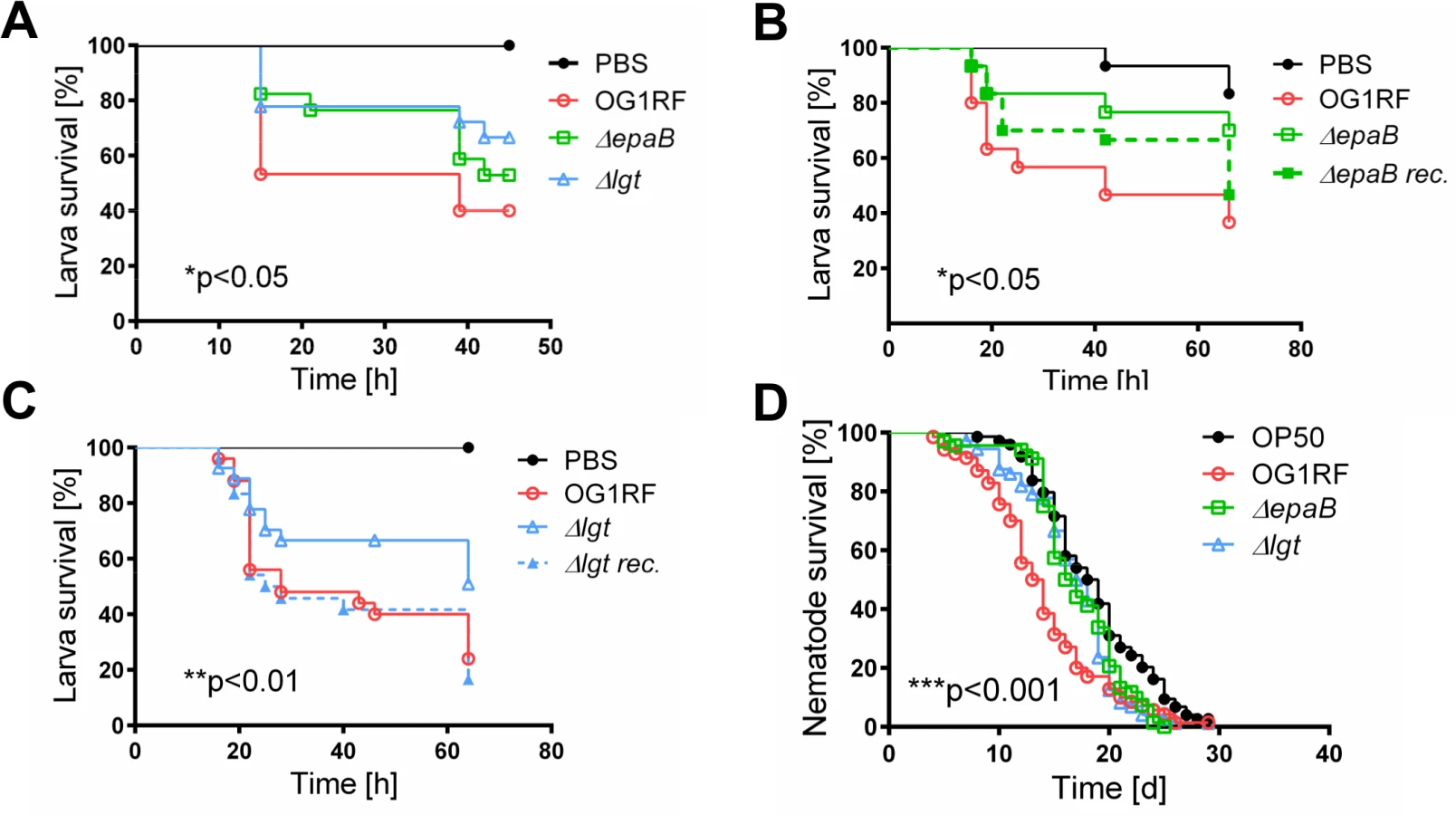
Similar to results in the IL-10-/ - mouse model of chronic T cell-mediated colitis, E. faecalis virulence in organisms with only innate immune system was also mediated by both epaB and lgt. Injection of E. faecalis ΔepaB or Δlgt into Galleria (G.) mellonella resulted in an increased survival of larvae (Fig 3A), while both reconstituted mutants exerted virulence comparable to wild type E. faecalis (Fig 3B and 3C). Oral administration of bacteria to Caenorhabditis (C.) elegans also revealed a significantly increased survival of nematodes in the presence of the two mutant strains compared to wild type E. faecalis OG1RF (Fig 3D).
E. faecalis ΔepaB show impaired adhesion to intestinal mucosa, attenuated formation of biofilm and associated bacterial microcolonies and reduced lysozyme resistance
Next, we performed co-staining of fluorescence in-situ hybridization (FISH)-labeled E. faecalis and MUC2 in the distal colon of monoassociated wild type and IL-10-/ - mice to visualize distribution of bacteria at inflammation-relevant mucosal sites (Fig 4A). In wild type mice, the bacterial distribution pattern and average distance to the epithelial surface was similar for all E. faecalis strains used in this study (Fig 4B). However, in IL-10-/ - mice the lack of epaB significantly increased the average distance of E. faecalis from the epithelial surface when compared to E. faecalis wild type OG1RF and Δlgt mutant strain (Fig 4C), suggesting attenuated bacterial penetration of the colonic mucus in the absence of EpaB. We then performed monocolonization experiments with germ-free Manduca (M.) sexta larvae demonstrating that both E. faecalis wild type OG1RF and Δlgt mutant strain were able to reach close proximity to the brush-border membrane of M. sexta larvae, whereas the ΔepaB mutant strain failed to localize in direct contact to the midgut epithelium (Fig 4D). Since E. faecalis is a natural commensal to M. sexta [39,40], this invertebrate represents a simplified model to study commensal-host interaction in vivo.

Finally, E. faecalis wild type OG1RF and all mutant strains were tested for their capacity to form biofilm associated multi-cellular aggregates (‘microcolonies’) measured as microcolony volume on a fixed monolayer of murine intestinal epithelial cells (Fig 5A). Immunofluorescence analysis demonstrated significantly reduced volume of visualized microcolonies for E. faecalis ΔepaB compared to wild type OG1RF, Δlgt or reconstituted ΔepaB mutant strains (Fig 5B). In addition, only the E. faecalis ΔepaB mutant strain showed significantly impaired capacity to form biofilm on polystyrene surfaces (Fig 5C) confirmed by reduced total biomass and average thickness of biofilms (S2 Fig).

In contrast to other strains, the growth of E. faecalis ΔepaB was inhibited when lysozyme was added to the Brain Heart Infusion (BHI) agar (S3 Fig), suggesting that the lack of EpaB contributes to lysozyme-mediated killing of E. faecalis. Immunofluorescence staining for lysozyme in the distal colon of monoassociated wild type and IL-10-/ - mice (S3 Fig) demonstrated an increased number of lysozyme-positive cells in IL-10-/ - mice. Comparing all groups of differently monoassociated IL-10-/ - mice, only IL-10-/ - mice monoassociated with E. faecalis Δlgt revealed lower amounts of lysozyme-positive cells resembling conditions found in wild type mice. In fact, the reduced number of lysozyme-producing cells in the distal colon of IL-10-/ - mice monoassociated with E. faecalis Δlgt can be explained by the decreased severity of colonic inflammation in these mice. Hence, the infiltration of lysozyme-positive cells may correlate with colonic inflammation and was demonstrated also for F4/80+ and Ly6G+ cells (Fig 2A and 2B) that are major subpopulations of cells containing cytoplasmic granules positive for lysozyme.
E. faecalis gelatinase E activity is not affected by epaB or lgt deficiency
Since GelE is known as a bacterial virulence factor relevant for the colitogenic activity of E. faecalis and upregulated under conditions of chronic colitis in IL-10-/ - mice, we analyzed how GelE activity is affected by epaB or lgt deficiency. As indicated by E-cadherin presence in a fixed monolayer of murine intestinal epithelial cells, E. faecalis ΔepaB and Δlgt mutant strains exerted a similar GelE activity compared to E. faecalis wild type OG1RF (Fig 6A). In the same assay, the quantification of immunofluorescence revealed a slightly reduced GelE activity for E. faecalis lacking epaB in vitro (Fig 6B), which may originate from the attenuated formation of microcolonies by E. faecalis ΔepaB in this setup (see also Fig 5A and 5B) resulting in less bacterial cells and less GelE present in close contact to epithelial cells. Taking into account the integrity of the extracellular domain of E-cadherin in the colonic epithelium of germ-free wild type and IL-10-/ - mice [20], we next investigated the degradation of E-cadherin in monoassociated mice. The immunofluorescence staining showed substantial degradation of extracellular parts of this junctional adhesion molecule for all three E. faecalis strains in the distal colon of monoassociated IL-10-/ - mice independently of the inflammation level (Fig 6C). This emphasizes that GelE activity of E. faecalis in vivo is not affected by the deletion of epaB or lgt and suggests that GelE-mediated cleavage of E-cadherin is not crucial for the induction of chronic colitis in IL-10-/ - mice.

E. faecalis deficient in lipoproteins show impaired TLR2-mediated activation of dendritic cells but reactivated colitogenic T cells
Since the E. faecalis Δlgt mutant was associated with significantly decreased inflammation in the distal colon of monoassociated IL-10-/ - mice, we further investigated the capacity of lipoprotein-deficient E. faecalis to trigger immune cell activation. First, bone marrow-derived dendritic cells (BMDC) from wild type mice secreted significantly less TNF (Fig 7A) and IL-6 (S4 Fig) in response to stimulation with lysates from E. faecalis Δlgt mutant strain vs. lysates from wild type OG1RF. The lipoprotein-dependent induction of TNF and IL-6 secretion was completely abrogated in BMDCs from TLR2-deficient (TLR2-/-) mice (Fig 7A) (S4 Fig). The secretion of IL-12p40 seen with wild type strain OG1RF was persistently reduced when BMDCs from IL-10-/ - mice were stimulated with E. faecalis Δlgt lysates for 3, 6, 12 and 24 hours (Fig 7B). By Western blot analysis using polyclonal antibodies recognizing either enterococcal lipoteichoic acid (LTA) or whole bacteria, we were able to show that both electrophoretic mobility and the amount of LTA as well as protein patterns are not affected in the lysates of the E. faecalis Δlgt mutant strain (S5 Fig) making it unlikely for this cell wall structure to modulate any of the effects observed in BMDCs. The phagocytic uptake of all E. faecalis strains by BMDCs was similar (S4 Fig), suggesting that the TLR2-mediated induction of pro-inflammatory cytokine secretion was not due to altered phagocytosis.

To investigate how differences in innate immune cell activation impact the potential to reactivate colitogenic T cells from inflamed IL-10-/ - mice, we performed dendritic cell-T cell co-cultures. BMDCs, isolated from IL-10-/ - mice and stimulated with lysates from the different E. faecalis strains, were co-cultured with CD4+ T cells isolated from mesenteric lymph nodes (MLN) of E. faecalis OG1RF monoassociated IL-10-/ - mice. As shown in Fig 7, there was no difference in IFN-γ (Fig 7C) and IL-12p40 (Fig 7D) secretion in the presence of T cells, suggesting that both E. faecalis mutant strains were able to stimulate an antigen-dependent reactivation of colitogenic T cells. However, in the absence of T cells there was a non-significant trend towards lower IL-12p40 secretion detectable, when BMDCs were stimulated with E. faecalis mutant lysates (Fig 7D). To determine whether the differences in colonic inflammation observed in IL-10-/ - mice monoassociated with the different E. faecalis strains correlate with altered antigen-specific T cells responses ex vivo, we isolated MLN cells from IL-10-/ - and wild type mice monoassociated with E. faecalis OG1RF, ΔepaB or Δlgt mutant strain and re-stimulated them with lysates of the corresponding E. faecalis strain (Fig 7E). IFN-γ secretion from reactivated MLN cells was similar for all IL-10-/ - mice, suggesting that independent of colonic pathology MLN cells from these mice were fully capable of responding to the bacterial antigens. Similarly, we detected no differences in IFN-γ in plasma from IL-10-/ - mice colonized with the different E. faecalis strains (Fig 7F).
Discussion
The colitogenic character of E. faecalis in genetically susceptible hosts was already demonstrated. However, the dynamic molecular relationship between disease-relevant host compartments and specific bacterial structures able to trigger intestinal inflammation remain unclear. Using E. faecalis as a model organism, we provide new insights regarding the significance of specific bacterial virulence factors in chronic colitis (Table 1). We identify a crucial role for bacterial cell wall-associated lipoproteins in the induction of experimental colitis, adding new knowledge into the complex interdependence of intestinal opportunistic pathogens and the genetically predisposed host.
Recent studies have emphasized the conceptual model of ‘pathobionts’ of the human commensal microbiota for disease conditions such as periodontitis [41] and chronic intestinal inflammation [6]. Opportunistic pathogens shown to exert detrimental effects in susceptible mouse models include: Helicobacter hepaticus [42,43] and Bilophila wadsworthia [44] that are capable of triggering colitis in IL-10-/ - mice in combination with specific microbiota composition or dietary exposure. Adherent-invasive strains of Escherichia coli (AIEC) occurring in ileal lesions of CD patients [45] adhere to intestinal epithelial cells [46] and exhibit invasive potential [47].
Analogous to the inflammation-promoting activity of AIEC mediated by adhesion to intestinal mucosal surfaces, we demonstrate that proximity of bacteria to the intestinal epithelium is a contributing prerequisite for E. faecalis-induced colitis in monoassociated IL-10-/ - mice. Resembling conditions in UC patients, the colonic mucus of IL-10-/ - mice is thicker than in wild type mice, but more penetrable to bacteria and allows gut microbes to attach to the epithelium [48]. While we observed no substantial penetration of the inner mucus layer in IL-10-/ - mice, the E. faecalis mutant lacking epaB fails to penetrate even the outer mucus area. Most importantly, this EpaB-mediated defective adhesion of E. faecalis to the outer colonic mucus occurred exclusively in the susceptible milieu of inflamed IL-10-/ - mice, where bacterial penetration of / adhesion to mucus is an important feature contributing to chronic colitis pathogenesis. Accordingly, the attenuated inflammation in IL-10-/ - mice monoassociated with E. faecalis ΔepaB most likely originated from impaired penetration of the colonic mucus and a defective adhesion to intestinal epithelial surfaces.
Of note, E. faecalis strains isolated from IBD patients exhibit higher adhesion capacity to Caco-2 cell monolayers compared to isolates from healthy controls [15] and showed increased mucosal growth in UC patients [13]. Enterococci also form biofilm structures with associated microcolonies in close proximity to the UC mucosa, whereas these multi-cellular aggregates are not found in crypts of colon biopsies from healthy controls [49]. Consistently, we here demonstrate an impaired formation of microcolonies on epithelial cell surfaces and attenuated biofilm generation for E. faecalis lacking epaB. In contrast to healthy controls, E. faecalis isolates from IBD patients exhibited higher biofilm formation [15]. Importantly, biofilm formation was also identified as a pathobiont feature of AIEC [50]. These results suggest that under conditions of experimental colitis the abilities to form biofilms and/or to grow in microcolonies adjacent to the intestinal mucosa are features shared by several pathobionts that are implicated in the pathogenesis of IBD and experimental colitis. In this context, the role of EpaB in lysozyme resistance is interesting, since E. faecalis lacking epaB is not only more susceptible to lysozyme, but also to neutrophil killing compared to E. faecalis OG1RF [28]. We might speculate that the lack of EpaB also facilitates the killing of E. faecalis cells translocating to or in close proximity to intestinal epithelium, via lysozyme produced by neutrophils infiltrating the inflamed colonic mucosa of IL-10-/ - mice.
In contrast to observations made from mouse models of transient colonization [22], in our study all monoassociated wild type and IL-10-/ - mice are similarly colonized with E. faecalis being not affected by epaB deletion. This might be explained by a difference in the experimental setup (transient colonization after pretreatment with antibiotics vs. long-term monoassociation of germ-free mice) or different E. faecalis strains (OG1RF vs. V583) and shows that the impaired colitogenic activity of E. faecalis ΔepaB in the IL-10-/ - mouse model is not dependent on colonization rates.
Still factors other than adhesion ability are required for full colitogenic activity of E. faecalis, since Epa-deficiency resulted only in partial reduction of intestinal inflammation. Noteworthy, the colitogenic activity of EpaB is GelE-independent. The impaired adhesion of E. faecalis ΔepaB to the intestinal epithelium does not substantially impact on GelE activity in the IL-10-/ - mouse model (as demonstrated by the loss of E-cadherin in all the groups of IL-10-/ - mice), suggesting that close proximity of E. faecalis to the intestinal epithelium is not a crucial prerequisite for GelE activity at inflammation-relevant sites. In this context, the diverging roles of GelE are remarkable and highlight the complexity of contributing and causal factors for colitogenic activity of E. faecalis. Both E. faecalis ΔepaB and Δlgt show substantial GelE activity without full colitogenic evolvement, but gelE is the only of these virulence factors overexpressed in E. faecalis OG1RF under inflammatory conditions. Recalling the intermediate effects of gelE deficiency in E. faecalis on chronic colitis in the IL-10-/ - mouse model [20], GelE can be considered as a factor contributing to, but not sufficient for colitogenic activity of E. faecalis. In contrast, the loss of E. faecalis cell surface-associated lipoproteins resulted in almost complete abrogation of intestinal pathology, despite the presence of both Epa and GelE. Hence, bacterial lipoproteins represent key structures in this complex of virulence factors exerting and limiting colitogenic activity of E. faecalis.
Similar to previous studies using Gram-positive bacterial mutants of the lgt gene, we also identified TLR2 as prime pattern recognition receptor responding to bacterial lipoproteins [32,35–38]. However, to our knowledge this is the first study demonstrating that one distinct group of structures related to bacterial virulence (bacterial lipoproteins) is almost solely responsible for inducing chronic intestinal inflammation in a susceptible mouse model. It remains unclear whether the specific interaction of E. faecalis lipoproteins with TLR2 is solely responsible for the exerted colitogenic effects, since about 40% of all predicted mature lipoproteins processed by Lgt have been linked to virulence of E. faecalis or Gram-positive bacteria in general [51]. This includes lipoproteins belonging to peptidylprolyl cis-trans isomerases [52], involved in capsular carbohydrate synthesis [53] or ABC transporters such as the endocarditis-specific antigen (EfaA) [54]. However, expression levels of none of these putatively virulence-relevant lipoproteins were substantially up-regulated in E. faecalis OG1RF in the chronically inflamed intestine of IL-10-/ - mice. This argues for a mechanism involving TLR2 signaling occurring after lipidation of immature lipoproteins via Lgt rather than a modified expression of individual lipoproteins that are relevant for E. faecalis virulence.
The numbers of F4/80+ macrophages and Ly6G+ granulocytes infiltrating the colon correlated with disease activity and were massively reduced in IL-10-/ - mice monoassociated with lipoprotein-deficient E. faecalis. This coincides with a similar observation for lysozyme-positive cells, since macrophages and granulocytes are major subpopulations of cells in the inflamed mucosa that contain lysozyme-positive granules. We observed similar numbers of CD11c+ cells in the colonic mucosa of E. faecalis OG1RF and Δlgt monoassociated IL-10-/ - mice despite of different tissue pathology. However, the number of CD3+ T cells infiltrating the colonic mucosa of IL-10-/ - mice was consistently reduced when E. faecalis lipoproteins were lacking. Most importantly, we showed that lipoprotein-deficient E. faecalis are fully capable of reactivating MLN-derived colitogenic T cells despite their significantly impaired potential to induce BMDC activation. In this context, it is remarkable that lipoprotein-deficient E. faecalis lysates are able to induce IFN-γ secretion in MLN cells isolated from minimally-inflamed IL-10-/ - mice colonized with the E. faecalis Δlgt mutant strain. Together with the observation that IFN-γ plasma levels were increased in all monoassociated IL-10-/ - mice, we might speculate that lipoprotein-deficient E. faecalis induce full maturation of colitogenic T cells, but obviously fail to activate and/or recruit a sustained inflammatory immune response in the colonic mucosa. The contribution of TLR2 in this context remains unclear.
Emerging evidence from genetic and experimental studies associate a TLR2-R753Q polymorphism with severe pancolitis in UC patients [55] and impaired intestinal barrier integrity [56,57]. Consistently, TLR2-deficient mice are more susceptible to dextran sodium sulfate (DSS) induced colitis [57]. However, TLR2 expression levels are significantly increased in the intestinal mucosa of UC patients [58,59]. Peripheral blood mononuclear cells isolated from CD or UC patients show increased TLR2 expression and secrete more TNF in response to TLR2 stimulation when compared to healthy controls [60], suggesting a heterogeneous functionality of TLR2 in the pathogenesis of IBD. Interestingly, the development of ovalbumin-induced colitis in mice deficient in nucleotide binding oligomerization domain 2 (NOD2), an intracellular pattern recognition receptor associated with increased susceptibility for CD, depends on TLR2-derived signals. Compared to NOD2-deficient mice, NOD2xTLR2 double-deficient mice showed attenuated ovalbumin-induced colitis with impaired CD4+ T cell infiltration into the mucosa and reduced production of IFN-γ in MLN cells [61]. Taken together, these findings emphasize the importance of a balanced TLR2 signaling in the mucosal innate immune compartment directing subsequent mucosal immunopathology.
In conclusion, this study demonstrates novel colitogenic function for two E. faecalis envelope structures that are also relevant for bacterial virulence. We provide the first evidence that bacterial cell surface-associated lipoproteins are essential in mounting colitogenic activity of antigen-primed T cells in the colonic mucosa of IL-10-/ - mice. Despite the fact that bacterial adhesion and close proximity to the intestinal epithelium as well as barrier disruption contribute to the colitogenic activity of E. faecalis, bacterial lipoprotein-mediated immune cell activation most likely through TLR2 is critical for the development of chronic intestinal inflammation (Fig 8A). Beyond the model organism E. faecalis, further characterization of bacterial structures relevant for the development of chronic inflammation will help to identify the most essential steps in IBD-related microbe-host interactions.

Materials and Methods
Ethics statement
Animal protocols used in this study were approved by the Institutional Animal Care and Use Committee of the University of North Carolina, Chapel Hill, NC (IACUC-ID: 12–300.0, accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care) and performed according to the guidelines in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Bacterial strains and growth conditions
E. faecalis strains used in this study (Table 3) were cultivated in Brain Heart Infusion (BHI) broth (BD, Sparks, MD, USA) or on BHI agar (Roth, Karlsruhe, Germany) at 37°C under aerobic conditions with shaking (160 rpm), unless otherwise indicated.

Generation of epaB deletion and reconstitution mutant
The epaB deletion mutant was created using the pHOU1 plasmid [62]. DNA fragments upstream (902 bp) and downstream (1,112 bp) of the epaB gene were amplified with primer pairs of 1F + 1Ra and 2Fa + 2Ra (Table 4), respectively. Amplified fragments were connected by cross-over PCR, digested with BamHI and EcoRI and then ligated into pHOU1 digested with the same restriction enzymes.

The construct, designated pJH132, was electrophorated into E. faecalis CK111 [63], which was then conjugated as described previously [62] with E. faecalis OG1RF. The first recombination was selected on BHI agar plates containing gentamicin (200 μg/mL), fusidic acid (25 μg/mL) and X-gal (200 μg/mL). Blue colonies resistant to gentamicin and fusidic acid were further characterized to verify recombination into the epaB region using outside primer pairs of upF + 2Ra and 1F + DnR (Table 4). The second crossover event was obtained by spreading a diluted culture of first cross-over cells onto BHI containing X-gal (200 μg/mL) agar. White colonies tested sensitive to gentamicin were isolated and further confirmed by DNA sequencing after PCR amplification (upF + DnR) (Table 4). The ORF of epaB is composed of 789 bp (encoding 262 amino acids); 726 bp starting from the start codon were deleted from the epaB mutant. The previously used counter selection medium MM9YEG supplemented with 10 mM p-CI-Phe was not successful in selecting for excision of pJH132, which we later found was due to severe inhibition of growth of the epaB deletion mutant by 10 mM p-CI-Phe.
For complementation of ΔepaB, the epaB region was amplified with the primers 1F + 2Ra (Table 4) and then subcloned into pCR-TOPO plasmid (Invitrogen, Carlsbad, UK). A silent mutation was introduced in the phenylalanine residue (TTT) of amino acid position 243 in the epaB to the same phenylalanine residue (TTC) using the primers, F243F for + F243F rev (Table 4). Phenylalanine is the most frequent amino acid (24 out of 292 amino acids) in EpaB. Among 24 phenylalanine codons, codon usage was as follow: 18 TTT and 6 TTC. Therefore, the complemented strain contains the same Phe-243, but has a silent nucleotide mutation to distinguish the complemented strain from wild type OG1RF. The complemented strain of the ΔepaB was created by allelic replacement using the pHOU1 plasmid [62]. DNA fragments containing epaB-F243F silent mutation were digested with BamHI and EcoRI, and then ligated into pHOU1 digested with the same restriction enzymes. The construct, designated pTEX5706 was electroporated into E. faecalis CK111 [63], which was then conjugated with E. faecalis ΔepaB. The first recombination event was selected on BHI plates as described above and blue colonies showing fusidic acid and gentamicin resistance were further characterized to verify recombination into the epaB region using outside primer pairs of upF + 2Ra and 1F + DnR (Table 4). The second recombination event was obtained by spreading the first recombinants on MM9YEG supplemented with 10 mM p-Cl-Phe. The complemented strain (TX5706) was confirmed by DNA sequencing and by PFGE patterns.
Generation of lgt deletion and reconstitution mutant
A non-polar deletion of a portion of gene lgt (EF_1748 in E.faecalis V583, GenBank ID accession number NP_815451) was created using the method described before [64] with the following modifications: primers pEF1748OER and pEF1748OEF (Table 4) were used to amplify a 503 bp fragment from the region upstream of gene lgt, and also the end part of EF1747. Primers pEF1748delR and pEF1748OER (Table 4) were used to amplify a 546 bp fragment downstream of the lgt gene, and at the beginning of EF1749. Primers pEF1748OEF and pEF1748delR contain a 21bp complementary sequence. Overlap extension PCR was used to create a PCR product lacking a portion of gene EF1748. The resulting fragment was cloned into Gram-negative cloning vector pCRII-TOPO (Invitrogen, Carlsbad, UK) and cut with the restriction enzyme EcoRI (Invitrogen, Carlsbad, UK); the resulting fragment was then inserted into shuttle vector pCASPER containing a temperature-sensitive origin of replication. The resulting plasmid, pCASPER/delta-lgt, was transformed into E. faecalis wild type OG1RF by electroporation, and integrants were selected at the non-permissive temperature (42°C) on TSA plates with kanamycin. A single colony was picked, and insertion of plasmid into the chromosome was confirmed by PCR. The integrant was passaged 10 times in liquid culture without antibiotics at the permissive temperature (30°C), and colonies were replica-plated to screen for loss of kanamycin resistance. The excision of the plasmid either creates a reconstituted wild type strain or leads to an allelic replacement with the deleted sequence in the chromosome. The deletion mutant created was designated E. faecalis OG1RF Δlgt, containing a 507 bp (169 amino acids) deletion. The genotype was confirmed by PCR and automated sequencing.
For reconstitution of the OG1RF Δlgt mutant, a DNA fragment of 1556 bp containing the entire lgt as well as upstream (403 bp) and downstream (316 bp) sequences was amplified by PCR using primer-pairs pEF1748delF and pEF1748delR (Table 4). The PCR product was cloned into the Gram-negative cloning vector pCRII-TOPO (Invitrogen, Carlsbad, UK) and cut with the restriction enzyme EcoRI (Invitrogen, Carlsbad, UK). The fragment was subsequently cloned into the Gram-positive vector pCASPER, which contains a temperature-sensitive origin of replication. The recombinant plasmid was electroporated into E. faecalis OG1RF Δlgt competent cells. Integrants were selected at the non-permissive temperature on TSA plates with kanamycin. Colony PCR was done to confirm the insertion of the plasmid into the chromosome. The integrants were passaged between 6 and 10 times in broth without antibiotics at the permissive temperature and screened by replica-plating for loss of kanamycin resistance. Kanamycin-susceptible clones were analyzed by PCR for the presence of the intact lgt gene.
Lysozyme sensitivity assay
Lysozyme sensitivity of the bacterial strains was assayed on BHI agar supplemented with chicken egg lysozyme (Sigma Aldrich, Taufkirchen, Germany). Overnight cultures were resuspended to an optical density (OD) of 1 measured at 600nm in saline water (0.9% [wt/vol] NaCl) and 2 μL of the 5−1 to 5−4 dilutions spotted on BHI agar supplemented with 12 mg/mL of lysozyme. Pictures were taken after 24 hours of incubation at 37°C.
Mice experiments
Germ-free 129S6/SvEv wild type and IL-10 - deficient (IL-10-/-) mice were maintained at the Division of Gastroenterology and Hepatology, University of North Carolina, Chapel Hill, USA. Germ-free mice (n = 8-10/group both females and males) were colonized at the age of 8–9 weeks with either E. faecalis wild type OG1RF or ΔepaB or Δlgt mutant strains by rectal swab and sacrificed after 16 weeks of monocolonization. To test colonization of mice luminal content was sampled from ileum, cecum and colon, serial dilutions were plated and colony forming units (CFU) were counted after incubation at standard conditions.
Microbial RNA-sequencing of virulence-related E. faecalis genes
Ribosomal RNA-depleted bacterial RNA was isolated from the colon content of wild type and IL-10-/ - mice (n = 8 mice/group) monoassociated with E. faecalis wild type OG1RF for 16 weeks as described previously with the following exceptions [65]. After the second round of DNase treatment, RNA was recovered using RNeasy mini columns (Qiagen, Venlo, Netherlands) and then depleted of rRNA using Ribo-Zero Magnetic kit (Epicentre, Madison, WI, USA) according to the manufacturer`s instructions prior to library construction. Samples were submitted to the Washington University at St. Louis Genome Technology Access Center for library preparation and sequencing on the Illumina HiSeq2000 instrument (Illumina, San Diego, CA, USA) to generate 190 million unidirectional 50nt reads. Each read was aligned to the E. faecalis OG1RF annotated genome available on NCBI using the Bowtie 2 algorithm and the counts per million reads per kilobase of each ORF were calculated.
The selection of virulence-relevant genes was focused on known virulence factors in E. faecalis OG1RF [2] including the epa cluster [24] and lgt-dependent (predicted) lipoproteins involved in E. faecalis or Gram-positive bacteria virulence [51]. The expression profiles of all virulent factors were first centered and scaled and then visualized as heatmaps using the package EMA (PMID: 21047405) in R. Samples and gene profiles were clustered using Ward’s method based on their Spearman rank correlation distance and each gene’s under - or over-expression per genotype was represented as the log2 of the groups mean expression fold changes.
Primary cell culture experiments and generation of dendritic cells
Mesenteric lymph nodes (MLN) were collected from wild type and IL-10-/ - mice (n = 3–4 mice) and non-fractionated cells were isolated by tissue homogenization and filtration through 70 μm cell strainers (Thermo Scientific, Waltham, MA, USA). MLN cells (5x105 cells/well) were then cultured in cell culture-treated 96-well plates (Thermo Scientific, Roskilde, Denmark) in RPMI-1640 medium (Invitrogen, Carlsbad, UK) containing 10% fetal calf serum (Biochrom, Berlin, Germany) and antibiotics. Cells were stimulated for 72 hours with corresponding bacterial lysates (15 μg/mL) from the strain that was used for monocolonization and supernatant collected and used for cytokines measurement, as indicated.
Dendritic cells were generated from bone-marrow (BMDC) of pooled wild type, IL-10-/ - or TLR2-/ - mice as described before [66]. BMDCs (5x105 cells/well) were stimulated in cell culture-treated 12-well plates (Thermo Scientific, Roskilde, Denmark) for 24 hours with bacterial lysates (3 μg/mL or 15 μg/mL) from E. faecalis wild type OG1RF, ΔepaB, Δlgt or reconstituted mutant strains or LPS (150 ng/mL) as control and supernatants taken for detection of cytokines.
For phagocytosis assays, BMDCs (106 cells/well) were incubated with the E. faecalis wild type strain OG1RF, ΔepaB, Δlgt or reconstituted mutant strains (MOI = 10 : 1) for 1 hour in antibiotic-free medium and further incubated for 1 hour in RPMI containing 250 μg/mL gentamycin. Subsequently, BMDCs were collected, washed three times with PBS to remove the antibiotic and the cells were lysed for 15 minutes using PBS containing 0.05% Triton-X 100. The cell lysates were plated in serial dilutions on BHI plates and CFUs were counted after overnight incubation at standard conditions.
For DC-T cell co-cultures, BMDCs were generated from pooled IL-10-/ - mice and cultured in 150 mm petri dishes with RPMI-1640 medium (containing 10 ng/mL GM-CSF (PeproTech, Hamburg, Germany), 5 ng/mL recombinant TNF (PeproTech, Hamburg, Germany), 0.055 mM 2-mercaptoethanol (Sigma Aldrich, Taufkirchen, Germany), 10% fetal calf serum and 1% antibiotics/antimycotics (Sigma Aldrich, Taufkirchen, Germany)) for 7 days with medium changed at day 3 and 5. At day 7 non-adherent BMDCs were harvested, and 2.24x106 cells per petri dish were pulsed with lysates from E. faecalis wild type OG1RF, ΔepaB or Δlgt mutant strain (15 μg/mL) for 24 hours. CD4+ T cells were isolated from pooled MLNs of IL-10-/ - mice (n = 5 monoassociated with E. faecalis OG1RF for 16 weeks) using a CD4+ T cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany). BMDCs pulsed with the E. faecalis lysates from different strains were washed three times with RPMI-1640 medium and adherent cells harvested. Finally, 8x104 BMDCs were co-cultured with 2x105 CD4+ T cells in triplicates in cell culture-treated 96-well plates (Thermo Scientific, Roskilde, Denmark) including appropriate controls in fresh medium (RMPI-1640, 10% fetal calf serum, 1% sodium pyruvate (Sigma Aldrich, Taufkirchen, Germany), 0.1% 2-mercaptoethanol, 100 μg/mL gentamycin). After 72 hours supernatants were collected for cytokine measurements.
Cytokine and chemokine quantification
Detection of IFN-γ, IL-12p40, TNF and IL-6 in murine plasma or cell culture-supernatant specimens was performed using commercially available ELISA kits (eBioscience, San Diego, CA, USA).
SDS-PAGE and immunoblotting
E. faecalis lysates were subjected to SDS-PAGE and analyzed by Western blot. Separated proteins were transferred onto a polyvinylidene difluoride membrane (Roth, Karlsruhe, Germany) and blocked at 20–22°C for 2 hours in TBST (0.05% Tween 20 in TBS), containing 3% skim milk (AppliChem, Darmstadt, Germany), then incubated for 18 hours at 4°C with the polyclonal rabbit antibodies raised either against enterococcal lipoteichoic acid (LTA) [67] or against whole bacteria (E. faecalis strain 12107) [68]. After washing in TBST the membrane was incubated at 20–22°C for 1 hour with goat anti-rabbit immunoglobulin G (IgG) (whole molecule) alkaline phosphatase conjugate (Sigma Aldrich, Taufkirchen, Germany) and then washed again. Binding of the antibodies conjugated to alkaline phosphatase was detected with nitro-blue tetrazolium (NBT) and 5-bromo-4-chloro-3'-indolyphosphate (BCIP) (both Sigma Aldrich, Taufkirchen, Germany).
Histopathological analysis
Tissue sections taken from distal colon and caecum were fixed in 4% formalin and embedded in paraffin. For histopathological analysis sections were cut (5 μm), deparaffinized and stained with hematoxylin and eosin. A histological scoring was performed by blindly assessing mononuclear cell infiltration into lamina propria, crypt hyperplasia, goblet cell depletion and ulcer formation resulting in a score from 0 (not inflamed) to 12 (highly inflamed) as previously described [69].
Immunofluorescence and fluorescence in-situ hybridization
For immunofluorescence staining formalin-fixed distal colon sections were cut in 5 μm sections, boiled 30 min in citrate buffer (for staining of Ly6G+ and CD3+ cells and lysozyme) or treated for 16 min at 37°C with proteinase K (Roth, Karlsruhe, Germany) (for staining of F4/80+ cells) for antigen-retrieval and stained with anti-Ly6G (BD, Sparks, MD, USA) or anti-CD3 (BD, Sparks, MD, USA) or anti-F4/80 (BMA Biomedicals, Augst, Switzerland) or anti-lysozyme (Dako, Glostrup, Denmark) and anti-E-cadherin (intracellular domain: Abcam, Cambridge, UK; extracellular domain: SantaCruz Biotechnology, Dallas, TX, USA) antibodies and DAPI for nuclei. Staining of CD11c+ cells was performed in fresh tissue embedded in OCT (Sakura Finetek, Alphen aan den Rijn, Netherlands), cut in 5 μm sections, fixed for 15 min in water-free methanol using anti-CD11c (BD, Sparks, MD, USA) antibodies. For co-staining of immunofluorescence and fluorescence in-situ hybridization (FISH), Carnoy’s solution-fixed samples embedded in paraffin were cut in 5 μm sections, manually deparaffinized, dehydrated and treated with buffer containing lysozyme (Sigma Aldrich, Taufkirchen, Germany) (45 min, 37°C) for antigen-retrieval. Specimens were again dehydrated and incubated with Cy5-labeled Eub338-probe [70] for hybridization (overnight, 46°C). Subsequently, mucus layer containing MUC2 and nuclei were visualized by anti-MUC2 (SantaCruz Biotechnology, Dallas, TX, USA) co-staining and DAPI, respectively. Pictures (n = 3–8 mice/group) were acquired using an Olympus FluoView FV-1000 confocal microscope (Olympus, Hamburg, Germany) and further analyzed using Volocity 3D Image Analysis software version 5.4.1 (PerkinElmer, Waltham, MA, USA). For distance measurements of single bacterial cells to the intestinal epithelial cell surface, an automated tracking and distance measurement protocol was generated in the Volocity 3D Image Analysis software.
Galleria mellonella infection model
G. mellonella larvae (HW-Terra KG, Wirtsgrund, Germany or Evergreen, Augsburg, Germany) were infected with the different E. faecalis wild type and mutant strains. E. faecalis strains were cultivated under routine conditions and collected by centrifugation after overnight incubation. G. mellonella larvae were sorted by weight and each larva infected at the second to last proleg with 5 μL bacterial suspension containing 2x105 CFU or 5 μL PBS as control using a 20 μL syringe and 0.210 x 51 mm needles (both from Hamilton Bonaduz AG, Bonaduz, Switzerland). Per group, 30 larvae were incubated at 37°C in petri dishes containing litter. Insect mortality was monitored at 3 to 6 hours intervals for 3 days post infection.
Caenorhabditis elegans infection model
C. elegans (wild type strain Bristol N2, which was a generous gift by Britta Spanier from the Chair for Molecular Nutrition of the Technical University of Munich) were maintained and cultivated as previously described [71]. For nematode infection, E. faecalis strains were cultivated overnight under routine conditions in 10 mL BHI broth. Bacteria cultures were 10-fold concentrated, spread on nematode growth medium (NGM) agar plates and incubated at 37°C overnight. Plates were equilibrated at room temperature before C. elegans L4 larvae (n = 3x30/group) were transferred individually onto the bacterial lawn of the respective E. faecalis strain or E. coli OP50 strain as food control. Nematodes were transferred to fresh NGM plates with the corresponding E. faecalis or E. coli OP50 strain every second day. Worms were considered dead, if they failed to respond to touch. The number of viable and killed worms was determined daily for about 25–30 days.
Manduca sexta infection model
The caterpillar M. sexta was used to investigate commensal-host interaction in vivo in a model system with simplified intestinal morphology [72–74], well described innate immunity (reviewed by [75]) and microbiota [76,77] with E. faecalis being one natural occurring bacterium [39,40]. To date M. sexta was predominantly used to study the impact of gut microbiota on Bacillus thuringiensis toxin insecticidal activity [78] and as infection model for insect pathogens such as Photorhabdus luminescens (reviewed by [79]). M. sexta eggs, which were a generous gift by Martina Kern from the Chair for Neurobiology of the Philipps-University in Marburg, were surface sterilized with 80% ethanol and in petri dishes transferred to a germ-free small isolator (at 26°C and controlled humidity). After hatching the larvae were maintained in groups in small containers and reared on a wheat germ-based artificial diet, which was autoclaved and regularly checked for contamination. Newly molted 3rd instar larvae were distributed to containers and received diet soaked with different E. faecalis strains or PBS as control (n = 5-6/group). Successful and similar monocolonization by all strains was tested and monitored regularly by plating serial dilutions of midgut content on BHI agar. All larvae were sacrificed during the 5th instar larva stadium and approximately 1 cm of the midgut was fixed in 4% phosphate-buffered formalin for histological analysis. Sections of 5 μm were prepared for immunofluorescence staining and pictures taken as described above. For immunofluorescence staining of E. faecalis an antibody raised in rabbit against purified enterococcal LTA was used [67].
Cell culture experiments for biofilm and adhesion assays
Biofilm formation on polystyrene surfaces was assessed as described before [80]. E. faecalis strains from stationary phase cultures were incubated overnight in tryptic soy broth (TSB) (Oxoid, Basingstoke, UK) supplemented with 1% glucose in 96-well tissue culture plates (n = 4 wells/strain). Growth rates were assessed by measuring the OD at 630 nm. After washing with PBS, plates were dried at 60°C for 1 hour and stained with 2% Hucker’s crystal violet (Sigma Aldrich, Taufkirchen, Germany) for 2 minutes. Excess stain was removed by rinsing the plates thoroughly under tap water and plates were dried for 10–20 minutes at 60°C. OD was measured at 630 nm and biofilm formation was normalized to growth with the biofilm index (ODbiofilm x (0.5/ODgrowth)) [81].
To determine biofilm thickness, E. faecalis strains were cultivated overnight in TSB supplemented with 1% glucose in collagen-IV-coated 8-well chambers (ibidi, Munich, Germany) at 37°C with gentle shaking. Bacteria and liquid broth were removed carefully and carbohydrate structures containing terminal α-D-glucose in the biofilm were stained for 1 hour using Alexa Fluor-488 labelled concavalin A (Invitrogen, Carlsbad, USA) in PBS. After carefully washing with PBS images of stained biofilm were acquired (40x magnification) using an Olympus IX81 inverted confocal microscope (Olympus, Hamburg, Germany). Biofilm thickness measurements (n = 5–7) and 3D-reassembling of acquired pictures were performed using Volocity 3D Image Analysis software version 5.4.1 (PerkinElmer, Waltham, MA, USA).
The murine colonic epithelial cell line Ptk6 has been previously described by Whitehead et al. [82]. Ptk6 cells were grown at 37°C in RPMI-1640 medium containing 5% fetal calf serum and 1 μg/mL Insulin-Transferrin-Selenium A (Invitrogen, Carlsbad, USA). Biofilm assays on biotic surface of fixed epithelial cell monolayer were performed in collagen-IV-coated 8-well plates (ibidi, Munich, Germany) with fully confluent and differentiated Ptk6 cells as described before [83] and adapted to E. faecalis routine growth conditions. E. faecalis microcolonies were stained by an antibody raised in rabbit against purified enterococcal LTA from E. faecalis [67] and Ptk6 cells were stained with anti-E-cadherin antibody (intracellular domain: Abcam, Cambridge, UK) and DAPI for nuclei. Pictures were acquired at 40x magnification using an Olympus IX81 inverted confocal microscope (Olympus, Hamburg, Germany). 3D-reassembling and volume quantifications (n = 3–4 pictures/strain, using a generated protocol that calculated the total volume of 3D-reassembled structures identified by a threshold signal) were performed using Volocity 3D Image Analysis software version 5.4.1 (PerkinElmer, Waltham, MA, USA).
Statistical analysis
Data was analyzed by using Prism 6.02 software (GraphPad Software, La Jolla, CA, USA). Data in figures are presented as mean with error bars indicating SD. Survival experiments with infection models were statistically analyzed by log-rank test performed on Kaplan-Meier curves. Statistical differences due to mouse genotype or different time points and E. faecalis strain were analyzed by Two-way ANOVA with Bonferroni post-test. Differences between E. faecalis strains were statistically analyzed by One-way ANOVA following Bonferroni post-test. Differences were considered significant for *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Unless otherwise stated all infection model (except for the mouse colonization) and in vitro experiments were repeated three times and representative results are shown.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, et al. (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464 : 59–65. doi: 10.1038/nature08821 20203603
2. Sava IG, Heikens E, Huebner J (2010) Pathogenesis and immunity in enterococcal infections. Clin Microbiol Infect Off Publ Eur Soc Clin Microbiol Infect Dis 16 : 533–540.
3. Arias CA, Murray BE (2012) The rise of the Enterococcus: beyond vancomycin resistance. Nat Rev Microbiol 10 : 266–278. doi: 10.1038/nrmicro2761 22421879
4. Pinholt M, Ostergaard C, Arpi M, Bruun NE, Schønheyder HC, et al. (2014) Incidence, clinical characteristics and 30-day mortality of enterococcal bacteraemia in Denmark 2006–2009: a population-based cohort study. Clin Microbiol Infect Off Publ Eur Soc Clin Microbiol Infect Dis 20 : 145–151.
5. Sievert DM, Ricks P, Edwards JR, Schneider A, Patel J, et al. (2013) Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect Control Hosp Epidemiol Off J Soc Hosp Epidemiol Am 34 : 1–14.
6. Round JL, Mazmanian SK (2009) The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 9 : 313–323. doi: 10.1038/nri2515 19343057
7. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, et al. (2012) Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491 : 119–124. doi: 10.1038/nature11582 23128233
8. Chassaing B, Darfeuille-Michaud A (2011) The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterology 140 : 1720–1728. doi: 10.1053/j.gastro.2011.01.054 21530738
9. Sartor RB (2008) Microbial influences in inflammatory bowel diseases. Gastroenterology 134 : 577–594. doi: 10.1053/j.gastro.2007.11.059 18242222
10. Kostic AD, Xavier RJ, Gevers D (2014) The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology 146 : 1489–1499. doi: 10.1053/j.gastro.2014.02.009 24560869
11. Shiga H, Kajiura T, Shinozaki J, Takagi S, Kinouchi Y, et al. (2012) Changes of faecal microbiota in patients with Crohn’s disease treated with an elemental diet and total parenteral nutrition. Dig Liver Dis Off J Ital Soc Gastroenterol Ital Assoc Study Liver. http://www.ncbi.nlm.nih.gov/pubmed/22622202. Accessed 25 June 2012.
12. Mondot S, Kang S, Furet JP, Aguirre de Carcer D, McSweeney C, et al. (2011) Highlighting new phylogenetic specificities of Crohn’s disease microbiota. Inflamm Bowel Dis 17 : 185–192. doi: 10.1002/ibd.21436 20722058
13. Furrie E, Macfarlane S, Cummings JH, Macfarlane GT (2004) Systemic antibodies towards mucosal bacteria in ulcerative colitis and Crohn’s disease differentially activate the innate immune response. Gut 53 : 91–98. 14684582
14. Fite A, Macfarlane S, Furrie E, Bahrami B, Cummings JH, et al. (2013) Longitudinal analyses of gut mucosal microbiotas in ulcerative colitis in relation to patient age and disease severity and duration. J Clin Microbiol 51 : 849–856. doi: 10.1128/JCM.02574-12 23269735
15. Golińska E, Tomusiak A, Gosiewski T, Więcek G, Machul A, et al. (2013) Virulence factors of Enterococcus strains isolated from patients with inflammatory bowel disease. World J Gastroenterol WJG 19 : 3562–3572. doi: 10.3748/wjg.v19.i23.3562 23801857
16. Barnett MPG, McNabb WC, Cookson AL, Zhu S, Davy M, et al. (2010) Changes in colon gene expression associated with increased colon inflammation in interleukin-10 gene-deficient mice inoculated with Enterococcus species. BMC Immunol 11 : 39. doi: 10.1186/1471-2172-11-39 20630110
17. Balish E, Warner T (2002) Enterococcus faecalis induces inflammatory bowel disease in interleukin-10 knockout mice. Am J Pathol 160 : 2253–2257. 12057927
18. Kim SC, Tonkonogy SL, Albright CA, Tsang J, Balish EJ, et al. (2005) Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology 128 : 891–906. 15825073
19. Ruiz PA, Shkoda A, Kim SC, Sartor RB, Haller D (2005) IL-10 gene-deficient mice lack TGF-beta/Smad signaling and fail to inhibit proinflammatory gene expression in intestinal epithelial cells after the colonization with colitogenic Enterococcus faecalis. J Immunol Baltim Md 1950 174 : 2990–2999. 15728512
20. Steck N, Hoffmann M, Sava IG, Kim SC, Hahne H, et al. (2011) Enterococcus faecalis metalloprotease compromises epithelial barrier and contributes to intestinal inflammation. Gastroenterology 141 : 959–971. doi: 10.1053/j.gastro.2011.05.035 21699778
21. Xu Y, Murray BE, Weinstock GM (1998) A Cluster of Genes Involved in Polysaccharide Biosynthesis from Enterococcus faecalis OG1RF. Infect Immun 66 : 4313–4323. 9712783
22. Rigottier-Gois L, Madec C, Navickas A, Matos RC, Akary-Lepage E, et al. (2014) The Surface Rhamnopolysaccharide Epa of Enterococcus faecalis is a Key Determinant for Intestinal Colonization. J Infect Dis.
23. Geiss-Liebisch S, Rooijakkers SHM, Beczala A, Sanchez-Carballo P, Kruszynska K, et al. (2012) Secondary Cell Wall Polymers of Enterococcus faecalis Are Critical for Resistance to Complement Activation via Mannose-binding Lectin. J Biol Chem 287 : 37769–37777. doi: 10.1074/jbc.M112.358283 22908219
24. Teng F, Singh KV, Bourgogne A, Zeng J, Murray BE (2009) Further characterization of the epa gene cluster and Epa polysaccharides of Enterococcus faecalis. Infect Immun 77 : 3759–3767. doi: 10.1128/IAI.00149-09 19581393
25. Xu Y, Singh KV, Qin X, Murray BE, Weinstock GM (2000) Analysis of a Gene Cluster of Enterococcus faecalis Involved in Polysaccharide Biosynthesis. Infect Immun 68 : 815–823. 10639451
26. Singh KV, Lewis RJ, Murray BE (2009) Importance of the epa locus of Enterococcus faecalis OG1RF in a mouse model of ascending urinary tract infection. J Infect Dis 200 : 417–420. doi: 10.1086/600124 19545208
27. Zeng J, Teng F, Weinstock GM, Murray BE (2004) Translocation of Enterococcus faecalis strains across a monolayer of polarized human enterocyte-like T84 cells. J Clin Microbiol 42 : 1149–1154. 15004067
28. Teng F, Jacques-Palaz KD, Weinstock GM, Murray BE (2002) Evidence that the enterococcal polysaccharide antigen gene (epa) cluster is widespread in Enterococcus faecalis and influences resistance to phagocytic killing of E. faecalis. Infect Immun 70 : 2010–2015. 11895965
29. Prajsnar TK, Renshaw SA, Ogryzko NV, Foster SJ, Serror P, et al. (2013) Zebrafish as a novel vertebrate model to dissect enterococcal pathogenesis. Infect Immun 81 : 4271–4279. doi: 10.1128/IAI.00976-13 24002065
30. Hutchings MI, Palmer T, Harrington DJ, Sutcliffe IC (2009) Lipoprotein biogenesis in Gram-positive bacteria: knowing when to hold ‘em, knowing when to fold ‘em. Trends Microbiol 17 : 13–21. doi: 10.1016/j.tim.2008.10.001 19059780
31. Reffuveille F, Serror P, Chevalier S, Budin-Verneuil A, Ladjouzi R, et al. (2012) The prolipoprotein diacylglyceryl transferase (Lgt) of Enterococcus faecalis contributes to virulence. Microbiol Read Engl 158 : 816–825.
32. Henneke P, Dramsi S, Mancuso G, Chraibi K, Pellegrini E, et al. (2008) Lipoproteins are critical TLR2 activating toxins in group B streptococcal sepsis. J Immunol Baltim Md 1950 180 : 6149–6158. 18424736
33. Bubeck Wardenburg J, Williams WA, Missiakas D (2006) Host defenses against Staphylococcus aureus infection require recognition of bacterial lipoproteins. Proc Natl Acad Sci U S A 103 : 13831–13836. 16954184
34. Petit CM, Brown JR, Ingraham K, Bryant AP, Holmes DJ (2001) Lipid modification of prelipoproteins is dispensable for growth in vitro but essential for virulence in Streptococcus pneumoniae. FEMS Microbiol Lett 200 : 229–233. 11425480
35. Wichgers Schreur PJ, Rebel JMJ, Smits MA, van Putten JPM, Smith HE (2011) Lgt processing is an essential step in Streptococcus suis lipoprotein mediated innate immune activation. PloS One 6: e22299. doi: 10.1371/journal.pone.0022299 21811583
36. Machata S, Tchatalbachev S, Mohamed W, Jänsch L, Hain T, et al. (2008) Lipoproteins of Listeria monocytogenes are critical for virulence and TLR2-mediated immune activation. J Immunol Baltim Md 1950 181 : 2028–2035. 18641340
37. Zähringer U, Lindner B, Inamura S, Heine H, Alexander C (2008) TLR2—promiscuous or specific? A critical re-evaluation of a receptor expressing apparent broad specificity. Immunobiology 213 : 205–224. doi: 10.1016/j.imbio.2008.02.005 18406368
38. Hashimoto M, Tawaratsumida K, Kariya H, Kiyohara A, Suda Y, et al. (2006) Not lipoteichoic acid but lipoproteins appear to be the dominant immunobiologically active compounds in Staphylococcus aureus. J Immunol Baltim Md 1950 177 : 3162–3169. 16920954
39. Singh S, Reese JM, Casanova-Torres AM, Goodrich-Blair H, Forst S (2014) Microbial Population Dynamics in the Hemolymph of Manduca sexta Infected with Xenorhabdus nematophila and the Entomopathogenic Nematode Steinernema carpocapsae. Appl Environ Microbiol 80 : 4277–4285. doi: 10.1128/AEM.00768-14 24814780
40. Mason KL, Stepien TA, Blum JE, Holt JF, Labbe NH, et al. (2011) From commensal to pathogen: translocation of Enterococcus faecalis from the midgut to the hemocoel of Manduca sexta. mBio 2: e00065–00011. doi: 10.1128/mBio.00065-11 21586646
41. Hajishengallis G (2014) Immunomicrobial pathogenesis of periodontitis: keystones, pathobionts, and host response. Trends Immunol 35 : 3–11. doi: 10.1016/j.it.2013.09.001 24269668
42. Yang I, Eibach D, Kops F, Brenneke B, Woltemate S, et al. (2013) Intestinal microbiota composition of interleukin-10 deficient C57BL/6J mice and susceptibility to Helicobacter hepaticus-induced colitis. PloS One 8: e70783. doi: 10.1371/journal.pone.0070783 23951007
43. Chow J, Mazmanian SK (2010) A pathobiont of the microbiota balances host colonization and intestinal inflammation. Cell Host Microbe 7 : 265–276. doi: 10.1016/j.chom.2010.03.004 20413095
44. Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, et al. (2012) Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/ - mice. Nature 487 : 104–108. doi: 10.1038/nature11225 22722865
45. Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, et al. (1998) Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology 115 : 1405–1413. 9834268
46. Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, et al. (2004) Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology 127 : 80–93. 15236175
47. Boudeau J, Glasser AL, Masseret E, Joly B, Darfeuille-Michaud A (1999) Invasive ability of an Escherichia coli strain isolated from the ileal mucosa of a patient with Crohn’s disease. Infect Immun 67 : 4499–4509. 10456892
48. Johansson MEV, Gustafsson JK, Holmén-Larsson J, Jabbar KS, Xia L, et al. (2013) Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut.
49. Macfarlane S, Furrie E, Cummings JH, Macfarlane GT (2004) Chemotaxonomic analysis of bacterial populations colonizing the rectal mucosa in patients with ulcerative colitis. Clin Infect Dis Off Publ Infect Dis Soc Am 38 : 1690–1699.
50. Martinez-Medina M, Naves P, Blanco J, Aldeguer X, Blanco JE, et al. (2009) Biofilm formation as a novel phenotypic feature of adherent-invasive Escherichia coli (AIEC). BMC Microbiol 9 : 202. doi: 10.1186/1471-2180-9-202 19772580
51. Reffuveille F, Leneveu C, Chevalier S, Auffray Y, Rincé A (2011) Lipoproteins of Enterococcus faecalis: bioinformatic identification, expression analysis and relation to virulence. Microbiol Read Engl 157 : 3001–3013. doi: 10.1099/mic.0.053314-0 21903750
52. Reffuveille F, Connil N, Sanguinetti M, Posteraro B, Chevalier S, et al. (2012) Involvement of Peptidylprolyl cis/trans Isomerases in Enterococcus faecalis Virulence. Infect Immun 80 : 1728–1735. doi: 10.1128/IAI.06251-11 22331431
53. Hancock LE, Gilmore MS (2002) The capsular polysaccharide of Enterococcus faecalis and its relationship to other polysaccharides in the cell wall. Proc Natl Acad Sci 99 : 1574–1579. 11830672
54. Singh KV, Coque TM, Weinstock GM, Murray BE (1998) In vivo testing of an Enterococcus faecalis efaA mutant and use of efaA homologs for species identification. FEMS Immunol Med Microbiol 21 : 323–331. 9753005
55. Pierik M, Joossens S, Van Steen K, Van Schuerbeek N, Vlietinck R, et al. (2006) Toll-like receptor-1, -2, and -6 polymorphisms influence disease extension in inflammatory bowel diseases. Inflamm Bowel Dis 12 : 1–8. 16374251
56. Podolsky DK, Gerken G, Eyking A, Cario E (2009) Colitis-associated variant of TLR2 causes impaired mucosal repair because of TFF3 deficiency. Gastroenterology 137 : 209–220. doi: 10.1053/j.gastro.2009.03.007 19303021
57. Cario E, Gerken G, Podolsky DK (2007) Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology 132 : 1359–1374. 17408640
58. Szebeni B, Veres G, Dezsofi A, Rusai K, Vannay A, et al. (2008) Increased expression of Toll-like receptor (TLR) 2 and TLR4 in the colonic mucosa of children with inflammatory bowel disease. Clin Exp Immunol 151 : 34–41. 17991289
59. Frolova L, Drastich P, Rossmann P, Klimesova K, Tlaskalova-Hogenova H (2008) Expression of Toll-like receptor 2 (TLR2), TLR4, and CD14 in biopsy samples of patients with inflammatory bowel diseases: upregulated expression of TLR2 in terminal ileum of patients with ulcerative colitis. J Histochem Cytochem Off J Histochem Soc 56 : 267–274.
60. Cantó E, Ricart E, Monfort D, González-Juan D, Balanzó J, et al. (2006) TNF alpha production to TLR2 ligands in active IBD patients. Clin Immunol Orlando Fla 119 : 156–165. 16480927
61. Watanabe T, Kitani A, Murray PJ, Wakatsuki Y, Fuss IJ, et al. (2006) Nucleotide binding oligomerization domain 2 deficiency leads to dysregulated TLR2 signaling and induction of antigen-specific colitis. Immunity 25 : 473–485. 16949315
62. Panesso D, Montealegre MC, Rincón S, Mojica MF, Rice LB, et al. (2011) The hylEfm gene in pHylEfm of Enterococcus faecium is not required in pathogenesis of murine peritonitis. BMC Microbiol 11 : 20. doi: 10.1186/1471-2180-11-20 21266081
63. Kristich CJ, Chandler JR, Dunny GM (2007) Development of a host-genotype-independent counterselectable marker and a high-frequency conjugative delivery system and their use in genetic analysis of Enterococcus faecalis. Plasmid 57 : 131–144. 16996131
64. Cieslewicz MJ, Kasper DL, Wang Y, Wessels MR (2001) Functional analysis in type Ia group B Streptococcus of a cluster of genes involved in extracellular polysaccharide production by diverse species of streptococci. J Biol Chem 276 : 139–146. 11027683
65. Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, et al. (2013) Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341 : 1241214. doi: 10.1126/science.1241214 24009397
66. Gruber L, Kisling S, Lichti P, Martin F - P, May S, et al. (2013) High fat diet accelerates pathogenesis of murine Crohn’s disease-like ileitis independently of obesity. PloS One 8: e71661. doi: 10.1371/journal.pone.0071661 23977107
67. Theilacker C, Kropec A, Hammer F, Sava I, Wobser D, et al. (2012) Protection against Staphylococcus aureus by antibody to the polyglycerolphosphate backbone of heterologous lipoteichoic acid. J Infect Dis 205 : 1076–1085. doi: 10.1093/infdis/jis022 22362863
68. Hufnagel M, Hancock LE, Koch S, Theilacker C, Gilmore MS, et al. (2004) Serological and genetic diversity of capsular polysaccharides in Enterococcus faecalis. J Clin Microbiol 42 : 2548–2557. 15184433
69. Katakura K, Lee J, Rachmilewitz D, Li G, Eckmann L, et al. (2005) Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J Clin Invest 115 : 695–702. 15765149
70. Amann RI, Krumholz L, Stahl DA (1990) Fluorescent-oligonucleotide probing of whole cells for determinative, phylogenetic, and environmental studies in microbiology. J Bacteriol 172 : 762–770. 1688842
71. Stiernagle T (2006) Maintenance of C. elegans. WormBook Online Rev C Elegans Biol: 1–11.
72. Baldwin KM, Hakim RS (1987) Change of form of septate and gap junctions during development of the insect midgut. Tissue Cell 19 : 549–558. 18620211
73. Cioffi M, Wolfersberger MG (1983) Isolation of separate apical, lateral and basal plasma membrane from cells of an insect epithelium. A procedure based on tissue organization and ultrastructure. Tissue Cell 15 : 781–803. 6648956
74. Cioffi M (1979) The morphology and fine structure of the larval midgut of a moth (Manduca sexta) in relation to active ion transport. Tissue Cell 11 : 467–479. 494237
75. Kanost MR, Jiang H, Yu X-Q (2004) Innate immune responses of a lepidopteran insect, Manduca sexta. Immunol Rev 198 : 97–105. 15199957
76. Brinkmann N, Martens R, Tebbe CC (2008) Origin and diversity of metabolically active gut bacteria from laboratory-bred larvae of Manduca sexta (Sphingidae, Lepidoptera, Insecta). Appl Environ Microbiol 74 : 7189–7196. doi: 10.1128/AEM.01464-08 18849461
77. Van der Hoeven R, Betrabet G, Forst S (2008) Characterization of the gut bacterial community in Manduca sexta and effect of antibiotics on bacterial diversity and nematode reproduction. FEMS Microbiol Lett 286 : 249–256. doi: 10.1111/j.1574-6968.2008.01277.x 18647359
78. Broderick NA, Raffa KF, Handelsman J (2006) Midgut bacteria required for Bacillus thuringiensis insecticidal activity. Proc Natl Acad Sci U S A 103 : 15196–15199. 17005725
79. Eleftherianos I, ffrench-Constant RH, Clarke DJ, Dowling AJ, Reynolds SE (2010) Dissecting the immune response to the entomopathogen Photorhabdus. Trends Microbiol 18 : 552–560. doi: 10.1016/j.tim.2010.09.006 21035345
80. Baldassarri L, Cecchini R, Bertuccini L, Ammendolia MG, Iosi F, et al. (2001) Enterococcus spp. produces slime and survives in rat peritoneal macrophages. Med Microbiol Immunol (Berl) 190 : 113–120. 11827199
81. Deighton M, Borland R (1993) Regulation of slime production in Staphylococcus epidermidis by iron limitation. Infect Immun 61 : 4473–4479. 8406839
82. Whitehead RH, Robinson PS, Williams JA, Bie W, Tyner AL, et al. (2008) Conditionally immortalized colonic epithelial cell line from a Ptk6 null mouse that polarizes and differentiates in vitro. J Gastroenterol Hepatol 23 : 1119–1124. doi: 10.1111/j.1440-1746.2008.05308.x 18205771
83. Chassaing B, Darfeuille-Michaud A (2013) The δE Pathway Is Involved in Biofilm Formation by Crohn’s Disease-Associated Adherent-Invasive Escherichia coli. J Bacteriol 195 : 76–84. doi: 10.1128/JB.01079-12 23104802
84. Gaspar F, Teixeira N, Rigottier-Gois L, Marujo P, Nielsen-LeRoux C, et al. (2009) Virulence of Enterococcus faecalis dairy strains in an insect model: the role of fsrB and gelE. Microbiology 155 : 3564–3571. doi: 10.1099/mic.0.030775-0 19696101
85. Sifri CD, Mylonakis E, Singh KV, Qin X, Garsin DA, et al. (2002) Virulence effect of Enterococcus faecalis protease genes and the quorum-sensing locus fsr in Caenorhabditis elegans and mice. Infect Immun 70 : 5647–5650. 12228293
86. Rigottier-Gois L, Alberti A, Houel A, Taly J-F, Palcy P, et al. (2011) Large-Scale Screening of a Targeted Enterococcus faecalis Mutant Library Identifies Envelope Fitness Factors. PLoS ONE 6: e29023. doi: 10.1371/journal.pone.0029023 22194979
87. Mohamed JA, Huang W, Nallapareddy SR, Teng F, Murray BE (2004) Influence of Origin of Isolates, Especially Endocarditis Isolates, and Various Genes on Biofilm Formation by Enterococcus faecalis. Infect Immun 72 : 3658–3663. 15155680
88. Murray BE, Singh KV, Ross RP, Heath JD, Dunny GM, et al. (1993) Generation of restriction map of Enterococcus faecalis OG1 and investigation of growth requirements and regions encoding biosynthetic function. J Bacteriol 175 : 5216–5223. 8349561
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 6
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- HIV Latency Is Established Directly and Early in Both Resting and Activated Primary CD4 T Cells
- Battling Phages: How Bacteria Defend against Viral Attack
- A 21st Century Perspective of Poliovirus Replication
- Adenovirus Tales: From the Cell Surface to the Nuclear Pore Complex
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy